

Abstract
Phenotypic high throughput screens are a valuable tool for identifying new chemical compounds with antimalarial activity. Traditionally, these screens have focused solely on the symptomatic asexual blood stage of the parasite's lifecycle; however, in order to discover new medicines for malaria's treatment and prevention, robust screening technologies against other parasite lifecycle stages are required. This review highlights recent advances and progress toward phenotypic screening methodologies over the past several years, with a focus on exoerythrocytic stage screens.
Keywords: malaria, drug discovery, phenotypic screens, high throughput screening
Current antimalarial therapies
While substantial advances have been made in its treatment and prevention, malaria continues to have a devastating global health and socioeconomic burden [1-3]. Over 200 million individuals are infected with Plasmodium parasites annually, resulting in nearly half a million deaths every year, of which the majority are young children [1]. These estimates place malaria among the three deadliest infectious diseases globally [4]. Five different parasite species are responsible for malaria infections in humans, with Plasmodium falciparum causing the most mortalities [1]. While not causing as much mortality as P. falciparum, Plasmodium vivax is the most widely distributed malaria parasite. Both P. vivax and P. ovale produce dormant forms, hypnozoites, that can reactivate, causing relapsing symptoms months to years after the initial infection [5].
Malaria parasites proceed through a complex, multistage lifecycle, involving development in both vertebrate and invertebrate hosts (Figure 1). Current antimalarial treatments rely primarily on small-molecule chemotherapies, targeting the intraerythrocytric (see Glossary) asexual blood stage [6]. Recent approval by the European Medicines Agency of GlaxoSmithKline's RTS,S vaccine has raised hopes that an effective and preventative malaria treatment is within reach [7]. RTS,S is affected by limited efficacy (18-36%) [8, 9], however, and immunity appears to quickly wane [9]. There are also questions about RTS,S's strain-specific immunity [8]. The recommended World Health Organization (WHO) standard of care for P. falciparum is treatment with small-molecule artemisinin combination therapies (ACTs). Recent reports of an increase in parasite clearance times by ACTs have raised concerns that resistance to artemisinin is beginning to emerge, underscoring the urgent need to identify new small molecule drug candidates with unique targets and mechanisms of action (MOA) [3, 10]. In this review, we will examine advancements made in high throughput screening technologies over the past few years, with an emphasis on exoerythrocytic stage screens.
Figure 1. Lifecycle of Plasmodium spp. parasites.
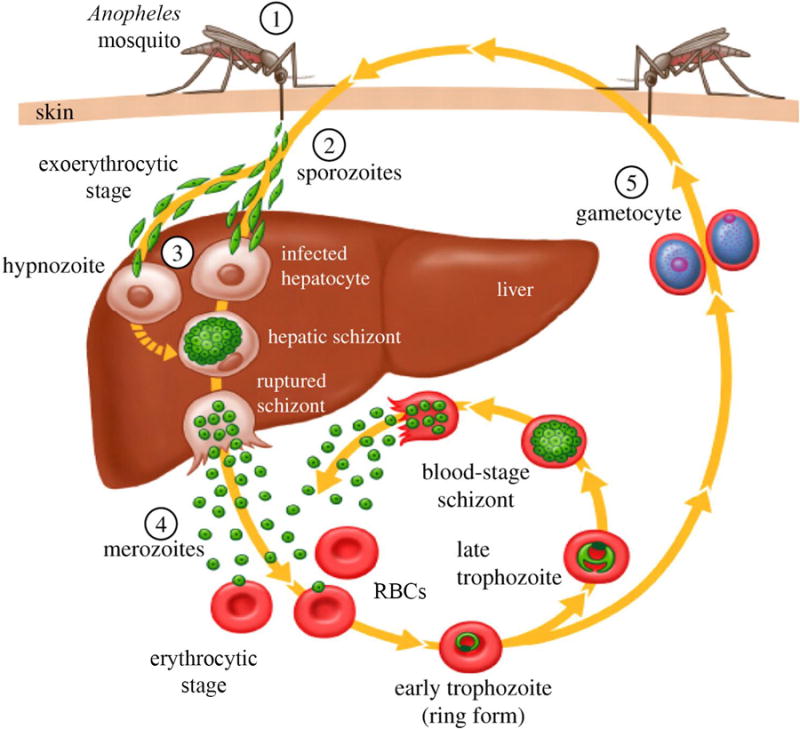
1) In the human host, infection is precipitated by the bite of an Anopheles spp. mosquito. 2) Sporozoites enter the bloodstream and quickly migrate to the liver. 3) In the liver, parasites undergo a replicative round within hepatic cells. The parasites then egress from the liver as merozoites, and rapidly invade circulating red blood cells (RBCs). For some species of Plasmodium, the liver stage parasites never fully mature, but rather transition into dormant forms of the parasite termed hypnozoites. These hypnozoites may re-activate months to years later, re-establishing productive infections in the human host. 4) Blood stage parasites proceed through several rounds of asexual replication. Each replication round culminates in the rupture of the host erythrocyte, leading to clinical symptoms in the host. The released merozoites then invade new RBCs, beginning the cycle over again. 5) A small percentage of blood stage merozoites leave the asexual replicative cycle, instead differentiating into male and female gametocytes. These sexual blood stage parasites are the only form of the parasite that can be transmitted from the human to the mosquito host. Figure reproduced from Hill et al [70].
