

Abstract
Phosphoinositides (PIs), a small fraction of the cellular lipids, function in almost all cellular physiological processes and especially in intracellular membrane trafficking events. PIs play a critical role in autophagy, but only PI(3)P is well studied. In this issue of The EMBO Journal, Hasegawa et al (2016) identified INPP5E, an inositol polyphosphate 5‐phosphatase, as a novel regulator of autophagy. INPP5E controls the level of PI(3,5)P2 at the lysosome and thereby locally regulates the actin cytoskeleton and autophagosome–lysosome fusion.
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Molecular Biology of Disease
Kinases and phosphatases alter the phosphorylation status of phosphatidylinositol in the 3‐, 4‐, and 5‐positions of the inositol ring to generate distinct phosphoinositides (PIs), widely known as the second messengers in transducing signals from cell surface receptors, like G protein‐coupled receptors (GPCRs) (Marinissen & Gutkind, 2001). PIs are also important for endomembrane identity, for shaping membranes, controlling vesicle trafficking, and organelle physiology. PI(3)P, for example, plays a significant role in endocytic trafficking and is a well‐known regulator of autophagy (Petiot et al, 2000). PI(4)P, PI(4,5)P2, and PI(3,4,5)P3 primarily localize to the plasma membrane, and PI(4)P is not only an intermediate of PI(4,5)P2 synthesis, but also has important functions in controlling plasma membrane ion channels (Hammond et al, 2012). PI(4,5)P2, a substrate of class I phosphoinositide‐3‐kinases and the precursor of PI(3,4,5)P3, is thought to be an important molecule for regulated exo‐ and endocytosis, while PI(3,4,5)P3 itself is critical for plasma membrane polarization and membrane traffic (Balla, 2013). All these signals suggest that PIs other than PI(3)P may also play a critical role in autophagy.
Polyphosphate‐5‐phosphatases, which can remove the phosphate from the 5‐position of PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3, are divided into four types, and INPP5E is the only type IV 5‐phosphatase with a high affinity toward dephosphorylating PI(4,5)P2 and PI(3,4,5)P3 (Kisseleva et al, 2000; Kong et al, 2000). This protein contains an N‐terminal proline‐rich segment with 13 PxxP motifs, followed by a putative immunoreceptor activation motif (ITAM) domain, two critical central phosphatase domains, and a C‐terminal farnesylation CAAX motif (Kisseleva et al, 2000). We used to think that this enzyme only localizes at the Golgi and partially at the plasma membrane, but Hasegawa et al (2016) now found that INPP5E also localizes to lysosomes in neuroblastoma cells.
Previous work has shown that INPP5E coordinates the initiation of ciliogenesis. It localizes to the ciliary axoneme and colocalizes with fluorescently tagged centrin‐2, which indicates the base of cilia. Defective primary cilia cause a broad spectrum of human genetic disorders, termed ciliopathies, and Joubert syndrome is one of them. Joubert syndrome is an autosomal recessive disorder presenting with psychomotor delay, hypotonia, ataxia, oculomotor apraxia, and neonatal breathing abnormalities. Joubert syndrome‐associated mutation of INPP5E causes an unstable cilium in response to stimulation (Bielas et al, 2009).
It is widely believed that Joubert syndrome is a cilia‐associated disorder, but in this issue of The EMBO Journal, Hasegawa et al (2016) show that INPP5E also plays a critical role in autophagy and that autophagy is dysfunctional in the presence of disease‐associated mutant INPP5E. The authors performed an siRNA screen for phosphatases of PIs to reveal the potential functions of PIs other than the well‐studied PI(3)P in autophagic flux regulation. The result showed that the knockdown of INPP5E significantly decreased autophagic flux. Further analyses indicated that INPP5E functions after the autophagosome is sealed and that it is required for autophagosome–lysosome fusion.
The authors also revealed that this feature is dependent on both phosphatase activity and its lysosome localization. However, by using LysoSensor, they found that INPP5E is not essential to maintain the luminal acidity of the lysosome, and its degradation activity is also not affected by INPP5E knockdown. Furthermore, after knocking down INPP5E, the lysosomes can still degrade DQ‐BSA and EGF normally, showing that the lysosome–endosome fusion and degradation pathways are not affected in INPP5E‐depleted cells. This indicates that the effect of INPP5E on fusion is specific to autophagosomal–lysosomal fusion. To understand how INPP5E affects this fusion process, it was essential to decipher the 5‐PI substrate of INPP5E in this context. Using phosphonate‐specific protein probes, surprisingly PI(3,5)P2 (but not PI(4,5)P2) was shown to be the specific substrate of INPP5E on the lysosome.
Figure 1. INPP5E is essential in neurons for autophagosome–lysosome fusion.
