

Abstract
Heme oxygenase (HO) is a ubiquitous enzyme with key roles in inflammation, cell signaling, heme disposal, and iron acquisition. HO catalyzes the oxidative conversion of heme to biliverdin (BV) using a conserved histidine to coordinate the iron atom of bound heme. This His-heme interaction has been regarded as essential for enzyme activity, since His-to-Ala mutants fail to convert heme to biliverdin in vitro. We probed a panel of proximal His mutants of cyanobacterial, human, and plant HO enzymes using a live-cell activity assay based on heterologous co-expression in E. coli of each HO mutant and a fluorescent biliverdin biosensor. In contrast to in vitro studies with purified proteins, we observed that multiple HO mutants retained significant activity within the intracellular environment of bacteria. X-ray crystallographic structures of human HO1 H25R with bound heme and additional functional studies suggest that HO mutant activity inside these cells does not involve heme ligation by a proximal amino acid. Our study reveals unexpected plasticity in the active site binding interactions with heme that can enable HO activity within cells, suggests important contributions by the surrounding active site environment to HO catalysis, and can guide efforts to understand the evolution and divergence of HO function.
Graphical abstract
In vitro enzymology with purified proteins is the workhorse of biochemistry and conforms with Arthur Kornberg’s ‘commandment’ to “not waste clean thinking on dirty enzymes”.1 Nevertheless, enzymes function natively within the complex milieu of living cells, and it remains an exciting yet formidable challenge to test whether the functional properties of enzymes deduced in vitro with purified proteins are modified by interactions inside living cells.2
Heme oxygenase is a conserved and ubiquitous enzyme that catalyzes the oxygen-dependent cleavage of heme to produce the linear tetrapyrrole biliverdin with concomitant release of iron and CO. HO is expressed by diverse organisms to serve a variety of central biological roles, including iron scavenging, heme disposal, phytochrome chromophore production, cell signaling, oxidative stress relief, and the inflammatory response.3–5 The vast majority of HO enzymes adopt a unique and highly conserved α-helical fold and utilize cytochrome P450 reductase or reduced ferredoxin as the electron donor to catalyze cleavage of heme at the α-meso position to produce biliverdin IXα (Fig. 1A & 1B).6
Figure 1. Heme oxygenase structure and reaction.

(A) Structure of human HO1 (PDB 1N45) highlighting the α-helical protein fold (green), bound heme (red), and proximal His (orange). (B) HO activity converts heme to biliverdin (BV), which can then be covalently bound by infrared fluorescent protein (IFP, cyan) to generate the fluorescent IFP-BV (bound BV in violet). The structure of IFP-BV was modeled using the BV-bound X-ray structure of the D. radiodurans chromophore-binding domain (PDB 3S7O), from which IFP is derived.
Although a variety of protein side-chains (e.g. Cys, His, Tyr, Lys, and Met) are utilized within the binding pockets of distinct classes of heme-binding proteins to coordinate the iron atom of heme,7 all known heme oxygenases, including the non-canonical heme-degrading enzymes from S. aureus (IsdG/I)8 and M. tuberculosis (MhuD),9 use a strictly conserved His residue as the proximal heme ligand.6, 10 Coordination of iron by the imidazole side-chain of histidine has been regarded as an essential interaction for HO catalysis, based on the observation that His-to-Ala mutants fail to convert heme to biliverdin in vitro despite still binding heme11–14 and, in the case of HmuO from C. diphtheriae, catalyzing formation of the verdoheme intermediate.15 HO activity in Ala mutants is restored, however, upon addition of imidazole, which can partition into the active site cavity created by the His-to-Ala mutation and functionally recapitulate the His-heme interaction of the WT enzyme.13, 15, 16 Furthermore, Cys and Tyr mutants of the proximal heme ligand have also been reported to lack HO activity in vitro,14 despite the ability of these residues to serve as heme ligands in other types of hemoproteins.7 These observations have suggested that the His-heme interaction is unique in its ability to support HO activity and is thus required for the conversion of heme to biliverdin within the HO active site.
Herein we use an in-cell bacterial model system to probe the functional dependence of diverse HO enzymes on the proximal His-heme interaction and test whether residues other than His at the proximal heme ligand position can support HO catalysis under live-cell conditions. Our results indicate that several His mutants of diverse HO enzymes retain significant activity within live bacteria, and X-ray crystallographic structures of ligand-bound human HO1 H25R suggest that such activity does not involve stable ligation of heme by a proximal amino acid. Our findings reveal unexpected plasticity in the proximal interactions with bound heme that can enable HO activity in live cells and suggest important contributions by the surrounding active site environment to HO catalysis.
MATERIALS AND METHODS
Materials
Biliverdin IXα was purchased from Frontier Scientific Inc. (Logan, UT). DMSO, reduced NADPH, spinach ferredoxin, spinach ferredoxin NADP+ reductase, heme (hemin chloride), sodium ascorbate, and bovine liver catalase were purchased from Sigma (St. Louis, MO). IPTG was purchased from Gold Biotechnology Inc. (St. Louis, MO).
Cloning and Site-directed Mutagenesis
Cloning of SynHO1 (Synechocystis sp. 6803, NCBI P72849), HuHO1 (NCBI NP_002124.1) residues 1-265, AtHO1 (NCBI AEC07872.1) residues 55-282, and IFP into pET21d (C-terminal hexa-His tag), pET22b (C-terminal hexa-His tag), or pET28a (N- or C-terminal hexa-His tag) vectors was described in a previous publication.17 The indicated mutations were introduced into the HO proteins using QuikChange II site-directed mutagenesis (Agilent Technologies, Santa Clara, CA). The gene for E. coli catalase (katE, NCBI NC_000913.3) was cloned by PCR from Top10 bacteria, and the PCR product was ligated into the NdeI/KpnI sites of a modified pBAD vector (chloramphenicol)18 in frame with an N-terminal hexa-His tag. Genes for E. coli flavodoxin (Genbank M59426) and flavodoxin NADP+ reductase (Uniprot 28861) were cloned by PCR from Top10 bacteria, and the PCR products were ligated into the NcoI/XhoI sites of pET21d or pET28a, respectively, in frame with a C-terminal hexa-His tag. The gene for rat outer membrane cytochrome b5 (NCBI NP_085075.1) residues 13-10319 was ordered as a gBlock® from IDT (www.idtdna.com) and sub-cloned by PCR into the NcoI/XhoI sites of pET28a in frame with a C-terminal hexa-His tag. The H39V mutation was introduced by PCR. Sequences of all clones and mutants were confirmed by Sanger sequencing of mini-prep DNA on Applied Biosciences 3130 and 3730 DNA Sequencers at the Protein and Nucleic Acid Chemistry Laboratory at the Washington University School of Medicine.
Protein Expression and Purification
Bacterial transformation, protein expression, and protein purification were performed as previously described.17 SynHO1, IFP, HuHO1, AtHO1, and rat cyt b5 were expressed in BL21/DE3 E. coli bacteria by transformation with mini-prep plasmid DNA and overnight growth and selection at 37° C in 10-ml LB cultures supplemented with 50 μg/ml carbenicillin (pET21d and pET22b) or 30 μg/ml kanamycin (pET28a). For co-expression of IFP and SynHO1, HuHO1, AtHO1, or rat cyt b5, bacteria were transformed with plasmid DNA encoding each desired protein but with orthogonal resistance cassettes (e.g., SynHO1/pET21d plus IFP/pET28a) and selected with both antibiotics. Overnight cultures were diluted 1:100 into 5 ml LB supplemented with appropriate antibiotics and grown at 37° C to an A600 ~0.5. Protein expression was induced by addition of 1 mM IPTG (and 1 mM 5-aminolevulinic acid for the IFP only experiment in Fig. 2), and bacteria were grown at 37° C and 250 rpm for 1–5 hours prior to harvest. Bacteria analyzed in Fig. 2A & 2B were harvested after 5 hours, whereas the bacteria analyzed in Fig. 2C were harvested at the stated time-points of 0–5 hours. Bacteria were harvested by centrifugation and resuspended in 1 ml 50 mM Tris•HCl (pH 8), 100 mM NaCl. For the western blot analyses, samples contained 2 μl of the 1 ml resuspended bacteria in Tris•HCl (Fig. 2A), 10 μl of the 5 ml LB growth medium (Fig. 2C), or 10 μl of the 100 μl reaction mix (Fig. 6C),
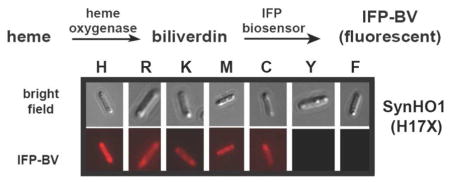
Figure 2. In vivo tests of catalytic activity by SynHO1 variants.