Identifying new drug candidates
With the technological developments of the past few decades, the ability to search for new drug candidates has rapidly accelerated. Advances in robotic automation and liquid handling, coupled with the ever-shrinking scale at which these assays are performed have facilitated the ultra high throughput screening (HTS) of very large compound libraries [11]. Furthermore, the continued development of cheminformatics, which encompasses the ability to analyze, filter, and interpret the large datasets generated from these screens, has greatly contributed to the establishment of HTS technology as a staple in the drug discovery process [12]. The pharmaceutical industry has relied on two different methods of HTS. Phenotypic or whole-cell screens look for changes in phenotype caused by exposing whole cells or microorganisms to potential drug candidates. This is in contrast to target-based screens, which examine the effect of compounds on purified target proteins [13].
Phenotypic screens have several intrinsic advantages over target-based screens. Non-membrane permeable drugs are naturally eliminated from further consideration. The whole-cell nature of the assay provides the opportunity to capture drugs acting with a cooperative effect or on multiple targets. Lastly, and perhaps most importantly, previous knowledge of the drug target is not required to execute the screen [13, 14]. Removing the restrictions on the sampled biological space increases the probability of identifying first-in-class drugs operating through new mechanisms of action [13]. The potential to identify drugs acting on new targets is especially important in light of emerging antimalarial drug resistance to all front-line clinical compounds.
Additionally, global eradication of malaria from the human population is predicated on finding drug therapies that target all stages of the causal parasite's lifecycle. Ultra high throughput campaigns involving million compound libraries have previously been conducted with asexual blood stage parasites [15-17]; however, the screening capacity against some of the other Plasmodium lifecycle stages is orders of magnitude lower than the asexual blood stage (ABS) screens (Figure 2) [18, 19]. Only recently has the antimalarial drug discovery community begun to develop assays that are compatible with screening for activity against other lifecycle stages at the same throughput as blood stage screens [20, 21]. Recent advances in high throughput technologies for exoerythrocytic stage screening will be discussed below.
Figure 2. Capacity of phenotypic drug screening at the indicated malaria lifecycle stages.

Asexual blood stage screens require the least amount of labor and specialized materials and thus can be used to evaluate very large libraries. Assays at the top of the pyramid are very labor intensive and can only be applied to a small number of compounds [15-21,42].
Previous asexual blood stage screens
Of all of the lifecycle stages, ABS P. falciparum parasites are the most easily maintained in vitro. Furthermore, since ABS parasites are maintained in suspensions of cultured erythrocytes, they are highly compatible with automated liquid handling procedures, making the ABS stage extremely amenable to HTS technologies [22]. As such, it is perhaps unsurprising that the first ultra high throughput screen looking for antimalarial activity was performed against asexual blood stage parasites [15]. This screen took advantage of the fact that erythrocytes naturally lack nuclei; a nucleic acid intercalating dye was used to specifically stain parasitic DNA, and any detected fluorescent signal was then attributed to parasite proliferation. Several subsequent high throughput ABS screens have been performed, using similar nucleic acid staining techniques [17, 23, 24]. Enzymatic methods, where parasite viability is linked to enzyme activity, have also been employed [16]. The results of these first ultra high throughput antimalarial screens are published in the public domain (www.ebi.ac.uk/chemblntd). Several compounds identified from these screens have been evaluated and optimized in hit-to-lead programs, with a few advancing into clinical trials. The spiroindolone KAE609, thought to target P-type cation-transporter ATPase4 (PfATP4, reviewed in [25]), works rapidly with a median parasite clearance half life of 0.9 hours [26]. In contrast, less than <1% of P. falciparum malaria patients treated with oral artesunate showed a parasite clearance half-life of less than 1 hour [26]. This molecule is now in further clinical trials, and has the potential to be the first new scaffold class to be introduced into the antimalarial chemical space in over 20 years [27].
The promising results of KAE609 highlight the utility and power of existing high throughput screening methodologies; however, creative new assays are needed to continue to successfully identify potent antimalarial drug candidates acting on novel targets and through previously undiscovered mechanisms. One such assay was proposed by Linares et al [28]. In light of emerging resistance to fast-acting artemisinin, the authors proposed a method to screen for rapidly parasiticidal compounds, positing that parasite proliferation and metabolism measurements, which are the traditional detection mechanisms for ABS screens, are insufficient for determining the killing kinetics of a drug. Instead, they established a method to screen for the ability of parasites to invade erythrocytes and re-establish infections. The invasion assay was performed by incubating P. falciparum ABS parasites with compounds of interest, followed by their sequential removal, and a second incubation with fresh, dye-labeled erythrocytes. Productive invasions were then identified by performing two-color flow cytometry to detect dye-labeled erythrocytes containing stained parasites. Using this assay, the authors were able to distinguish between antimalarial drugs displaying rapid, artemisinin-like killing kinetics and those with a slower onset of action. This medium throughput assay (96 well assay format) is dependent on recent advances in high capacity flow cytometry technologies [29] and, as reported, only gives parasite killing at 24 hour intervals [28]. Assays using genetically modified parasites bearing a luciferase reporter give similar rate of kill kinetics with less effort, although at the expense of relying on a recombinant parasite line [30].
