

Abstract
Autophagy is a multistep membrane traffic pathway. In contrast to autophagosome formation, the mechanisms underlying autophagosome–lysosome fusion remain largely unknown. Here, we describe a novel autophagy regulator, inositol polyphosphate‐5‐phosphatase E (INPP5E), involved in autophagosome–lysosome fusion process. In neuronal cells, INPP5E knockdown strongly inhibited autophagy by impairing the fusion step. A fraction of INPP5E is localized to lysosomes, and its membrane anchoring and enzymatic activity are necessary for autophagy. INPP5E decreases lysosomal phosphatidylinositol 3,5‐bisphosphate (PI(3,5)P2), one of the substrates of the phosphatase, that counteracts cortactin‐mediated actin filament stabilization on lysosomes. Lysosomes require actin filaments on their surface for fusing with autophagosomes. INPP5E is one of the genes responsible for Joubert syndrome, a rare brain abnormality, and mutations found in patients with this disease caused defects in autophagy. Taken together, our data reveal a novel role of phosphoinositide on lysosomes and an association between autophagy and neuronal disease.
Keywords: autophagosomes, Joubert syndrome, lysosomes, phosphoinositides
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Molecular Biology of Disease
Introduction
Autophagy is an intracellular membrane traffic pathway that delivers cargos in the cytoplasm to lysosomes for the purpose of degradation (Mizushima & Komatsu, 2011; Mizushima et al, 2011; Lamb et al, 2013). During autophagy, flattened membrane compartments called isolation membranes or phagophores form de novo as needed, elongate to surround a portion of the cytoplasm, and finally close to form sealed double‐membranous autophagosomes. The completed autophagosomes then fuse with lysosomes. In hybrid organelles called autolysosomes, the sequestered contents are degraded. The autophagy‐related (Atg) proteins, which are conserved from yeast to mammals, have been identified as essential components of the autophagic machinery that tightly regulates the process of autophagy. The primary function of the pathway is to protect cells against stress, including nutrient starvation, making it possible for them to recycle nutrients such as amino acids and lipids from digested organelles. Furthermore, accumulating evidence indicates that autophagy plays pivotal roles in suppression of various human disorders including infectious diseases, inflammatory diseases, neurodegeneration, cancer, diabetes, and heart failure. Thus, autophagy is a potential therapeutic target for treatment of these diseases (Mizushima & Levine, 2010; Singh & Cuervo, 2011; Schneider & Cuervo, 2014).
In addition to Atg and other protein components, a specific phospholipid, phosphatidylinositol 3‐phosphate (PI(3)P), is also involved in autophagy (Kihara et al, 2001; Backer, 2008; Simonsen & Tooze, 2009; Dall'Armi et al, 2013). Whereas some PI(3)P localizes to endosomes and controls membrane trafficking along the endocytic pathway, some of it localizes to isolation membranes, forming autophagosomes (also called phagophores). PI(3)P in isolation membranes is generated by the class III PI3‐kinase Vps34, and impairment of its activity results in a defect in autophagosome formation. PI(3)P recruits its effectors, such as DFCP1 and WIPI proteins (Atg18 in yeast), to the membranes. Although the precise role of these proteins in autophagy remains elusive, recent work showed that WIPI2 recruits the Atg12–5–16L1 complex, which is essential for autophagosome formation, to the membrane by binding to Atg16L1 (Dooley et al, 2014). Furthermore, several groups including ours have reported that myotubularin‐related protein 3 (MTMR3) and MTMR14/Jumpy, both of which dephosphorylate PI(3)P to phosphatidylinositol, associate with autophagosomes and negatively control autophagy by reducing the level of PI(3)P (Vergne et al, 2009; Taguchi‐Atarashi et al, 2010). By contrast, MTMR6 promotes autophagosome formation (Mochizuki et al, 2013). Because this protein localizes to the Golgi apparatus, reduction in PI(3)P on that organelle may indirectly promote autophagy.
PI(3)P is one of eight phosphoinositides (PIs) present in animal cells. Each PI has its own specific distribution pattern in the cytoplasmic leaflet of the plasma and organelle membranes. Like PI(3)P, other PIs also play crucial roles in a wide range of cellular events including cytoskeletal dynamics, intracellular signaling, and vesicular trafficking (Vicinanza et al, 2008; Balla, 2013). For instance, PI(4,5)P2 is mainly localized to the plasma membrane and is involved in trafficking in the endocytic pathway. PI(4)P is concentrated in the trans‐Golgi network and recruits its effectors, such as oxysterol‐binding protein, to control the secretory pathway (Di Paolo & De Camilli, 2006). Therefore, there is a possibility that PIs other than PI(3)P also function in other steps of autophagy. Indeed, the decrease in PI(3,5)P2 level results in the inhibition of autophagy albeit the mechanism has been unclear (Ferguson et al, 2009; de Lartigue et al, 2009). Because the level and localization of PIs are controlled by phosphoinositide phosphatases (Sasaki et al, 2009; Balla, 2013), we decided to perform siRNA screening of phosphoinositide phosphatases other than MTMRs by assaying the effect of knockdown on autophagy.
The screen revealed inositol polyphosphate‐5‐phosphatase E (INPP5E), a PI 5‐phosphatase, as a positive regulator of autophagy. The enzyme localizes to lysosomes and is required for fusion between autophagosome and lysosomes. Because mutations in INPP5E cause Joubert syndrome, a rare disorder affecting the cerebellum, our findings suggest that autophagic defects in the brain could cause human disease.
