

Abstract
Termed volumetric muscle loss (VML), the bulk loss of skeletal muscle tissue either through trauma or surgery overwhelms the capacity for repair, leading to the formation of non-contractile scar tissue. The myogenic potential, along with other factors that influence wound repair are known to decline with age. In order to develop effective treatment strategies for VML injuries that are effective across a broad range of patient populations, it is necessary to understand how the response to VML injury is affected by aging. Towards this end, this study was conducted to compare the response of young and aged animal groups to a lower extremity VML injury. Young (3 months, n=12) and aged (18 months, n=8) male Fischer 344 rats underwent surgical VML injury of the tibialis anterior muscle. Three months after VML injury it was found that young TA muscle was on average 16% heavier than aged muscle when no VML injury was performed and 25% heavier when comparing VML treated young and aged animals (p<0.0001, p<0.0001). Peak contractile force for both the young and aged groups was found to decrease significantly following VML injury, producing 65% and 59% of the contralateral limbs’ peak force, respectively (p<0.0001). However, there were no differences found for peak contractile force based on age, suggesting that VML affects muscle’s ability to repair, regardless of age. In this study, we used the ratio of collagen I to MyoD expression as a metric for fibrosis vs. myogenesis. Decreasing fiber cross-sectional area with advancing age (p<0.005) coupled with the ratio of collagen I to MyoD expression, which increased with age, supports the thought that regeneration is impaired in the aged population in favor of fibrosis (p=0.0241). This impairment is also exacerbated by the contribution of VML injury, where a 77-fold increase in the ratio of collagen I to MyoD was observed in the aged group (p<0.0002). The aged animal model described in this study provides a tool for investigators exploring not only the development of VML injury strategies but also the effect of aging on muscle regeneration.
Keywords: orthopedics, musculoskeletal, animal model, tibialis anterior, aging, VML
1. Introduction
Our multidisciplinary team is exploring the development of implantable biomaterials targeting the repair of damaged skeletal muscle (Hurd and others 2015; Kasukonis and others 2016). Towards that end we are particularly interested in understanding the response of muscle to volumetric muscle loss injury (Corona and others 2015; Grogan and others 2011). In response to strains, contusions, and crush injuries, in which the myocytes and satellite cell pool are damaged but the underlying muscle structure is largely preserved, muscle has a strong capacity for regeneration (Hill and others 2003; Mauro 1961; Stratos and others 2013). Muscle regeneration is characterized by progressive and coordinated inflammation, repair, and remodeling phases (Charge and Rudnicki 2004; Mann and others 2011; Turner and Badylak 2012). During the first hours and days after injury, phagocytic neutrophils and macrophages are recruited to the wound site to clear the injury of dead cells and necrotic tissue during what is termed the inflammatory phase. Transition from the inflammatory phase to the repair phase, is characterized by a shift in the phenotype of the macrophages from the inflammatory M1 phenotype, responsible for the digestion of necrotic tissues and promotion of satellite cell proliferation, to tissue remodeling M2 macrophages responsible for myoblast proliferation, growth and differentiation (Tidball 2005; Tidball and Villalta 2010). During the repair phase, satellite cells proliferate and differentiate into myoblasts that fuse with other myoblasts or with existing fibers to form new skeletal muscle (Turner and Badylak 2012). The remodeling phase is a continuation of the repair phase where myofibers mature and start to adhere to both native and newly remodeled extracellular matrix (ECM). In muscle, the regenerative process peaks anywhere from 7 to 30+ days depending on the type and severity of the injury (Benoit and Belt 1970; Nikolaou and others 1987; Warren and others 2007).
Alternatively, when significant skeletal muscle volume is lost (trauma or surgical resection) the regenerative cues provided by the underlying ECM structure is missing, and regeneration is poor. Termed volumetric muscle loss (VML), the bulk loss of muscle tissue (>20% by mass) overwhelms the capacity for regeneration, leading to the formation of non-contractile scar tissue at the defect site and functional impairment for the patient (Grogan and others 2011; Terada and others 2001). The poor clinical outcome following VML injury combined with a lack of effective treatment options motivates our exploration of VML regeneration. To better understand the sequence of events that follow VML injury and aid in the development of effective treatment strategies, various pre-clinical animal models have been developed (Chen and Walters 2013; Hou and others 2016; Turner and others 2012; Wu and others 2012; Zhang and others 2016). Among these, the hind limb muscles of the rat are a reliable pre-clinical model that recapitulates healing with non-contractile scar tissue observed in humans following VML injury (Wu and others 2012). In particular, the rat hind limb has become a standard model for military research (Aurora and others 2014; Aurora and others 2015; Corona and others 2013a; Corona and others 2014). This group of investigators is seeking treatments for VML injuries caused by devastating battlefield trauma. With the active military population in mind, the response to VML injury has typically been explored in young (<50% of the median lifespan = 24 months) rat models. However, in addition to traumatic injuries, the surgical removal of muscle as a treatment for soft tissue sarcoma is another anticipated target for VML repair strategies (Fischer and others 2015). This group of patients is typically older, and therefore an improved understanding of the response to VML injury in aged muscle may provide unique insights that could guide future treatment strategies.
With increasing age, both animals and humans develop pathological changes to muscle tissue which includes decreased muscle volume and increased adipose infiltration which reduces muscle strength and can increase the risk of injury (Freemont and Hoyland 2007). These aged-associated changes in muscle physiology can act as obstacles to muscle regeneration (Grounds 1998). We suspect that age related changes in muscle physiology might significantly influence the response to VML injury. It is well established that muscle resident satellite cells account for the majority of skeletal muscle’s regenerative capacity following injury (Conboy and Rando 2005). However, aging leads to an overall decrease in myogenic capacity mostly in part due to reduced satellite cell activity (Brack and Rando 2007b; Conboy and Rando 2005; Snow 1977). Specifically, it appears there is an age-related loss in satellite cell functionality, possibly indicating that the “aging” of the satellite cells renders them less responsive to regenerative cues (Brack and Rando 2007a; Charge and Rudnicki 2004; Gopinath and Rando 2008). In addition, a decreased responsiveness to the stimuli needed to induce activation and proliferation of satellite cells has been observed with aging (Brack and others 2007; Gopinath and Rando 2008; Kuang and others 2008; Vinciguerra and others 2010). When taken as a whole, the literature supports the conclusion that aging diminishes the regenerative capacity of muscle.
While aging associated changes in muscle regeneration are known to exist, the effects of aging on VML recovery has yet to be explored to our knowledge. With that gap in mind, an aged animal model of VML injury was developed to investigate and compare the regenerative response following VML injury of the hind limb tibialis anterior (TA) muscle. Two populations of Fisher 344 rats aged 3 months and 18 months were used in this study. These ages are representative of adolescent (25% of mean lifespan) and early elderly (75% of mean lifespan) rat lifespans. In order to assess the response to injury; muscle strength, mass, histology, and gene expression profiles were measured for both aged and young animals three-months post VML injury, a commonly explored VML recovery time point (Aurora and others 2015; Corona and others 2013a; Corona and others 2014; Merritt and others 2010; Wu and others 2012). Overall, the study was designed to compare the response between the two age groups and test the hypothesis that aged animals recover differently from VML injury when compared to young animals as might be evidenced by differences in myogenesis, contractile force, muscle atrophy, and tissue fibrosis
The results generated by this study not only provide some of the first insights into recovery from VML injury in aged muscle, but also provide baseline recovery measures from which to compare future VML injury strategies targeting aged patients.