Asexual blood stage apicoplast screens
The apicoplast (Box 1) is a relict plastid found in malaria parasites that is essential for parasite survival, and it presents a potential source of new, druggable targets. In blood stage parasites, drugs targeting apicoplast housekeeping processes produce a death-phenotype with unusual kinetics; parasites continue to develop normally through a single 48-hour lifecycle, however, the resultant progeny fail to develop and eventually die [31, 32]. This phenomenon is termed “delayed death”, since parasite growth inhibition occurs in the second generation. Although delayed death inhibitors might not give rapid symptomatic relief, they could be very useful in combination therapies, to prevent survival of parasites that are resistant to the co-administered, fast-acting drug. Ekland and coworkers capitalized on this unique phenotype in an attempt to screen for compounds acting on drug targets in the apicoplast [33]. Using a luciferase-based parasite viability assay, they examined the effect of drugs at 48 and 96 hour time points. Compounds that exhibited potency at 96 hours, were considered to potentially be acting through an apicoplast-mediated delayed death mechanism.
Box 1. Phenotypic screens to identify apicoplast-targeting compounds.
The apicoplast is an organelle which contains its own circular genome, and encodes approximately 35 proteins; however, the majority of apicoplast-proteins are encoded by the host-nucleus. The four main metabolic processes attributed to the apicoplast are isoprenoid precursor synthesis, fatty acid synthesis, heme synthesis, and iron-sulfur cluster biogenesis. Additionally, the apicoplast executes the standard housekeeping functions (genome replication, transcription and translation, post-translational modification, and protein homeostasis) associated with cellular maintenance [31, 32]. Screening campaigns to identify apicoplast-targeting drugs represent a unique niche in the malaria drug discovery landscape. Since they are performed against the entire P. falciparum parasite, they are still considered phenotypic screens; however, the methodologies employed are tailored to search for drugs acting against a specific organelle within the parasite. This makes these screens a hybrid between phenotypic and target-based techniques. A potential benefit to this approach is the narrowing in the amount of possible molecular targets, which could simplify any subsequent target identification.
This assay was a first step towards successfully identifying apicoplast-targeting compounds; however, since the assay only searches for compounds exhibiting a delayed death mechanism, thus presumably targeting apicoplast general housekeeping pathways, it cannot identify drugs acting on other metabolic pathways carried out in the apicoplast. Furthermore, the generation of transfected lines is not trivial and does not lend itself to easily testing other parasite strains besides the canonical 3D7 and Dd2 (which would be important in the case of combating artemisinin resistance). It has previously been shown that isoprenoid precursor biosynthesis is the only essential function of the apicoplast during blood stage infections. Simply supplementing the isoprenoid precursor isopentenyl pyrophosphate (IPP) into the growth media of cultured parasites is sufficient to reverse the growth inhibition effects of known apicoplast-targeting inhibitors [34]. This IPP chemical rescue phenomenon was used to directly screen for compounds acting on the apicoplast [28, 35]. After the addition of test compounds, parasites were incubated in the presence or the absence of IPP. Drugs that lost growth inhibition abilities upon addition of IPP were considered to be apicoplast inhibitors. This assay more directly searches for compounds acting against the apicoplast. Using this methodology, a chemical screen was executed against the Medicines for Malaria Venture (MMV) Malaria Box, a publically available library with previously demonstrated asexual blood stage activity. From this screen, the candidate compound MMV08138 was identified. MMV08138 appears to target IspD, an enzyme in the apicoplast's metabolic pathways [36]. While this chemical rescue assay holds promise for identifying apicoplast-targeting drugs with new targets and mechanisms of action, the number of replicates required, and the use of expensive reagents make this assay cost prohibitive for high throughput screening campaigns of large libraries.
Synergy screens
Any new antimalarial drug that is developed will ultimately be delivered as a combination therapy to delay the likely emergence of parasite resistance. Compounds in combination should ideally act against different cellular targets to offset the likelihood that a single parasite genome amplification event could render parasites resistant to both drugs. To identify compounds that might act synergistically, Mott et al. performed an ABS screen using a small library of 2317 compounds including known and investigational antimalarial compounds [37]. They then performed eleven iterative combination screens, with compounds selected from the single agent screen, identifying compounds pairs that act antagonistically or synergistically, such as the artemisinin and the phosphatidylinositide 3-kinase inhibitors, NVP-BGT226. Although combination assays are likely to inform how drugs are paired with one another, properties that are introduced during medicinal chemistry optimization (e.g. pharmacokinetics/pharmacodynamics/toxicity/delivery) tend to be the most important features in designing combination therapies. In this respect, co-screening of compounds may not be as useful before hit to lead optimization has been performed. On the other hand, the approach could provide details on a compound's mechanism of action—compounds acting against the same pathway or target (e.g. the cytochrome bc1 inhibitors, atovaquone and decoquinate) tend to synergize [37].
Liver stage screens
Initial HTS efforts were heavily focused on the asexual blood stage, however, there is increasing interest in identifying drugs with activity at different or multiple stages of the parasite lifecycle [38]. The liver stage is particularly attractive, since the number of parasites infecting the host at that point is orders of magnitude lower than in the asexual blood stage [5]. Furthermore, targeting parasites in the liver stage presents unique opportunities for discovering prophylactic medicines (e.g. medicines that will protect the user from being infected in the first place). The ability to establish and maintain in vitro Plasmodium sporozoite infections in hepatic cells was a key breakthrough for studying liver stage parasites [39, 40]. Using this methodology, the first in vitro liver stage screens were performed with antibody staining of malaria parasites in hepatic cells, followed by infrared [41] or high content imaging (HCI) [42]. The latter method was used to screen ∼4000 compounds comprised of positives from a previous 1.7 million compound ABS screen. From this screen, the imidazolopiperazine scaffolds were identified, and the optimized lead compound KAF156 was developed [43, 44]. Resistance to KAF156, is mediated by mutations in the P. falciparum cyclic amine resistance locus (PfCARL [45]), an uncharacterized transmembrane protein responsible for resistance to unrelated compounds [46]. KAF156's mechanism of action remains obscure although it has also shown good blood stage potency, and is currently in clinical trials [47].