(A) Bright field and fluorescence images (Zeiss filter set 50: excitation 640/30 and emission 690/50 nm) of live E. coli bacteria (5 hours post-induction) expressing IFP alone (±1 mM 5-aminolevulinic acid, ALA) or in combination with the indicated SynHO1 variant. Fluorescence images used identical acquisition times, brightness, and contrast settings. Below each fluorescence image is an anti-polyhistidine western blot probing expression of the His-tagged IFP and SynHO1 variant in samples harvested 5 hours post-induction. (B) Fluorescence excitation and emission spectra of clarified bacterial lysates expressing IFP alone or co-expressed with the indicated SynHO1 variant. Spectra for each variant (harvested 5 hours post-induction) were normalized to the maximum intensity for the WT enzyme. Spectra in gray were offset from each other to avoid overlap. (C) Fluorescence emission spectra of resuspended intact bacteria harvested at the indicated time points post-induction. Spectra are plotted with the raw fluorescence intensities. Below each spectrum is an anti-polyhistidine western blot probing expression of the His-tagged IFP and SynHO1 variant in samples harvested at the indicated time points post-induction. The right-most panel is a plot of the fluorescence intensity at 708 nm as a function of time for each variant, fit with a linear function (slopes given in text).
Figure 6. UV-Vis absorbance spectra and HO activity assays with purified SynHO1-heme complexes.

(A) UV-Vis absorbance spectra of 20 μM purified SynHO1-heme complexes. Inset shows a y-axis expansion for the WT and H17R spectra. (B) Time-dependent (min.) UV-Vis absorbance changes for 20 μM WT and H17R SynHO1-heme complexes after addition of 10 mM ascorbate and 1 μM catalase. (C) Time-dependent (min.) fluorescence emission spectra for 20 μM WT and H17R SynHO1-heme complexes after addition of 10 mM ascorbate, 1 μM catalase, and 20 μM apo IFP. The inset is a plot of the fluorescence intensity at 708 nm as a function of time for WT and H17R.
For co-expression of katE, IFP, and SynHO1 (WT and His17 mutants), Top10 bacteria were transformed with mini-prep plasmid DNA of the pBAD (chloramphenicol) vector encoding katE with N-terminal hexa-His tag (described above) and a dual-expression pBAD vector (ampicillin) that encoded both IFP (with C-terminal hexa-His tag) and untagged SynHO1 (WT or mutants), each with its own ribosome binding site. Bacteria were selected overnight with both chloramphenicol and ampicillin, diluted 1:100 into fresh LB media supplemented with antibiotics, and grown at 37° C for ~3 hours to an OD600 ~0.4. Protein expression was induced by addition of 0.04% arabinose, and bacteria were grown a further 5 hours at 37° C and then harvested. In all cases, protein expression was confirmed by Coomassie-stained SDS-PAGE and/or α-polyhistidine Western blot.
To test whether in vivo HO activity by SynHO1 mutants depended on the presence of a hexa-His tag on either SynHO1 or IFP, we modified the IFP/SynHO1 dual-expression pBAD vector described above to remove the hexa-His tag from the IFP cassette and to encode either WT or H17R SynHO1. Top10 bacteria were transformed with this plasmid, and protein expression was induced with 0.04% arabinose. Lysates from bacteria expressing IFP and either WT or H17R SynHO1 gave the expected fluorescence peaks indicative of IFP-BV formation in both cases, indicating that in vivo HO activity by SynHO1 H17R did not depend on the presence of a hexa-His tag.
Heme-bound complexes of the purified SynHO1 WT and mutants were prepared by addition of concentrated hemin chloride (alkaline aqueous solution) to the clarified bacterial lysates, centrifugation (30 min. × 20,000 g) and syringe filtration (0.45 μM), and passage over a HisTrap HP metal chelation column. Purification was then continued as previously described,17 using nickel-chelation and size-exclusion chromatography.
Western blots were performed with a rabbit anti-polyhistidine antibody conjugated to an IR800 fluorescent dye (Rockland Inc.) used at 1:10,000 dilution. Blots were imaged on an Odyssey Imaging System (LI-COR Biosciences) using an intensity setting of 5 and quantified using Image Studio Lite (LI-COR Biosciences).
Microscopy
Images of live E. coli bacteria were acquired on an Axio Imager.M1 epifluorescence microscope (Carl Zeiss Microimaging, Inc.) equipped with a Hamamatsu ORCA-ER digital CCD camera and running Axiovision 4.8 software. Fluorescence images were acquired with the Zeiss filter set 50 (excitation 625–655 nm, emission 665–715 nm) using identical acquisition times and processed with identical brightness and contrast settings.
UV-Vis Absorbance and Fluorescence Spectroscopy
Fluorescence and absorbance spectra were acquired as previously described.17 For analysis of bacterial lysates, bacteria were harvested 5 hours post-induction, resuspended as describe above, lysed by sonication, and then centrifuged 10 min. at 16,000 g to clarify. Fluorescence excitation (emission 740 nm) and emission (excitation 630 nm) spectra were acquired on a Cary Eclipse fluorescence spectrometer (Varian Inc.) using 10-nm slit widths and a PMT detector voltage setting of 800. Spectra in Fig. 2B, Fig. 3, and Fig. 5 were normalized to the maximum intensity observed for WT, which was set to a value of 1. Fluorescence emission spectra were obtained for intact bacteria in Fig. 2C by diluting 30 μl of the LB growth culture at each time point into 30 μl PBS and acquiring an emission scan (excitation 630 nm) using the settings above.
Figure 3. Testing in-cell HO activity by HO1 and rat outer membrane cytochrome b5 variants.

(A) UV-Vis absorbance spectra of clarified lysates from uninduced bacteria or bacteria expressing SynHO1 WT or H17R) 4 hours after IPTG induction. (B & C) Fluorescence excitation and emission spectra of clarified bacterial lysates expressing IFP alone or co-expressed with the indicated HuHO1 (B) or AtHO1 (C) variant. Spectra for each variant (harvested and lysed 5 hours post-induction) were normalized to the maximum intensity for the WT enzyme. Spectra in gray were offset from each other to avoid overlap. (D) Fluorescence excitation and emission spectra of clarified bacterial lysates co-expressing IFP and either SynHO1 WT or rat OM cyt b5 H39V. Spectra for both proteins (5 hours post-induction) were normalized to the maximum intensity for SynHO1. Also shown for each species is an anti-polyhistidine western blot probing expression of the His-tagged proteins in samples harvested at the indicated time points.
Figure 5. Catalase over-expression does not ablate in vivo activity of SynHO1 proximal His mutants.

(A) Anti-polyhistidine Western blot of soluble bacterial lysates showing expression of E. coli katE, IFP, and SynHO1. All proteins contained an N-terminal (katE, 85 kDa) or C-terminal (IFP, 36 kDa; SynHO1, 28 kDa) hexa-His tag. (B) Fluorescence excitation and emission spectra of soluble lysates from E. coli bacteria expressing katE, IFP, and SynHO1. Spectra for each variant (harvested 5 hours post-induction) were normalized to the maximum intensity for the WT enzyme.
UV-Vis absorbance spectra were acquired on a DU 640 spectrophotometer (Beckman Coulter) using 1-cm path length quartz cuvettes and 100 μl samples. Absorbance spectra of clarified bacterial lysates in Fig. 3A were background corrected by subtracting the absorbance spectrum of an equivalent empty vector culture. Fluorescence scans of resuspended bacterial pellets in 1.7-ml eppendorf tubes were acquired on the 700-nm channel of an Odyssey Imaging System (LI-COR Biosciences) using an intensity setting of 1 and a focal offset of 3 mm.
In vitro HO Activity Assays
Assays were performed as previously described 17. For reactions using ascorbate, 100-μl samples contained 20 μM reconstituted WT or H17R SynHO1-heme complex, 10 mM ascorbate, 1 μM bovine catalase, 100 μM NADPH, 50 mM Tris•HCl (pH 8), and 100 mM NaCl. For reactions using spinach ferredoxin (Fd) and ferredoxin NADP+ reductase (FNR) or E. coli flavodoxin (Fld) and flavodoxin NADP+ reductase (FNR), 100-μl samples contained 20 μM reconstituted WT or H17R SynHO1-heme complex, 20 μM spinach Fd or E. coli Fld, 0.025 U/ml spinach FNR or 40 μM E. coli FNR, 1 μM bovine catalase, 100 μM NADPH, 50 mM Tris•HCl (pH 8), and 100 mM NaCl. Reactions were initiated by adding the ascorbate, ferredoxin, or flavodoxin to sample mixtures containing the other components. The complete reaction mix was then transferred to a cuvette, and spectra were acquired at the indicated time points. For the experiments in Fig. 6C, samples also included 20 μM apo IFP, and fluorescence emission spectra were acquired at the indicated time points using the instrument settings given above and plotted using the raw fluorescence intensities. In vitro activity assays were repeated multiple (≥2) times using independently purified protein samples. Similar results were observed in all cases, and representative spectra have been included herein.
HPLC Analysis of Purified SynHO1 Variants
SynHO1 WT or His17 mutants were expressed in E. coli (without IFP), isolated using affinity and size-exclusion chromatography as described above, and concentrated to 500 μl using Amicon spin concentrators (Millipore) with a 10-kDa molecular mass cutoff. Protein concentrations were determined by absorbance at 280 nm using a molar extinction coefficient of 21,890 M−1 cm−1. Protein purity was confirmed by the appearance of a single band at the expected molecular weight using Coomassie-stained SDS-PAGE, and protein masses were confirmed by LC-MS as described below. 20 μl of the final concentrated solution for each SynHO1 WT or His17 mutant protein isolated from bacteria was analyzed by C18 reverse-phase HPLC, using a previously described HPLC system, column, and solvent system.17 Briefly, the 20-μl sample was injected onto the column and eluted using a constant flow rate of 0.3 ml/min., with absorbance monitored at 400 nm. The resulting chromatogram for analysis of each variant shown in Fig. 4 was normalized to the maximum absorbance for each variant.