Results
INPP5E, a new component of a later stage of autophagy
Using indirect immunofluorescence microscopy to observe LC3 and p62 dot formation, we tested the effect of siRNA knockdown of ten phosphoinositide phosphatases on the number of autophagosomes in HeLa cells. The results revealed that INPP5E knockdown causes massive accumulation of both LC3 and p62 dots, which represent autophagosomes (Appendix Fig S1).
There are two possible causes for the accumulation of autophagosomes: induction of autophagy or inhibition of a later stage of autophagy that follows autophagosome formation. To determine which of these was responsible, we next performed autophagic flux assays in which we measured degradation of LC3‐II (a lipidated form of LC3 localized to the autophagosome membrane) and p62, specific substrates of autophagy (Klionsky et al, 2008). Prior to the autophagic flux assay, we added two different siRNAs that significantly knocked down INPP5E expression in cells (Fig 1B). Treatment with INPP5E siRNA (siINPP5E) strongly suppressed autophagic flux in mouse N1E‐115 neuroblastoma cells (Fig 1C and D). This inhibitory effect was also observed, albeit moderately, in mouse embryonic fibroblasts (Appendix Fig S2A–C) and HeLa cells (data not shown). INPP5E may be more important for execution of autophagy in neuronal cells than in other cell types. Indeed, expression of INPP5E is high in brain (Kisseleva et al, 2000; Kong et al, 2000). The decrease in autophagic flux was rescued by expression of mStrawberry (mSt)‐INPP5E, confirming the specificity of the effect (Fig 3). In addition, as in the initial screening, siINPP5E significantly increased the number of autophagosomes labeled by LC3, as well as Atg5‐labeled autophagosomes in these cells (Figs 1E and F and Appendix Fig S3A–C). Consistent with the data, electron microscopy analysis revealed the accumulation of autophagosomes in INPP5E‐depleted cells (Fig 1G).
Figure 1. INPP5E, an essential factor for autophagy.
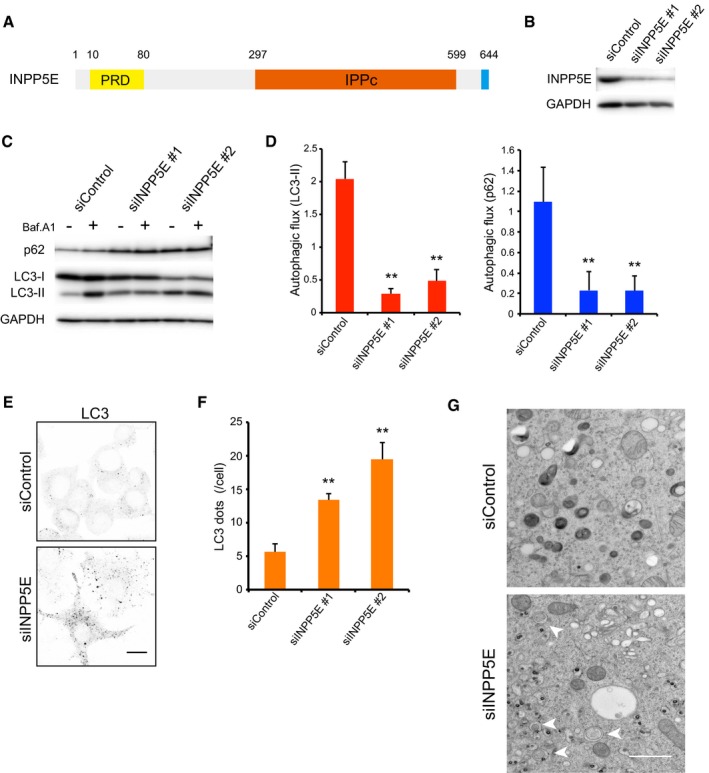
- Schematic diagram of human INPP5E. PRD, proline‐rich domain; IPPc, inositol polyphosphate phosphatase domain.
- Levels of INPP5E protein 72 h after transfection of N1E‐115 cells with control (siControl) or INPP5E‐specific siRNAs (siINPP5E #1 or #2), as determined by immunoblot.
- N1E‐115 cells treated with siControl or siINPP5Es were cultured for 2 h in growth medium with or without 125 nM bafilomycin A1 (Baf.A1) and then analyzed by immunoblot using anti‐p62, anti‐LC3, and anti‐GAPDH antibodies.
- Quantitation of protein signal intensities from immunoblots in (C) showing LC3‐II (left) or p62 (right) levels, following normalization to the control protein GAPDH. Because Baf.A1 inhibits autophagosome–lysosome fusion and degradation within lysosomes via inhibition of vacuolar‐type H+‐ATPase, it is possible to measure autophagic flux, that is, the degradation of autophagic substrates such as p62 and LC3‐II, by calculating the difference of the signal intensities of these proteins in the presence and absence of Baf.A1. Results represent the mean ± s.d. of three independent experiments. **P < 0.01.
- N1E‐115 cells treated with siControl or siINPP5Es were cultured in growth medium. Cells were fixed and stained with anti‐LC3 antibodies and analyzed by immunofluorescence microscopy. Scale bar, 10 μm.
- Quantitation of the number of LC3 puncta per cell as described in (E) (mean ± s.d.; n > 100 cells from three independent experiments). **P < 0.01.
- Electron micrographs in N1E‐115 cells treated with siControl or siINPP5Es were cultured in growth medium. Arrowheads indicate autophagosomes. Scale bar, 1 μm.
Source data are available online for this figure.
Figure 3. Phosphatase activity and lysosomal localization of INPP5E are needed for autophagy.