While antibody staining has seen success in HTS screening campaigns, ultimately, the use of antibodies is less than ideal as it requires several liquid handling steps. Additionally, the high cost of antibodies is prohibitive for sustainably screening large compound libraries. An alternative to staining parasites with fluorescent antibodies is to use transgenic parasites that stably express a reporter protein. Cruz et al used a transgenic Plasmodium berghei parasite line expressing green fluorescent protein (GFP), coupled with high content imaging to screen a commercially available compound library [48]. While use of a fluorescent reporter protein eliminated the need for antibodies, the requirement for high content imaging is still a rate-limiting step in the screening process. Another step toward an even more robust and higher throughput liver stage screen was realized in a report by Derbyshire et al [49]. Their assay utilized a luciferase-expressing transgenic P. berghei line, where luciferase activity was correlated to parasite viability. Whole-well luminescence readout after incubation with drugs was used to determine the drug compounds' effect. This technology was used to screen the large, asexual blood stage active Tres Cantos Antimalarial Compound Set (TCAMS) from GlaxoSmithKline [16], and led to the identification of several candidate compounds with potent liver stage activity [50]. An ultra high throughput version of this screen was recently reported, and used to screen the MMV Malaria Box, as well as the Broad Diversity-Oriented Synthesis (DOS) Library for liver stage activity [20]. Several of the potent compounds identified from the MMV Malaria Box had been shown previously to possess exoerythrocytic activity. Screening of the unbiased Broad DOS library identified several scaffolds with liver stage activity, and interestingly, a handful of those compounds did not have asexual blood stage activity. This work illustrates the importance of continuing to develop HTS techniques for exoerythrocytic lifecycle stages; as these screening methodologies reach the same throughput as ABS screens, comparison of a drug's activity across all the lifecycle stages will be possible. This will not only enable the identification of compounds with activity across multiple lifecycle stages, but will also discover drugs active solely against exoerythrocytic stages. Compounds lacking blood stage activity hold great promise in prophylactic and radical cure treatments, as well as potentially deterring or completely avoiding the acquirement of drug resistance. Furthermore, if these compounds could be formulated to possess extended clearance times, it could be imagined that these prophylactic treatments could be implemented as “chemical vaccines”. A minor drawback is that all high throughput liver assays depend on the use of rodent malaria parasites. False negatives and false positives may arise because rodent parasites may not show the same response as human parasites to treatment with a given compound.
While these assays have significantly improved the ability to screen large compound libraries for activity against liver stage malaria parasites, new innovative methods are still needed to increase the throughput of these assays. Continued miniaturization to make these methods compatible with ultra high throughput 1536-well microwell plate format is a promising starting point. Additionally, another screening bottleneck in liver stage assays is their dependence on a supply of freshly harvested sporozoites from live mosquitoes. Methods to accelerate or even completely bypass this step could drastically improve the current capacity of liver stage screens.
Hypnozoite ‘radical cure’ screens
While the five species of malaria that infect humans share a similar lifecycle, P. vivax and Plasmodium ovale parasites do not exclusively egress from the liver into the blood stage, but can also transition into dormant forms called hypnozoites. These hypnozoites can reactivate, through mechanisms that are not currently well understood, months to years later, causing a recrudescence of symptoms in the absence of a new infection [5, 51]. If complete malaria eradication is to be accomplished, effective drugs are needed to clear these latent parasite reservoirs. Primaquine is currently the only clinically approved drug that can eliminate these latent forms of the parasite. Unfortunately, primaquine causes acute hemolysis in patients with glucose-6-phosphate dehydrogenase deficiency (G6PD), a common genetic disorder in malaria endemic regions, and has a short in vivo half-life [5, 51, 52]. There is also growing concern over the development of primaquine resistance [51, 53], underscoring the continued need to search for new drugs that can completely clear latent forms of the malaria parasite from human hosts, and thus provide a radical cure.
Robust assays to identify compounds that are active against dormant hypnozoites have remained elusive. The first screening campaign that attempted to identify drugs with radical curative properties was conducted using in vivo methodologies in the 1940s [54]. In this screen, hypnozoite-forming simian Plasmodium cynomolgi sporozoites where used to infect rhesus monkeys, providing a model of hypnozoite infections. Compounds that prevented relapse in malaria-infected monkeys were considered to be active against hypnozoites. While this screen successfully identified primaquine as a hypnozoiticidal drug, assays of this nature would, nowadays, be hindered by their low throughput and ethical concerns.
A significant step toward a hynpozoite high throughput screen was made with the report of an in vitro model of hypnozoite infection, which utilized P. cynomolgi sporozoites to infect primary monkey hepatocytes [55]. The authors observed a distinct, non-dividing population of P. cynomolgi parasites that were resistant to all antimalarial drugs, except for hypnozoiticidal primaquine. This suggested that these uninucleate “small forms” were, in fact, the quiescent hypnozoites. This assay was modified for compatibility with microwell plates, and a low throughput screen was then performed against a focused panel of compounds already possessing blood and/or liver-stage activity. This small screen identified the imidazopyrazine KAI407 as having hypnozoiticidal activity [18]. KAI407 and the related KDU691 scaffold are the first non-8-aminoquinoline compounds identified that potentially possesses the ability to clear quiescent hypnozoites [18, 56].