2. Methods
2.1. Volumetric Muscle Loss Injury
In vivo VML injury studies were performed with young (3 month) and aged (18 months) male Fisher 3 44 rats. The young rats (n=12) were purchased commercially from Harlan Laboratories (Indianapolis, IN) and the aged rats (n=8) were obtained through the National Institute on Aging (Bethesda, MD). Surgical procedures and implant preparation methods were performed in accordance with protocols approved by the University of Arkansas IACUC (Protocol #14044) and guided by published methods (Wu and others 2012). Anesthesia was induced using isoflurane (1–3%) in oxygen. The implant site was surgically exposed through a 1–2 cm incision running parallel to the tibia. The TA was identified and a partial thickness VML defect (8mm diameter × 3mm deep) was created using a sterile biopsy punch (Figure 1). Data that was previously collected correlated animal body weight to TA mass for each animal group (Figure 2). From this relationship, the percentage of total TA mass excised during surgery was calculated for each animal, to help insure consistent defect creation across all animals. The average size of the VML defect was 18.7±0.6% and 19.5±0.6% for the young and aged animal groups respectively (Corona and others 2014). The difference in defect size between groups was not statistically significant (p=0.36). The contralteral limb served as an internal control for comparison and is denoted as the normal group. The deeper periosteum and surface skin layers were separately closed using an interrupted stich with a 5-0 absorbable suture (Vicryl, Ethicon, Summerville, MA). Postoperative analgesia consisted of 0.1mg/kg buprenorphine administered subcutaneously via injection (neck scruff) twice daily for two days. Animals also received anti-inflammatory medication (Carprophan) via a dietary gel cup (Medigel CPF, ClearH2O, Westbrook, ME) added as needed to each cage out to 7 days. Following surgery, animals were housed in standard-sized rat cages with unrestricted movement. The animals were allowed to bear weight on the operative extremity as tolerated. All animals were housed for a 12-week recovery period.
Figure 1.

Surgical VMLs are gross defect creations in the left TA muscle of the rat. Surgical site was cleaned and disinfected prior to a vertical incision that was made to expose underlying muscle (A). An 8mm biopsy punch was used to create a defect to a depth of 2mm and the amount of muscle removed is approximately equivalent to 20% of the TA weight (B). Surgical defects were created in the rat left hindlimb and the contralateral limbs were untouched, serving as internal controls. The surgical site was then sutured with 5-0 absorbable sutures (C).
Figure 2.

Correlation of animal weight and TA weight. (A) Aged animal (n=8) terminal weight vs TA weight. (B) Young animal (n=12) terminal weight vs TA weight. Linear regression analysis performed for all cases provided information to approximate TA weight given animal weight. From the TA weight, the size of the defect (20% of TA weight) can be determined.
2.2. In-situ Contractile Force Measurement
Peak tetanic contraction produced by the TA muscle of both young (n=12) and aged (n=8) rats were measured isometrically in vivo using similar procedures previously described18. After rats were anesthetized (2–2.5% isoflurane), the left hindlimb was prepared for measurement. To eliminate force contributions from the synergist muscle, distal tenotomies were performed on the extensor digitorum longus (EDL) and extensor halluces longus (EHL). The knee was stabilized in place using a custom mounting system and the knee and ankle were positioned at a right angle. The foot was attached to a foot-plate with surgical tape, which was attached to the shaft of a servomotor (Aurora Scientific, Inc., Mod. 305b) and was in turn controlled by a computer. Percutaneous needle electrodes were inserted into the anterior compartment of the TA to stimulate the peroneal nerve. Optimal voltage (2 – 5 V) was determined using a series of tetanic contractions (150Hz, 0.1 ms pulse width, 400 ms train). Average peak tetanic force for each animal was calculated from the average of 5 contractions. To minimize muscle fatigue, 1-minute rest periods were taken between contraction cycles. Commercial muscle physiology software (Aurora Scientific, Inc., Ontario, Canada) was used to collect contractile force versus time data. At the conclusion of electrophysiological testing, all animals were euthanized through carbon dioxide inhalation in accordance with guidelines provided by the 2013 AVMA Panel on Euthanasia of Animals.
2.3. Tissue Histology and Image Analysis
VML treated TA muscles along with contralateral untreated TA muscles were harvested from young (n=12) and aged (n=8) and trimmed to remove fascia and tendon, then weighed. EDL muscles were also collected and weighed for comparison between groups. Muscles were rinsed in sterile PBS, dabbed dry, and then weighed. Harvested muscles were flash frozen in liquid nitrogen, and stored at −80°C for histological analysis. Tissue cross-sections were obtained with the aid of a cryostat (Leica BioSystems) and maintained at a temperature between −25 to −20°C and thickness of 7 μm. Cross-sectional slices were taken from the top third of the TA muscle, the region where the surgical VML injury was created. Prior to immunostaining, slides were permeabilized in 0.1% 10X Triton then rinsed in phosphate-buffered solution (PBS, pH 7.4). Slides were then blocked in PBS containing 4% goat serum and 0.05% sodium azide for 1h at room temperature prior to incubation in primary antibodies including mouse-anti-collagen I IgG (1:500, Sigma Aldrich), rabbit polyclonal anti-collagen III (1:1000, Abcam), and mouse-anti-myosin IgG2B (MF-20, 1:10, Developmental Studies Hybridoma Bank, Iowa City, IA) for 4h at 4°C. Following PBS washes, slides were incubated in the appropriate corresponding Alexa Fluor 488 and 596 (1:500, Life Technologies) labeled secondary antibodies for 30 minutes at room temperature. Additional tissue sections were stained using commercially available Hematoxylin and Eosin (H&E) and Masson’s Trichrome staining kits following manufacturers’ guidelines (Sigma). All sections were mounted onto microscope slides and digitally imaged. Treated sections were evaluated for myofiber average cross-sectional area, formation, and organization as well as the formation and extent of VML site repair tissue for comparison to normal contralateral muscle tissue sections.
Collagen type I and III accumulation, a potential indicator of diffuse muscle fibrosis, in tissue regions away from the VML defect area was estimated using measures of collagen I (Sigma Aldrich, St. Louis, MO) and collagen III (Abcam, Cambridge, MA) immunoreactivity and guided by published methods (Bedair and others 2007). Tissue immunoreactivity to collagen I was calculated from fluorescent microscopic images (200X) using a custom algorithm developed for publically available image analysis software (Image J, NIH). Using the algorithm, images were converted to 8-bit, gray scale, and uniformly thresholded across all samples to isolate collagen type I and III positive tissue regions. From these images, the software was used to determine the percent collagen type I and III positive tissue area for all tissue sections. Similar image analysis methods were used to compute muscle fiber cross-sectional area from magnified (200X) images. Images were converted to gray scale and thresholded to isolate individual muscle fibers. Fiber cross sectional area (um2) for all fibers within an individual image (typically 50+ fibers) were calculated. Representative tissue sections collected from four animals per group were used for all calculations. Within each section, three separate regions were imaged and analyzed to calculate the average collagen type I and III percent area as well as average fiber cross-sectional area for each sample. A total of twelve images were analyzed for each treatment group.
2.4. Gene Expression
Real-time PCR was performed using the protocol reported by Washington et al (Washington and others 2013). In preparation for RT-PCR, samples of muscle from the defect site of the right and left TA (n=4 animals/experimental group) were homogenized with Trizol (Ambion, Carlsbad, CA)/choloroform (Sigma Aldrich, St. Louis, MO). Samples were treated with DNase (Invitrogen, Carlsbad, CA) and then RNA was extracted using an RNAeasy kit (Invitrogen, Carlsbad, CA). After quantification of RNA with a plate reader (BioTek, Winooski, VT), RNA was converted to cDNA with a kit (Invitrogen, Carlsbad, CA). TAQMAN primers (Invitrogen, Carlsbad, CA) for MyoD, Collagen I, Collagen III, ratios of Collagen I to MyoD and Collaen I to Collagen III, and 18S rRNA housekeeping were used to quantify the expression of desired genes. Experimental sample group expressions were normalized to 18S rRNA and then referenced to the contralateral normal limb. Gene expression levels are reported as fold change using the 2−(ΔΔCt) method.
