

Abstract
Because detergents are commonly used to solvate membrane proteins for structural evaluation, much attention has been devoted to assessing the conformational bias imparted by detergent micelles in comparison to the native environment of the lipid bilayer. Here, we conduct six 500-ns simulations of a system with >600,000 atoms to investigate the spontaneous self assembly of dodecylphosphocholine detergent around multiple molecules of the integral membrane protein PagP. This detergent formed equatorial micelles in which acyl chains surround the protein’s hydrophobic belt, confirming existing models of the detergent solvation of membrane proteins. In addition, unexpectedly, the extracellular and periplasmic apical surfaces of PagP interacted with the headgroups of detergents in other micelles 85 and 60% of the time, respectively, forming complexes that were stable for hundreds of nanoseconds. In some cases, an apical surface of one molecule of PagP interacted with an equatorial micelle surrounding another molecule of PagP. In other cases, the apical surfaces of two molecules of PagP simultaneously bound a neat detergent micelle. In these ways, detergents mediated the non-specific aggregation of folded PagP. These simulation results are consistent with dynamic light scattering experiments, which show that, at detergent concentrations ≥600 mM, PagP induces the formation of large scattering species that are likely to contain many copies of the PagP protein. Together, these simulation and experimental results point to a potentially generic mechanism of detergent-mediated protein aggregation.
Keywords: Molecular dynamics, Simulation, Membrane protein, Dynamic light scattering
1. Introduction
All cells and their compartments are surrounded by lipid membranes to which embedded proteins bestow biological activity. These membrane proteins are crucial for signal transduction (Cantrell, 1996; Schlessinger, 2000; Wettschureck and Offermanns, 2005; Bezanilla, 2008; Gilman, 1987), molecular transport (Pohorille et al., 2005; Ackerman and Clapham, 1997; Gould and Holman, 1993), catalysis (Bishop, 2005; Zhou et al., 2008), as well as membrane fusion (Ungar and Hughson, 2003) and biosynthesis (Bishop et al., 2000). Accordingly, they are implicated in a wide variety of diseases (Sanders and Myers, 2004) and are the targets of a large portion of approved drugs (Yildirim et al., 2007).
Although integral membrane proteins adopt a variety of topologies (Bowie, 2005; von Heijne, 2006; Popot and Engelman, 2000), they are all anchored in the membrane by apolar transmembrane segments and polar flanking regions (von Heijne, 2006; Segrest and Feldmann, 1974; von Heijne, 1989; Eisenberg et al., 1984). The clustering of apolar and polar/charged residues on the surface of integral membrane proteins complements the transmembrane profile of partial charge density in their native environment (Nagle et al., 1996; Nagle and Tristram-Nagle, 2000): integral membrane proteins expose a belt of hydrophobic residues to the apolar bilayer core, and expose hydrophilic apical surfaces to zwitterionic/ionic lipid headgroups and aqueous solution (Schulz, 2002; Sipos and Von Heijne, 1993).
Three-dimensional structures offer invaluable insight into the molecular basis of protein function (Ahn et al., 2004; Hwang et al., 2002, 2004; Cuesta-Seijo et al., 2010) and provide a starting point for the rational design or enhancement of drugs that bind membrane proteins and alter their activities (Congreve and Marshall, 2010; Wacker et al., 2010; Giacomini et al., 2010; Durdagi et al., 2011). Unfortunately, the two most commonly used methods of high-resolution structural determination, solution NMR and X-ray crystallography, are not well suited to study proteins embedded in lipid bilayers. One the one hand, the large size of lipid bilayers leads to long rotational correlation times and therefore low signal resolution in solution NMR (Opella et al., 2002). On the other hand, target protein concentrations in lipid bilayers are usually low, presenting a bottleneck to the production of high-quality crystals (Schulz, 2002; Bill et al., 2011). These problems can be circumvented by replacing the lipid membrane with a mimetic whose properties are more suitable to the experimental approach. A membrane mimetic is necessary because the structure of a membrane protein depends on its environment (White et al., 2001; White and Wimley, 1999) and the native tertiary and/or quaternary structure is therefore not generally maintained in aqueous solution.
One popular membrane mimetic strategy is to replace lipids with detergents, which can solvate membrane proteins by forming expanded micelles around their hydrophobic belts (Tamm and Liang, 2006). For example, the β-barrel Gram-negative bacterial outer membrane enzyme PagP (Bishop, 2005) (Fig. 1) adopts similar folds when solvated by detergents such as dodecylphosphocholine (DPC) (Hwang et al., 2002; Hwang and Kay, 2005), lauryldimethy-lamineoxide (LDAO) (Ahn et al., 2004; Khan et al., 2007), CYFOS-7 (Hwang et al., 2004; Hwang and Kay, 2005), n-octyl-β-D-glucoside (OG) (Hwang et al., 2002; Hwang and Kay, 2005), and a mixture of sodium dodecylsulfate (SDS) and 2-methyl-2,4-pentanediol (MPD) (Cuesta-Seijo et al., 2010). Furthermore, the specific activity of PagP after denaturation, purification, and refolding into DPC and CYFOS-7 micelles is indistinguishable (Hwang et al., 2002) from that of native PagP purified from membranes (Bishop et al., 2000), indicating that the conserved fold of PagP observed in complexation with detergents is a native state.
Fig. 1.
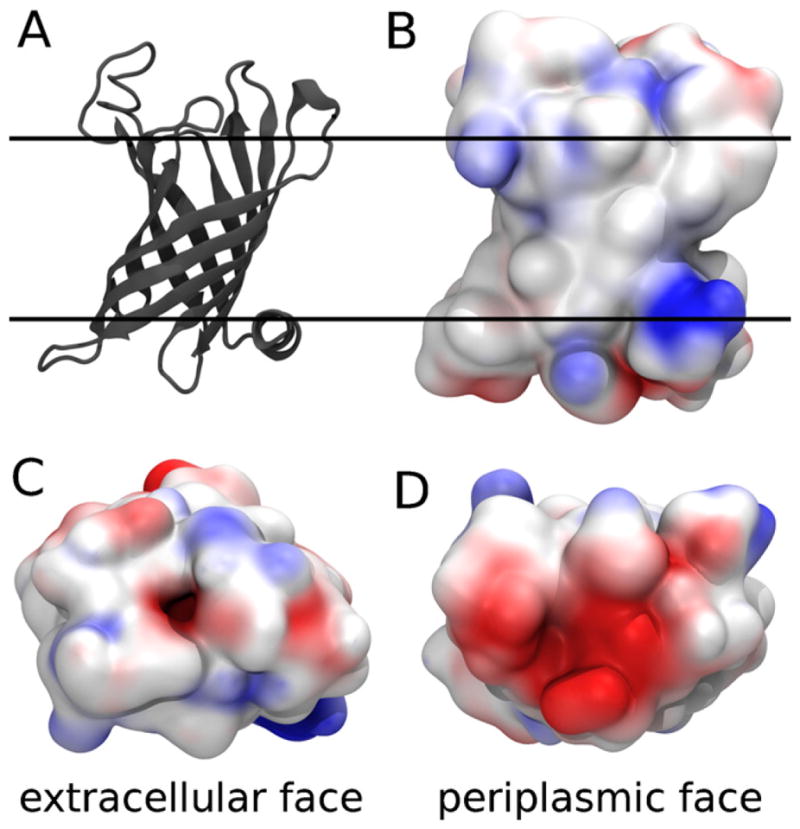
The Gram negative bacterial outer membrane enzyme PagP. (A) Cartoon representation of the crystal structure of PagP in LDAO (PDB ID 1THQ) (Ahn et al., 2004). The proposed (Ahn et al., 2004) bilayer interfaces are depicted as solid lines. (B–D) Electrostatic potential of PagP, computed by the adaptive Poisson-Boltzmann solver (Baker et al., 2001), mapped onto its van der Waals surface by VMD (Humphrey et al., 1996) showing (B) side view along the proposed (Ahn et al., 2004) bilayer plane, and (C) extracellular and (D) periplasmic apical surfaces. (Red) Electronegative and (blue) electropositive regions of the protein surface are highlighted. For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.
PagP is an enzyme of lipid metabolism whose role is to catalyze the transfer of a palmitate group from the sn-1 position of a phospholipid to the N-linked hydroxymyristate on the proximal unit of lipid A (Bishop, 2005; Bishop et al., 2000). This modification protects the Gram-negative bacterial outer membrane against disruption by cationic antimicrobial peptides and thus promotes intracellular infection and virulence (Robey et al., 2001). The binding pocket for the donor acyl chain is a deep hydrophobic furrow in the extra-cellular face of PagP, which acts as a hydrocarbon ruler to select 16-carbon chains for the acylation reaction (Ahn et al., 2004; Khan et al., 2007). As such, detergents that mimic fatty acids can interfere with PagP’s activity by binding to the interior palmitate recognition pocket. Indeed, a detergent molecule was observed deep inside the hydrocarbon ruler in crystal structures of PagP obtained in LDAO (Ahn et al., 2004) and SDS/MPD (Cuesta-Seijo et al., 2010). Specific activity is therefore measured using dodecylmaltoside (DDM) (Cuesta-Seijo et al., 2010; Khan et al., 2010) or CYFOS-7 (Hwang et al., 2004), which possess bulky tail groups that exclude them from the hydrocarbon ruler.