A limitation of this in vitro assay is that it only screens for compounds that are active against the developing hypnozoite. Since true relapsing malaria occurs from the reactivation of dormant hypnozoites over a timeframe of weeks to months after the initial infection, an assay that monitors activity against established, mature hypnozoites over longer periods of time is desired. A recently proposed assay by Dembélé et al makes significant strides toward this goal [57]. In this report, cultures of P. cynomolgi infected primary hepatic cells were established and maintained in vitro for over 40 days. Furthermore, it was shown that over the course of these long-term cultures, small form non-dividing parasites reactivate, and are functionally identical to hypnozoites. While these results represent an important addition to the toolbox of methods for studying P. vivax and hypnozoite biology, significant advances are still needed in order to apply this technology to a high throughput screening endeavor.
Thus far, all hypnozoite screens have been performed in primary hepatic cells. Growth and maintenance of these cells is labor intensive and requires specialized cell-substrate materials, rendering the assay incompatible with true high throughput screening efforts [58]. Because there are no known radical cure targets that could be used in biochemical screens, the search for new radical cure drugs will remain dependent on phenotypic screens and a supply of sporozoites harvested from live mosquitoes. Compounding this issue are the traditionally low sporozoite infection rates of primary hepatic cells [5]. Improvements in these areas are necessary to perform a high throughput-compatible screen for compounds with hypnozoiticidal activity. A bright spot has been the development of human liver-chimeric (huHep) FRG knockout mice that can be used as a model for P. vivax infection [59]. The use of these chimeric mice may allow researchers to develop activity profiles for possible radical cure drugs.
Sexual blood stage and transmission screens
Gametocytogenesis, the transition of asexual blood stage parasites into their sexual forms, is necessary for successful transmission of the parasite from the human to the mosquito host. The process occurs after a limited number of asexual parasites undergo sexual differentiation. The resultant gametocytes remain in the erythrocyte, and progress through five distinct stages (stages I to V). Only mature stage V gametocytes are able to survive uptake and incubation in the mosquito vector, making these late, sexual blood stage forms of the parasite a key step in productive transmission [60]. In addition, stage V gametocytes may circulate for weeks in humans, making them more abundant and attractive targets than, for example, exflagellating male gametes, which may exist for only minutes. Because of this, gametocytocidal activity against late stage V gametocytes is likely to be the best indicator of whether a compound series will exhibit transmission-blocking activity in a clinical setting where drug exposure may not be consistent. Primaquine is the only drug currently approved that displays radical transmission-blocking abilities [5]. Unfortunately, as discussed previously, primaquine is plagued by safety concerns.
A challenge with gametocyte assays is that DNA replication is stalled and only a single gametocyte is produced during gametocytogenesis, which precludes the use of monitoring DNA or cell proliferation markers. However, over the last several years, several methods for assessing activity against sexual stage gametocytes have been implemented. These methods include the use of metabolic indicators [60-63], antibody detection of stage-specific markers [64], and genetically engineered parasites lines with gametocyte-specific reporters [60, 65, 66]. One such recent assay was developed by Plouffe et al and aimed to assess stage V gametocyte activity using cost effective methods that were compatible with high throughput screening techniques [21]. Sexual blood stage gametocytes were stained with a dye that only produces a signal in the presence of mitochondria with an active membrane potential, an indicator of parasite viability. A fluorescent signal, measured by high-content imaging, was then correlated to viable gametocytes, with less signal indicating parasite death.
A key step in this assay was using highly synchronized parasites for the subsequent production of gametocytes. This tight synchronization produced gametocytes at each of the five stages in very high purities, rather than mixed populations of several different sexual stages [21]. Another key step in this assay was the addition of an in situ red blood cell saponin-lysis step before the addition of the mitochondrial-labeling dye. By lysing the RBCs directly in the well, a second liquid transfer step is eliminated. This facilitates compound exposure and imaging in the same assay plate, increasing the screening throughput and making the assay amenable to ultra HTS efforts. This Saponin-lysis Sexual Stage Assay (SaLSSA) was used to screen three different compound libraries. A general trend was observed in that compounds displaying activity in asexual blood stage or early stage I-III gametocytes appeared to lose efficacy in later gametoctye stages IV and V. Compounds active against stage V gametocytes showed good correlation with activity in complementary transmission block studies, indicating that SaLSSA is a HTS compatible alternative to identify compounds with transmission-blocking potential.
An alternative approach to monitoring late stage gametocyte viability as a predictor of transmission-blocking potential was proposed by Lucatoni et al [67]. They used high-content imaging, coupled with a custom computer script to monitor the morphological change of elongated stage V gametocytes into spherical gametes. The gametocyte to gamete transition, or ‘rounding up’ events, provided a functional readout of gametocyte activation, which is a necessary step for successful transmission to occur. This assay capitalizes on the physically distinct morphologies of parasites at different developmental stages (e.g. round, versus elongated). A nucleic acid intercalating dye, acridine orange (AO) was used to stain the parasites for HCI visualization. The MMV malaria box was screened using the acridine orange gamete (AO-GMT) assay, and the assay successfully identified previously reported inhibitors of both gametocytes and female gametes. A key strength of this assay is the ability to visualize both gametocytes and gametes in the same well. This allows for the identification and potential differentiation of transmission-blocking compounds acting through a gametocytocidal mechanism from those acting through a sterilizing mechanism of action.