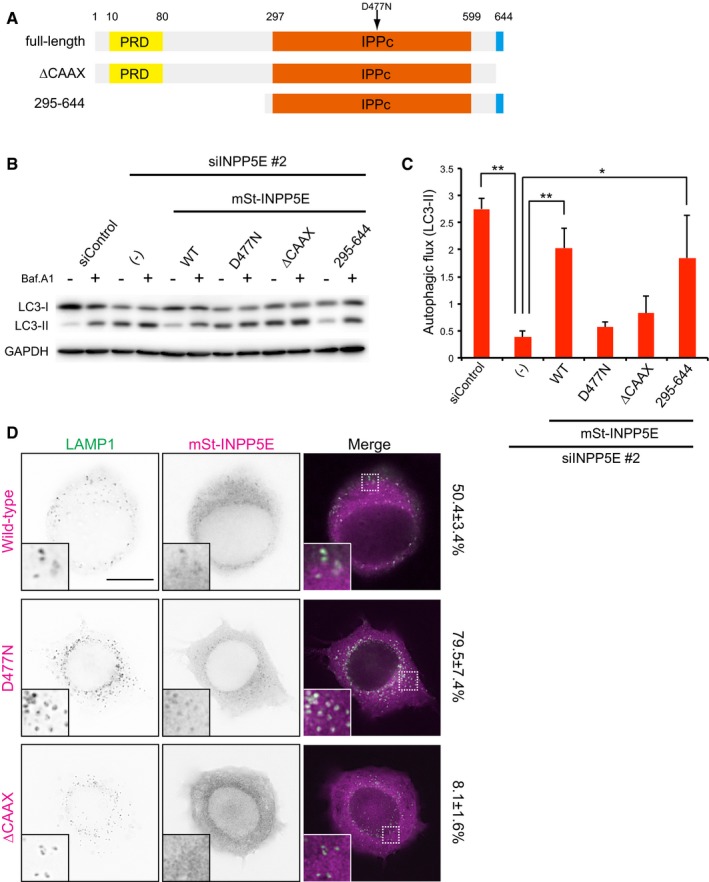
- Schematic diagram of human INPP5E and mutants used in this study. D477N, phosphatase‐inactive mutant.
- N1E‐115 cells stably expressing mSt‐INPP5E (WT, D477N, ∆CAAX, or 295–644) treated with siControl or siINPP5E were cultured in growth medium with or without 125 nM Baf.A1 for 2 h and then analyzed by immunoblot using anti‐LC3 and anti‐GAPDH antibodies. The mSt‐INPP5Es used in this study were resistant to mouse siINPP5E because target sequence of the siRNA is different from the human correspondent sequence.
- Quantitation of protein signal intensities from immunoblots in (B), showing differences in LC3‐II levels in the presence and absence of Baf.A1 following normalization to the control protein GAPDH. Results represent means ± s.d. of three independent experiments. **P < 0.01; *P < 0.05.
- NIE‐115 cells stably expressing mSt‐INPP5E (WT, D477N, or ∆CAAX) were cultured in growth medium. Cells were fixed and stained with anti‐LAMP1 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. The number showing to the right side indicates the colocalization rate of LAMP1 with mSt‐INPP5E. Scale bar, 10 μm.
Source data are available online for this figure.
To further verify that siINPP5E impairs autophagic flux, we used mRFP‐GFP tandem fluorescent‐tagged LC3 (tfLC3) (Kimura et al, 2007). mRFP(+) GFP(+)‐LC3 dots represent autophagosomes or their precursors, whereas mRFP(+) GFP(−) dots indicate autolysosomes, because GFP fluorescence, but not mRFP fluorescence, is attenuated by lysosomal acidic pH and hydrolases. In INPP5E‐depleted N1E‐115 cells, the ratio of the number of autolysosomes (mRFP(+) GFP(−) dots) to that of autophagosomes (mRFP(+) GFP(+)‐LC3 dots) obviously decreased (Appendix Fig S4A and B), confirming that autophagic flux is hampered by INPP5E knockdown. On the basis of these findings, we concluded that INPP5E is required for the later stage of autophagy in neuronal cells.
INPP5E functions in fusion between autophagosomes and lysosomes
INPP5E seems to play a role in autophagy after initiation of autophagosome formation. Hence, we next sought to determine the exact step in which INPP5E is involved: closure of isolation membrane, fusion between autophagosomes and lysosomes, or degradation in autolysosomes. First, we performed a proteinase K protection assay to determine whether the LC3 puncta that accumulated upon INPP5E knockdown are enclosed autophagosomes or not; if they are, the LC3 and p62 inside the completed autophagosomes would not be degraded by proteinase K. We found that the proteinase K sensitivity to p62 and LC3‐II in fractionated samples was not altered by INPP5E knockdown, whereas Atg2a/2b knockdown increased in the sensitivity as previously reported (Fig 2A; Velikkakath et al, 2012) after confirming the suppression of autophagic flux in Atg2a/2b‐depleted cells (Appendix Fig S5A and B), suggesting that autophagosomes are properly enclosed in INPP5E‐depleted neuronal cells. Next, we investigated whether INPP5E is required for autophagosome–lysosome fusion. In siControl‐treated N1E‐115 cells, approximately 35% of LC3 puncta colocalized with the lysosome marker LAMP1, whereas the colocalization rate decreased to approximately 15% in INPP5E‐depleted N1E‐115 cells (Fig 2B and C). In this experiment, autophagy was induced by Torin1, and degradation of LC3 in autolysosomes was suppressed by protease inhibitors (Fig 2B and C). The result clearly demonstrates that autophagosome–lysosome fusion was inhibited in INPP5E‐depleted cells. Recently, it has reported that INPP5E regulates the transcriptional levels of aurora kinase A, which has an important role in mitosis (Plotnikova et al, 2015). Thus, we investigated the effect of INPP5E RNAi on the transcriptional level of genes involved in this fusion step, such as syntaxin 17, SNAP29, and VAMP8 (Itakura et al, 2012; Takáts et al, 2013). However, the transcriptional level of such genes was not affected by INPP5E knockdown (Appendix Fig S6), indicating that the suppression of fusion between autophagosomes and lysosomes observed in INPP5E knockdown is not caused by transcriptional reduction of the genes involved in fusion machinery. In addition, as shown later, lysosomal degradation activity was not affected by INPP5E knockdown.