2.5. Data Analysis
All data is presented as the mean ± standard error. Data was tested for normality using the Shapiro-Wilk Test. The key outcome measures, muscle mass, peak tetanic force, percent collagen I / III areas, fiber cross-sectional area, and gene expression fold changes were analyzed with two-way ANOVA. Post hoc comparison testing was performed using Tukey’s HSD test. All statistical analyses were performed using commercial statistical analysis software (JMP 11 Pro). Animal growth and defect size comparisons were made using a two-sided Students t-test. A standard p<0.05 level of significance was used for all statistical tests.
3. Results
3.1. Animal Body Weight
Both aged and young animals tolerated the surgery well and there were no signs of post-surgical infection. At one week post-implantation, all animals were fully ambulatory with no noticeable gait differences between groups. At the end of the study period, the young animals increased in mass by an average of 36% (312±3 vs. 424±7 g, p<0.0001) whereas the aged animals showed no significant change in body weight (Figure 3). Also, by the end of the 12-week study period the difference in average mass between the aged and young groups had narrowed to 3%. The difference in mass between young and aged animals at the end of the study period was not statistically significant (p=0.38). Over the study period, the young animals put on mass at a growth rate of 9.3±0.4 g/week while the aged animals lost mass at a negative growth rate of 2.3±0.9 g/week. Age (young vs. old) was shown to have a statistically significant effect on animal growth rate (p<0.05).
Figure 3.

Young and aged Fisher 344 rat weight (g) prior to surgery and at the end of the study period. The younger animals experienced significant weight gain post-surgery, whereas the aged animals lost weight. Values are mean+SEM. * represents p<0.05 Student’s t-test for the young group.
3.2. TA Peak Tetanic Contractile Force
Tetanic contraction plots for all muscles were characterized by a sharp rise in force, followed by a plateau at the peak force, and then a return to a no force resting state (Figure 4). There was a main effect of treatment with VML injury to decrease peak contractile force, regardless of age (p<0.0001). Based on age, peak contractile force showed a decreasing trend moving from the young to the aged group (p=0.08). The young and aged VML groups produced 65% (2.16±0.07 N/kg vs. 1.39±0.11 N/kg, p<0.0001) and 59% (2.02±0.05 N/kg vs 1.19±0.12 N/kg, p<0.0001) of their respective contralateral normal muscles in response to VML injury (Figure 4A).
Figure 4.

Peak tetanic force measurements of Young and Aged rats via electrophysiology. (A) Percutaneous needle electrodes were placed in the anterior compartment of the TA to stimulate the peroneal nerve. The knee was stabilized and the foot was attached to a muscle lever system for isometric measurements. (B) In order to account for normal growth over the 3-month period post-surgery, contractile force measurements were normalized to body weight (N/kg). The contralateral normal limbs for both age groups yielded similar values as well as showing a similar decrease in force production after VML injury. Values are mean+SEM. * represents p<0.05 two-way ANOVA for normal vs VML injury.
3.3. TA and EDL Muscle Mass
Upon gross examination the defect site was visible on TA muscle samples collected from both young and aged VML treated animals. Morphologically, there is a drastic difference in overall muscle appearance and size following VML injury (Figure 5). EDL muscles were not surgically disrupted during the TA VML defect treatment. Superficially, the EDL muscles associated with both the normal and VML treated TA muscles appeared normal, with the VML side EDL appearing slightly larger in size than the normal EDL. However, treated muscles were noticeably larger in size. There was a main effect of age and treatment to decrease TA muscle mass normalized to body weight, but no interaction effect was observed (p<0.0001, p<0.0001, Figure 5). Based on age, the young normal group compared to the aged normal group exhibited a 16% increase in normalized TA weight (1.61±0.04 mg/g vs. 1.37±0.05 mg/g, p<0.0001). A similar response to age was exhibited by the young VML group compared to the aged VML group with a 25% increase in normalized TA weight (1.27±0.02 mg/g vs. 0.99±0.02 mg/g, p<0.0001). In response to treatment within the young group, young normal TA mass was on average 24% heavier than young VML TA muscle (1.61±0.04 mg/g vs. 1.27±0.04 mg/g, p<0.0001, Figure 5). Similarly, within the aged group, aged normal TA mass on average was 32% heavier than aged VML TA muscle (1.37±0.05 mg/g vs 0.99±0.02 mg/g, p<0.0001, Figure 5). While VML injury decreased TA muscle mass for both young and aged animals, no interaction effect was detected, suggesting that the decrease was not disproportionate between age groups.
Figure 5.
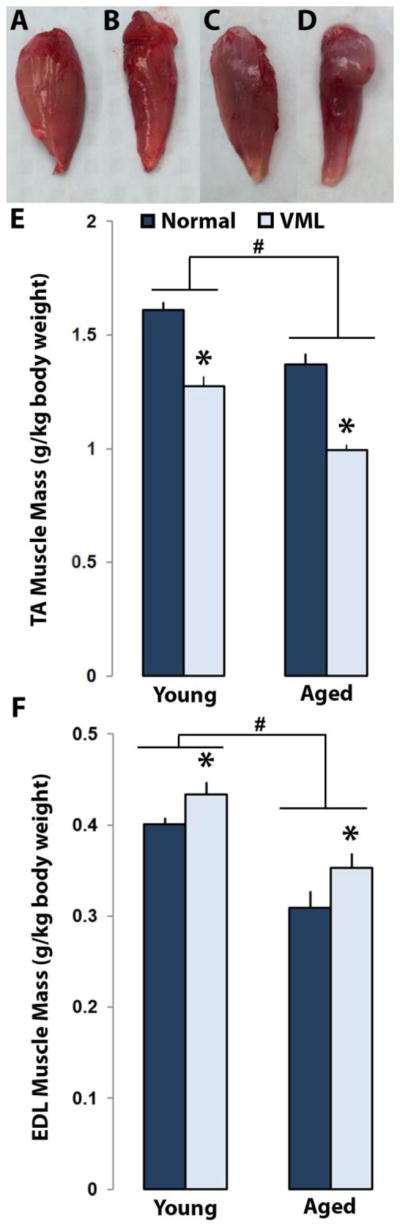
Comparison of TA and EDL muscle weight between normal and VML injury groups between Young (n=12) and Aged (n=8) rats. Gross morphological images of young normal (A), young VML (B), aged normal (C), and aged VML (D) TA muscles. At the end of the 3 month study period, terminal weights for TA (E) and EDL (F) of both young and aged normal and VML groups were measured. Values are mean+SEM. # represents p<0.05 two-way ANOVA for young vs aged. * represents p<0.05 two-way ANOVA for normal vs VML injury.
Interestingly for the normalized EDL weights, the main effect of age decreased EDL mass whereas treatment was found to have an effect to increase EDL mass (p<0.0001 and p<0.008, respectively). However, an interaction effect was not observed. Based on age, the aged normal group compared to the young normal group exhibited a 25% decrease in normalized EDL weight (0.4±0.01 mg/g vs. 0.31±0.02 mg/g, p<0.0001, Figure 5). Similarly, the aged VML group compared to the young VML group exhibited a 20% decrease in normalized EDL weight (0.43±0.01 mg/g vs. 0.35±0.02 mg/g, p<0.0001). Within the young group, there was an increase of 7% in EDL mass in response to VML injury (0.4±0.01 mg/g vs. 0.43±0.01 mg/g, p<0.008). Similarly, within the aged group in response to VML injury, there was an increase of 12% in EDL weight (0.31±0.02 mg/g vs. 0.35±0.02 mg/g, p<0.008).