While these types of screens show good promise for identifying compounds with transmission-blocking potential, they can be prone to false negatives since they only capture drugs affecting gametocyte viability, or the initial stages of gamete formation. While these metrics are good indicators of transmission block potential, they do not represent a direct measurement of transmission reducing activity, and therefore, many candidates may be missed which could successfully be employed in transmission control. For this reason, the standard membrane feeding assay (SFMA) remains the current biological gold standard for assessing transmission reducing activity of a drug candidate [68]. In the traditional SFMA, mature stage V gametoctyes are incubated with compounds of interest for 24 hours. The compounds are then fed to Anopheles spp. mosquitoes through an artificial membrane. One week following the blood meal, mosquito midguts are dissected, and the rate of parasite infection is determined by counting the number of oocysts using light microscopy.
No truly high throughput methodology exists for SFMA; however, a recent study reported a higher capacity, semi-automated approach [19]. This assay relied on an engineered P. falciparum strain expressing a firefly luciferase reporter protein. Mosquitoes were fed compounds through a blood meal, as in traditional SFMAs, and following the blood meal, the mosquitoes were frozen to increase flexibility in timing of the assay. Direct mechanical disruption and homogenization of whole mosquitoes played a key role in increasing the screening capacity of this assay, as it eliminated the need for manual mosquito midgut dissections. Following mechanical disruption, establishment of infections in mosquito vectors was determined by luminescent readout, which replaced more subjective, microscopy-based oocyst counts. This semi-automated SFMA assay was used to screen a panel of known antimalarial compounds. Results from the screen showed that the assay was able to identify several compounds with transmission-blocking activity that had been missed by gametocytocidal and gametogenesis assays. The SFMA is intrinsically low-throughput, due to its reliance on live mosquito vector feedings, and does not lend itself to HTS technologies. While this assay is still not amenable to screening large compound libraries, it represents an important step toward increasing the robustness and throughput of SFMAs. It is envisioned that this approach could be used as a high capacity validation method of compounds identified by other transmission blocking screens, or even to screen smaller, focused libraries of compounds.
Concluding remarks
The promising results of new antimalarial candidates identified from phenotypic screens highlights the power of these methods; however, significant questions about the future direction of these screening technologies exist (see Outstanding Questions). While these types of screens will continue to be an effective tool for identifying new antimalarial compounds, it is also expected that they will prove to be a rich source of new, druggable targets. Although antimalarial drug screens have historically focused on the asexual blood stage, the field is currently witnessing a shift from that paradigm, and new, innovative techniques are emerging to identify compounds acting on exoerythrocytic stages of the parasite lifecycle. So far, these screens have largely been implemented to test compounds already known to possess blood stage activity; however, it is envisioned that drugs which do not possess ABS activity could be of even greater value, since the opportunity to develop resistance against these molecules is much lower [69]. Furthermore, the liver stage and host transmission stages represent important bottlenecks in the parasite lifecycle, and compounds targeting these stages will be a necessary addition to the repertoire of antimalarial drugs if global malaria eradication is to be achieved.
Trends Box.
The field of antimalarial drug discovery has seen a shift to phenotypic screening from target-based screening.
The goal of achieving complete malaria eradication has prompted the development of new and higher throughput screening methods to look for drugs that are active in exoerythrocytic and transmission stages.
The success of high throughput screening is founded on the implementation of robust, scalable, reproducible, and inexpensive assays.
Phenotypic screens are not only a rich source for identifying potent drug candidates, but also for discovering new potential drug targets.
Outstanding Questions.
Is there limited chemical diversity in the compound libraries that have been and are currently being employed in high throughput drug discovery campaigns, and can new sources be implemented to expand the sampled chemical space and find new, potent chemotypes?
Is activity against liver stage hypnozoites of Plasmodium vivax predictive of a radical cure?
Can these screening methodologies be more broadly applied to drug development for other parasitic diseases?
Will there be a return to target based drug discovery once phenotypic screening has helped to identify the complete set of druggable targets?
Can narrowly-focused screens that selectively look at a transient and specific period in the parasite's lifecycle (i.e. invasion, fertilization, or egress) translate into effective drugs?
Acknowledgments
The authors would like to thank Dr. Gregory LaMonte for helpful input and Dr. Stephan Meister for graphical suggestions.
Glossary
- Eradication
The complete, global elimination of malaria parasites from the human population.
- First-in-class drugs
Drugs that act against previously unprecedented targets or through new mechanisms of action.
- High content imaging
A process by which automated fluorescence microscopy images are collected and then analyzed by advanced computer scripts to identify metrics such as spatial or morphological characteristics.
- High throughput screening
A process in the field of drug discovery by which large numbers of compounds are interrogated for their biological activity. It generally implements small test volumes, automation, and advanced computation.
- Hit-to-lead
A process in early stage drug discovery programs where an initial hit obtained from a primary screen is evaluated and optimized into a promising lead compound or a series of structurally related compounds.
- Intraerythrocytic
The asexual blood stage of the malaria parasite.