Figure 4. Spectroscopic and chromatographic analysis of SynHO1 variants isolated from bacteria.

(A) UV-Vis absorbance spectra of purified proteins. (B) Reverse-phase C18 HPLC chromatograms (absorbance at 400 nm) of SynHO1 variants isolated from bacteria and tetrapyrrole standards. The y-axis in each chromatogram was normalized to the maximum absorbance for each variant or standard (C) Mass spectra of SynHO1 variants isolated from bacteria, corresponding to an LC column retention time of 24–25 min. Each spectrum was normalized to the 583.3 m/z peak for each variant.
Mass Spectrometry
Mass spectrometry analysis of purified WT and H17 mutant (K, R, M and A) SynHO1 proteins was performed at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (http://mass-spec.stanford.edu). Samples were analyzed by liquid chromatography-mass spectrometry (LC-MS) on an Agilent 1100 binary pump HPLC system coupled to a Thermo-Fisher LTQ XL ion trap mass spectrometer. The LC column was a 30 × 2.1 mm Zorbax SB-C8, with initial conditions of 98% A buffer (0.1% formic acid in water)/2% B buffer (0.1% formic acid in acetonitrile) ramped to 95% B buffer in 20 min. at a flow rate of 0.3 ml/min. Samples were analyzed by ESI in the positive ion mode with a mass range of 400–2000 m/z. The injection volume was 20 μl for all samples. The Biomass Deconvolution algorithm was used to determine the intact molecular weight of each protein. All proteins had deconvoluted masses within 1–4 Da of the expected molecular weight for the specified WT or mutant protein minus the start Met residue, consistent with N-terminal processing of these proteins in E. coli by methionine aminopeptidase.20
Mass spectrometry analysis of affinity-isolated SynHO1 was performed at the Fingerprints Proteomics Facility, College of Life Sciences, University of Dundee, Dundee, United Kingdom. Ni-CAM™ HC Resin (Sigma-Aldrich, St. Louis, MO) was used to isolate WT SynHO1 bearing a C-terminal hexahistidine tag and expressed from BL21/DE3 E. coli. Resin was incubated with clarified bacterial lysates, washed five times with potassium phosphate buffer (pH 7.5, 0.5M NaCl, 10 mM imidazole), and eluted with identical buffer containing 0.5 M imidazole. The eluted protein was separated by 10% SDS-PAGE. A gel slice spanning the entire lane was excised, trypsinized, and analyzed by tandem mass spectrometry on an LTQ Orbitrap and the Mascot search engine to identify E. coli proteins.
X-ray Crystallography: Protein expression and purification of HuHO1 H25R
Guided by prior structural studies of HuHO1,21, 22 the truncated HuHO1 H25R gene encoding residues 1-233 was cloned into a pET28a vector in frame with the N-terminal hexa-His tag and thrombin cleavage site. This plasmid was transformed into BL21 DE3 E. coli cells, and bacteria were selected overnight in kanamycin. Cells were grown to OD600 = 0.5, and expression was induced with 1 mM IPTG for 18 hours at 30 °C. Cells were lysed by sonication, and the clarified supernatant was subsequently loaded onto a His-Trap HP metal chelating column (GE Healthcare). Protein was eluted with a linear gradient of imidazole between 0 and 500 mM. Relevant fractions were pooled and then dialyzed against 25 mM HEPES, 150 mM NaCl pH 7.2 and simultaneously treated overnight with thrombin to cleave off the hexa-His tag. The protein was passed again over a His-Trap column to recover the untagged protein, although this time only the flow-through was recovered. Protein was passed through a PD-10 desalting column (Sephadex G-25) to exchange the buffer to 10 mM KH2PO4 (pH 7.4). The protein was then applied to a Hi-Trap Q XL anion-exchange column (GE Healthcare) and eluted with a gradient of 0–250 mM KCl to remove bound biliverdin from the protein. The heme-HO1 and biliverdin-HO1 complexes were prepared by mixing the respective ligand with purified protein at a molar ratio of 2:1 (ligand:protein). Excess ligand was removed by passage through a PD-10 column in 10 mM KH2PO4 (pH 7.4) buffer. Homogeneous samples for crystallization trials were obtained after a chromatographic step in a Superdex-75 size-exclusion column.
Protein Crystallization
Suitable crystals of H25R HuHO1 in complex with heme or biliverdin were obtained by hanging drop vapor diffusion. Crystals of HuHO1 in complex with biliverdin were obtained in the reported crystallization solution of HuHO1 in complex with heme23 by mixing 5 μl of protein solution (9 mg/ml) with 5 μl of a solution composed of 100 mM HEPES (pH 7.5), 2.08 M ammonium sulfate, and 0.9% 1,6-hexanediol at 20 °C. In contrast, we failed to obtain single crystals of H25R in complex with heme under these conditions. We therefore explored modified conditions by using commercial screens containing additives (Hampton) in an ORYX8 instrument (Douglas Instruments, UK). Single crystals of HuHO1 (13 mg ml−1) in complex with heme were obtained by mixing 0.45 μl of protein solution with 0.45 μl of a solution composed of 100 mM HEPES (pH 7.5), 2.08 M ammonium sulfate, and 0.9% 1,6-hexanediol, and with 0.10 μl of a detergent solution of 1.4 mM Big-CHAP at 20 °C. Suitable crystals grew to full size within a few weeks at 20 °C, after which they were transferred to mother liquor supplemented with 20% glycerol and subsequently frozen and stored in a vessel containing liquid N2.
Diffraction Data Collection and Structure Refinement
Diffraction data from single crystals of HuHO1 complexes were collected at beamline BL5A of the Photon Factory (Tsukuba, Japan) under cryogenic conditions (100 K). Diffraction images recorded by the CCD detector were processed with the program MOSFLM and merged and scaled with the program SCALA or AIMLESS of the CCP4 suite.24 The structures were determined by molecular replacement using the coordinates of wild-type HuHO1 in complex with heme (PDB code, 1N45) with the program PHASER.25 The models were refined with the programs REFMAC526 and manually built with COOT.27 Validation was carried out with PROCHECK.28 Data collection and structure refinement statistics are given in Table S1. Structure factors and model coordinates of ligand-bound HuHO1 H25R were deposited with the RCSB Protein Data Bank under accession codes 5BTQ (biliverdin) and 4WD4 (heme).
RESULTS
In-cell enzymology of HO catalysis by proximal His ligand mutants
Nearly all studies of HO activity have been performed in vitro using purified enzymes, reconstituted HO-heme complexes, and cytochrome P450 reductase, ferredoxin, or ascorbate as the electron donor. Indeed, in vitro studies have enabled sophisticated detailed investigations of HO structure and reaction mechanism. We sought to test and extend the conclusions from prior in vitro studies regarding the dependence of HO activity on the proximal His-heme interaction, using a live-cell model system that involves heterologous co-expression in E. coli of an HO enzyme together with a fluorescent biliverdin biosensor.
E. coli strains B and K-12 do not express an HO and thus do not degrade endogenous heme to biliverdin.29 However, HO enzymes from many diverse organisms are enzymatically active when heterologously expressed in E. coli,17, 30–35 providing a model system for HO activity in living cells. The endogenous bacterial electron donor that supports heterologous HO activity within E. coli remains unknown but is thought to be flavodoxin.36 We previously showed that HO co-expression in E. coli with infrared fluorescent protein (IFP),37 an engineered version of the chromophore-binding domain of the D. radiodurans phytochrome that specifically binds biliverdin IXα,38, 39 provides a sensitive fluorescent reporter of HO activity by cyanobacterial (Synechocystis sp. 6803, SynHO1), human (HuHO1), and plant (A. thaliana, AtHO1) HO1 enzymes.17 In this live-cell system (Fig. 1B), recombinant HO and IFP expression are induced in E. coli, and HO activity is monitored by testing the intact live bacteria or clarified bacterial lysates (typically 3–5 hours post-induction) for the unique fluorescence excitation (686 nm) and emission (708 nm) peaks characteristic of mature biliverdin-bound IFP. Consistent with the absence of HO activity in E. coli, expression of IFP alone gave no detectable fluorescence peaks for IFP-BV, including growth conditions in which endogenous heme biosynthesis was stimulated with exogenous 5-aminolevulinic acid (Fig. 2A & 2B and Fig. S1).
We first tested a broad panel of His17 mutants of SynHO1, since this residue was previously identified as the proximal ligand for bound heme.40, 41 In contrast to prior in vitro studies of diverse HO enzymes that failed to detect conversion of heme to BV by proximal His mutants, we observed that bacteria expressing IFP and six different SynHO1 His17 mutants (K, R, M, C, Q, and A) gave the expected fluorescence indicative of BV-bound IFP (Fig. 2 and Fig. S1). These results suggested that the proximal His-heme interaction, despite conservation in all known heme-degrading enzymes, might not be strictly required for HO activity within live cells. Nevertheless, three mutants (Y, E, and F) showed little or no evidence of BV formation, suggesting that bulky or negatively charged side-chains at this position may inhibit HO activity (see below).