Although there are now multiple crystallographic (Ahn et al., 2004; Cuesta-Seijo et al., 2010) and NMR (Hwang et al., 2002, 2004) structures of PagP, it is generally difficult to obtain detergent–protein complexes that are stable and monodisperse, and in which the protein is correctly folded (Bill et al., 2011; Privé, 2007). This difficulty is in part due to the propensity of membrane proteins to aggregate in solution.
The non-specific aggregation of proteins in solution with detergents is generally thought to be mediated by interactions between protein surfaces that are exposed upon protein unfolding (Privé, 2007). However, there are no experimentally-derived structures of disordered aggregates because the formation of such aggregates precludes their high-resolution experimental evaluation. Even experimental methods that primarily produce medium- and low-resolution structural representations, such as cryo-electron microscopy and small angle X-ray or neutron scattering, are poorly suited to evaluate the conformational preferences of heterogeneous aggregates because these methods provide ensemble-averaged structural depictions that cannot be deconvoluted.
To rationally facilitate the solvation of membrane proteins for experimental structure determination, we must first understand the interactions between membrane proteins and the detergents and/or lipids that are used for their solubilization. In this perspective, interactions between proteins and detergents or lipids are revealed in some high-resolution X-ray structures (Lee, 2003) and have been investigated by NMR (Fernández et al., 2002; Hilty et al., 2004; Roosild et al., 2005; Chill et al., 2006; Lee et al., 2008). To complement these experimental approaches, computer simulations can provide atomistic details of the conformational preferences of both soluble and insoluble species as well as direct insight to the mechanisms of solvation and aggregation.
The first computer simulation of a peptide in a detergent micelle was published in 1999 (Wymore and Wong, 1999). Since then, simulations have been used to characterize the conformations and interactions of many other proteins and peptides in various detergents. While most of these studies have been initiated with a preformed detergent micelle (Wymore and Wong, 1999; Rodríguez-Ropero and Fioroni, 2012; Friemann et al., 2009; Cuthbertson et al., 2006; Lagüe et al., 2005; Khandelia and Kaznessis, 2005a,b; Löw et al., 2008; Langham et al., 2007; Chevalier et al., 2006; Patargias et al., 2005; Bond and Sansom, 2003; Khao et al., 2011; Renthal et al., 2011; Cox and Sansom, 2009; Choutko et al., 2011; Krishnamani and Lanyi, 2012; Sands et al., 2006; Psachoulia et al., 2006), simulations have also evaluated the self-assembly of detergents around a protein or peptide using atomistic (Psachoulia et al., 2006; Bond et al., 2004; Böckmann and Caflisch, 2005; Braun et al., 2004; Jalili and Akhavan, 2011) and coarse-grained (Jalili and Akhavan, 2011; Friedman and Caflisch, 2011; Bond et al., 2007; Bond and Sansom, 2006) models. These simulation systems, however, have almost exclusively contained a single protein molecule or preformed dimer (or crystal lattice (Bond et al., 2006)). To our knowledge, only one simulation study has addressed the self-assembly of detergents and multiple protein molecules. In that study, Friedman and Caflisch probed the influence of surfactants on the kinetics of amyloid aggregation by conducting simulations of a system containing 125 simplified peptides and up to 1000 coarse-grained detergent molecules (Friedman and Caflisch, 2011). While their pioneering simulation study provided an explanation for the fluorescence maximum sometimes observed before the plateau in thioflavin-T kinetic traces, the peptide model employed by Friedman and Caflisch is too simple to provide sequence- or structure-specific information (Friedman and Caflisch, 2011).
Our goal is to identify mechanisms of membrane protein aggregation in the presence of detergents. To this end, we conducted atomistic molecular dynamics (MD) simulations of a system containing four molecules of PagP in explicit aqueous solution with hundreds of initially dispersed detergent molecules. PagP has an experimental thermal denaturation temperature of 88 °C (Khan et al., 2007), suggesting that its β-barrel fold is likely to remain stable during the initial phases of detergent self-assembly. To our knowledge, this is the first report of MD simulations comprising multiple membrane protein molecules in solution with detergents. In these simulations, PagP retained its native structure while an expanded micelle of detergent molecules formed around its equatorial hydrophobic belt. Concurrently, the polar surfaces of PagP that protrude from the membrane into the extracellular and periplasmic spaces (the apical surfaces of PagP) interacted with the headgroups of detergents in other micelles, forming complexes that were stable for hundreds of nanoseconds. In some cases, an apical surface of one molecule of PagP interacted with an equatorial micelle that surrounded another molecule of PagP. In other cases, the apical surfaces of two molecules of PagP simultaneously bound an intervening neat detergent micelle. In these ways, detergents mediated the non-specific aggregation of folded PagP. To test the validity of these theoretical predictions, we conducted dynamic light scattering experiments and confirmed that at high DPC concentrations PagP induces the formation of substantially larger, more slowly diffusing species in solution that are likely to contain many copies of the PagP protein.
2. Methods
2.1. System setup
The simulation system consisted of 4 molecules of PagP in aqueous solution with 720 molecules of DPC detergent. The concentration of DPC in these simulations was 190 mM, well above the experimental concentration at which DPC forms micelles (critical micelle concentration (CMC) = 1.1 mM) (Lauterwein et al., 1979). The concentration of PagP was 1 mM. These concentrations are similar to those used by Hwang et al. (Hwang et al., 2002) in NMR experiments (0.8 mM PagP, 410 mM DPC, and 50 mM sodium phosphate at pH 6.0). Coordinates for PagP, including the single molecule of LDAO in the protein’s extracellular acyl-chain binding pocket (the hydrocarbon ruler (Ahn et al., 2004)) and crystallographic water molecules within 0.3 nm of the protein, were taken from the crystal structure of PagP in LDAO (PDB ID 1THQ) (Ahn et al., 2004). The six N-terminal residues of PagP, which are not present in the LDAO crystal structure, were omitted. The two C-terminal residues L162 and E163, which are cloning artifacts that are resolved in the LDAO crystal structure, were included. The PagP sequence in these simulations is therefore T7-E163. We modeled L1 loop residues 38-47 as random coil using the program Loopy (Xiang et al., 2002). Four molecules of PagP, each with a different conformation of the L1 loop, were subjected to 500 steps of steepest descent energy minimization and then placed in a rhombic dodecahedral unit cell. In the starting conformations, the minimum distance between any pair of atoms of two protein molecules was 5 nm. One molecule of LDAO was placed in the hydrocarbon ruler of each molecule of PagP (according to the 1THQ crystal structure) and the system was neutralized with 8 sodium ions. There was no excess salt. This procedure was repeated to generate 6 independent starting conformations of the simulation system, which is large by contemporary standards, having a total of 6.4 × 105 atoms in a box of 6.7 × 103 nm3. The initial conformation of one of the six simulation systems is depicted in Fig. 2. Each of these systems was simulated for 500 ns. In addition to the aforementioned simulations, we conducted one 500-ns simulation in the absence of DPC detergents, but under otherwise identical conditions.
Fig. 2.

One of 6 initial conformations of the simulation system. Four molecules of PagP are shown as red cartoons and periodic images are shown in blue. Detergent molecules are shown as gray lines. Water molecules and ions are omitted for clarity. For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.
2.2. Simulation protocol
MD simulations were conducted with version 4.5.5 of the GROMACS simulation package (Hess et al., 2008). PagP and sodium/chloride ions were modeled by the OPLS-AA/L parameters (Jorgensen et al., 1996; Kaminski et al., 2001), DPC was modeled by the Berger parameters (Berger et al., 1997; Tieleman et al., 2000), and the water model was TIP4P (Jorgensen et al., 1983). We constructed parameters for LDAO by analogy to Berger lipid parameters (Berger et al., 1997), as described in the supplementary material. Berger and OPLS-AA/L parameter sets were combined self-consistently using the half-ε double-pairlist method (Chakrabarti et al., 2010). Water molecules were rigidified with SETTLE (Miyamoto and Kollman, 1992) and protein and detergent bond lengths were constrained with P-LINCS (Hess, 2008). Lennard-Jones interactions were evaluated using a group-based twin-range cutoff (van Gunsteren and Berendsen, 1990) calculated every step for separation distances less than 0.9 nm and every 10 steps for distances between 0.9 and 1.4 nm, when the nonbonded list was updated. Coulomb interactions were calculated using the smooth particle-mesh Ewald method (Darden et al., 1993; Essmann et al., 1995) with a real-space cutoff of 0.9 nm and a Fourier grid spacing of 0.12 nm. Simulation in the NpT ensemble was enforced by isotropic coupling to a Berendsen barostat (Berendsen et al., 1984) at 1 bar with a coupling constant of 4 ps and temperature-coupling the simulation system using velocity Langevin dynamics (van Gunsteren and Berendsen, 1988) at 300 K with a coupling constant of 1 ps. The integration time step was 2 fs. The nonbonded pairlist was updated every 20 fs.
2.3. Stability of protein structure
Protein Cα root-mean-squared deviations (RMSD) were computed with the GROMACS g rms tool after rotational and translational fitting of each trajectory frame to a reference structure based on the Cα positions of the β-barrel core (residues 23–32, 50–59, 66–75, 83–92, 104–113, 123–132, 135–144, and 152–161). RMSD values were recomputed without the N-terminal helix (residues 7–22) and without the L1 loop (residues 33–51).