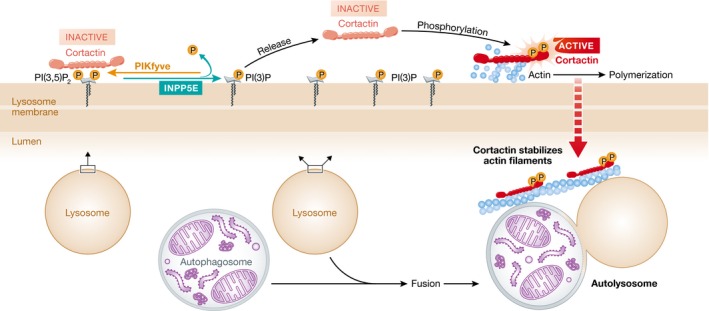
INPP5E partially localizes on lysosomes where it converts PI(3,5)P2 to PI(3)P. Cortactin can directly bind PI(3,5)P2, and INPP5E decreases PI(3,5)P2 levels to potentially release cortactin. Released cortactin then can be phosphorylated by kinases to become activated. Activated cortactin functions to promote actin polymerization and to maintain the stability of actin filaments on lysosomes. Hasegawa et al (2016) suggest that decreased actin filament abundance due to PI(3,5)P2‐bound cortactin is responsible for the compromised autophagosome–lysosome fusion observed in INPP5E‐deficient and disease‐associated mutant‐expressing neuronal cells.
To uncover the functional mechanism of INPP5E in autophagy, Hasegawa et al (2016) tested the effect of INPP5E on actin polymerization at the lysosome. They found that INPP5E can enhance actin polymerization at the lysosome, a process necessary for autophagosome–lysosome fusion. They also provided a plausible mechanism. It was previously reported that cortactin binds to actin via its actin filament‐binding region, and PI(3,5)P2 competes with actin filaments for cortactin binding (Hong et al, 2015). Hasegawa et al (2016) found that INPP5E decreases the PI(3,5)P2 levels on the lysosome and therefore releases cortactin from PI(3,5)P2 trapping. Cortactin is then further activated by phosphorylation, and the activated cortactin binds to actin, which improves and stabilizes actin polymerization on the lysosome, an event that is essential for autophagosomal fusion with lysosomes.
As mentioned above, INPP5E is well known for its role in ciliogenesis and is mutated in the ciliopathy Joubert syndrome. In this study, Hasegawa et al (2016) use the Joubert syndrome‐related INPP5E mutation to attempt rescue of the autophagy defect in INPP5E‐knockdown cells. None of the disease‐associated mutations can rescue the autophagy defect caused by the depletion of INPP5E. Thus, the results indicate that Joubert syndrome may not only be a ciliopathy, but also a disease linked to autophagy deficiency, raising the possibility to treat Joubert syndrome by targeting autophagy. The study cannot exclude the possibility that INPP5E regulates autophagy through its effects on primary cilia in ciliated cells and likely centrosomes in non‐ciliated cells. It will be helpful in the future to decipher whether autophagy is directly affected or whether INPP5E‐regulated cilia modulate autophagy.
Taken together, the study reveals that the phosphatase INPP5E, and the turnover of PI(3,5)P2 mediated by it, are necessary for actin polymerization to allow autophagosome–lysosome fusion. However, it remains unclear how INPP5E activates cortactin. Is there a direct interaction or an indirect interaction via PI(3,5)P2? INPP5E is well known for its effect on primary cilia (Bielas et al, 2009; Jacoby et al, 2009), and ciliogenesis is highly related to autophagy (Pampliega et al, 2013; Tang et al, 2013). Future investigations will reveal whether and how INPP5E‐dependent PI(3,5)P2 conversion into PI(3)P, autophagy, and ciliogenesis are potentially all interconnected.
See also: J Hasegawa et al (September 2016)
References
- Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93: 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al‐Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel‐Aleem A, Rosti RO (2009) Mutations in INPP5E, encoding inositol polyphosphate‐5‐phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41: 1032–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, Fischer MJ, Anderson KE, Holdich J, Koteci A, Balla T, Irvine RF (2012) PI4P and PI (4, 5) P2 are essential but independent lipid determinants of membrane identity. Science 337: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa J, Iwamoto R, Otomo T, Nezu A, Hamasaki M, Yoshimori T (2016) Autophagosome‐lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J 35: 1853–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong NH, Qi A, Weaver AM (2015) PI(3, 5)P2 controls endosomal branched actin dynamics by regulating cortactin‐actin interactions. J Cell Biol 210: 753–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Kisseleva MV, Wilson MP, Majerus PW (2000) The isolation and characterization of a cDNA encoding phospholipid‐specific inositol polyphosphate 5‐phosphatase. J Biol Chem 275: 20110–20116 [DOI] [PubMed] [Google Scholar]
- Kong AM, Speed CJ, O'Malley CJ, Layton MJ, Meehan T, Loveland KL, Cheema S, Ooms LM, Mitchell CA (2000) Cloning and characterization of a 72‐kDa inositol‐polyphosphate 5‐phosphatase localized to the Golgi network. J Biol Chem 275: 24052–24064 [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS (2001) G‐protein‐coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: 368–376 [DOI] [PubMed] [Google Scholar]
- Pampliega O, Orhon I, Patel B, Sridhar S, Diaz‐Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM (2013) Functional interaction between autophagy and ciliogenesis. Nature 502: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Ogier‐Denis E, Blommaart EF, Meijer AJ, Codogno P (2000) Distinct classes of phosphatidylinositol 3′‐kinases are involved in signaling pathways that control macroautophagy in HT‐29 cells. J Biol Chem 275: 992–998 [DOI] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]