3.4. VML Site Histology
Normal TA muscle histology for both young and aged groups (Figure 6A/B, E/F) was characterized by ordered circular bundles of MHC positive myofibers surrounded by collagen positive perimysium extending completely out to the anterior surface of the muscle. Twelve-weeks following VML injury, tissue sections prepared from young animals were characterized by collagen-dense repair layer located at the muscle surface. For the young VML muscles, MHC positive muscle fibers were observed up to the border of the repair layer, but did not extend into the repair tissue itself (Figure 6C, D). A similar distinct layer of collagen enriched repair tissue was not typically observed within the aged animal groups. Aged animal sections prepared from VML site tissue samples were frequently characterized by a region of diffuse collagen I deposition, as well as an overall appearance indicating increased MHC positive myofibers disorganization in the repair region (Figure 6G, H). While MHC positive muscle fibers were present they were generally less closely packed than in normal TA muscle sections.
Figure 6.

Comparison of histological cross-sections of young normal (A, B) and VML (C, D), aged normal (E, F) and VML (G, H) TA muscles. The inset depicting the TA shows from where the histological cross-sections were taken. Masson’s trichrome stained sections imaged at 100X magnification (A, C, E, G) depict collagen in blue/purple colored regions whereas muscle is shown in red/pink. Sections stained for collagen I (green) and myosin heavy chain (MHC, red) at 100X magnification (B, D, F, H). * indicates the collagen I rich scar region in the young muscles treated with VML (D). ** indicates the VML region for the aged group which is characterized by diffuse collagen I deposition and disorganized myofiber bundles compared to the young VML group (H). Masson’s trichrome was stained using a commercially available kit. Primary antibodies used were anti-collagen I mouse IgG (1:500) and anti-myosin mouse IgG2B (1:10). Secondary antibodies used were goat anti-mouse IgG (H+L) Alexa Fluor 488 and goat anti-mouse IgG2B Alexa Fluor 594.
3.5. Collagen Percent Area and Fiber Cross-sectional Area
Percent area positively stained for collagen I and III provided a quantitative measure of collagen content in the TA muscle regions outside the defect area (Figure 7A, B). There were no changes in percent area stained for collagen I between young normal and VML muscles (7.6±1.6% vs. 8.3±1.0%, Figure 7C). Similarly, aged normal and VML treated muscles showed no changes in percent collagen I area (6.0±1.3% vs. 8.3±0.9%).
Figure 7.

Representative TA cross-sections immunostained for Collagen I (A, green) and Collagen III (B, red). Stained images were then analyzed in ImageJ software to calculate % area of the stains as a function of cross-sectional area. No difference was found for collagen I area (C) but a decrease in collagen III area was found for the young group after VML injury (D). On the contrary, the aged group had an increase in collagen III following VML. Primary antibody stains used were anti-collagen I mouse IgG (1:500) and anti-collagen III rabbit polyclonal (1:500). Secondary antibody stains used were goat anti-mouse IgG (H+L) Alexa Fluor 488 and goat anti-rabbit IgG (H+L) Alexa Fluor 594. * represents p<0.05 two-way ANOVA for normal vs VML injury.
There was a main effect of VML injury to decrease collagen III percent area in the young group and increase in the aged group (p=0.011, Figure 7D). Collagen III area decreased by 13% (19.2%±0.2% and 16.8%±1.6%) for the young normal group compared to the young VML group. Collagen III percent area increased by 10% (20.8%±1.6% and 23%±3.0%) in the aged VML group compared to the aged normal group.
Similarly, age had a significant effect to decrease average TA fiber cross-sectional area (p=0.0365, Figure 8C). Average cross-sectional area decreased by 17% (2299±154 μm2 and 1926±122 μm2) for the aged normal group compared to the young normal group. For the aged VML group compared to the young VML group, fiber cross-sectional area decreased by 28% (2123±100 μm2 and 1585±318 μm2).
Figure 8.
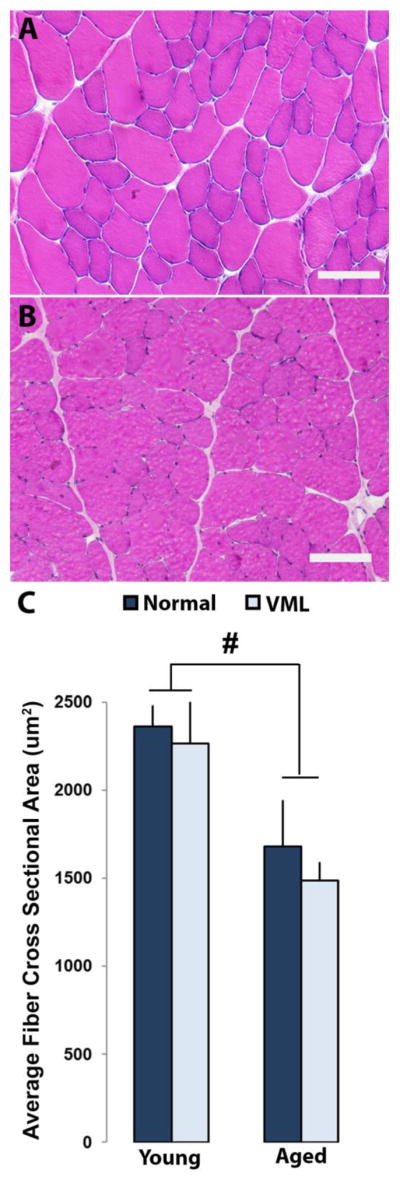
Rat TA muscle cross-sections of young (A) and aged (B) rats stained with H&E and imaged at 200X magnification. Average fiber cross-sectional areas were calculated using ImageJ software for young normal, young VML, aged normal, and aged VML groups (n=3, C). It was observed that fiber cross-sectional area decreases significantly with age, regardless of VML injury. # represents p<0.05 two-way ANOVA for young vs aged.
3.6. Gene Expression
There was a main effect of VML injury to decrease MyoD gene expression regardless of age in rat TA muscle (p=0.0231, Figure 9A). There was approximately a 13-fold decrease (1±0.58 and 0.08±0.04) in MyoD expression for the young VML group compared to the young normal group (Figure 9A). Comparing the aged VML group to the aged normal group, there was a 10-fold decrease in MyoD expression (1±0.21 vs. 0.1±0.03). For collagen I an interaction effect of age and VML injury was found to increase collagen I expression (p<0.003, Figure 9B). There were no differences found in collagen I expression within the young group, however, between the aged normal (1±0.33) and aged VML group (14±4), a 14-fold increase in collagen I expression was found (p<0.0009) exhibiting a treatment effect. Between the young VML (1.55±0.14) and aged VML (14±4) groups, a 9-fold increase in collagen I expression in the aged VML group compared to the young VML group was observed (p<0.0014, Figure 9B). Regarding collagen III gene expression, there was an interaction effect of age and VML injury on expression levels (p=0.0164, Figure 9C). Post-hoc comparison revealed that the young VML group responded with a 2-fold increase in collagen III expression over the young normal group to VML injury (1±0.14 vs. 2.14±0.25, p=0.034). While not exhibiting significance for VML injury within the aged group, the aged VML group exhibited a significantly lower response compared to the young VML group with a 3.5-fold decrease in collagen III expression (2.14±0.25 vs. 0.61±0. 26, p=0.013, Figure 9C).
Figure 9.