- Mechanism of action
The manner in which a drug elicits its pharmacological effect.
- Radical cure
The complete elimination of all forms of the Plasmodium parasite, including latent hypnozoites, from the human host.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.World Health Organization. World malaria report. Geneva, Switzerland: World Health Organization; 2015. [Google Scholar]
- 2.Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 3.Wells TN, et al. Malaria medicines: a glass half full? Nature Reviews Drug Discovery. 2015;14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 4.Bourzac K. Infectious disease: beating the big three. Nature. 2014;507:S4–S7. doi: 10.1038/507s4a. [DOI] [PubMed] [Google Scholar]
- 5.Campo B, et al. Killing the hypnozoite–drug discovery approaches to prevent relapse in Plasmodium vivax. Pathogens and global health. 2015;109:107–122. doi: 10.1179/2047773215Y.0000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrows JN, et al. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12 doi: 10.1186/1475-2875-12-187. doi:10.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morrison C. Landmark green light for Mosquirix malaria vaccine. Nature biotechnology. 2015;33:1015–1016. doi: 10.1038/nbt1015-1015. [DOI] [PubMed] [Google Scholar]
- 8.Neafsey DE, et al. Genetic Diversity and Protective Efficacy of the RTS,S/AS01 Malaria Vaccine. N Engl J Med. 2015;373:2025–2037. doi: 10.1056/NEJMoa1505819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rts S. Efficacy and safety of RTS, S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. The Lancet. 2015;386:31–45. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashley EA, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lackovic K, et al. A Perspective on 10-Years HTS Experience at the Walter and Eliza Hall Institute of Medical Research–Eighteen Million Assays and Counting. Combinatorial chemistry & high throughput screening. 2014;17:241–252. doi: 10.2174/1386207317666140109122450. [DOI] [PubMed] [Google Scholar]
- 12.Duffy BC, et al. Early phase drug discovery: cheminformatics and computational techniques in identifying lead series. Bioorganic & medicinal chemistry. 2012;20:5324–5342. doi: 10.1016/j.bmc.2012.04.062. [DOI] [PubMed] [Google Scholar]
- 13.Swinney D. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin Pharmacol Ther. 2013;93:299–301. doi: 10.1038/clpt.2012.236. [DOI] [PubMed] [Google Scholar]
- 14.Katsuno K, et al. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nature Reviews Drug Discovery. 2015;14:751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 15.Plouffe D, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gamo FJ, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 17.Guiguemde WA, et al. Chemical genetics of Plasmodium falciparum. Nature. 2010;465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeeman AM, et al. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob Agents Chemother. 2014;58:1586–1595. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vos MW, et al. A semi-automated luminescence based standard membrane feeding assay identifies novel small molecules that inhibit transmission of malaria parasites by mosquitoes. Scientific reports. 2015;5 doi: 10.1038/srep18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swann J, et al. High-Throughput Luciferase-Based Assay for the Discovery of Therapeutics That Prevent Malaria. ACS Infectious Diseases. 2016 doi: 10.1021/acsinfecdis.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Plouffe DM, et al. High-Throughput Assay and Discovery of Small Molecules that Interrupt Malaria Transmission. Cell Host Microbe. 2016;19:114–126. doi: 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 23.Avery VM, et al. Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum. Malar J. 2014;13:190. doi: 10.1186/1475-2875-13-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baragana B, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist. 2015;5:149–162. doi: 10.1016/j.ijpddr.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White NJ, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med. 2014;371:403–410. doi: 10.1056/NEJMoa1315860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flannery EL, et al. Antimalarial drug discovery - approaches and progress towards new medicines. Nat Rev Microbiol. 2013;11:849–862. doi: 10.1038/nrmicro3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linares M, et al. Identifying rapidly parasiticidal anti-malarial drugs using a simple and reliable in vitro parasite viability fast assay. Malaria journal. 2015;14:1–8. doi: 10.1186/s12936-015-0962-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Black CB, et al. Cell-based screening using high-throughput flow cytometry. Assay and drug development technologies. 2011;9:13–20. doi: 10.1089/adt.2010.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasenkamp S, et al. Evaluation of bioluminescence-based assays of anti-malarial drug activity. Malar J. 2013;12:58. doi: 10.1186/1475-2875-12-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman CD, McFadden GI. Targeting apicoplasts in malaria parasites. Expert opinion on therapeutic targets. 2013;17:167–177. doi: 10.1517/14728222.2013.739158. [DOI] [PubMed] [Google Scholar]
- 32.MacRae JI, et al. The apicoplast: a key target to cure malaria. Current pharmaceutical design. 2012;18:3490–3504. [PubMed] [Google Scholar]
- 33.Ekland EH, et al. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9:e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowman JD, et al. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box. Antimicrobial agents and chemotherapy. 2014;58:811–819. doi: 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu W, et al. A chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis. Antimicrob Agents Chemother. 2015;59:356–364. doi: 10.1128/AAC.03342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mott BT, et al. High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Sci Rep. 2015;5:13891. doi: 10.1038/srep13891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alonso PL, et al. A Research Agenda to Underpin Malaria Eradication. PLoS Med. 2011;8:e1000406. doi: 10.1371/journal.pmed.1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuster FL. Cultivation of Plasmodium spp. Clin Microbiol Rev. 2002;15:355–364. doi: 10.1128/CMR.15.3.355-364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollingdale MR, et al. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in irradiated hepatoma cells. Am J Trop Med Hyg. 1985;34:21–23. doi: 10.4269/ajtmh.1985.34.21. [DOI] [PubMed] [Google Scholar]