We observed a hierarchy of fluorescence intensities across the variants, despite similar protein expression levels, with the largest IFP-BV signal observed for the WT enzyme in all cases (Fig. 2 & Fig. S2). A time-dependent analysis of BV production in live bacteria confirmed that the activity (kobs) of the WT enzyme (35 fluorescence units/hr) is at least 3 to 5-fold greater than the most active H17M (12 units/hr) and H17R mutants (7 units/hr) (Fig. 2C). We emphasize, however, that this comparison provides only a crude estimate of the relative in-cell activities of the WT and mutant enzymes. Indeed, the specific rate-limiting step or steps that underpin the observed rate of increase in IFP-BV fluorescence within bacteria remain obscure and could include rate-limiting contributions that are independent of turnover by the WT or mutant HO enzymes, such as heme biosynthesis or IFP expression. We note, however, that absorbance spectra of clarified lysates from bacteria expressing WT or H17R SynHO1 without IFP (Fig. 3A) displayed relative biliverdin absorbance intensities (H17R/WT absorbance ratio at 390 nm = 0.5) that qualitatively matched the relative IFP-BV fluorescence intensities in Fig. 2B (H17R/WT fluorescence ratio at 708 nm = 0.3). We also observed that IFP quantitatively forms IFP-BV within the dead time (5–10 s) required to mix apo IFP with either free or HO-bound BV and acquire fluorescence spectra. These observations suggest that expression of IFP and formation of IFP-BV do not limit the detection of BV production by WT or mutant HO enzymes in bacteria. Overall, we conclude that the WT His-heme interaction maximizes HO activity but that several H17 mutants retain significant (but lower) in-cell activity, in contrast to prior in vitro studies of proximal His mutants that reported no activity. Additional activity tests for SynHO1 proximal His mutants confirmed the absence of detectable activity in vitro (see below).
To test the generality of our findings with SynHO1, we next tested whether proximal His mutants of HuHO121, 32 and AtHO142 were active when expressed within E. coli. Co-expression of IFP with HuHO1 His25 mutants (R, K, M, and C) but not AtHO1 His86 mutants (R, M, and K) resulted in fluorescence peaks indicative of BV-bound IFP (Fig. 3B & 3C). The HuHO1 results extend our observations with SynHO1 that some HO mutants can retain activity within live cells in the absence of a proximal His-heme interaction. The AtHO1 data, however, indicate that this functional plasticity within E. coli bacteria is not a general property of all HO enzymes and that proximal His mutations can differentially affect the activity of discrete HO proteins, possible due to differing interactions with redox partners (see additional discussion below).
To calibrate the level of in-cell HO activity by proximal His mutants of SynHO1 and HuHO1 relative to other heme-binding proteins previously reported to produce BV from heme in the presence of reducing agents, we co-expressed IFP in bacteria with the H39V mutant of rat outer membrane cytochrome b5. This axial His mutant of rat cyt b5 was previously shown to convert heme to biliverdin in vitro in a catalase-insensitive manner, indicating low-level HO activity.43 In contrast to the significant in-cell BV production observed by IFP-BV fluorescence (Fig. 2 and Fig. 3B) or BV absorbance (Fig. 3A) for SynHO1 or HuHO1 mutants, we were unable to detect BV production in bacteria co-expressing IFP and rat cyt b5 H39V (Fig. 3D). This observation supports our conclusion that several (but not all) proximal His mutants of SynHO1 and HuHO1 retain significant HO activity much greater than that expected for low-level or non-HO specific background activity.
Proximal His mutants of SynHO1 contain bound biliverdin when purified from E. coli
To further test our conclusion that several SynHO1 proximal His mutants remain active in E. coli and to confirm production of the expected biliverdin IXα, we expressed WT and each of four His17 mutants (R, M, K, and F) of SynHO1 in E. coli (without IFP) and purified each protein using affinity and size-exclusion chromatography. Prior work has suggested that product release from HO enzymes is slow in the absence of HO-interacting proteins that bind biliverdin (e.g., biliverdin reductase).44 We therefore reasoned that expression of each active HO mutant in E. coli without IFP would lead to a stable HO-BV complex that could be chromatographically isolated and subsequently analyzed for bound product.
We first acquired UV-Vis absorbance spectra of each HO protein purified from bacteria to assess the presence of bound biliverdin based on its characteristic absorbance spectrum. Spectra of WT and three active His17 mutants (R, M, and K) displayed absorbance maxima at 390 and 685 nm (Fig. 4A), as expected for the presence of bound biliverdin and as previously reported for WT HO from diverse organisms.15, 35, 44, 45 The spectrum of the H17F mutant showed no evidence of bound BV, consistent with the IFP fluorescence data in Fig. 2 that suggested the absence of HO activity.
We next analyzed the isolated proteins by reverse-phase C18 HPLC and confirmed that biliverdin IXα co-purified with the WT and the R, M, and K mutants, based on observation of a 400 nm-absorbing peak in the chromatogram of each injected protein sample that migrated with a retention time identical to a commercial biliverdin IXα standard (Fig. 4B). Purified SynHO1 WT, H17R, and H17K were further analyzed by liquid chromatography-mass spectrometry, which identified ion peaks in each spectrum with identical chromatographic retention times and m/z 583.3, as expected for biliverdin IXα (Fig. 4B & 4C). HPLC analysis of the H17F mutant revealed the presence of bound heme but not biliverdin, supporting the conclusion that this mutant is inactive in bacteria but suggesting that it retains the ability to bind heme (Fig. 4B). We note that the experiments in Fig. 4 were carried out in order to confirm product identity only and were not intended to assess relative amounts of BV production.
Over-expression of E. coli catalase does not ablate in-cell activity of SynHO1 mutants
Prior work has indicated that certain heme-bound proteins can display HO-like activity called coupled oxidation that differs from canonical HO catalysis in that it depends on unbound hydrogen peroxide and can be inhibited by catalase.43, 46 E. coli produces endogenous hydrogen peroxide as a by-product of aerobic metabolism and growth.47 Although bacteria produce anti-oxidant enzymes such as catalase (katG and katE) and alkyl hydroperoxide reductase (ahpCF) that scavenge hydrogen peroxide and thus protect E. coli during aerobic growth,48 we considered whether in-cell production of biliverdin by proximal His mutants of SynHO1 and HuHO1 might reflect coupled oxidation from endogenous hydrogen peroxide rather that canonical HO activity.
Catalase is often included with in vitro HO activity assays to distinguish true HO activity (catalase insensitive) from coupled oxidation (catalase sensitive).46, 49 To perform an analogous test of the in-cell activity of proximal His mutants of SynHO1, we cloned the E. coli catalase gene katE and over-expressed katE in bacteria along with IFP and either WT, H17K, or H17R SynHO1. If BV production by SynHO1 proximal His mutants were due to coupled oxidation, then we expected katE over-expression to substantially reduce or ablate activity by the mutants relative to WT activity. We observed robust katE expression similar to IFP and SynHO1 (Fig. 5A). Nevertheless, the SynHO1 proximal His mutants remained active (Fig. 5B), with IFP-BV fluorescence intensities relative to WT that were indistinguishable from those in the absence of katE (Fig. 2B). This result suggests that catalysis by WT HO and proximal His mutants in bacteria proceeds by similar reaction mechanisms that do not involve coupled oxidation by hydrogen peroxide.
Proximal His mutants of SynHO1 are inactive in vitro
Prior in vitro studies of proximal His-to-Ala HO mutants from multiple organisms,12, 16, 50 and His-to-Cys or Tyr mutants in HuHO1,14 have reported little or no conversion of heme to biliverdin by the purified proteins. Because our in-cell results contrast with these prior in vitro studies and because in vitro activity measurements with several of mutants herein (e.g., K, R, and M) have not been reported previously, we reconstituted heme-bound complexes with several of our purified SynHO1 mutants and assessed their activity in vitro.
We first acquired UV-Vis absorbance spectra of the ferric heme complexes of WT and H17 mutants of SynHO1. A prominent Soret peak centered near 404 nm was observed in the spectrum of WT and each of the four purified mutants of SynHO1 (Fig. 6A), and both the WT and H17R complexes had absorption bands at 500 and 630 nm (Fig. 6A, inset), consistent with a high-spin complex in all cases.40 The Soret peaks of the mutants had broader line widths than WT, which may suggest a mixture of coordination states.14
We first tested the HO activity of the WT and mutant enzymes using 10 mM ascorbate as the reducing agent, and we included 1 μM bovine catalase to prevent heme degradation via coupled oxidation. Addition of ascorbate to the WT SynHO1-heme complex resulted in a time-dependent decrease in Soret peak intensity and concomitant rise in UV-Vis absorbance at 686 nm (Fig. 6B), as expected for HO-catalyzed conversion of heme to BV and as previously reported for WT SynHO1.40 In contrast, ascorbate addition to the SynHO1 H17R-heme complex resulted in no significant decrease in Soret peak intensity and no detectable increase in 686-nm absorbance over a similar 90-min. timescale (Fig. 6B), providing no evidence for conversion of heme to BV (see below). Very similar results were observed for the H17K and H17M mutants with ascorbate (Fig. S3A), and exogenous imidazole did not stimulate activity by H17R (Fig. S3B). Since SynHO1 is thought to use ferredoxin as its cognate redox partner in vivo,30 we also tested the ability of spinach ferredoxin and ferredoxin NADP+ reductase to support activity of the H17R mutant in vitro. As with ascorbate, we observed no significant changes in the Soret peak intensity with no evidence for biliverdin production (Fig. S3C).