Normal gene expression compared to expression in response to VML. Expression of MyoD (A), Collagen I (B), Collagen III (C), ratio of Collagen I to MyoD (D), and ratio of Collagen I to Collagen III (E) are presented as fold-changes obtained using RT-PCR. Except for MyoD, which experienced only a treatment effect, interaction effects of age and treatment were found for all other genes/ratios tested. Values are mean+SEM. * represents p<0.05 two-way ANOVA for normal vs VML injury and # represents p<0.05 two-way ANOVA for young vs aged. Post-hoc analysis was performed upon finding significant interaction effects.
There was an interaction effect of age and treatment on the ratio of collagen I to MyoD (p<0.011). Post-hoc comparison revealed that the aged VML group responded with a 77-fold increase in the collagen I to MyoD ratio over the aged normal group to VML injury (0.95±0.25 vs 74.5±18.8, p<0.0002, Figure 9D). While not exhibiting significant differences for VML injury, the young group did exhibit significantly lower ratios of collagen I to MyoD compared to the aged VML group. Compared to the young VML group, the aged VML group exhibited a 2-fold increase in collagen I to MyoD ratio over the young VML group exhibiting an age effect (32.9±10.5 vs. 74.5±18.8, p=0.0241, Figure 9D).
Similarly, there was an interaction effect of age and VML injury on the ratio of collagen I to collagen III gene expression. Post-hoc comparison revealed that the aged VML group responded with an 8-fold increase in the collagen I to collagen III ratio over the aged normal group to VML injury (3.33±2.18 vs 27.8±5.21, p<0.0001, Figure 9E). While not exhibiting significant differences for VML injury, the young group did exhibit significantly lower ratios of collagen I to collagen III compared to the aged VML group. Compared to the young VML group, the aged VML group exhibited a 38-fold increase in collagen I to collagen III ratio over the young VML group exhibiting an age effect (0.736±0.044 vs. 27.8±5.2, p<0.0001, Figure 9D). A summary of the key outcome measures (mean ± standard deviation) for both aged and young animals is provided in Table 1.
Table 1.
Growth rate, final rat body weight, tibialis anterior muscle weight, EDL muscle weight, tibialis anterior muscle weight normalized to final body weight, peak contractile force, fiber cross-sectional area, and collagen I and III percent areas at the end of the 3 month study period for normal and VML treatment groups of both young and aged animals.
| Animal Group | Growth Rate (g/wk) | Final Body Weight (g) | TA weight (mg) | EDL weight (mg) | TA/Final BW (mg/g) | Peak Contractile Force (N/kg) | Fiber Cross-sectional Area (um2) | Collagen 1 Area (%) | Collagen 3 Area (%) |
|---|---|---|---|---|---|---|---|---|---|
| Young | 9.33±1.48 | 424.8±25.3 | |||||||
| Normal | 681.7±46.5 | 168.7±7.9 | 1.61±0.13 | 2.16±0.25 | 2299±308 | 7.6±3.3 | 19.2±0.4 | ||
| VML | 542.1±76.7 | 182.2±17.3 | 1.27±0.15 | 1.39±0.39 | 2123±200 | 6.0±2.5 | 16.8±3.2 | ||
| Aged | 2.25±2.39 | 436.4±33.2 | |||||||
| Normal | 598.3±68.3 | 134.2±22.5 | 1.37±0.13 | 2.02±0.15 | 1926±243 | 8.3±2.1 | 20.8±3.2 | ||
| VML | 432.6±38.0 | 153.0±17.7 | 0.99±0.07 | 1.19±0.34 | 1585±636 | 6.6±1.8 | 23.0±6. 0 |
Values are mean ± SD.
4. Discussion
Extracellular matrix (ECM) remodeling is a critical aspect of successful resolution of the regenerative response in skeletal muscle. Skeletal muscle has a robust capacity to recover from injury in which there is not a significant loss of the underlying ECM blueprint. However, when there is traumatic muscle injury and more than 20% of the muscle mass is lost (VML), the regenerative process will not resolve and there will be a functional deficit (Corona and others 2015; Garg and others 2014a; Grasman and others 2015; Grogan and others 2011). Though VML itself has been investigated, the response to VML in the aged population has yet to be explored. To our knowledge this is the first study to investigate how aging modulates the response to VML. The current study reports that aging muscle modifies the ECM response post-VML. The current study reports that the ECM response post-VML is modified in muscle from aged rats. It is of vital importance to understand how aged muscle response to VML so effective tissue engineering treatment strategies can be developed for this population to aid in skeletal muscle regeneration. Although not explored in this study, it is important to also recognize that VML injury is not limited to muscle fibers alone. VML injury destroys not only the contractile muscle tissue, but also nervous system tissue. The loss of nervous system tissue is potentially significant when one considers repair strategies, since the pool of progenitor cells needed for nerve regeneration may be distinct from those required for myogenesis (satellite cells). Like satellite cells, nerve progenitor cells are less numerous and robust in aged tissue (Verdu and others 2000), and therefore tissue engineering repair strategies targeting aged patients may need to carefully consider schemes that target both myofiber and nerve regeneration.
Sarcopenia is associated with decreased muscle mass and force deficits (Blough and Linderman 2000; Carson and others 2002; Washington and others 2014). Our data is consistent with the literature in that we demonstrate an aged-dependent decrease in muscle mass in both the TA and the EDL. Additionally we demonstrate a trend for an age-dependent decrease in peak tetanic force. VML is associated with incomplete skeletal muscle regeneration. We are the first to demonstrate how aging affects skeletal muscle post-VML. 3 months post-VML skeletal muscle mass had only returned to 75% of the contralateral control limb regardless of age. Additionally, muscle force only returned to approximately 60% of the contralateral control limb 3 months post-VML in young and aged rats. Taken together, the similar muscle mass loss and force deficit between young and aged rats, suggests age does not exacerbate the post-VML functional response.
Skeletal muscle regeneration is a complex process that is characterized by inflammation, ECM remodeling, and myofiber growth (Brown and others 2015; Washington and others 2011). Impairments to any one of these processes can lead to incomplete skeletal muscle regeneration (Washington and others 2011). The ECM plays an important role during skeletal muscle regeneration and acts as a functional link between skeletal muscle and bone. In skeletal muscle, the ECM is composed primarily of collagen I and collagen III (Kovanen 2002). Previous research has demonstrated that 28 days post-VML there is a larger induction of collagen III compared to collagen I gene expression in young rats (Garg and others 2014a). Our data is in agreement with this observation. We demonstrate an increase in collagen III gene expression with no change in collagen I gene expression 3 months post-VML. However, muscle from aged rats had a different response to VML. In muscle from aged rats, there was an increase in collagen I gene expression with no change in collagen III gene expression 3 months post-VML. The increased expression of collagen I gene expression may contribute to the impaired regenerative response associated with aged muscle. The mdx mouse is a model for Duchenne muscular dystrophy (DMD). DMD is a degenerative muscular disease associated with accumulation of ECM material (Fadic 2005). It has been demonstrated that collagen I is elevated during the lifespan in this animal (McDonald and others 2015). Collagen I inhibits muscular regeneration and supports the production of more collagen (Alexakis and others 2007). It has been demonstrated that type I collagen inhibits differentiation of C2C12 myoblasts (Alexakis and others 2007). Differentiation of C2C12 myoblasts and myotubes is associated with a reduction in transcription of collagen I mRNA (Alexakis and others 2007). The increased collagen I gene expression in aged muscle post-VML could negatively affect the ability of myoblasts to proliferate and differentiate and contribute to regeneration of aged skeletal muscle.