2.4. Evaluating aggregates
We used custom-built software to identify protein and detergent molecules that were part of the same molecular aggregate. Briefly, molecular aggregates were defined to include protein or detergent molecules for which at least one pair of non-hydrogen atoms was separated by <0.4 nm. In this identification, contacts involving detergents were computed based only on the 12-carbon acyl chains. To identify the equatorial micelle that surrounds the β-barrel core of PagP, we repeated this procedure but excluded protein residues that were not in the central region of the β-barrel (for this evaluation we only used protein residues 26–29, 53–56, 69–72, 86–89, 107–110, 126–129, 138–141, and 155–158). Spatial distribution functions (SDFs) were computed with the GROMACS tool g spatial with a bin width of 0.2 nm after Cα fitting as described above. To generate a single SDF based on the four protein molecules in each of six simulations, we created four separate copies of each trajectory file (using only the last 145 ns of each simulation) and, prior to concatenating them, reordered the protein molecules such that each copy of the molecular trajectory had a different protein listed first. To identify protein residues that were in contact with detergent molecules, we used a non-hydrogen atom distance cutoff of 0.435 nm, the same distance that we used in our earlier studies of amino acid side chain analogs partitioning into lipid bilayers (Neale et al., 2011). For this contact definition, we included all DPC non-hydrogen atoms.
2.5. Simulations with excess salt
In Section 3, we show that detergent micelles not only surrounded the β-barrel core of PagP, but also made stable contacts with the protein’s apical surfaces. To test whether these apical interactions were promoted by the absence of excess salt, we conducted an additional set of 250-ns simulations at varying concentrations of NaCl (0, 50, 100, 250, 500, and 1000 mM). Salt concentrations were restricted to ≤1 M because the headgroup region of a Berger lipid bilayer can catalyze the formation of large, stable OPLS-AA/L salt crystals in TIP4P water on the microsecond timescale when the NaCl concentration is ≥2 M (unpublished results). These simulation systems contained a single molecule of PagP, 200 molecules of detergent (at 625 mM), and 19,340 water molecules in a dodecahedral box of 714 nm3. Salt ions and detergent molecules were initially distributed as monomers throughout the simulation system. Salt concentration was determined in reference to the number of water molecules, not by system volume.
2.6. Dynamic light scattering experiments
DPC was purchased from Anatrace. Dynamic light scattering (DLS) measurements were performed on a Protein Solutions DynaPro801 instrument. Escherichia coli PagP was expressed in BL21 (DE3) bacteria as inclusion bodies and refolded in DPC as described by Hwang et al. (Hwang et al., 2002). The refolded protein was purified by size exclusion chromatography in 50 mM sodium phosphate pH 6.0 buffer containing 0.1% DPC. Peak fractions were pooled and concentrated by ultrafiltration using Vivaspin 30 kDa cutoff membranes to a protein concentration of 0.25 mM. Samples for DLS were prepared by mixing the protein stock solution with a stock solution of 1 M DPC and were centrifuged at 14,000 × g for 10 min immediately prior to making the measurements. The final concentration of PagP for all DLS measurements was 0.055 mM and all experiments were performed at 20 °C. Translational diffusion coefficients and hydrodynamic radii were obtained from the DLS correlation curves using the Dynamics software package supplied with the instrument (Protein Solutions Inc.).
3. Results
To test the hypothesis that detergent inhibits protein aggregation, we conducted two sets of simulations with 4 PagP molecules, first in pure water and then in a mixture of water and detergent. In this section we begin by verifying the stability and the aggregation propensity of PagP in pure water, before examining the effect of detergent in detail
3.1. Aggregation in pure water
We begin by assessing the aggregation of 4 molecules of the integral membrane enzyme PagP in pure water. In a 500-ns simulation, the protein molecules remained folded while 3 of them formed a stable aggregate that buried a considerable portion of PagP’s apolar belt (Fig. 3), which, in PagP’s native environment, is exposed to the membrane’s hydrophobic core (Fig. 1). This aggregate formed after 260 ns and was stable for the remainder of the simulation.
Fig. 3.
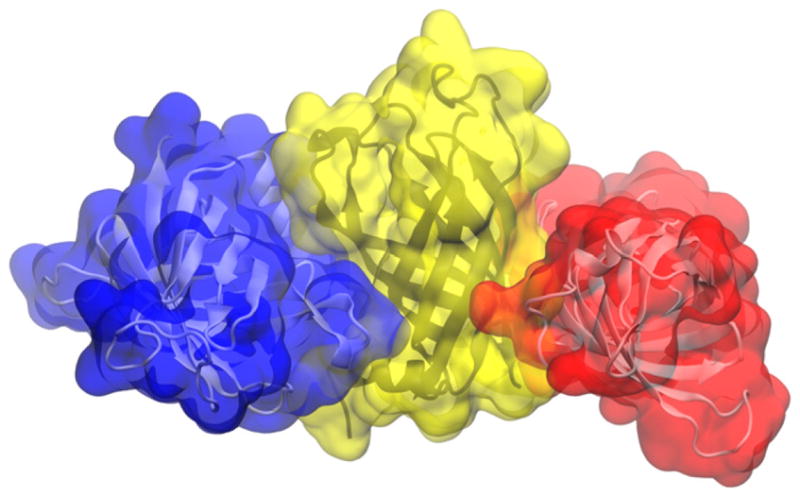
PagP aggregate in pure aqueous solution.
3.2. Aggregation in detergent
Having verified that PagP aggregates in water, we proceeded to characterize the self-assembly of DPC detergents around PagP and to assess their ability to inhibit protein aggregation. To this end, we conducted six 500-ns simulations of a system comprising 4 molecules of PagP in aqueous solution with 720 molecules of DPC, which were initially dispersed. The starting conformation of one of the 6 simulations is depicted in Fig. 2.
At the start of our simulations in detergent, prior to the self-assembly of detergent molecules, each molecule of PagP was surrounded by water. Because this represents a highly non-native environment, it is important to ensure that the protein retained its native fold. To this end, we computed the average Cα RMSD of PagP to its starting conformations as a function of time. This analysis shows that structural drift was present throughout the six 500-ns simulations (Fig. 4A), but was predominantly localized in the L1 loop (Fig. 4B). In contrast, the N-terminal α-helix and the β-barrel core stopped drifting away from their initial configurations after 100 ns (Fig. 4B–D). The extensive conformational heterogeneity of the long extracellular L1 loop is consistent with the fact that this loop is disordered in crystal structures obtained in LDAO (Ahn et al., 2004) and SDS/MPD (Cuesta-Seijo et al., 2010), is highly mobile when PagP is solvated by DPC (Hwang et al., 2002), and undergoes a large conformational rearrangement in CYFOS-7 (Hwang et al., 2002).
Fig. 4.

Protein stability. Average Cα root mean squared deviations (RMSD) of PagP to its starting conformations as a function of simulation time (A) for the entire protein, (B) without the L1 loop residues, (C) without the L1 loop and N-terminal helix residues, and (D) just for the core of the β-barrel. Vertical bars show the standard deviation of these values among the 24 simulated molecules of PagP. Numerical Figures were created with gnuplot (Williams and Kelly, 2012).
As the simulations progressed, the protein and detergent formed aggregates that became larger (Fig. 5A) and fewer (Fig. 5B). In the last 100 ns of each simulation, the minimum, average, and maximum numbers of molecules per aggregate were, respectively, 15 ± 15, 125 ± 36, and 414 ± 141 (Fig. 5A). Concurrently, the average number of aggregates was 6.3 ± 1.5 (Fig. 5B). Here, the ± sign indicates the standard deviation of the mean values among the 6 simulations and is unrelated to the standard deviation of aggregate size. Throughout these simulations, LDAO molecules remained bound to PagP’s hydrocarbon ruler.
Fig. 5.

Time series of aggregation. (A) Number of molecules per aggregate showing the (thick solid line with triangles) maximum, (dashed line) average, and (thin solid line) minimum size as a function of simulation time. Vertical bars show the standard deviation of these values among the six simulations. (B) Number of molecular aggregates per simulation. Gray shading indicates the last 145 ns, over which time-averaged values were evaluated (see Section 3).
The above analysis is dominated by the aggregation of detergents, which, in our simulation systems, far outnumbered the number of proteins. We therefore computed the probability that a given number of protein molecules were part of the same aggregate as a function of simulation time. The number of aggregates containing only one protein molecule decreased (Fig. 6A) as the number of multi-protein aggregates increased (Fig. 6B–D). This process is depicted in Movies S1 and S2. After 500 ns per simulation, a given molecule of PagP was more likely to be involved in a molecular aggregate of 4 protein molecules than in an aggregate of any other size, and only 4 of 24 PagP molecules did not aggregate with other proteins (Fig. 6).
Fig. 6.

Number of proteins per aggregate. For each protein molecule, the probability at which it was aggregated with (A) no, (B) one, (C) two, or (D) three other protein molecules as a function of simulation time. Gray shading indicates the last 145 ns, over which time-averaged values were evaluated (see Section 3).
3.3. Aggregated states
Snapshots of aggregates containing different numbers of protein molecules, obtained after 500 ns of simulation, are depicted in Fig. 7. These snapshots are representative in that there was always an equatorial detergent micelle coating the hydrophobic belt of PagP. Furthermore, the apical surfaces of PagP often bound neat micelles (Fig. 7A–C) or an equatorial micelle surrounding another molecule of PagP (Fig. 7B, C).
Fig. 7.