We extended the in vitro tests of WT and H17R catalysis with ascorbate by adding purified apo IFP and measuring the time-dependent rise in IFP-BV fluorescence. WT SynHO1 showed a steady increase in IFP-BV fluorescence (Fig. 6C) over a similar time-scale as the loss of Soret peak intensity observed by UV-Vis absorbance in Fig. 6B. However, no IFP-BV fluorescence was observed in vitro for the H17R mutant over 5 hours (Fig. 6C), in contrast to the steady increase in IFP-BV fluorescence observed for this mutant in live bacteria over a similar time-scale and at similar protein levels (Fig. 2C).
Prior study of proximal His mutants of HuHO1 suggested that these proteins have more negative redox potentials compared to WT HuHO1, resulting in a diminished driving force for reduction of the mutants compared to WT by a common redox partner.14 We therefore considered whether the more negative redox potential of E. coli flavodoxin (−455 mV)51 compared to commonly used in vitro electron donors such as spinach ferredoxin (−420 mV)52, cytochrome P450 reductase (−270 mV)14, or ascorbate (−282 mV)53 might account for our observation of enhanced HO activity by proximal His mutants within live E. coli compared to in vitro conditions. As noted above, the endogenous E. coli flavodoxin has been proposed as the in vivo redox partner for heterologous HO activity.36
We cloned the E. coli genes for flavodoxin and flavodoxin NADP+ reductase (FNR), expressed these proteins in E. coli, purified them to homogeneity using affinity and size-exclusion chromatography, and then tested their ability to support HO activity in vitro. The purified proteins had the same SDS-PAGE migrations and UV-Vis absorbance spectra as previously reported.36 We observed a time-dependent decrease in the Soret peak for the heme complex of WT SynHO1 in the presence of E. coli flavodoxin, FNR, NADPH, and catalase, as expected for HO-catalyzed heme degradation (Fig. S3D). The heme complex of SynHO1 H17R, however, showed no evidence of heme degradation (Fig. S3D), suggesting that the more negative E. coli flavodoxin redox potential alone does not account for the enhanced in-cell mutant activity.
X-ray crystallographic structures of HuHO1 H25R with bound heme or biliverdin
To directly assess the consequences of mutating the proximal His on HO active site architecture and interactions with bound ligands, we determined X-ray crystallographic structures of HuHO1 H25R bound to either heme or BV. Data collection and refinement statistics are given in Table S1. Although crystallographic studies have been described for the HuHO1 H25A mutant,54 the previously reported structure did not contain bound ligand, and coordinates and structure factors were not deposited with the RCSB Protein Data Bank (PDB). Our work thus presents the first comprehensive structural study of an HO proximal His mutant with bound heme substrate and biliverdin product.
The overall structure that we obtained of heme-bound HuHO1 H25R at 2.95 Å resolution is nearly identical to that previously determined for the WT HuHO1-heme complex (PDB code 1N45),21, 22 with a root mean square deviation (RMSD) between backbone atoms for the two structures of 0.33 Å. The electron density map of the HuHO1 H25R-heme structure shows well-defined density to support the modeled atomic positions for bound heme and active site residues, with no indication of alternative ligand or side-chain conformations (Fig. 7A).
Figure 7. X-ray structural studies of HuHO1 H25R.

(A) Sigma-A-weighted 2Fo − Fc electron density map (contoured at 1σ) from the 2.95-Å resolution structure of heme (orange) bound to HuHO1 H25R (determined herein, PDB 4WD4). (B) Superposition of the HuHO1 H25R•heme structure (protein in cyan, heme in orange) and the previously published WT HuHO1•heme structure (PDB 1N45, protein backbone in green, side-chains in white, heme in white). (C) Sigma-A-weighted 2Fo − Fc electron density map (contoured at 1.2σ) from the 2.08-Å resolution structure of biliverdin IXα (yellow) bound to HuHO1 H25R (determined herein, PDB 5BTQ). (D) Superposition of the heme- and biliverdin-bound structures of HuHO1 H25R (heme in orange, biliverdin in yellow, residues from heme-bound structure in orange, residues from BV-bound structure in yellow). See Table S1 for data acquisition and refinement statistics
In contrast to heme-bound WT HuHO1 in which His25 is positioned to coordinate the iron atom of heme (Fe-Nε2 distance, 2.0 Å), the Arg25 side-chain in the H25R mutant is oriented away from the parent His25 position such that it does not serve as an axial heme ligand and instead adopts a conformation within hydrogen-bonding distance (3.0–3.4 Å) of the Thr21 side-chain and Lys18 backbone oxygen atoms (Fig. 7B). This change is accompanied by three additional rearrangements within the H25R active site: 1) a 22° tilt in the orientation of bound heme relative to WT, 2) a ~1 Å displacement of the C-terminal half of the proximal backbone helix toward the bound heme, and 3) reorientation of the Glu29 side-chain, which in WT HuHO1 is hydrogen-bonded to the Nδ1 atom of His25, to place a carboxylate oxygen atom within 3.0 Å of the heme iron (Fig. 7B). This 3-Å distance, however, is at least 1 Å longer than expected for coordination of iron by a carboxylate group.55 Outside of these changes, the positions of the remaining residues appear to be conserved between the active sites of H25R and WT HuHO1 with bound heme. Although the WT structure reported a water molecule as a distal axial ligand of heme,22 at the 2.95-Å resolution of our structure we did not observe electron density for a distal water molecule.
We considered whether the reorientation of Glu29 in the H25R mutant and the possible interactions between this residue and bound heme might be a common feature of HuHO1 His25 mutants and contribute to their in-cell activity (Fig. 3A). We prepared the H25M/E29A and H25R/E29A double mutants and co-expressed them in E. coli with IFP. Lysates from these bacteria displayed the expected fluorescence peak for IFP-BV formation (Fig. S4A), indicating that the double mutants retain HO activity and suggesting that in-cell catalysis by His25 mutants does not depend critically on interactions between heme and Glu29.
To assess the effect of proximal His mutation on the orientation of bound product and its interactions with the surrounding active site, we determined a 2.08 Å resolution structure of biliverdin-bound HuHO1 H25R. The overall structure was very similar to heme-bound WT HuHO1 (backbone RMSD of 0.38 Å), with well-defined electron density to support the modeled orientations of active site residues and bound BV and no indication of alternative conformations (Fig. 7C). The orientation of BV bound to H25R is very similar to that of bound heme, although the terminal pyrrole moieties are offset in a helical conformation that avoids steric clashes between the lactam oxygen atoms (Fig. 7D). This configuration is nearly identical to previous structures of BV bound to rat HO156 (Fig. S4B) and HmuO57 from C. diphtheriae. In addition, one of the propionate groups of BV bound to H25R has adopted an alternate conformation compared to bound heme that places it within hydrogen-bonding distance (3.2 Å) of the side-chain of Arg25, which is repositioned relative to the heme-bound structure (Fig. 7D). Glu29 also adopts an altered position with bound BV that differs from the H25R-heme complex (Fig. 7D) and is closer to that in the WT H25R-heme complex.
We note that the BV position observed in our HuHO1 H25R structure is in striking contrast to the very different position and orientation described previously for BV bound to WT HuHO145, in which BV was suggested to partition into a deeper and more sequestered binding pocket within the HO protein core. The prior study, however, reported limited electron density to support the modeled conformation and suggested that BV might be disordered between multiple conformations. We did not observe electron density for this alternative, deeper orientation of BV. The conformation of BV modeled in our H25R HuHO1 structure is supported by clear electron density and, as noted above, is very similar to the reported positions of BV bound to WT rat HO1 and C. diphtheriae HmuO.
DISCUSSION
Role of His-heme coordination and additional active site interactions in HO catalysis
Understanding the active site features of heme oxygenase that underpin its ability to catalyze conversion of heme to biliverdin remains an on-going challenge. Mutagenesis studies of HO catalysis in vitro have supported the view that the proximal His-heme interaction makes an essential contribution to the chemical conversion of heme to biliverdin within the HO active site.6, 58 This role has been proposed to reflect the unique electronic effects of the His-heme interaction that tune the redox potential of bound heme and prevent O-O bond cleavage in the ferric heme-hydroperoxy intermediate, as occurs in peroxidases, favoring hydroxylation of the α-meso carbon of the bound heme.58–60 A second model also posits that the His-heme interaction may contribute to positioning the bound heme relative to active site groups on the distal porphyrin face that are critical for catalysis.14, 15
In contrast to in vitro studies, we observed significant HO activity by diverse proximal His mutants of SynHO1 and HuHO1 within live bacteria. Our work provides direct evidence that the His-heme interaction is not strictly required for HO activity by cyanobacterial and mammalian enzymes under intracellular conditions within bacteria and uncovers surprising plasticity in the proximal active site interactions with heme that support HO catalysis in live cells. This conclusion is consistent with prior in vitro work that suggested that D. melonogaster HO retains the `conserved proximal His residue but catalyzes heme degradation in the absence of stable His-heme coordination.61 A corollary of our findings is that the surrounding active site environment also makes important contributions to HO catalysis, a conclusion supported by extensive in vitro work that has revealed critical interactions between heme and active site groups and water molecules on the distal face of heme.6, 59, 62, 63 The lack of in-cell activity by a subset of SynHO1 His 17 mutants (Y, E, and F) may reflect unfavorable interactions between these bulky residues and heme and/or propagated effects that disrupt favorable interactions within the remainder of the active site or with redox partners. The inactivity of the H17F mutant, despite still binding heme (Fig. 4B), supports the conclusion that in-cell BV production by R, M, and K mutants reflects specific HO activity within the active sites of these mutants, rather than non-specific heme degradation due to remnant heme association with a protein binding pocket, as has been reported for other heme binding proteins in vitro.43, 46 If such non-specific activity were the general explanation for remnant in-cell activity by R, M, and K mutants, then we would have most simply expected to have observed in-cell activity for all of the mutants tested herein, which is not what we observed (see summary of models and tests in Supporting Information).