Figure 2. INPP5E functions in autophagosome–lysosome fusion.

- N1E‐115 cells treated with siControl, siINPP5E, or siAtg2a/2b were cultured in growth medium. The postnuclear fraction (PNS) was separated into LSP, HSP, and HSS fractions and then analyzed by immunoblots using anti‐p62, anti‐LC3, and anti‐GAPDH antibodies. The subfractions were treated with proteinase K (Pro. K) with or without Triton X‐100. Quantitation of protein signal intensities from immunoblots showing LC3‐II (left) or p62 (right) levels, following normalization to the control protein GAPDH. Results represent the mean ± s.d. of three independent experiments. **P < 0.01.
- NIE‐115 cells stably expressing LAMP1‐mCherry treated with siControl or siINPP5Es were cultured for 2 h in growth medium containing 200 nM Torin1 with protease inhibitors (10 μg/ml pepstatin A and 10 μg/ml E‐64‐d). Because the treatment of protease inhibitors inhibits lysosomal degradation, LC3 puncta persist even if autophagosomes fuse with lysosomes. Cells were fixed and stained with anti‐LC3 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm.
- Percentages of colocalization of LC3 dots with LAMP1 dots (mean ± s.d.; n > 20 cells from three independent experiments). **P < 0.01.
Source data are available online for this figure.
Phosphatase activity and lysosomal localization of INPP5E are needed for autophagy
INPP5E is a 5‐phosphatase that dephosphorylates PI(3,4,5)P3, PI(3,5)P2, and PI(4,5)P2 to PI(3,4)P2, PI(3)P, and PI(4)P in vitro. The protein contains a proline‐rich domain (PRD) at the N‐terminus, followed by an inositol polyphosphate phosphatase catalytic domain (IPPc) and a CAAX motif (Fig 1A). To determine which domains and/or motifs of INPP5E are required for autophagy in N1E‐115 cells, we generated a set of deletion and point mutants, as shown in Fig 3A. Suppression of autophagic flux by INPP5E knockdown was efficiently rescued by stable expression of siRNA‐resistant wild‐type and 295–644 constructs, but not by D477N (phosphatase‐inactive mutant) and ∆CAAX constructs (Fig 3B and C), suggesting that phosphatase activity and the CAAX motif of INPP5E are necessary for autophagy in N1E‐115 cells.
CAAX motif provides membrane‐anchoring ability to proteins. Therefore, we sought to identify the membrane to which INPP5E is recruited. Most mSt‐INPP5E was diffusely distributed in the cytoplasm, but some of the protein was distributed in a faint punctate pattern. These puncta were colocalized with LAMP1 (Fig 3D, upper panel), suggesting that a fraction of INPP5E is localized to lysosomes. This localization pattern was not altered even under autophagy induced condition in both MEFs and N1E‐115 cells (Appendix Fig S7A and B). Catalytically inactive variants of phosphatases such as MTMR14/Jumpy and MTMR3, which are thought to behave as substrate‐locked mutants, exhibit a punctate localization, in contrast to the diffuse cytoplasmic localization of the respective wild‐type proteins (Vergne et al, 2009; Taguchi‐Atarashi et al, 2010). Hence, we also examined a catalytically inactive mutant of INPP5E, D477N. In comparison with INPP5E (WT), D477N was more prominently localized to lysosomes (Fig 3D, middle panel). Therefore, we concluded that INPP5E associates with the lysosomal membrane, perhaps transiently. Importantly, INPP5E ∆CAAX, which lacks the ability to promote autophagy, was not recruited to lysosomes (Fig 3D, bottom panel), suggesting that the lysosomal localization of INPP5E is essential for autophagy.
INPP5E knockdown influences neither lysosomal integrity nor fusion between endosomes and lysosomes
Given the localization of INPP5E in lysosomes, we investigated whether lysosomal integrity is compromised in siINPP5E‐treated N1E‐115 cells. By using LysoSensor, we assessed lysosomal pH in INPP5E‐depleted N1E‐115 cells. INPP5E knockdown did not alter the ratio of LysoSensor (Yellow/Blue) (Appendix Fig S8). The data indicate that INPP5E is not essential to maintain the luminal acidity of lysosomes. Moreover, the activity of each lysosomal glycosidase was unchanged in between siControl‐ and siINPP5E‐treated N1E‐115 cells (Appendix Fig S9).