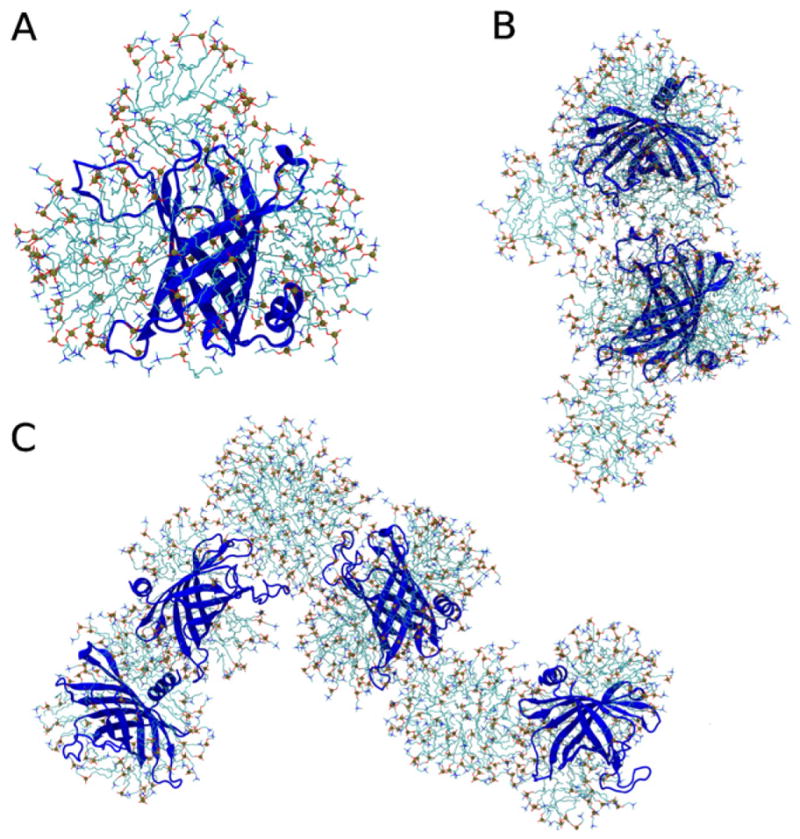
Representative molecular aggregates obtained after 500 ns of simulation. PagP is shown as a blue cartoon and detergent molecules are shown as sticks that are colored for (cyan) carbon, (blue) nitrogen, (red) oxygen, and (brown with sphere) phosphorus atoms. (A) Single molecule of PagP with an expanded equatorial micelle of detergent molecules between PagP’s aromatic belt residues and a neat apical micelle bound to the extracellular loops. (B) Aggregate containing two molecules of PagP that are bridged by interactions between the equatorial micelle surrounding one protein molecule and the apical extracellular loops of another protein molecule. (C) Aggregate containing four molecules of PagP. For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.
To assess the time- and ensemble-averaged properties of the aggregates, we used the data from the last 145 ns of each simulation. In this range, there was a relatively constant average number of aggregates (Fig. 5B) and protein molecules per aggregate (Fig. 6), and the average number of detergent molecules per aggregate only drifted slightly (Fig. 5A). Our decision to use the last 145 ns of the trajectories for analysis was qualitative and was based on visual inspection of the time-series. However, even though further states of aggregation may be reached with more sampling, the conclusions of this study would not change. Fig. 8A shows the probability distributions of the number of detergent molecules in aggregates that contained different numbers of protein molecules. The near linear relationship between the number of detergents and the number of protein molecules in a given aggregate (approximately 133 detergents/protein, Fig. 8B) suggests that protein aggregation does not displace bound detergent and is consistent with detergent-mediated protein aggregation, examples of which are provided in Fig. 7.
Fig. 8.

Number of detergent molecules in aggregates with different numbers of protein molecules. (A) Probability distributions. (B) Heat map of the data presented in part A. The data points that were used to construct the heat map are shown as × symbols. Large diamonds with plus signs in the middle mark the average number of detergent molecules for each number of proteins per aggregate. A line of best fit to these average values, computed with gnuplot (Williams and Kelly, 2012), is superimposed on the heat map.
To assess the time- and ensemble-averaged arrangement of detergents and other protein molecules around a central molecule of PagP, we computed spatial distribution functions (SDFs), which are depicted in Fig. 9 and Movie S3. In addition to the presence of detergent around the hydrophobic belt of PagP (Figs. 7 and 9), the high density of detergent molecules at the apical surfaces of PagP (Fig. 9) demonstrates that this type of protein-detergent interaction was common in our simulations. In these SDFs, the detergent headgroups of apical micelles are delocalized, especially distal to the central molecule of PagP (Fig. 9D). This delocalization is a consequence of the varying size and precise location of apical micelles, which sometimes were neat and sometimes contained another molecule of PagP.
Fig. 9.

Distribution of detergent and other protein atoms around a central molecule of PagP. (A, B) Spatial distribution function (SDF) of DPC detergents shown as a yellow isodensity surface around a black cartoon representation of PagP. The SDF was obtained from all 4 protein molecules in the last 145 ns of all 6 simulations. (C, D) Two slices through the SDF. Darker color represents increased time- and ensemble-averaged sampling density for (blue) phosphocholine (PC) detergent headgroups, (cyan) acyl chains, and (red) other protein molecules. For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.
The SDFs depicted in Fig. 9 also reveal regions of other protein density near PagP’s apical surfaces, which are a consequence of the detergent-mediated protein aggregation shown in Fig. 7.
Our simulations revealed two distinct types of detergent-protein interactions: the equatorial micelle that surrounds the hydrophobic core of PagP, and the micelles that are bound to PagP’s apical surfaces. The time evolution of the number of detergent molecules in the equatorial micelle is shown in Fig. 10A. Although the average size of the equatorial micelle continues to grow with increasing simulation time (Fig. 10A) and other estimates indicate that the simulations have not yet reached equilibrium (Figs. 5 and 6), computational limitations precluded extending these simulations substantially. The distribution of equatorial micelle sizes over the last 145 ns of each simulation is depicted in Fig. 10B. In this time range, equatorial micelles contained an average of 80 detergent molecules (σ = 18). Importantly, the acyl-chain detergent tails do not form a continuous greasy surface connecting the equatorial and apical micelles that are bound to a given molecule of PagP. Furthermore, detergent headgroups are present at the interface between PagP and apical micelles (Fig. 9D).
Fig. 10.

Number of detergents in the equatorial micelle surrounding each molecule of PagP. (A) Time-evolution of the average. Vertical bars represent the standard deviation among 24 molecule of PagP at selected timepoints. (B) Histogram of the data from the last 145 ns of each simulation (bin with of 2 molecules).
To identify the PagP residues that interacted with detergents in equatorial and apical micelles, these two types of micelle-protein contacts are each depicted on a per-residue basis in Fig. 11. The β-barrel core of PagP frequently made contact with detergent in the equatorial micelle (Fig. 11A, D), whereas the protein’s loops and turns interacted more frequently with detergents in apical micelles (Fig. 11B, E). These apical interactions occurred more often at the protein’s extracellular surface than at its periplasmic surface (Fig. 11B, E), and most commonly involved the long extracellular L1 loop (Fig. 11B). To identify residues that were particularly prone to interact with apical, rather than equatorial, micelles, we computed the ratio of apical to equatorial micelle DPC contacts for each PagP residue (Fig. 11C). Residues for which apical micelle interactions were more than twice as common as equatorial micelle interactions are listed in Table 1. Here, there is a strong preference for charged and polar residues, which represent 11 of the 15 residues identified in Table 1 to favor interactions with apical micelles.
Fig. 11.

Equatorial and apical micelle binding by PagP residue. Fraction of the last 145 ns of each simulation for which each PagP residue made contact with a detergent molecule in an (A) equatorial and (B) apical micelle. Contacts were defined as protein-detergent non-hydrogen atom distances <0.435 nm. PagP’s N-terminal α-helix and β-sheet strands A–H are marked above the plot, as are extracellular loops L1–L4 and periplasmic turns T1–T3, using the definitions of Ahn et al. (Ahn et al., 2004). (C) Per-residue ratio of apical to equatorial micelle detergent contact fraction. (D, E) Surface of the LDAO crystal structure of PagP colored by fraction of time that each residue made (D) equatorial and (E) apical contacts. Color indicates (red) infrequent or (blue) frequent contacts. For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.
Table 1.
Residues that make more contacts with apical micelles than with the equatorial micelle. All residues for which Papical /Pequatorial > 2.0 are listed.
| Residue
|
|
|||
|---|---|---|---|---|
| # | Type | Class | ||
| 59 | Arg | Charged | 5.8 | |
| 77 | Ser | Polar | 5.3 | |
| 16 | Thr | Polar | 4.8 | |
| 40 | Lys | Charged | 4.5 | |
| 78 | Trp | Aromatic | 4.5 | |
| 152 | Val | Apolar | 4.1 | |
| 150 | Gly | Apolar | 3.7 | |
| 42 | Lys | Charged | 3.5 | |
| 62 | Asp | Charged | 3.3 | |
| 41 | Glu | Charged | 3.0 | |
| 39 | Asp | Charged | 2.7 | |
| 76 | Asp | Charged | 2.2 | |
| 15 | Gln | Polar | 2.2 | |
| 60 | Trp | Aromatic | 2.2 | |
| 43 | Thr | Polar | 2.1 | |
Histograms of the number of DPC molecules that made contacts with the apical surfaces of a given molecule of PagP but were not part of its equatorial micelle are shown in Fig. 12. More often than not, the extracellular and periplasmic surfaces of PagP were in contact with apical DPC molecules, these interactions being absent in only 15 and 40%, respectively, of the configurations sampled in the last 145 ns of each simulation (Fig. 12). The extracellular face of PagP tended to make contacts with more of these DPC molecules than the periplasmic face (Fig. 12). On average, the contacts between these apical micelles and PagP were mediated by 9 ± 1 and 4 ± 1 detergent molecules at the extracellular and periplasmic protein surfaces, respectively.