Although our results indicate that proximal His mutants of multiple HO enzymes can remain active within the intracellular environment, the proximal His is nevertheless strictly conserved as an axial ligand in all known heme-degrading enzymes. Indeed, all proximal His mutants had diminished activity relative to the WT enzyme (Fig. 2 and Fig. 3). This observation suggests that the proximal His-heme interaction optimizes enzyme function and that Nature has consistently selected for this optimal arrangement. As noted above, our results provide only a crude estimate of the in-cell activity of SynHO1 mutants relative to WT. In our view, the most telling observation of our study is that several mutants retain significant activity in live cells (irrespective of whether mutant activity is 25% or 5% of WT), whereas no activity can be detected with purified proteins in vitro. This remnant activity by several proximal His mutants is consistent with the general view that enzymatic catalysis arises from many enzyme-substrate interactions within the active site environment, rather than from a single dominant interaction.64, 65 Although the conserved His-heme interaction may optimize positioning and redox potential within the HO active site, our results indicate that HO activity can persist in the absence of His-heme coordination under intracellular conditions within E. coli. It remains possible that certain proximal His mutants could also show detectable activity in vitro at reaction times substantially longer than those sampled herein (≤5 hours, Fig. 6B & 6C). Nevertheless, the central observation remains that certain HO mutants display enhanced in-cell activity relative to the in vitro conditions explored herein.
Our failure to detect HO activity by proximal His mutants of AtHO1 provides evidence that HO enzymes can display differing in-cell dependences on the proximal His-heme interaction. This difference between AtHO1 versus SynHO1 and HuHO1, despite conservation of additional active site features such as the distal GG motif,17 may reflect the structural and functional effects of other active site residues that differ between these enzymes. No crystallographic structures have been reported for AtHO1, and it remains a future challenge to understand the differences in AtHO1 active site architecture that result in the observed functional differences from SynHO1 and HuHO1.
Recent works have identified HO-like proteins in several organisms, including A. thaliana (AtHO2) and P. falciparum (PfHO), that show overall sequence homology to known heme oxygenases but lack the conserved His residue and instead feature an Arg (AtHO2) or Lys (PfHO) at the position of the proximal heme ligand.42, 66 Both AtHO2 and PfHO bind porphyrins but fail to convert heme to biliverdin in vitro or within E. coli, and mutation of the proximal Arg (AtHO2) or Lys (PfHO) to His does not confer HO activity in either protein.17, 31, 67 These observations suggest that sequence divergence in these proteins beyond loss of the proximal His ligand also contributes to the observed inactivity, underscoring the important roles contributed by additional active site interactions beyond the His-heme coordination to HO catalysis.
Why do proximal His mutants of SynHO1 and HuHO1 show activity in bacterial cells but no activity in vitro?
As noted above, neither coupled oxidation nor differences in reduction potentials appear to explain the observed activity differences between these two environments. Based on reports that exogenous imidazole could rescue the activity of His-to-Ala mutants in vitro, we also considered whether free histidine or the His side-chain of another protein not present in vitro might partition into the mutant HO active site within E. coli and enable catalysis, akin to prior reports of imidazole rescue of Src kinase mutants inside cells.68, 69 Two observations suggest that this mechanism is not the origin of the observed activity differences: exogenous imidazole does not stimulate activity by SynHO1 H17R in vitro (Fig. S3B), and our X-ray structure of HuHO1 H25R suggests insufficient cavity volume beneath the bound heme to accommodate an imidazole group as an axial ligand (Fig. 7A). Finally, to identify protein-protein interactions that might be critical for HO activity in vivo, we affinity-isolated WT SynHO1 from bacteria and used mass spectrometry to identify more than 200 co-associating proteins (Table S2). Multiple redox proteins, but not E. coli flavodoxin or ferredoxin, were among the identified candidates, and we have on-going experiments to test the possible role of these proteins in supporting HO catalysis.
It thus remains a future challenge to elucidate the biochemical mechanism that enhances HO activity by proximal His mutants within E. coli compared to in vitro conditions. We speculate that there may be specific adaptor proteins in bacteria that enhance the interaction and transfer of electrons between HO and its redox partners. Indeed, recent work has suggested that HuHO2 and cytochrome P450 reductase (CPR) interact only weakly and transiently in vitro,70 and scaffolding proteins in other systems have been shown to increase both enzyme specificity and reaction rate.71 Our hypothesis is consistent with a prior study of the HuHO1-CPR fusion protein that reported that covalently tethering CPR and HuHO1, akin to how a scaffolding protein might facilitate interactions between these proteins via non-covalent binding interactions, accelerated the rate of biliverdin formation in vitro compared to the reaction involving intermolecular association between the separate HO and CPR proteins.32 Prior work has also suggested that electron transfer reactions can show heightened sensitivity to the effects of covalent tethering or physical scaffolds that enhance interactions between the electron donor and acceptor groups.72 Alternatively or in addition, there may be synergistic interactions between multiple bacterial reductases that cooperatively enhance HO activity compared to solution conditions in vitro. Finally, it remains an important future challenge to test and extend the conclusions of our study of HO activity in bacteria to other cell types, including the native host cells of specific HO enzymes. Future work can also test whether the activity of non-canonical HO enzymes such IsdG/I or MhuD can persist in live cells without His-heme coordination.
Closing perspective
Several recent perspectives have highlighted the importance of developing tools and approaches to test our understanding of enzyme function inside cells and the heterogeneous environment of stable and transient protein-protein interactions within which enzymes operate natively.2, 73, 74 Indeed, we note other recent examples of enzymes lacking key residues that appear inactive in solution but show modest activity in the presence of additional intracellular interaction partners or in reconstituted systems in vitro.75, 76 In our case, use of a fluorescent biliverdin biosensor was critical to detecting the lower activity of HO mutants compared to WT. Our results can guide on-going efforts to fully understand active site contributions to HO catalysis,6 to understand the evolution and functional divergence of HO enzymes in biology,6, 10, 17, 31, 67 to engineer novel heme proteins with desired functionalities,77 and to develop in-cell probes of enzymatic function in other systems.78
Supplementary Material
Acknowledgments
We thank Maurizio Splendore, Ludmila Alexandrova, and Allis Chien of the Vincent Coates Foundation Mass Spectrometry Facility (Stanford University) and the staff of the Photon Factory (Tsukuba, Japan) for technical support. Access to beamline BL-5A was granted by the Photon Factory Advisory Committee (Proposals 2013G738, and 2014G190).
The abbreviations used are
- HO
heme oxygenase
- BV
biliverdin IXα
- IFP
infrared fluorescent protein
- SynHO1
cyanobacterial heme oxygenase-1 from Synechocystis sp. 6803
- HuHO1
human heme oxygenase-1
- AtHO1
heme oxygenase-1 from Arabidopsis thaliana
- ALA
5-aminolevulinic acid
Footnotes
Funding sources:
This work was supported by National Institutes of Health grant R21 AI110712-01 (to D.E.G.); a Burroughs Wellcome Fund Career Award at the Scientific Interface (to P.A.S.); the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan; and a Grant-in-Aid for Scientific Research A (grant 25249115) from the Japan Society for the Promotion of Science (to K.T.)
Atomic coordinates and structure factors have been deposited in the RCSB Protein Data Bank (http://www.rcsb.org/pdb) under accession codes 4WD4 and 5BQT.