Next, we examined the effect of INPP5E knockdown on lysosomal degradation of the endocytosed cargos. DQ Red BSA is intramolecularly quenched under normal condition, but produces bright fluorescence by enzyme‐based hydrolysis under acidic conditions (Settembre et al, 2013). If it is added to cultured cells, it is endocytosed and delivered to lysosomes, where it becomes fluorescent. The intensity of the DQ‐BSA signal colocalized with LAMP1 was not altered in either siControl‐ or siINPP5E‐treated N1E‐115 cells (Fig 4A and B). We also checked epidermal growth factor (EGF) receptor (EGFR) degradation. Upon stimulation of the ligand, EGFR on the cell surface is internalized and degraded in lysosomes after transport along the endocytic pathway. As shown in Fig 4C and quantitated in Fig 4D, in INPP5E‐depleted N1E‐115 cells, EGFR degradation by EGF stimulation was not significantly affected, whereas the depletion of CHMP5, which has an important role in multivesicular body formation (Ward et al, 2005), clearly suppressed EGFR degradation. Because both of the extracellular cargos, DQ‐BSA and EGF, were degraded almost normally in INPP5E‐depleted cells, we concluded that the effect of INPP5E knockdown on autophagy is not due to a defect in lysosomal degradation activity. In addition, these observations also revealed that INPP5E is specific to lysosome–autophagosome fusion, which is not involved in lysosome–endosome fusion.
Figure 4. INPP5E knockdown influences neither lysosomal integrity nor fusion between endosomes and lysosomes.

- N1E‐115 cells treated with siControl or siINPP5Es were cultured in growth medium with 20 μg/ml DQ‐BSA for 12 h and chased for 2 h. Cells were fixed and stained with anti‐LAMP1 antibodies and analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm.
- Quantitation of signal intensities in (A) showing colocalization of DQ‐BSA with LAMP1 (means ± s.d.; n > 50 cells from three independent experiments). Baf.A1 was used as a control for inhibition of delivery and degradation of DQ‐BSA via the endocytic pathway. **P < 0.01; n.s., non‐significant.
- N1E‐115 cells treated with siControl, siINPP5Es, or siCHMP5 were stimulated with 100 ng/ml EGF for the indicated time periods and then analyzed by immunoblot using anti‐EGFR and anti‐α‐tubulin antibodies. Levels of CHMP5 mRNA 72 h after transfection of N1E‐115 cells with siControl or siCHMP5 as analyzed by RT–PCR.
- Quantitation of EGFR degradation ratio in (C). Results represent means ± s.d. of three independent experiments. **P < 0.01.
Source data are available online for this figure.
PI(3,5)P2 localizes to lysosomes and is catalyzed by INPP5E
In vitro, INPP5E dephosphorylates PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3 to generate PI(3)P, PI(4)P, and PI(3,4)P2 (Sasaki et al, 2009). To identify the endogenous substrate of INPP5E in lysosomes, we examined the intracellular localization of each candidate PI using protein probes. We observed that mSt‐2xML1N, which binds to PI(3,5)P2, was colocalized with LAMP1, but not LC3, as in a previous study (Li et al, 2013), whereas mSt‐2xPLCδ1‐PH (a PI(4,5)P2 marker) was localized to the plasma membrane, but not lysosomes (Fig 5A). Btk‐PH, which binds specifically to PI(3,4,5)P3, displayed diffuse localization in the cytosol (date not shown). INPP5E knockdown increased the intensity of ML1N‐positive puncta colocalizing with lysosomes (Fig 5B and C). To validate the INPP5E‐dependent suppression of PI(3,5)P2 level on lysosomes, we took three independent approaches. First, we tested effect of the suppression of PIKfyve, a phosphoinositide kinase on the intensity of ML1N on lysosomes, because PI(3,5)P2 is generated from PI(3)P by the enzymatic activity of PIKfyve (McCartney et al, 2014). The fluorescence intensity of mSt‐2xML1N on LAMP1 was almost suppressed by PIKfyve knockdown as well as treatment of YM201636, a potent inhibitor of PIKfyve (Appendix Fig S10A and B) (Jefferies et al, 2008), suggesting that we could actually observe PI(3,5)P2 by mSt‐2xML1N. Next, we performed the rescue experiment. The PI(3,5)P2 increment by INPP5E knockdown was restored by the expression of siRNA‐resistant wild type, but not by D477N (phosphatase‐dead mutant) (Fig 5D). Moreover, we observed the 5‐phosphatase activity of INPP5E against PI(3,5)P2 in vitro (Appendix Fig S11). These results demonstrate that lysosomal PI(3,5)P2 level is suppressed by INPP5E.
Figure 5. PI(3,5)P2 is localized to lysosomes and catalyzed by INPP5E.

- NIE‐115 cells stably expressing mSt‐2xML1N or ‐2xPLCδ1 PH were cultured in growth medium with or without 200 nM Torin1. Cells were fixed and stained with anti‐LAMP1 or anti‐LC3 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm.
- NIE‐115 cells stably expressing mSt‐2xML1N treated with siControl or siINPP5Es were cultured in growth medium. Cells were fixed and stained with anti‐LAMP1 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm.
- Quantitation of signal intensities in (B) showing mSt‐2xML1N colocalizing with LAMP1 (means ± s.d.; n > 100 cells from three independent experiments). **P < 0.01; *P < 0.05.
- N1E‐115 cells stably expressing GFP‐2xML1N treated with siControl or siINPP5E were transiently transfected with mSt‐INPP5E (WT, D477N). Quantitation of signal intensities showing GFP‐2xML1N colocalizing with LAMP1 (means ± s.d.; n > 20 cells from three independent experiments). **P < 0.01; *P < 0.05.
- NIE‐115 cells stably expressing mCherry‐2xFYVE treated with siControl or siINPP5Es were cultured in growth medium. Cells were fixed and stained with anti‐LAMP1 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm.
- Quantitation of signal intensities in (E) showing mCherry‐2xFYVE colocalizing with LAMP1 (means ± s.d.; n > 100 cells from three independent experiments). **P < 0.01.