There was a difference in gene expression and protein expression, as determined by immunofluorescence, of collagen I and III post-VML. The collagen constitutive rate of synthesis is very low due to the long half-life (45–70 days) (Rucklidge and others 1992). Therefore, changes in gene expression are a more accurate representation of the acute effects of VML. Collagen I and collagen III are produced from 1–2 different genes and coalesce into a triple-helix pro-collagen molecule. Further processing is done leading to the mature collagen molecule. Collagen is degraded by a family of enzymes referred to as matrix metalloproteinases (MMPs). There is evidence that muscle MMP expression differs as a function of age (Dennis and others 2008; Robert and others 1997; Sharples and others 2012). Therefore, several factors can regulate collagen expression post-transcriptionally and explain the discrepancy between gene expression and protein expression.
In order to employ quantitative measures to assess myogenic capability and fibrosis, we calculated two ratios: the ratios of collagen I to MyoD and collagen I to collagen III expression. Due to MyoD’s role in regulating muscle differentiation and myogenesis, exploring the proportion of a pro-regenerative marker to a fibrotic marker such as collagen I can assist in assessing the degree of regeneration. Satellite cells/myoblasts are critical to optimal skeletal muscle regeneration. Upon activation satellite cells proliferate and differentiate into myoblasts and begin expressing MyoD (Hawke and Garry 2001). In aged animals, satellite cell number as well as activation and proliferation are depressed (Barani and others 2003; Kadi and others 2004), which should reduce the regenerative capacity of aged skeletal muscle. Our findings, indicating no difference in muscle strength between aged and young animals following VML injury would at first appear to contradict the published evidence. However, we suggest that our findings suggest the response to VML injury is not significantly different between aged and young animals, yet the response to repair strategies may still be. As we progress towards VML repair schemes (biomaterials, cell implantation, etc) the reduced regenerative capacity of aged muscle may be more evident.
VML was associated with an increase in the Collagen I to MyoD ratio regardless of age. However, there was a much larger increase in this ratio in the aged VML group. The increase is suggestive of a switch from myogenesis to fibrosis. There are numerous studies that demonstrate increased collagen deposition and fibrosis following VML with limited skeletal muscle regeneration (Corona and others 2014; Corona and others 2013b; VanDusen and others 2014). In addition, increased intermuscular adipose tissue (IMAT) has been found to be related to decreased whole body glucose metabolism, particularly in individuals who have experienced an injury resulting in loss of muscle function (Addison and others 2014). As a result, the decrease in muscle function that we observed in aged muscle could in part be attributed to fatty muscle degeneration occurring at both the VML injury site as well as within the surrounding uninjured muscle.
The ratio of collagen I to collagen III gene expression can provide insight in post-VML ECM composition changes. As skeletal muscle regeneration involves the migration of satellite cells and inflammatory cells through the ECM to the site of injury, the composition of the ECM is critical for an efficient regenerative response (Keely and Nain 2015). Garg et. al. demonstrated an impaired ability of satellite cells to migrate into the VML site (Garg and others 2014b). Collagen I is a triple helix that polymerizes into fibrils that are stabilized by covalent cross-links contributing to its high tensile strength. Collagen III forms a looser network and is ideal for maintaining structural integrity and distensibility of the collagen network. Higher ratios of collagen I to collagen III is indicative of a stiffer scar region due to the mechanical properties of collagen I whereas a lower ratio could mean greater flexibility and a greater tensility during recovery leading to a more well-defined tissue reformation. 3 months post-VML, muscle from aged rats had an increased collagen I to collagen III ratio whereas in young rats there was no difference. This increased ratio has been demonstrated in skin from aged individuals (Goldspink and others 1994). Due to the long half-life of collagen molecules (45–70 days) this sustained response 3 months post-VML could lead to an altered collagen I and collagen III protein expression at a later time point. This would suggest an altered ECM composition that could ultimately affect the regenerative capacity of skeletal muscle. Aged tissue is associated with accumulation of advanced glycation end products (AGEs) and their receptors (RAGEs) (Odetti and others 1998). AGEs have been demonstrated to induce expression of collagen I and inhibit collagen III expression (Tang and others 2008). AGEs and RAGE signaling have been implicated in chronic inflammation which is a characteristic associated with aging (Chuah and others 2013; Goto and others 2004; Kislinger and others 2001). Taken together, AGEs and chronic inflammation could alter the muscle microenvironment and contribute to the altered collagen III to collagen I ratio observed post-VML in aging skeletal muscle. As the functional outcome was similar between young and aged rats, the altered ECM response has implications for treatment strategies. This data implies treatment strategies in a young animal may not be effective in an older animal possibly due to the altered ECM composition post-VML.
5. Conclusion
In summary, this study is the first to describe the response to VML in muscle from aged rats. By characterizing and understanding the responses to VML by both young and aged muscle, various VML injury strategies can target areas that have a direct impact on myogenesis. With the age range of the young and aged rats being equivalent to the human age range of 9–18 years old and 54–63 years old, our aged model is effective in discriminating young vs aged responses to VML (Andreollo and others 2012). We clearly demonstrate an altered response to VML between young and aged rats. The aged rats had a much different ECM response as compared to the young rats. This would suggest that tissue engineering strategies aimed at treating VML should account for the age of the host.
Highlights.
Comparison between young and aged rats to volumetric muscle loss injury (VML).
Age does not exacerbate the post-VML functional response.
Age affects ECM composition and results in a more fibrotic post-VML environment.
Acknowledgments
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R15AR064481 as well as the Arkansas Biosciences Institute. We would also like to thank Dr. Thomas J. Walters of the US Army Institute of Surgical Research for his assistance during the study.
Footnotes
Disclosure Statement
No competing financial interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addison O, Marcus RL, Lastayo PC, Ryan AS. Intermuscular fat: a review of the consequences and causes. Int J Endocrinol. 2014;2014:309570. doi: 10.1155/2014/309570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexakis C, Partridge T, Bou-Gharios G. Implication of the satellite cell in dystrophic muscle fibrosis: a self-perpetuating mechanism of collagen overproduction. Am J Physiol Cell Physiol. 2007;293:C661–669. doi: 10.1152/ajpcell.00061.2007. [DOI] [PubMed] [Google Scholar]
- Andreollo NA, Santos EF, Araujo MR, Lopes LR. Rat’s age versus human’s age: what is the relationship? Arq Bras Cir Dig. 2012;25:49–51. doi: 10.1590/s0102-67202012000100011. [DOI] [PubMed] [Google Scholar]