SUPPORTING INFORMATION AVAILABLE
Summary of models and tests regarding the origin of in-cell activity, fluorescence images and photos of intact bacteria expressing SynHO1 variants (Fig. S1), quantification of protein expression in Western blots in Fig. 2 (Fig. S2), in vitro activity assays of SynHO1 variants with different redox partners (Fig. S3), in-cell studies of HO activity by H17R/E29A and E17M/E29A SynHO1 mutants (Fig. S4A), structural overlay of HuHO1 H25R•BV and rat HO1•BV X-ray structures (Fig. S4B), X-ray crystallographic data collection and refinement statistics (Table S1), and E. coli proteins identified by mass spectrometry as associated with WT SynHO1 affinity-purified from bacterial lysates (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kornberg A. Ten commandments: Lessons from the enzymology of DNA replication. J Bacteriol. 2000;182:3613–3618. doi: 10.1128/jb.182.13.3613-3618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khosla C. Quo vadis, enzymology? Nat Chem Biol. 2015;11:438–441. doi: 10.1038/nchembio.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirkby KA, Adin CA. Products of heme oxygenase and their potential therapeutic applications. Am J Physiol. 2006;290:F563–571. doi: 10.1152/ajprenal.00220.2005. [DOI] [PubMed] [Google Scholar]
- 4.Beale SI. Biosynthesis of Phycobilins. Chem Rev. 1993;93:785–802. [Google Scholar]
- 5.Wilks A, Burkhard KA. Heme and virulence: how bacterial pathogens regulate, transport and utilize heme. Nat Prod Rep. 2007;24:511–522. doi: 10.1039/b604193k. [DOI] [PubMed] [Google Scholar]
- 6.Unno M, Matsui T, Ikeda-Saito M. Structure and catalytic mechanism of heme oxygenase. Nat Prod Rep. 2007;24:553–570. doi: 10.1039/b604180a. [DOI] [PubMed] [Google Scholar]
- 7.Fufezan C, Zhang J, Gunner MR. Ligand preference and orientation in b- and c-type heme-binding proteins. Proteins. 2008;73:690–704. doi: 10.1002/prot.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP. The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol Microbiol. 2010;75:1529–1538. doi: 10.1111/j.1365-2958.2010.07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chim N, Iniguez A, Nguyen TQ, Goulding CW. Unusual Diheme Conformation of the Heme-Degrading Protein from Mycobacterium tuberculosis. J Mol Biol. 2010;395:595–608. doi: 10.1016/j.jmb.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilks A, Ikeda-Saito M. Heme Utilization by Pathogenic Bacteria: Not All Pathways Lead to Biliverdin. Acc Chem Res. 2014;47:2291–2298. doi: 10.1021/ar500028n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chu GC, Couture M, Yoshida T, Rousseau DL, Ikeda-Saito M. Axial ligation states of five-coordinate heme oxygenase proximal histidine mutants, as revealed by EPR and resonance Raman spectroscopy. J Am Chem Soc. 2000;122:12612–12613. [Google Scholar]
- 12.Sun J, Loehr TM, Wilks A, Demontellano PRO. Identification of Histidine-25 as the Heme Ligand in Human Liver Heme Oxygenase. Biochemistry. 1994;33:13734–13740. doi: 10.1021/bi00250a026. [DOI] [PubMed] [Google Scholar]
- 13.Wilks A, Sun J, Loehr TM, Demontellano PRO. Heme Oxygenase His25ala Mutant - Replacement of the Proximal Histidine Iron Ligand by Exogenous Bases Restores Catalytic Activity. J Am Chem Soc. 1995;117:2925–2926. [Google Scholar]
- 14.Liu Y, Moenne-Loccoz P, Hildebrand DP, Wilks A, Loehr TM, Mauk AG, de Montellano PRO. Replacement of the proximal histidine iron ligand by a cysteine or tyrosine converts heme oxygenase to an oxidase. Biochemistry. 1999;38:3733–3743. doi: 10.1021/bi982707s. [DOI] [PubMed] [Google Scholar]
- 15.Wilks A, Moenne-Loccoz P. Identification of the proximal ligand His-20 in heme oxygenase (Hmu O) from Corynebacterium diphtheriae - Oxidative cleavage of the heme macrocycle does not require the proximal histidine. J Biol Chem. 2000;275:11686–11692. doi: 10.1074/jbc.275.16.11686. [DOI] [PubMed] [Google Scholar]
- 16.Chu GC, Katakura K, Tomita T, Zhang XH, Sun DY, Sato M, Sasahara M, Kayama T, Ikeda-Saito M, Yoshida T. Histidine 20, the crucial proximal axial heme ligand of bacterial heme oxygenase Hmu O from Corynebacterium diphtheriae. J Biol Chem. 2000;275:17494–17500. doi: 10.1074/jbc.M000830200. [DOI] [PubMed] [Google Scholar]
- 17.Sigala PA, Crowley JR, Hsieh S, Henderson JP, Goldberg DE. Direct Tests of Enzymatic Heme Degradation by the Malaria Parasite Plasmodium falciparum. J Biol Chem. 2012;287:37793–37807. doi: 10.1074/jbc.M112.414078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feissner RE, Richard-Fogal CL, Frawley ER, Loughman JA, Earley KW, Kranz RG. Recombinant cytochromes c biogenesis systems I and II and analysis of haem delivery pathways in Escherichia coli. Mol Microbiol. 2006;60:563–577. doi: 10.1111/j.1365-2958.2006.05132.x. [DOI] [PubMed] [Google Scholar]
- 19.Rivera M, Barillas-Mury C, Christensen KA, Little JW, Wells MA, Walker FA. Gene synthesis, bacterial expression, and 1H NMR spectroscopic studies of the rat outer mitochondrial membrane cytochrome b5. Biochemistry. 1992;31:12233–12240. doi: 10.1021/bi00163a037. [DOI] [PubMed] [Google Scholar]
- 20.Xiao Q, Zhang FR, Nacev BA, Liu JO, Pei DH. Protein N-Terminal Processing: Substrate Specificity of Escherichia coli and Human Methionine Aminopeptidases. Biochemistry. 2010;49:5588–5599. doi: 10.1021/bi1005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuller DJ, Wilks A, de Montellano PRO, Poulos TL. Crystal structure of human heme oxygenase-l. Nat Struct Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 22.Lad L, Schuller DJ, Shimizu H, Friedman J, Li HY, de Montellano PRO, Poulos TL. Comparison of the heme-free and -bound crystal structures of human heme oxygenase-1. J Biol Chem. 2003;278:7834–7843. doi: 10.1074/jbc.M211450200. [DOI] [PubMed] [Google Scholar]
- 23.Schuller DJ, Wilks A, Ortiz de Montellano P, Poulos TL. Crystallization of recombinant human heme oxygenase-1. Protein Sci. 1998;7:1836–1838. doi: 10.1002/pro.5560070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans P. Scaling and assessment of data quality. Acta Crystallogr D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 29.Suits MD, Pal GP, Nakatsu K, Matte A, Cygler M, Jia Z. Identification of an Escherichia coli O157:H7 heme oxygenase with tandem functional repeats. Proc Natl Acad Sci U S A. 2005;102:16955–16960. doi: 10.1073/pnas.0504289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornejo J, Willows RD, Beale SI. Phytobilin biosynthesis: cloning and expression of a gene encoding soluble ferredoxin-dependent heme oxygenase from Synechocystis sp. PCC 6803. Plant J. 1998;15:99–107. doi: 10.1046/j.1365-313x.1998.00186.x. [DOI] [PubMed] [Google Scholar]
- 31.Emborg TJ, Walker JM, Noh B, Vierstra RD. Multiple heme oxygenase family members contribute to the biosynthesis of the phytochrome chromophore in Arabidopsis. Plant Physiol. 2006;140:856–868. doi: 10.1104/pp.105.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilks A, Black SM, Miller WL, Ortiz de Montellano PR. Expression and characterization of truncated human heme oxygenase (hHO-1) and a fusion protein of hHO-1 with human cytochrome P450 reductase. Biochemistry. 1995;34:4421–4427. doi: 10.1021/bi00013a034. [DOI] [PubMed] [Google Scholar]
- 33.Gambetta GA, Lagarias JC. Genetic engineering of phytochrome biosynthesis in bacteria. Proc Natl Acad Sci U S A. 2001;98:10566–10571. doi: 10.1073/pnas.191375198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa K, Sato M, Yoshida T. Expression of Rat Heme Oxygenase in Escherichia-Coli as a Catalytically Active, Full-Length Form That Binds to Bacterial-Membranes. Eur J Biochem. 1991;202:161–165. doi: 10.1111/j.1432-1033.1991.tb16357.x. [DOI] [PubMed] [Google Scholar]
- 35.Wegele R, Tasler R, Zeng Y, Rivera M, Frankenberg-Dinkel N. The heme oxygenase(s)-phytochrome system of Pseudomonas aeruginosa. J Biol Chem. 2004;279:45791–45802. doi: 10.1074/jbc.M408303200. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins CM, Waterman MR. Flavodoxin and NADPH-flavodoxin reductase from Escherichia coli support bovine cytochrome P450c17 hydroxylase activities. J Biol Chem. 1994;269:27401–27408. [PubMed] [Google Scholar]
- 37.Shu X, Royant A, Lin MZ, Aguilera TA, Lev-Ram V, Steinbach PA, Tsien RY. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner JR, Brunzelle JS, Forest KT, Vierstra RD. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–331. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 39.Bhoo SH, Davis SJ, Walker J, Karniol B, Vierstra RD. Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature. 2001;414:776–779. doi: 10.1038/414776a. [DOI] [PubMed] [Google Scholar]