If INPP5E dephosphorylates PI(3,5)P2 on lysosomes, PI(3)P, a product of the reaction should decrease in INPP5E‐depleted cells. Indeed, the intensity of mCherry‐2xFYVE, a well‐known PI(3)P probe, colocalizing with LAMP1 significantly decreased in INPP5E‐depleted cells (Fig 5E and F). On the other hand, the number and localization of PI(3)P effector WIPI2 on the LC3‐positive autophagosomes were not changed (data not shown), suggesting that INPP5E is not involved in control of autophagosomal PI(3)P level. In addition, the intensity of OSBP‐PH‐mCherry, which binds specifically to PI(4)P, was unchanged in between siControl‐ and siINPP5E‐treated cells (Appendix Fig S12A and B). On the basis of these findings, we suggest that dephosphorylation of PI(3,5)P2 on lysosomes by INPP5E is necessary for fusion between autophagosomes and lysosomes.
We also examined the effect of depletion of PIKfyve on the fusion defect in INPP5E‐depleted cells. Interestingly, double knockdown of PIKfyve and INPP5E abolished PI(3,5)P2 staining of lysosomes (Appendix Fig S13A), suggesting that effect of PIKfyve depletion is stronger and dominant to INPP5E depletion. Since LC3 dots were accumulated in the double KD cells, autophagy seemed to be compromised after formation of autophagosomes (Appendix Fig S13B). This result consists with the previous study demonstrating that treatment of cells with PIKfyve inhibitor causes defect in autophagy, which is possibly due to suppression of degradation of autophagosomal contents in autolysosomes by loss of lysosomal activity (de Lartigue et al, 2009). Therefore, both excess and less PI(3,5)P2 level on lysosomes hamper autophagy at late stages (fusion or lysosomal degradation), that is, the optimal level of lysosomal PI(3,5)P2 (or PI(3)P) is essential to the process.
INPP5E enhances actin polymerization on lysosomes through cortactin activation, followed by autophagosome–lysosome fusion
If INPP5E‐mediated downregulation of PI(3,5)P2 level or upregulation of PI(3)P level on lysosomes is required for autophagosome–lysosome fusion, what is the underlying mechanism? Hong et al (2015) recently reported that PI(3,5)P2 controls the endosomal actin dynamics by regulating cortactin–actin interactions. They show that PI(3,5)P2 binds to actin filament‐binding region of cortactin, causing release of actin filaments from the Rab7‐positive late endosomes. Therefore, they propose that decrease in PI(3,5)P2 level promotes stabilization of actin filaments on endosomes. In addition, Lee et al demonstrated that cortactin knockdown inhibits autophagosome–lysosome fusion (Lee et al, 2010). They and other group also showed that the inhibition of actin polymerization suppresses the fusion as well (Lee et al, 2010; Zhuo et al, 2013). Given these results, we envisaged that cortactin and actin filaments also exist on lysosomes and decrease in lysosomal PI(3,5)P2 by INPP5E enhances cortactin‐dependent actin filament stabilization on lysosome, followed by fusion between autophagosome and lysosome.
We firstly confirmed colocalization of F‐actin representing actin filament with LAMP1 (Fig 6A). Furthermore, as predicted, INPP5E knockdown decreased the intensity of F‐actin on lysosomes to 40% compared to control cells (Fig 6A). Next, we detected cortactin on lysosomes. While INPP5E knockdown did not affect the amount of total lysosomal cortactin (Appendix Fig S14A), we found cortactin activation by INPP5E. The activity of cortactin is known to be regulated by its phosphorylation (Oser et al, 2010). Tyr421 and Tyr466 phosphorylations activate cortactin. We found that phosphorylated (activated) cortactin localized to lysosome by using the anti‐phospho‐Tyr421 or Tyr466 cortactin antibodies. INPP5E knockdown apparently reduces both Tyr421 (Fig 6B) and Tyr466 (Appendix Fig S14B) phosphorylated cortactin intensity on lysosome (c. 60% reduction). Finally, we demonstrated that treatment of cells with latrunculin A (Lat A), an inhibitor of actin polymerization, showed the accumulation of LC3 dots (Fig 6C) and the reduction in the colocalization rate between LC3 and LAMP1 (Fig 6D), indicating that actin polymerization is required for autophagosome–lysosome fusion as reported previously.
Figure 6. INPP5E enhances actin polymerization on lysosomes through cortactin activation.

- N1E‐115 cells treated with siControl or siINPP5Es were cultured in growth medium. Cells were fixed and stained with anti‐LAMP1 antibodies and phalloidin and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm. Quantitation of signal intensities showing phalloidin colocalizing with LAMP1 (means ± s.d.; n > 40 cells from three independent experiments). **P < 0.01.
- N1E‐115 cells treated with siControl or siINPP5Es were cultured in growth medium. Cells were fixed and stained with anti‐phospho‐Y421 cortactin and anti‐LAMP1 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm. Quantitation of signal intensities showing cortactin (pY421) colocalizing with LAMP1 (means ± s.d.; n > 40 cells from three independent experiments). **P < 0.01.
- N1E‐115 cells were treated with 0.5 μM latrunculin A (Lat A) in growth medium for 6 h. Cells were fixed and stained with anti‐LC3 antibodies and then analyzed by immunofluorescence microscopy. Scale bar, 10 μm. Quantitation of the number of LC3 puncta per cell (mean ± s.d.; n > 100 cells from three independent experiments). **P < 0.01.