Fig. 12.
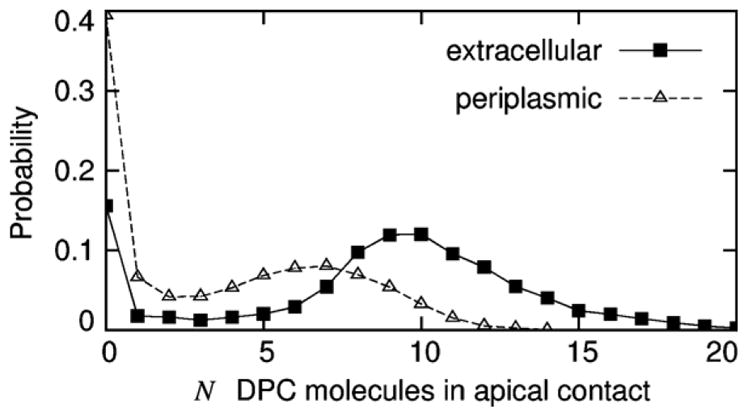
Number of DPC molecules making contact with the apical surfaces of PagP. Data, based on the last 145 ns of each simulation, are shown for (solid line with filled squares) extracellular and (dashed line with open triangles) periplasmic surfaces.
To identify which apical micelle detergent atoms were most often in contact with PagP, the number of non-equatorial DPC molecules making contact with each apical surface of PagP was computed as a function of DPC atom type (Fig. 13). The interactions between DPC molecules and the apical surfaces of PagP were more often mediated by interactions with detergent headgroups than with acyl chains, especially at PagP’s periplasmic face (Fig. 13). Note that these numbers of contacts are averages that are influenced by the number and size of the bound micelle(s), the interacting surface area, and the fraction of PagP molecules that bound apical micelles. The general features of Fig. 13 do not depend on the particular distance criterion used to identify contacts (data not shown).
Fig. 13.
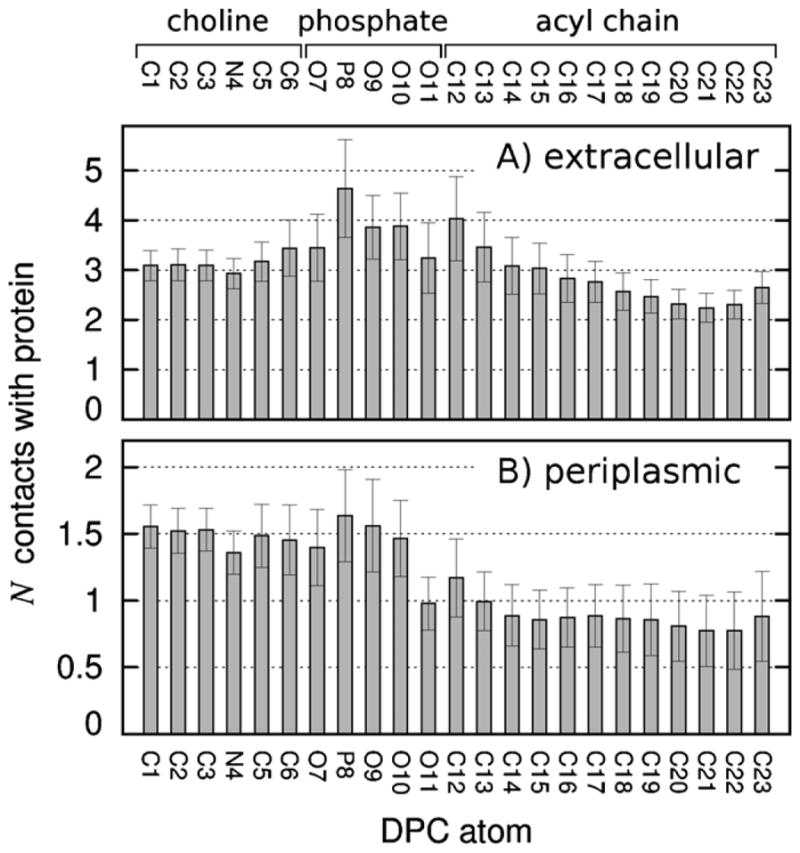
Number of DPC molecules making contact with the apical surfaces of PagP as a function of DPC atom type. Data, based on the last 145 ns of each simulation, are shown separately for (A) extracellular and (B) periplasmic apical surfaces of PagP.
Finally, given that Hwang et al. (Hwang et al., 2002) purified PagP in the presence of 50 mM sodium phosphate, we considered the possibility that the detergent-mediated protein aggregation observed in these simulations was due to the absence of salt. This is not the case, as the binding of detergent micelles at the apical surfaces of PagP was also observed in simulations with up to 1 M NaCl (Fig. S1). Therefore, our simulation data support the existence of detergent-mediated protein aggregation in the presence of salt, although we note that chloride ions (used in these simulations) differ from phosphate ions (used by Hwang et al. (Hwang et al., 2002) for NMR), and salt concentrations up to 7 M affect the critical micelle concentration of DPC (Palladino et al., 2010).
3.4. Experimental assessment of aggregation
To evaluate the validity of our simulation results, we used dynamic light scattering (DLS) to measure the effect of detergent concentrations on PagP aggregation at 20 °C. Specifically, we measured DLS correlation functions for a series of solutions with and without PagP at increasing DPC concentrations. The protein concentration was fixed at 0.055 mM in these experiments.
At DPC concentrations of 50, 100 and 200 mM, we observed particles with hydrodynamic radii of 2.4 ± 0.4 nm irrespective of whether PagP was present in the solution. Given a DPC CMC of 1.1 mM, (Lauterwein et al., 1979) a mean aggregation number of 56 for neat DPC micelles, (Lauterwein et al., 1979) and 77–102 bound DPC molecules in a monomeric PagP/detergent complex (see Section 4 and Hwang et al., 2002), we expect approximately 16, 32 or 64 unoccupied DPC micelles for every PagP/detergent complex at 50, 100 and 200 mM DPC, respectively. The resolution of a DLS experiment is not sufficient to resolve the scattering from unoccupied micelles and monomeric PagP/detergent complexes, and scattering in these cases is well described by a single species.
At detergent concentrations above 200 mM, the refractive index and viscosity of the solutions were clearly affected (data not shown). Because of this, we did not attempt to derive hydrodynamic radii from the DLS data at these DPC concentrations, choosing instead to directly compare the effect of PagP on the DLS correlation functions. At 200 and 400 mM DPC, the presence of PagP had very little effect on the shape of the correlation curves (Fig. 14A, B), consistent with the similar values of hydrodynamic radii obtained at 200 mM DPC in the presence and absence of PagP (see preceding paragraph). However, at 600 mM DPC, the addition of PagP shifts the scattering curve significantly to the right (Fig. 14C), which can be interpreted as the presence of additional larger scattering particles that exist only in the presence of the protein. Both DLS curves show different scattering behavior at 800 mM DPC (Fig. 14D), but again, the scattering curve obtained in the presence of PagP shows a shift to the right at correlation times less than 1 ms. Given that solutions of ≤400 mM DPC produced very little correlated scattering with time values larger than 0.1 ms despite the presence of PagP/detergent complexes, which we presume contain 1 protein molecule per complex, the PagP-dependent shifts in the DLS correlation curves at 600 and 800 mM DPC, with time values up to 1 ms, indicate the presence of very large more slowly diffusing scattering species that are likely to contain many copies of the PagP protein.
Fig. 14.

Dynamic light scattering correlation functions. Data are shown for solutions of DPC (solid lines) without protein and (dashed lines) with 0.055 mM PagP. Correlation curves for samples at DPC concentrations of 50 and 100 mM were very similar to those shown for the 200 mM DPC data.
4. Discussion
This work represents the first simulation study of the spontaneous self-assembly of detergent molecules in the context of multiple molecules of a membrane protein. To this end, we conducted extensive sampling (seven 500-ns simulations) of a large simulation system (>600,000 atoms).
In each of the six 500-ns simulations conducted in the presence of DPC detergent, the four molecules of the PagP protein retained the native fold (Fig. 4) while expanded micelles of detergent molecules spontaneously self-assembled around their central apolar belts (Figs. 7 and 9). These equatorial micelles presented a chemical environment broadly consistent with the profile of charge density across a lipid bilayer (Nagle et al., 1996; Nagle and Tristram-Nagle, 2000) (Fig. 9C, D). Detergent acyl chains covered the β-barrel core of PagP between its aromatic girdles (Ahn et al., 2004) (Fig. 9D), which tend to reside in the interfacial regions of a lipid bilayer (Schulz, 2002). Concurrently, detergent headgroups of the equatorial micelles made contacts with the protein’s apical surfaces (Fig. 9D). Additional evidence for the correct positioning of PagP in the equatorial micelle is provided by the interfacial detergent acyl-chain/headgroup environment that, in our simulations, formed around the N-terminal amphipathic α-helix of PagP (Fig. 9D). Experimental measurements of the destabilizing effects of N-terminal mutations suggests that this helix is disposed at the bilayer interface, where it “acts as a clamp, locking the protein in the native, active conformation once folding and insertion are complete” (Huysmans et al., 2007). DPC micelle solvation thus mimics bilayer solvation for PagP in these and other (Cox and Sansom, 2009) simulations.