- 41.Gego A, et al. New approach for high-throughput screening of drug activity on Plasmodium liver stages. Antimicrob Agents Chemother. 2006;50:1586–1589. doi: 10.1128/AAC.50.4.1586-1589.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meister S, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science. 2011;334:1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T, et al. Imidazolopiperazines: hit to lead optimization of new antimalarial agents. J Med Chem. 2011;54:5116–5130. doi: 10.1021/jm2003359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagle A, et al. Imidazolopiperazines: lead optimization of the second-generation antimalarial agents. J Med Chem. 2012;55:4244–4273. doi: 10.1021/jm300041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhen KL, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58:5060–5067. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magistrado PA, et al. Plasmodium falciparum Cyclic Amine Resistance Locus, PfCARL: a Resistance Mechanism for Two Distinct Compound Classes. ACS Infectious Diseases. 2016 doi: 10.1021/acsinfecdis.6b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leong FJ, et al. A First-in-Human Randomized, Double-Blind, Placebo-Controlled, Single- and Multiple-Ascending Oral Dose Study of Novel Imidazolopiperazine KAF156 To Assess Its Safety, Tolerability, and Pharmacokinetics in Healthy Adult Volunteers. Antimicrob Agents Chemother. 2014;58:6437–6443. doi: 10.1128/AAC.03478-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.da Cruz FP, et al. Drug Screen Targeted at Plasmodium Liver Stages Identifies a Potent Multistage Antimalarial Drug. Journal of Infectious Diseases. 2012;205:1278–1286. doi: 10.1093/infdis/jis184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derbyshire ER, et al. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci U S A. 2012;109:8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raphemot R, et al. Discovery of Dual Stage Malaria Inhibitors with New Targets. Antimicrobial agents and chemotherapy, AAC. 2015:02110–02115. doi: 10.1128/AAC.02110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernando D, et al. Primaquine in vivax malaria: an update and review on management issues. Malar J. 2011;10:351. doi: 10.1186/1475-2875-10-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rajapakse S, et al. Tafenoquine for preventing relapse in people with Plasmodium vivax malaria. Cochrane Database of Systematic Reviews. 2015;4:CD010458. doi: 10.1002/14651858.CD010458.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goller JL, et al. Regional differences in the response of Plasmodium vivax malaria to primaquine as anti-relapse therapy. Am J Trop Med Hyg. 2007;76:203–207. [PubMed] [Google Scholar]
- 54.Schmidt L, et al. Radical cure of infections with Plasmodium cynomolgi: a function of total 8-aminoquinoline dose. American journal of tropical medicine and hygiene. 1977;26:1116–1128. doi: 10.4269/ajtmh.1977.26.1116. [DOI] [PubMed] [Google Scholar]
- 55.Dembele L, et al. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One. 2011;6:e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNamara CW, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504:248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dembélé L, et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nature medicine. 2014;20:307–312. doi: 10.1038/nm.3461. [DOI] [PubMed] [Google Scholar]
- 58.Lin C, et al. The application of engineered liver tissues for novel drug discovery. Expert opinion on drug discovery. 2015;10:519–540. doi: 10.1517/17460441.2015.1032241. [DOI] [PubMed] [Google Scholar]
- 59.Mikolajczak SA, et al. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe. 2015;17:526–535. doi: 10.1016/j.chom.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duffy S, Avery VM. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J. 2013;12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Almela MJ, et al. A New Set of Chemical Starting Points with Plasmodium falciparum Transmission-Blocking Potential for Antimalarial Drug Discovery. PLoS One. 2015;10:e0135139. doi: 10.1371/journal.pone.0135139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D'Alessandro S, et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013;68:2048–2058. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 63.Tanaka TQ, et al. A quantitative high throughput assay for identifying gametocytocidal compounds. Mol Biochem Parasitol. 2013;188:20–25. doi: 10.1016/j.molbiopara.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miguel-Blanco C, et al. Imaging-based high-throughput screening assay to identify new molecules with transmission-blocking potential against Plasmodium falciparum female gamete formation. Antimicrob Agents Chemother. 2015;59:3298–3305. doi: 10.1128/AAC.04684-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peatey CL, et al. A high-throughput assay for the identification of drugs against late-stage Plasmodium falciparum gametocytes. Mol Biochem Parasitol. 2011;180:127–131. doi: 10.1016/j.molbiopara.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Adjalley SH, et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci U S A. 2011;108:E1214–1223. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lucantoni L, et al. A simple and predictive phenotypic High Content Imaging assay for Plasmodium falciparum mature gametocytes to identify malaria transmission blocking compounds. Sci Rep. 2015;5:16414. doi: 10.1038/srep16414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Kolk M, et al. Evaluation of the standard membrane feeding assay (SMFA) for the determination of malaria transmission-reducing activity using empirical data. Parasitology. 2005;130:13–22. doi: 10.1017/s0031182004006067. [DOI] [PubMed] [Google Scholar]
- 69.Sinden RE. The cell biology of malaria infection of mosquito: advances and opportunities. Cell Microbiol. 2015;17:451–466. doi: 10.1111/cmi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hill AV. Vaccines against malaria. Philos Trans R Soc Lond B Biol Sci. 2011;366:2806–2814. doi: 10.1098/rstb.2011.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]