- NIE‐115 cells stably expressing LAMP1‐mCherry were treated with DMSO or 0.5 μM Lat A in growth medium for 4 h, subsequently cultured with 200 nM Torin1 and protease inhibitors (10 μg/ml pepstatin A and 10 μg/ml E‐64‐d) for 2 h (Lat A treatment for total 6 h). Cells were fixed and stained with anti‐LC3 antibodies and then analyzed by immunofluorescence microscopy. Insets show the boxed areas at high magnification. Scale bar, 10 μm. Percentages of colocalization of LC3 dots with LAMP1 dots (mean ± s.d.; n > 20 cells from three independent experiments). **P < 0.01.
Taken together, it is very likely that suppression of PI(3,5)P2 level on lysosome by INPP5E‐dependent conversion of PI(3,5)P2 to PI(3)P stabilizes actin filaments on the organelle, followed by the autophagosome–lysosome fusion.
Joubert syndrome‐associated INPP5E mutations compromise autophagy
INPP5E is one of the genes in which mutations are responsible for Joubert syndrome (Bielas et al, 2009). The syndrome is a congenital cerebellar ataxia characterized by dysplasia of the cerebellum and brainstem (Romani et al, 2013). Patients with the syndrome exhibit several symptoms including hypotonia, abnormal eye and tongue movements, tachypnea, and sleep apnea. Although Joubert syndrome is thought to be a ciliopathy, the development of this disease remains poorly understood. On the basis of the findings in this paper, we sought to test the possibility that mutations in INPP5E present in Joubert syndrome patients hamper autophagy. Disease‐causing mutations in INPP5E are contained within IPPc (Fig 7A), and they decrease phosphatase activity of INPP5E in vitro (Bielas et al, 2009). To determine the effect of these mutations on autophagy, we performed rescue experiments in INPP5E‐depleted cells stably expressing the corresponding siRNA‐resistant mutants of mSt‐INPP5E. The results clearly demonstrated that none of the mutants could rescue the defect in autophagic flux in INPP5E‐depleted cells (Fig 7B and C). In vitro phosphatase activity was strongly inhibited in the R435Q, R512/515W, and K580E mutants, but only mildly suppressed in the R378C and R563H mutants (Bielas et al, 2009). Accordingly, N1E‐115 cells expressing the R435Q, R512/515W, or K580E mutant exhibited strong inhibition of autophagy relative to cells expressing R378C or R563H, suggesting that there is a correlation between the loss of enzymatic activity and the degree of the defect in autophagy. Therefore, a defect in autophagy could be a cause of Joubert syndrome.
Figure 7. Joubert syndrome‐associated INPP5E mutations compromise autophagy.
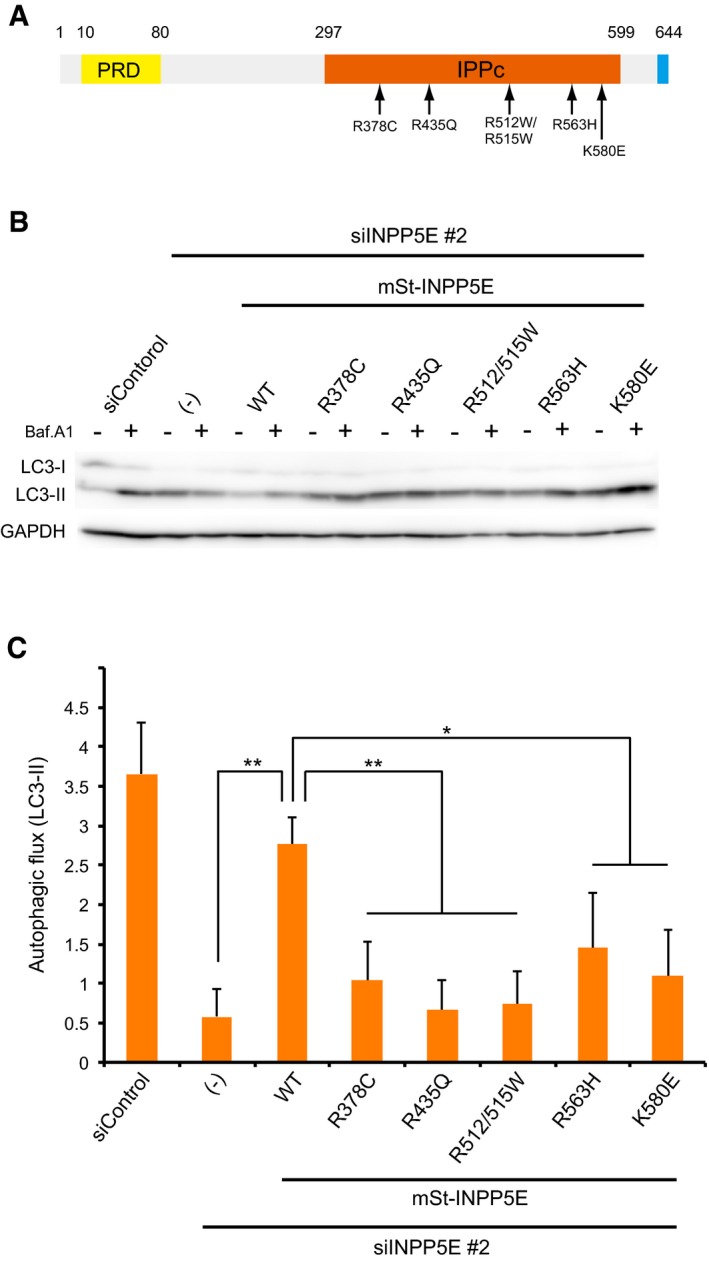
- Joubert syndrome‐associated INPP5E mutations (Bielas et al, 2009) used in this study.