In spite of the similarity between PagP solvation by DPC micelles and lipid bilayers, there are important differences. Primarily, in addition to the formation of the equatorial micelle around each molecule of PagP, the apical surfaces of PagP concurrently interacted with the headgroups of detergent molecules that were part of either a neat micelle or another equatorial micelle surrounding a different molecule of PagP (Figs. 7 and 9). This feature has not been evident in previous simulations of PagP (Cox and Sansom, 2009) or other membrane proteins (Rodríguez-Ropero and Fioroni, 2012; Friemann et al., 2009; Patargias et al., 2005; Bond and Sansom, 2003; Choutko et al., 2011; Sands et al., 2006; Psachoulia et al., 2006; Bond et al., 2004; Böckmann and Caflisch, 2005; Bond and Sansom, 2006), in whole or in part because of the small number of detergent and/or protein molecules in previous simulations. Unexpectedly, the above results suggest that these interactions can mediate protein aggregation at sufficiently high detergent concentration. It is presently unclear if this detergent-mediated protein aggregation can drive the formation of amorphous aggregates, regular crystals, or both.
The interpretation of our results was facilitated by the generation of 6 independent assembly simulations. Each simulation reached a different extent of aggregation and it is only because we conducted multiple simulations that it was possible to evaluate the precision of our numerical estimates. Nevertheless, these simulations have not attained equilibrium (Figs. 5, 6 and 10A). We are unaware of any method that can be used to judge how close brute-force MD simulations are to attaining convergence if the equilibrium value is not known in advance. In spite of this limitation, we have used the last portion of the trajectories to provide our best estimates of the equilibrium properties of this system. The major conclusions of this study – that detergents can mediate the aggregation of membrane proteins in solution – would not change with increased simulation time.
4.1. Comparison to other simulation studies
Böckmann and Caflisch (2005) simulated the self-assembly of 105–200 dihexanoylphosphatidylcholine (DHPC) short-chain lipids around a single molecule of the β-barrel membrane protein OmpX. They reported that, in addition to forming a monolayer ring around the apolar β-barrel core, DHPC molecules also bound a predominantly polar β-sheet protruding on the extracellular surface of the membrane and posited that interactions between zwitterionic lipid headgroups and charged protein residues seeded the formation of micelles (Böckmann and Caflisch, 2005). They went on to conclude that the stable interaction of DHPC micelles with the extracellular surface of OmpX were consistent with Vogt and Schulz’s hypothesis that OmpX’s protruding β-sheet, in analogy to a fishing rod, “functions in cell adhesion and invasion and that it inhibits the complement system by binding one of its essential proteins” (Vogt and Schulz, 1999).
We observed similar interactions in our simulations of PagP (Figs. 7, 9 and 11–13). Detergent binding to PagP’s extracellular surface may be attributed to the molecular properties of the region surrounding the acyl-chain binding pocket, which, given its biological role, are likely to favor interactions with lipids. It is thus possible to provide a biologically relevant explanation for the stable interaction of detergent micelles with the extracellular face of PagP. However, we also observed the complexation of detergent micelles with the periplasmic face of PagP (Figs. 7B, C, 9, 11B, E and 13B), which has no known lipid binding sites. The presence of excess detergent in our simulation system together with extensive sampling allowed us to assess the heterogeneity of PagP-DPC aggregate morphology. Given that DPC micelles formed stable interactions with both apical surfaces of PagP, our results suggest that the apical interactions of detergent micelles with membrane proteins may be more generic than previously thought.
4.2. Comparison to experiment
The macromolecular concentrations in our simulations (1 mM PagP, 190 mM DPC) were similar to those under which PagP-DPC has been previously evaluated by NMR (0.8 mM PagP, 410 mM DPC) (Hwang et al., 2002). Hwang et al. (2002) reported that the overall isotropic rotational correlation time for PagP-DPC is 20 ns at 45 °C and, based on this result, estimated that PagP-DPC micelles have a molecular mass of 50–60 kDa. Given that the masses of PagP and fully deuterated DPC are, respectively, 20.175 (Cuesta-Seijo et al., 2010) and 0.389 kDa, this NMR result indicates that monomeric PagP is surrounded by 77–102 detergent molecules. This experimental estimate matches the number of detergent molecules that formed the equatorial micelle around the hydrophobic belt of PagP in our simulations, 80, σ = 18 (Fig. 10B). However, our estimate of the total number of detergent molecules that interact with each protein molecule (approximately N = 133, Fig. 8B) is larger than the NMR estimate. This disagreement may, in part, be due to assumptions underlying the conversion of backbone 15N T1, T1ρ, and 1H–15N NOE values to a molecular mass by way of an overall isotropic rotational correlation time (Hwang et al., 2001, 2002). Nevertheless, in our simulations, apical micelle interactions led to large-scale detergent-mediated protein aggregation, which has not been observed in previous experiments (Hwang et al., 2002) (although there may be a low population of protein aggregates that are not visible in the NMR spectra (Baldwin and Kay, 2009; Korzhnev et al., 2004, 2010; Vallurupalli et al., 2008)).
To resolve this apparent discrepancy, we used DLS experiments to assess the size distribution profile of particles in solutions of increasing DPC concentration in the presence and absence of PagP. PagP did not significantly perturb these profiles for DPC concentrations ≤400 mM (Fig. 14A, B) but at DPC concentrations ≥600 mM the presence of PagP led to the formation of larger particles (Fig. 14C, D) that likely contain multiple protein molecules. Despite this experimental corroboration, it seems probable that our simulations overestimated the stability of interactions between DPC micelles and the apical surfaces of PagP, which we observed at 190 mM DPC (Fig. 6), because NMR experiments conducted at 45 °C did not detect substantial protein aggregation at 410 mM DPC (Hwang et al., 2002) and DLS experiments conducted at 20 °C indicate that detergent-mediated aggregation of PagP occurs at ≥600 mM DPC (Fig. 14). Because the NMR spectra of integral membrane proteins are sensitive to temperature (Sanders and Sönnichsen, 2006; Dötsch, 2003; Cavanagh et al., 1996), future studies should evaluate the temperature dependence of detergent-mediated protein aggregation.
Whereas previous simulations have shown that detergent-protein interactions are sufficiently favorable in comparison to the interactions between water and detergents or proteins to incite the formation of stable detergent-protein complexes (Psachoulia et al., 2006; Bond et al., 2004, 2007; Böckmann and Caflisch, 2005; Braun et al., 2004; Jalili and Akhavan, 2011; Friedman and Caflisch, 2011; Bond and Sansom, 2006), to our knowledge nobody has investigated whether the interactions between detergents and proteins in atomistic force fields are, in fact, too favorable. The above results suggest that this may be the case, at least for the combination of OPLS-AA/L protein parameters and the so-called Berger lipid/detergent parameters. The new type of protein-detergent interactions identified in this study will be useful in the development and evaluation of new and existing detergent force field parameters.
5. Conclusions
In this study, we used large-scale molecular simulations and dynamic light scattering measurements to examine the effect of detergent on the solvation and the aggregation of a membrane protein. The simulations showed that in pure aqueous solution, the integral membrane protein PagP aggregates via interactions between the apolar surfaces that constitute the protein’s central hydrophobic belt. DPC detergent molecules abolish this type of protein aggregation by forming an equatorial micelle around each molecule of PagP that excludes water and other protein molecules from the protein’s hydrophobic belt. Concurrently, the simulations predict that detergent molecules bind to the predominantly polar apical surfaces of PagP and mediate protein aggregation either via direct binding between the equatorial micelle of one protein and an apical surface of another protein, or by the simultaneous binding of a neat micelle by the apical surfaces of two protein molecules. Importantly, these simulation results are consistent with dynamic light scattering experiments, which confirmed that at high DPC concentrations PagP induces the formation of substantially larger, more slowly diffusing species in solution that are likely to contain many copies of the PagP protein. The mechanisms of detergent-mediated protein aggregation identified in this work provide potential reasons why membrane proteins are so difficult to solubilize as monodisperse folded entities for experimental evaluation.
Supplementary Material
Acknowledgments
We thank Peter M. Hwang for assistance interpreting previous NMR results. Computations were performed on the resources of the WestGrid (www.westgrid.ca), Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca), and SciNet (Loken et al., 2010) HPC Consortia of Compute Canada/Calcul Québec. C.N. is funded by the Research Training Center at the Hospital for Sick Children and by the University of Toronto. R.E.B. is supported by CIHR Operating Grant MOP-84329. G.G.P. was supported in part by an NSERC Discovery Grant and CIHR grant MOP-246884. R.P. was supported in parts by CIHR Operating Grants MOP-43998 and MOP-43949. R.P. was a Canada Research Chairs Program (CRCP) chair holder.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.chemphyslip.2013.02.005.