- 40.Migita CT, Zhang X, Yoshida T. Expression and characterization of cyanobacterium heme oxygenase, a key enzyme in the phycobilin synthesis. Properties of the heme complex of recombinant active enzyme. Eur J Biochem. 2003;270:687–698. doi: 10.1046/j.1432-1033.2003.03421.x. [DOI] [PubMed] [Google Scholar]
- 41.Sugishima M, Migita CT, Zhang X, Yoshida T, Fukuyama K. Crystal structure of heme oxygenase-1 from cyanobacterium Synechocystis sp. PCC 6803 in complex with heme. Eur J Biochem. 2004;271:4517–4525. doi: 10.1111/j.1432-1033.2004.04411.x. [DOI] [PubMed] [Google Scholar]
- 42.Davis SJ, Bhoo SH, Durski AM, Walker JM, Vierstra RD. The heme-oxygenase family required for phytochrome chromophore biosynthesis is necessary for proper photomorphogenesis in higher plants. Plant Physiol. 2001;126:656–669. doi: 10.1104/pp.126.2.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avila L, Huang HW, Damaso CO, Lu S, Moenne-Loccoz P, Rivera M. Coupled oxidation vs heme oxygenation: Insights from axial ligand mutants of mitochondrial cytochrome b(5) J Am Chem Soc. 2003;125:4103–4110. doi: 10.1021/ja029311v. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Ortiz de Montellano PR. Reaction intermediates and single turnover rate constants for the oxidation of heme by human heme oxygenase-1. J Biol Chem. 2000;275:5297–5307. doi: 10.1074/jbc.275.8.5297. [DOI] [PubMed] [Google Scholar]
- 45.Lad L, Friedman J, Li HY, Bhaskar B, de Montellano PRO, Poulos TL. Crystal structure of human heme oxygenase-1 in a complex with biliverdin. Biochemistry. 2004;43:3793–3801. doi: 10.1021/bi035451l. [DOI] [PubMed] [Google Scholar]
- 46.Sigman JA, Wang X, Lu Y. Coupled oxidation of heme by myoglobin is mediated by exogenous peroxide. J Am Chem Soc. 2001;123:6945–6946. doi: 10.1021/ja015776u. [DOI] [PubMed] [Google Scholar]
- 47.Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J Bacteriol. 2001;183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lansky IB, Lukat-Rodgers GS, Block D, Rodgers KR, Ratliff M, Wilks A. The cytoplasmic heme-binding protein (PhuS) from the heme uptake system of Pseudomonas aeruginosa is an intracellular heme-trafficking protein to the delta-regioselective heme oxygenase. J Biol Chem. 2006;281:13652–13662. doi: 10.1074/jbc.M600824200. [DOI] [PubMed] [Google Scholar]
- 50.Kim D, Yukl ET, Moenne-Loccoz P, de Montellano PRO. Fungal heme oxygenases: Functional expression and characterization of Hmx1 from Saccharomyces cerevisiae and CaHmx1 from Candida albicans. Biochemistry. 2006;45:14772–14780. doi: 10.1021/bi061429r. [DOI] [PubMed] [Google Scholar]
- 51.Blaschkowski HP, Neuer G, Ludwigfestl M, Knappe J. Routes of Flavodoxin and Ferredoxin Reduction in Escherichia-Coli - Coa-Acylating Pyruvate - Flavodoxin and Nadph - Flavodoxin Oxidoreductases Participating in the Activation of Pyruvate Formate-Lyase. Eur J Biochem. 1982;123:563–569. [PubMed] [Google Scholar]
- 52.Cammack R, Rao KK, Bargeron CP, Hutson KG, Andrew PW, Rogers LJ. Midpoint redox potentials of plant and algal ferredoxins. Biochem J. 1977;168:205–209. doi: 10.1042/bj1680205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Creutz C. Complexities of ascorbate as a reducing agent. Inorg Chem. 1981;20:4449–4452. [Google Scholar]
- 54.Zhou WP, Zhong WW, Zhang XH, Ding JP, Zhang ZL, Xia ZW. Comparison of the crystal structure and function to wild-type and His25Ala mutant human heme oxygenase-1. Int J Mol Med. 2009;23:379–387. doi: 10.3892/ijmm_00000142. [DOI] [PubMed] [Google Scholar]
- 55.Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr D. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- 56.Sugishima M, Sakamoto H, Higashimoto Y, Noguchi M, Fukuyama K. Crystal structure of rat heme oxygenase-1 in complex with biliverdin-iron chelate - Conformational change of the distal helix during the heme cleavage reaction. J Biol Chem. 2003;278:32352–32358. doi: 10.1074/jbc.M303682200. [DOI] [PubMed] [Google Scholar]
- 57.Unno M, Ardevol A, Rovira C, Ikeda-Saito M. Structures of the Substrate-free and Product-bound Forms of HmuO, a Heme Oxygenase from Corynebacterium diphtheriae X-RAY CRYSTALLOGRAPHY AND MOLECULAR DYNAMICS INVESTIGATION. J Biol Chem. 2013;288:34443–34458. doi: 10.1074/jbc.M113.486936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montellano PR. The mechanism of heme oxygenase. Curr Opin Chem Biol. 2000;4:221–227. doi: 10.1016/s1367-5931(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 59.Matsui T, Unno M, Ikeda-Saito M. Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc Chem Res. 2010;43:240–247. doi: 10.1021/ar9001685. [DOI] [PubMed] [Google Scholar]
- 60.Chen H, Moreau Y, Derat E, Shaik S. Quantum mechanical/molecular mechanical study of mechanisms of heme degradation by the enzyme heme oxygenase: The strategic function of the water cluster. J Am Chem Soc. 2008;130:1953–1965. doi: 10.1021/ja076679p. [DOI] [PubMed] [Google Scholar]
- 61.Zhang XH, Sato M, Sasahara M, Migita CT, Yoshida T. Unique features of recombinant heme oxygenase of Drosophila melanogaster compared with those of other heme oxygenases studied. Eur J Biochem. 2004;271:1713–1724. doi: 10.1111/j.1432-1033.2004.04077.x. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, Koenigs Lightning L, Huang H, Moenne-Loccoz P, Schuller DJ, Poulos TL, Loehr TM, Ortiz de Montellano PR. Replacement of the distal glycine 139 transforms human heme oxygenase-1 into a peroxidase. J Biol Chem. 2000;275:34501–34507. doi: 10.1074/jbc.M004245200. [DOI] [PubMed] [Google Scholar]
- 63.Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid Redox Signal. 2002;4:603–614. doi: 10.1089/15230860260220102. [DOI] [PubMed] [Google Scholar]
- 64.Fersht AR. Structure and Mechanism in Protein Science. 2. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 65.Herschlag D, Natarajan A. Fundamental challenges in mechanistic enzymology: progress toward understanding the rate enhancements of enzymes. Biochemistry. 2013;52:2050–2067. doi: 10.1021/bi4000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okada K. The novel heme oxygenase-like protein from Plasmodium falciparum converts heme to bilirubin IX alpha in the apicoplast. FEBS Lett. 2009;583:313–319. doi: 10.1016/j.febslet.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 67.Gisk B, Yasui Y, Kohchi T, Frankenberg-Dinkel N. Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions. Biochem J. 2010;425:425–434. doi: 10.1042/BJ20090775. [DOI] [PubMed] [Google Scholar]
- 68.Qiao YF, Molina H, Pandey A, Zhang J, Cole PA. Chemical rescue of a mutant enzyme in living cells. Science. 2006;311:1293–1297. doi: 10.1126/science.1122224. [DOI] [PubMed] [Google Scholar]
- 69.Muratore KE, Seeliger MA, Wang ZH, Fomina D, Neiswinger J, Havranek JJ, Baker D, Kuriyan J, Cole PA. Comparative Analysis of Mutant Tyrosine Kinase Chemical Rescue. Biochemistry. 2009;48:3378–3386. doi: 10.1021/bi900057g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spencer ALM, Bagai I, Becker DF, Zuiderweg ERP, Ragsdale SW. Protein/Protein Interactions in the Mammalian Heme Degradation Pathway HEME OXYGENASE-2, CYTOCHROME P450 REDUCTASE, AND BILIVERDIN REDUCTASE. J Biol Chem. 2014;289:29836–29858. doi: 10.1074/jbc.M114.582783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Good MC, Zalatan JG, Lim WA. Scaffold Proteins: Hubs for Controlling the Flow of Cellular Information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Agapakis CM, Silver PA. Modular electron transfer circuits for synthetic biology: insulation of an engineered biohydrogen pathway. Bioeng Bugs. 2010;1:413–418. doi: 10.4161/bbug.1.6.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zalatan JG, Herschlag D. The far reaches of enzymology. Nat Chem Biol. 2009;5:516–520. doi: 10.1038/nchembio0809-516. [DOI] [PubMed] [Google Scholar]
- 74.Ringe D, Petsko GA. How enzymes work. Science. 2008;320:1428–1429. doi: 10.1126/science.1159747. [DOI] [PubMed] [Google Scholar]
- 75.Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinkamp MP, Low-Nam ST, Yang S, Lidke KA, Lidke DS, Wilson BS. erbB3 is an active tyrosine kinase capable of homo- and heterointeractions. Mol Cell Biol. 2014;34:965–977. doi: 10.1128/MCB.01605-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu FT, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Protein Design: Toward Functional Metalloenzymes. Chem Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Razgulin A, Ma N, Rao J. Strategies for in vivo imaging of enzyme activity: an overview and recent advances. Chem Soc Rev. 2011;40:4186–4216. doi: 10.1039/c1cs15035a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.