- Aurora A, Garg K, Corona BT, Walters TJ. Physical rehabilitation improves muscle function following volumetric muscle loss injury. BMC Sports Sci Med Rehabil. 2014;6:41. doi: 10.1186/2052-1847-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora A, Roe JL, Corona BT, Walters TJ. An acellular biologic scaffold does not regenerate appreciable de novo muscle tissue in rat models of volumetric muscle loss injury. Biomaterials. 2015;67:393–407. doi: 10.1016/j.biomaterials.2015.07.040. [DOI] [PubMed] [Google Scholar]
- Barani AE, Durieux AC, Sabido O, Freyssenet D. Age-related changes in the mitotic and metabolic characteristics of muscle-derived cells. J Appl Physiol (1985) 2003;95:2089–2098. doi: 10.1152/japplphysiol.00437.2003. [DOI] [PubMed] [Google Scholar]
- Bedair H, Liu TT, Kaar JL, Badlani S, Russell AJ, Li Y, Huard J. Matrix metalloproteinase-1 therapy improves muscle healing. J Appl Physiol (1985) 2007;102:2338–2345. doi: 10.1152/japplphysiol.00670.2006. [DOI] [PubMed] [Google Scholar]
- Benoit PW, Belt WD. Destruction and regeneration of skeletal muscle after treatment with a local anaesthetic, bupivacaine (Marcaine) J Anat. 1970;107:547–556. [PMC free article] [PubMed] [Google Scholar]
- Blough ER, Linderman JK. Lack of skeletal muscle hypertrophy in very aged male Fischer 344 x Brown Norway rats. J Appl Physiol (1985) 2000;88:1265–1270. doi: 10.1152/jappl.2000.88.4.1265. [DOI] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science (New York, NY) 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Brack AS, Rando TA. Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem cell reviews. 2007a;3:226–237. doi: 10.1007/s12015-007-9000-2. [DOI] [PubMed] [Google Scholar]
- Brack AS, Rando TA. Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev. 2007b;3:226–237. doi: 10.1007/s12015-007-9000-2. [DOI] [PubMed] [Google Scholar]
- Brown LA, Lee DE, Patton JF, Perry RA, Jr, Brown JL, Baum JI, Smith-Blair N, Greene NP, Washington TA. Diet-induced obesity alters anabolic signalling in mice at the onset of skeletal muscle regeneration. Acta Physiol (Oxf) 2015;215:46–57. doi: 10.1111/apha.12537. [DOI] [PubMed] [Google Scholar]
- Carson JA, Lee WJ, McClung J, Hand GA. Steroid receptor concentration in aged rat hindlimb muscle: effect of anabolic steroid administration. J Appl Physiol (1985) 2002;93:242–250. doi: 10.1152/japplphysiol.01212.2001. [DOI] [PubMed] [Google Scholar]
- Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiological Reviews. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- Chen XK, Walters TJ. Muscle-derived decellularised extracellular matrix improves functional recovery in a rat latissimus dorsi muscle defect model. J Plast Reconstr Aesthet Surg. 2013;66:1750–1758. doi: 10.1016/j.bjps.2013.07.037. [DOI] [PubMed] [Google Scholar]
- Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam. 2013;2013:403460. doi: 10.1155/2013/403460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy IM, Rando TA. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle. 2005;4:407–410. doi: 10.4161/cc.4.3.1518. [DOI] [PubMed] [Google Scholar]
- Corona BT, Garg K, Ward CL, McDaniel JS, Walters TJ, Rathbone CR. Autologous minced muscle grafts: a tissue engineering therapy for the volumetric loss of skeletal muscle. Am J Physiol Cell Physiol. 2013a;305:C761–775. doi: 10.1152/ajpcell.00189.2013. [DOI] [PubMed] [Google Scholar]
- Corona BT, Rivera JC, Owens JG, Wenke JC, Rathbone CR. Volumetric muscle loss leads to permanent disability following extremity trauma. J Rehabil Res Dev. 2015;52:785–792. doi: 10.1682/JRRD.2014.07.0165. [DOI] [PubMed] [Google Scholar]
- Corona BT, Ward CL, Baker HB, Walters TJ, Christ GJ. Implantation of in vitro tissue engineered muscle repair constructs and bladder acellular matrices partially restore in vivo skeletal muscle function in a rat model of volumetric muscle loss injury. Tissue engineeringPart A. 2014;20:705–715. doi: 10.1089/ten.tea.2012.0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona BT, Wu X, Ward CL, McDaniel JS, Rathbone CR, Walters TJ. The promotion of a functional fibrosis in skeletal muscle with volumetric muscle loss injury following the transplantation of muscle-ECM. Biomaterials. 2013b;34:3324–3335. doi: 10.1016/j.biomaterials.2013.01.061. [DOI] [PubMed] [Google Scholar]
- Dennis RA, Przybyla B, Gurley C, Kortebein PM, Simpson P, Sullivan DH, Peterson CA. Aging alters gene expression of growth and remodeling factors in human skeletal muscle both at rest and in response to acute resistance exercise. Physiol Genomics. 2008;32:393–400. doi: 10.1152/physiolgenomics.00191.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadic R. Cell surface gene expression regulation molecules in dystrophinopathy: mdx vs Duchenne. Biol Res. 2005;38:375–380. doi: 10.4067/s0716-97602005000400010. [DOI] [PubMed] [Google Scholar]
- Fischer S, Soimaru S, Hirsch T, Kueckelhaus M, Seitz C, Lehnhardt M, Goertz O, Steinau HU, Daigeler A. Local tendon transfer for knee extensor mechanism reconstruction after soft tissue sarcoma resection. J Plast Reconstr Aesthet Surg. 2015;68:729–735. doi: 10.1016/j.bjps.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Freemont AJ, Hoyland JA. Morphology, mechanisms and pathology of musculoskeletal ageing. The Journal of pathology. 2007;211:252–259. doi: 10.1002/path.2097. [DOI] [PubMed] [Google Scholar]
- Garg K, Corona BT, Walters TJ. Losartan administration reduces fibrosis but hinders functional recovery after volumetric muscle loss injury. J Appl Physiol (1985) 2014a;117:1120–1131. doi: 10.1152/japplphysiol.00689.2014. [DOI] [PubMed] [Google Scholar]
- Garg K, Ward CL, Rathbone CR, Corona BT. Transplantation of devitalized muscle scaffolds is insufficient for appreciable de novo muscle fiber regeneration after volumetric muscle loss injury. Cell Tissue Res. 2014b;358:857–873. doi: 10.1007/s00441-014-2006-6. [DOI] [PubMed] [Google Scholar]
- Goldspink G, Fernandes K, Williams PE, Wells DJ. Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscular disorders : NMD. 1994;4:183–191. doi: 10.1016/0960-8966(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7:590–598. doi: 10.1111/j.1474-9726.2008.00399.x. [DOI] [PubMed] [Google Scholar]
- Goto K, Mizutani A, Shingu C, Hasegawa A, Hidaka S, Ito K, Iwasaka H, Noguchi T. Colforsin daropate does not affect the cerebral blood-flow in cardiac surgery patients under cardiopulmonary bypass. Masui. 2004;53:385–390. [PubMed] [Google Scholar]
- Grasman JM, Zayas MJ, Page RL, Pins GD. Biomimetic scaffolds for regeneration of volumetric muscle loss in skeletal muscle injuries. Acta Biomater. 2015;25:2–15. doi: 10.1016/j.actbio.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grogan BF, Hsu JR Skeletal Trauma Research C. Volumetric muscle loss. The Journal of the American Academy of Orthopaedic Surgeons. 2011;19(Suppl 1):S35–37. doi: 10.5435/00124635-201102001-00007. [DOI] [PubMed] [Google Scholar]
- Grounds MD. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Annals of the New York Academy of Sciences. 1998;854:78–91. doi: 10.1111/j.1749-6632.1998.tb09894.x. [DOI] [PubMed] [Google Scholar]
- Hawke TJ, Garry DJ. Myogenic satellite cells: physiology to molecular biology. J Appl Physiol (1985) 2001;91:534–551. doi: 10.1152/jappl.2001.91.2.534. [DOI] [PubMed] [Google Scholar]
- Hill M, Wernig A, Goldspink G. Muscle satellite (stem) cell activation during local tissue injury and repair. J Anat. 2003;203:89–99. doi: 10.1046/j.1469-7580.2003.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Gong C, Zhu Y. In vitro construction and in vivo regeneration of esophageal bilamellar muscle tissue. J Biomater Appl. 2016;30:1373–1384. doi: 10.1177/0885328215627585. [DOI] [PubMed] [Google Scholar]