References
- Ackerman MJ, Clapham DE. Ion channels – basic science and clinical disease. New England Journal of Medicine. 1997;336(22):1575–1586. doi: 10.1056/NEJM199705293362207. [DOI] [PubMed] [Google Scholar]
- Ahn VE, Lo EI, Engel CK, Chen L, Hwang PM, Kay LE, Bishop RE, Privé GG. A hydrocarbon ruler measures palmitate in the enzymatic acylation of endotoxin. EMBO Journal. 2004;23(15):2931–2941. doi: 10.1038/sj.emboj.7600320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nature Chemical Biology. 2009;5(11):808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. Journal of Chemical Physics. 1984;81(8):3684–3690. [Google Scholar]
- Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophysical Journal. 1997;72(5):2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. How membrane proteins sense voltage. Nature Reviews Molecular Cell Biology. 2008;9(4):323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- Bill RM, Henderson PJF, Iwata S, Kunji ERS, Michel H, Neutze R, Newstead S, Poolman B, Tate CG, Vogel H. Overcoming barriers to membrane protein structure determination. Nature Biotechnology. 2011;29(4):335–340. doi: 10.1038/nbt.1833. [DOI] [PubMed] [Google Scholar]
- Bishop RE. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Molecular Microbiology. 2005;57(4):900–912. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CRH. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. EMBO Journal. 2000;19(19):5071–5080. doi: 10.1093/emboj/cdd507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckmann RA, Caflisch A. Spontaneous formation of detergent micelles around the outer membrane protein OmpX. Biophysical Journal. 2005;88(5):3191–3204. doi: 10.1529/biophysj.105.060426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, Sansom MSP. Membrane protein dynamics versus environment: simulations of OmpA in a micelle and in a bilayer. Journal of Molecular Biology. 2003;329(5):1035–1053. doi: 10.1016/s0022-2836(03)00408-x. [DOI] [PubMed] [Google Scholar]
- Bond PJ, Sansom MSP. Insertion and assembly of membrane proteins via simulation. Journal of the American Chemical Society. 2006;128(8):2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, Cuthbertson JM, Deol SS, Sansom MSP. MD simulations of spontaneous membrane protein/detergent micelle formation. Journal of the American Chemical Society. 2004;126(49):15948–15949. doi: 10.1021/ja044819e. [DOI] [PubMed] [Google Scholar]
- Bond PJ, Faraldo-Gómez JD, Deol SS, Sansom MSP. Membrane protein dynamics and detergent interactions within a crystal: a simulation study of OmpA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(25):9518–9523. doi: 10.1073/pnas.0600398103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MSP. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. Journal of Structural Biology. 2007;157(3):593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Bowie JU. Solving the membrane protein folding problem. Nature. 2005;438(7068):581–589. doi: 10.1038/nature04395. [DOI] [PubMed] [Google Scholar]
- Braun R, Engelman DM, Schulten K. Molecular dynamics simulations of micelle formation around dimeric glycophorin A transmembrane helices. Biophysical Journal. 2004;87(2):754–763. doi: 10.1529/biophysj.104.040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell D. T cell antigen receptor signal transduction pathways. Annual Review of Immunology. 1996;14(1):259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AGI, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Elsevier Academic Press; San Diego, California, USA: 1996. [Google Scholar]
- Chakrabarti N, Neale C, Payandeh J, Pai EF, Pomès R. An iris-like mechanism of pore dilation in the CorA magnesium transport system. Biophysical Journal. 2010;98(5):784–792. doi: 10.1016/j.bpj.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier F, Lopez-Prados J, Groves P, Perez S, Martín-Lomas M, Nieto PM. Structure and dynamics of the conserved protein GPI anchor core inserted into detergent micelles. Glycobiology. 2006;16(10):969–980. doi: 10.1093/glycob/cwl015. [DOI] [PubMed] [Google Scholar]
- Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Science. 2006;15(4):684–698. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choutko A, Glättli A, Fernández C, Hilty C, Wüthrich K, van Gunsteren WF. Membrane protein dynamics in different environments: simulation study of the outer membrane protein X in a lipid bilayer and in a micelle. European Biophysics Journal. 2011;40(1):39–58. doi: 10.1007/s00249-010-0626-7. [DOI] [PubMed] [Google Scholar]
- Congreve M, Marshall F. The impact of GPCR structures on pharmacology and structure-based drug design. British Journal of Pharmacology. 2010;159(5):986–996. doi: 10.1111/j.1476-5381.2009.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox K, Sansom MSP. One membrane protein, two structures and six environments: a comparative molecular dynamics simulation study of the bacterial outer membrane protein PagP. Molecular Membrane Biology. 2009;26(4):205–214. doi: 10.1080/09687680902788967. [DOI] [PubMed] [Google Scholar]
- Cuesta-Seijo JA, Neale C, Khan MA, Moktar J, Tran CD, Bishop RE, Pomès R, Privé GG. PagP crystallized from SDS/cosolvent reveals the route for phospholipid access to the hydrocarbon ruler. Structure. 2010;18(9):1210–1219. doi: 10.1016/j.str.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson JM, Bond PJ, Sansom MSP. Transmembrane helix–helix interactions: comparative simulations of the glycophorin A dimer. Biochemistry. 2006;45(48):14298–14310. doi: 10.1021/bi0610911. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. Journal of Chemical Physics. 1993;98(12):10089–10092. [Google Scholar]
- Dötsch V. NMR strategies for protein assignments. In: Zerbe O, editor. BioNMR in Drug Research. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2003. pp. 79–94. [Google Scholar]
- Durdagi S, Zhao C, Cuervo JE, Noskov SY. Atomistic models for free energy evaluation of drug binding to membrane proteins. Current Medicinal Chemistry. 2011;18(17):2601–2611. doi: 10.2174/092986711795933641. [DOI] [PubMed] [Google Scholar]
- Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. Journal of Molecular Biology. 1984;179(1):125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. Journal of Chemical Physics. 1995;103(19):8577–8593. [Google Scholar]
- Fernández C, Hilty C, Wider G, Wüthrich K. Lipid–protein interactions in DHPC micelles containing the integral membrane protein OmpX investigated by NMR spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13533–13537. doi: 10.1073/pnas.212515099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R, Caflisch A. Surfactant effects on amyloid aggregation kinetics. Journal of Molecular Biology. 2011;414(2):303–312. doi: 10.1016/j.jmb.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Friemann R, Larsson DSD, Wang Y, van der Spoel D. Molecular dynamics simulations of a membrane protein–micelle complex in vacuo. Journal of the American Chemical Society. 2009;131(46):16606–16607. doi: 10.1021/ja902962y. [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Lei Z. Membrane transporters in drug development. Nature Reviews Drug Discovery. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annual Review of Biochemistry. 1987;56(1):615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochemical Journal. 1993;295(2):329–341. doi: 10.1042/bj2950329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. Journal of Chemical Theory and Computation. 2008;4(1):116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS: 4 algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hilty C, Wider G, Fernández C, Wüthrich K. Membrane protein–lipid interactions in mixed micelles studied by NMR spectroscopy with the use of paramagnetic reagents. ChemBioChem. 2004;5(4):467–473. doi: 10.1002/cbic.200300815. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Huysmans GHM, Radford SE, Brockwell DJ, Baldwin SA. The N-terminal helix is a post-assembly clamp in the bacterial outer membrane protein PagP. Journal of Molecular Biology. 2007;373(3):529–540. doi: 10.1016/j.jmb.2007.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Kay LE. Solution structure and dynamics of integral membrane proteins by NMR: a case study involving the enzyme PagP. Methods in Enzymology. 2005;394:335–350. doi: 10.1016/S0076-6879(05)94013-5. [DOI] [PubMed] [Google Scholar]
- Hwang PM, Skrynnikov NR, Kay LE. Domain orientation in β-cyclodextrin-loaded maltose binding protein: diffusion anisotropy measurements confirm the results of a dipolar coupling study. Journal of Biomolecular NMR. 2001;20(1):83–88. doi: 10.1023/a:1011226512421. [DOI] [PubMed] [Google Scholar]
- Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CRH, Privé GG, Bishop RE, Kay LE. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(21):13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(26):9618–9623. doi: 10.1073/pnas.0402324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili S, Akhavan M. Study of the Alzheimer’s Aβ40 peptide in SDS micelles using molecular dynamics simulations. Biophysical Chemistry. 2011;153(2–3):179–186. doi: 10.1016/j.bpc.2010.11.007. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79(2):926–935. [Google Scholar]
- Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society. 1996;118(45):11225–11236. [Google Scholar]
- Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B. 2001;105(28):6474–6487. [Google Scholar]
- Khan MA, Neale C, Michaux C, Pomès R, Privé GG, Woody RW, Bishop RE. Gauging a hydrocarbon ruler by an intrinsic exciton probe. Biochemistry. 2007;46(15):4565–4579. doi: 10.1021/bi602526k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Moktar J, Mott PJ, Bishop RE. A thiolate anion buried within the hydrocarbon ruler perturbs PagP lipid acyl chain selection. Biochemistry. 2010;49(11):2368–2379. doi: 10.1021/bi901669q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia H, Kaznessis YN. Molecular dynamics simulations of helical antimicrobial peptides in SDS micelles: What do point mutations achieve? Peptides. 2005a;26(11):2037–2049. doi: 10.1016/j.peptides.2005.03.058. [DOI] [PubMed] [Google Scholar]
- Khandelia H, Kaznessis YN. Molecular dynamics simulations of the helical antimicrobial peptide ovispirin-1 in a zwitterionic dodecylphosphocholine micelle: insights into host-cell toxicity. Journal of Physical Chemistry B. 2005b;109(26):12990–12996. doi: 10.1021/jp050162n. [DOI] [PubMed] [Google Scholar]
- Khao J, Arce-Lopera J, Sturgis J, Duneau JP. Structure of a protein–detergent complex: the balance between detergent cohesion and binding. European Biophysics Journal. 2011;40(10):1143–1155. doi: 10.1007/s00249-011-0745-9. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430(6999):586–590. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329(5997):1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- Krishnamani V, Lanyi JK. Molecular dynamics simulation of the unfolding of individual bacteriorhodopsin helices in sodium dodecyl sulfate micelles. Biochemistry. 2012;51(6):1061–1069. doi: 10.1021/bi201770y. [DOI] [PubMed] [Google Scholar]
- Lagüe P, Roux B, Pastor RW. Molecular dynamics simulations of the influenza hemagglutinin fusion peptide in micelles and bilayers: conformational analysis of peptide and lipids. Journal of Molecular Biology. 2005;354(5):1129–1141. doi: 10.1016/j.jmb.2005.10.038. [DOI] [PubMed] [Google Scholar]
- Langham A, Waring A, Kaznessis Y. Comparison of interactions between beta-hairpin decapeptides and SDS/DPC micelles from experimental and simulation data. BMC Biochemistry. 2007;8(11):1–13. doi: 10.1186/1471-2091-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterwein J, Bösch C, Brown LR, Wüthrich K. Physicochemical studies of the protein–lipid interactions in melittin-containing micelles. BBA-Biomembranes. 1979;556(2):244–264. doi: 10.1016/0005-2736(79)90046-4. [DOI] [PubMed] [Google Scholar]
- Lee AG. Lipid–protein interactions in biological membranes: a structural perspective. BBA-Biomembranes. 2003;1612(1):1–40. doi: 10.1016/s0005-2736(03)00056-7. [DOI] [PubMed] [Google Scholar]
- Lee D, Walter KFA, Bru ckner AK, Hilty C, Becker S, Griesinger C. Bilayer in small bicelles revealed by lipid–protein interactions using NMR spectroscopy. Journal of the American Chemical Society. 2008;130(42):13822–13823. doi: 10.1021/ja803686p. [DOI] [PubMed] [Google Scholar]
- Loken C, Gruner D, Groer L, Peltier R, Bunn N, Craig M, Henriques T, Dempsey J, Yu CH, Chen J, Dursi LJ, Chong J, Northrup S, Pinto J, Knecht N, Van Zon R. SciNet: lessons learned from building a power-efficient top-20 system and data centre. Journal of Physics: Conference Series. 2010;256(1):012026. [Google Scholar]
- Löw C, Weininger U, Lee H, Schweimer K, Neundorf I, Beck-Sickinger AG, Pastor RW, Balbach J. Structure and dynamics of helix-0 of the N-BAR domain in lipid micelles and bilayers. Biophysical Journal. 2008;95(9):4315–4323. doi: 10.1529/biophysj.108.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Kollman PA. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. Journal of Computational Chemistry. 1992;13(8):952–962. [Google Scholar]
- Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. BBA-Reviews on Biomembranes. 2000;1469(3):159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle JF, Zhang R, Tristram-Nagle S, Sun W, Petrache HI, Suter RM. X-ray structure determination of fully hydrated L alpha phase dipalmitoylphosphatidylcholine bilayers. Biophysical Journal. 1996;70(3):1419–1431. doi: 10.1016/S0006-3495(96)79701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale C, Bennett WFD, Tieleman DP, Pomès R. Statistical convergence of equilibrium properties in simulations of molecular solutes embedded in lipid bilayers. Journal of Chemical Theory and Computation. 2011;7(12):4175–4188. doi: 10.1021/ct200316w. [DOI] [PubMed] [Google Scholar]
- Opella SJ, Nevzorov A, Mesleh MF, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Biochemistry and Cell Biology. 2002;80(5):597–604. doi: 10.1139/o02-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino P, Rossi F, Ragone R. Effective critical micellar concentration of a zwitterionic detergent: a fluorimetric study on n-dodecyl phosphocholine. Journal of Fluorescence. 2010;20(1):191–196. doi: 10.1007/s10895-009-0537-0. [DOI] [PubMed] [Google Scholar]
- Patargias G, Bond PJ, Deol SS, Sansom MSP. Molecular dynamics simulations of GlpF in a micelle vs in a bilayer: conformational dynamics of a membrane protein as a function of environment. Journal of Physical Chemistry B. 2005;109(1):575–582. doi: 10.1021/jp046727h. [DOI] [PubMed] [Google Scholar]
- Pohorille A, Schweighofer K, Wilson MA. The origin and early evolution of membrane channels. Astrobiology. 2005;5(1):1–17. doi: 10.1089/ast.2005.5.1. [DOI] [PubMed] [Google Scholar]
- Popot JL, Engelman DM. Helical membrane protein folding, stability, and evolution. Annual Review of Biochemistry. 2000;69(1):881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- Privé GG. Detergents for the stabilization and crystallization of membrane proteins. Methods. 2007;41(4):388–397. doi: 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Psachoulia E, Bond PJ, Sansom MSP. MD simulations of Mistic: conformational stability in detergent micelles and water. Biochemistry. 2006;45(30):9053–9058. doi: 10.1021/bi0608818. [DOI] [PubMed] [Google Scholar]
- Renthal R, Brancaleon L, Peña I, Silva F, Chen LY. Interaction of a two-transmembrane-helix peptide with lipid bilayers and dodecyl sulfate micelles. Biophysical Chemistry. 2011;159(2–3):321–327. doi: 10.1016/j.bpc.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey M, O’Connell W, Cianciotto NP. Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infection and Immunity. 2001;69(7):4276–4286. doi: 10.1128/IAI.69.7.4276-4286.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ropero F, Fioroni M. Structural and dynamical analysis of an engineered FhuA channel protein embedded into a lipid bilayer or a detergent belt. Journal of Structural Biology. 2012;177(2):291–301. doi: 10.1016/j.jsb.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of Mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307(5713):1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Myers JK. Disease-related misassembly of membrane proteins. Annual Review of Biophysics and Biomolecular Structure. 2004;33(1):25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Sönnichsen F. Solution NMR of membrane proteins: practice and challenges. Magnetic Resonance in Chemistry. 2006;44(S1):S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- Sands ZA, Grottesi A, Sansom MSP. The intrinsic flexibility of the Kv voltage sensor and its implications for channel gating. Biophysical Journal. 2006;90(5):1598–1606. doi: 10.1529/biophysj.105.072199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schulz GE. The structure of bacterial outer membrane proteins. BBA-Reviews on Biomembranes. 2002;1565(2):308–317. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Feldmann RJ. Membrane proteins: amino acid sequence and membrane penetration. Journal of Molecular Biology. 1974;87(4):853–858. doi: 10.1016/0022-2836(74)90090-4. [DOI] [PubMed] [Google Scholar]
- Sipos L, Von Heijne G. Predicting the topology of eukaryotic membrane proteins. European Journal of Biochemistry. 1993;213(3):1333–1340. doi: 10.1111/j.1432-1033.1993.tb17885.x. [DOI] [PubMed] [Google Scholar]
- Tamm LK, Liang B. NMR of membrane proteins in solution. Progress in Nuclear Magnetic Resonance Spectroscopy. 2006;48(4):201–210. doi: 10.1016/j.pnmrs.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman DP, van der Spoel D, Berendsen HJC. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: micellar structure and chain relaxation. Journal of Physical Chemistry B. 2000;104(27):6380–6388. [Google Scholar]
- Ungar D, Hughson FM. SNARE protein structure and function. Annual Review of Cell and Developmental Biology. 2003;19(1):493–517. doi: 10.1146/annurev.cellbio.19.110701.155609. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P, Hansen DF, Kay LE. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(33):11766–11771. doi: 10.1073/pnas.0804221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren WF, Berendsen HJC. A leap-frog algorithm for stochastic dynamics. Molecular Simulation. 1988;1(3):173–185. [Google Scholar]
- van Gunsteren WF, Berendsen HJC. Computer simulation of molecular dynamics: methodology, applications, and perspectives in chemistry. Angewandte Chemie International Edition. 1990;29:992–1023. [Google Scholar]
- Vogt J, Schulz GE. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure. 1999;7(10):1301–1309. doi: 10.1016/s0969-2126(00)80063-5. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Control of topology and mode of assembly of a poly-topic membrane protein by positively charged residues. Nature. 1989;341(6241):456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Membrane-protein topology. Nature Reviews Molecular Cell Biology. 2006;7(12):909–918. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human β2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. Journal of the American Chemical Society. 2010;132(33):11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiological Reviews. 2005;85(4):1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annual Review of Biophysics and Biomolecular Structure. 1999;28(1):319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- White SH, Ladokhin AS, Jayasinghe S, Hristova K. How membranes shape protein structure. Journal of Biological Chemistry. 2001;276(35):32395–32398. doi: 10.1074/jbc.R100008200. [DOI] [PubMed] [Google Scholar]
- Williams T, Kelly C. [accessed 20.3.12];Gnuplot 4.4: In Interactive Plotting Program. 2012 http://gnuplot.sourceforge.net/
- Wymore T, Wong TC. Molecular dynamics study of substance P peptides partitioned in a sodium dodecylsulfate micelle. Biophysical Journal. 1999;76(3):1213–1227. doi: 10.1016/S0006-3495(99)77285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Soto CS, Honig B. Evaluating conformational free energies: the colony energy and its application to the problem of loop prediction. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(11):7432–7437. doi: 10.1073/pnas.102179699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim MA, Goh KI, Cusick ME, Barabási AL, Vidal M. Drug-target network. Nature Biotechnology. 2007;25(10):1119–1126. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Cierpicki T, Jimenez RHF, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Molecular Cell. 2008;31(6):896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.