- Hurd SA, Bhatti NM, Walker AM, Kasukonis BM, Wolchok JC. Development of a biological scaffold engineered using the extracellular matrix secreted by skeletal muscle cells. Biomaterials. 2015;49:9–17. doi: 10.1016/j.biomaterials.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadi F, Charifi N, Denis C, Lexell J. Satellite cells and myonuclei in young and elderly women and men. Muscle Nerve. 2004;29:120–127. doi: 10.1002/mus.10510. [DOI] [PubMed] [Google Scholar]
- Kasukonis B, Kim J, Washington T, Wolchok J. Development of an infusion bioreactor for the accelerated preparation of decellularized skeletal muscle scaffolds. Biotechnol Prog. 2016 doi: 10.1002/btpr.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P, Nain A. Capturing relevant extracellular matrices for investigating cell migration. F1000Res. 2015;4 doi: 10.12688/f1000research.6623.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ, Jr, Taguchi A, Olson K, Bucciarelli L, Goova M, Hofmann MA, Cataldegirmen G, D’Agati V, Pischetsrieder M, Stern DM, Schmidt AM. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2001;21:905–910. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- Kovanen V. Intramuscular extracellular matrix: complex environment of muscle cells. Exerc Sport Sci Rev. 2002;30:20–25. doi: 10.1097/00003677-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Kuang S, Gillespie MA, Rudnicki MA. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell stem cell. 2008;2:22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, Munoz-Canoves P. Aberrant repair and fibrosis development in skeletal muscle. Skeletal muscle. 2011;1:21-5040-5041-5021. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AA, Hebert SL, Kunz MD, Ralles SJ, McLoon LK. Disease course in mdx:utrophin+/− mice: comparison of three mouse models of Duchenne muscular dystrophy. Physiol Rep. 2015;3 doi: 10.14814/phy2.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt EK, Cannon MV, Hammers DW, Le LN, Gokhale R, Sarathy A, Song TJ, Tierney MT, Suggs LJ, Walters TJ, Farrar RP. Repair of traumatic skeletal muscle injury with bone-marrow-derived mesenchymal stem cells seeded on extracellular matrix. Tissue engineeringPart A. 2010;16:2871–2881. doi: 10.1089/ten.TEA.2009.0826. [DOI] [PubMed] [Google Scholar]
- Nikolaou PK, Macdonald BL, Glisson RR, Seaber AV, Garrett WE., Jr Biomechanical and histological evaluation of muscle after controlled strain injury. Am J Sports Med. 1987;15:9–14. doi: 10.1177/036354658701500102. [DOI] [PubMed] [Google Scholar]
- Odetti P, Aragno I, Garibaldi S, Valentini S, Pronzato MA, Rolandi R. Role of advanced glycation end products in aging collagen, A scanning force microscope study. Gerontology. 1998;44:187–191. doi: 10.1159/000022008. [DOI] [PubMed] [Google Scholar]
- Robert V, Besse S, Sabri A, Silvestre JS, Assayag P, Nguyen VT, Swynghedauw B, Delcayre C. Differential regulation of matrix metalloproteinases associated with aging and hypertension in the rat heart. Lab Invest. 1997;76:729–738. [PubMed] [Google Scholar]
- Rucklidge GJ, Milne G, McGaw BA, Milne E, Robins SP. Turnover rates of different collagen types measured by isotope ratio mass spectrometry. Biochim Biophys Acta. 1992;1156:57–61. doi: 10.1016/0304-4165(92)90095-c. [DOI] [PubMed] [Google Scholar]
- Sharples AP, Player DJ, Martin NR, Mudera V, Stewart CE, Lewis MP. Modelling in vivo skeletal muscle ageing in vitro using three-dimensional bioengineered constructs. Aging Cell. 2012;11:986–995. doi: 10.1111/j.1474-9726.2012.00869.x. [DOI] [PubMed] [Google Scholar]
- Snow MH. The effects of aging on satellite cells in skeletal muscles of mice and rats. Cell Tissue Res. 1977;185:399–408. doi: 10.1007/BF00220299. [DOI] [PubMed] [Google Scholar]
- Stratos I, Li Z, Herlyn P, Rotter R, Behrendt AK, Mittlmeier T, Vollmar B. Vitamin D increases cellular turnover and functionally restores the skeletal muscle after crush injury in rats. The American journal of pathology. 2013;182:895–904. doi: 10.1016/j.ajpath.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Tang M, Zhong M, Shang Y, Lin H, Deng J, Jiang H, Lu H, Zhang Y, Zhang W. Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell Mol Life Sci. 2008;65:2924–2932. doi: 10.1007/s00018-008-8255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada N, Takayama S, Yamada H, Seki T. Muscle repair after a transsection injury with development of a gap: an experimental study in rats. Scand J Plast Reconstr Surg Hand Surg. 2001;35:233–238. doi: 10.1080/028443101750523131. [DOI] [PubMed] [Google Scholar]
- Tidball JG. Inflammatory processes in muscle injury and repair. American journal of physiologyRegulatory, integrative and comparative physiology. 2005;288:R345–353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. American journal of physiologyRegulatory, integrative and comparative physiology. 2010;298:R1173–1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner NJ, Badylak JS, Weber DJ, Badylak SF. Biologic scaffold remodeling in a dog model of complex musculoskeletal injury. J Surg Res. 2012;176:490–502. doi: 10.1016/j.jss.2011.11.1029. [DOI] [PubMed] [Google Scholar]
- Turner NJ, Badylak SF. Regeneration of skeletal muscle. Cell and tissue research. 2012;347:759–774. doi: 10.1007/s00441-011-1185-7. [DOI] [PubMed] [Google Scholar]
- VanDusen KW, Syverud BC, Williams ML, Lee JD, Larkin LM. Engineered skeletal muscle units for repair of volumetric muscle loss in the tibialis anterior muscle of a rat. Tissue Eng Part A. 2014;20:2920–2930. doi: 10.1089/ten.tea.2014.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu E, Ceballos D, Vilches JJ, Navarro X. Influence of aging on peripheral nerve function and regeneration. Journal of the peripheral nervous system : JPNS. 2000;5:191–208. doi: 10.1046/j.1529-8027.2000.00026.x. [DOI] [PubMed] [Google Scholar]
- Vinciguerra M, Musaro A, Rosenthal N. Regulation of muscle atrophy in aging and disease. Advances in Experimental Medicine and Biology. 2010:211–233. doi: 10.1007/978-1-4419-7002-2_15. [DOI] [PubMed] [Google Scholar]
- Warren GL, Summan M, Gao X, Chapman R, Hulderman T, Simeonova PP. Mechanisms of skeletal muscle injury and repair revealed by gene expression studies in mouse models. The Journal of physiology. 2007;582:825–841. doi: 10.1113/jphysiol.2007.132373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington TA, Brown L, Smith DA, Davis G, Baum J, Bottje W. Monocarboxylate transporter expression at the onset of skeletal muscle regeneration. Physiological reports. 2013;1:e00075. doi: 10.1002/phy2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington TA, Healey JM, Thompson RW, Lowe LL, Carson JA. Lactate dehydrogenase regulation in aged skeletal muscle: Regulation by anabolic steroids and functional overload. Exp Gerontol. 2014;57:66–74. doi: 10.1016/j.exger.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Washington TA, White JP, Davis JM, Wilson LB, Lowe LL, Sato S, Carson JA. Skeletal muscle mass recovery from atrophy in IL-6 knockout mice. Acta Physiol (Oxf) 2011;202:657–669. doi: 10.1111/j.1748-1716.2011.02281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Corona BT, Chen X, Walters TJ. A Standardized Rat Model of Volumetric Muscle Loss Injury for the Development of Tissue Engineering Therapies BioResearch Open Access. 2012;1:280–290. doi: 10.1089/biores.2012.0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Hu ZQ, Turner NJ, Teng SF, Cheng WY, Zhou HY, Zhang L, Hu HW, Wang Q, Badylak SF. Perfusion-decellularized skeletal muscle as a three-dimensional scaffold with a vascular network template. Biomaterials. 2016;89:114–126. doi: 10.1016/j.biomaterials.2016.02.040. [DOI] [PubMed] [Google Scholar]