- N1E‐115 cells stably expressing the indicated mutants treated with siControl or siINPP5E were cultured in growth medium with or without 125 nM Baf.A1 for 2 h and analyzed by immunoblot using anti‐LC3 and anti‐GAPDH antibodies. The mutants were resistant to mouse siINPP5E because target sequence of the siRNA is different from the human correspondent sequence.
- Quantitation of protein signal intensities from immunoblots in (B) showing differences in LC3‐II levels in the presence and absence of Baf.A1 following normalization to the control protein GAPDH. Results represent means ± s.d. of three independent experiments. **P < 0.01; *P < 0.05.
Source data are available online for this figure.
Discussion
In this study, we identified the novel autophagy regulator INPP5E, inositol polyphosphate 5‐phosphatase. INPP5E localizes to lysosomes and promotes autophagosome–lysosome fusion, but is not involved in lysosomal degradation and the endocytic trafficking. Our findings provide new mechanistic insights into a later autophagic process, which remains poorly understood in contrast to the process of autophagosome formation. We propose a cascade of reactions that regulates the fusion process; (i) INPP5E catalyzes PI(3,5)P2 to PI(3)P on lysosomes, (ii) the resulted decrease in lysosomal PI(3,5)P2 level promotes activation of cortactin on lysosomes and their binding to actin filament, (iii) activated cortactin stabilizes actin filaments on lysosomes, which is required for autophagosome–lysosome fusion. Intriguingly, PI(3)P binds to Wiskott–Aldrich syndrome protein and Scar homologue (WASH) complex (Jia et al, 2010). Since WASH complex is known to promote actin polymerization through Arp2/3 complex activation, both decrease in PI(3,5)P2 and increase in PI(3)P by INPP5E possibly contribute the fusion process via actin polymerization.
Mi et al (2015) have recently reported that actin polymerization is required for the autophagosomal membrane shaping. They found CapZ, an actin‐capping protein, localizes to both isolation membranes and autophagosomes and regulates the formation of autophagosomes. In the meanwhile, we and other groups have shown that actin polymerization is important in autophagosome–lysosome fusion (Lee et al, 2010; Zhuo et al, 2013). Therefore, actin plays pivotal roles in different steps in autophagy. A remaining question is how actin filaments promote the fusion process.
Reduction in the level of PI(3)P is important in autophagosome–vacuole fusion in yeast (Cebollero et al, 2012). Deletion of YMR1, the yeast gene that encodes PI(3)P phosphatase, inhibited the fusion of autophagosomes with vacuoles, which are the equivalent of lysosomes in yeast. In this case, however, PI(3)P was accumulated not in vacuoles but in autophagosomes, thereby inhibiting release of Atg proteins from the autophagosomal membrane. Therefore, PI(3)P removal from autophagosomes is essential for fusion with vacuoles. By contrast, our study shows that increase in PI(3)P (or decrease in PI(3,5)P2) in lysosomes is required for fusion.
This and the previous study (de Lartigue et al, 2009; Martin et al, 2013) showed that severe reduction in lysosomal PI(3,5)P2 level by depletion of PIKfyve impaired lysosomal integrity and degradation of autophagosomal contents. Consistent with this, neurons and astrocytes from Fig4‐deficient mice, which contain significantly reduced levels of PI(3,5)P2, exhibit accumulation of LC3‐II and p62, indicating that a reduction in PI(3,5)P2 causes autophagic defects in the brain (Ferguson et al, 2009). On the other hand, we found here that increase in PI(3,5)P2 by INPP5E depletion inhibits fusion between autophagosome and lysosomes. Presumably, there is a “optimal window” of the amount of PI(3,5)P2 on lysosomes to proceed autophagy.
INPP5E is reportedly also localized to primary cilia. Kidney cells and MEFs from INPP5E‐depleted mice exhibit normal cilia biogenesis, although the resultant cilia are short and unstable (Jacoby et al, 2009). It has been also reported the accumulation of PI(4,5)P2 at cilia by INPP5E knockdown, resulting in the suppression of cilia‐mediated Hedgehog signaling with the phenotype that cilia length is slightly decreased (Chávez et al, 2015; Garcia‐Gonzalo et al, 2015). Moreover, a recent study showed that disruption of IFT20, which is essential for ciliogenesis, decreased autophagic activity (Pampliega et al, 2013). The inhibition occurred not at autophagosome–lysosome fusion but autophagosome formation. In our case, ciliogenesis was not observed in N1E‐115 cells under ciliated condition even without INPP5E knockdown (data not shown). Although this may imply that INPP5E can function in autophagosome–lysosome fusion without ciliogenesis, future works are needed to elucidate the exact relationship between role of INPP5E in ciliogenesis and in autophagosome–lysosome fusion.
The present study not only adds a new item to the list of biological functions of PIs but also illuminated that the INPP5E mutations in patients with Joubert syndrome cause defects in autophagy. This observation suggests that a neuronal autophagic defect may be involved in the development of this disease; thus, we may have discovered a new genetic autophagic or lysosomal disease. In mice, a strong defect in autophagy causes death shortly after birth (Kuma et al, 2004; Komatsu et al, 2005). Cells expressing INPP5E mutations associated with Joubert syndrome exhibited a mild reduction in autophagic flux, allowing the patients to be born alive but with abnormalities in brain development. Another study suggested that autophagy triggers cilia formation (Tang et al, 2013), in contrast to the aforementioned linkage between autophagy and cilia. Because a variety of genes responsible for Joubert syndrome, including INPP5E, are involved in ciliogenesis (Romani et al, 2013), the autophagic defect may cause disease also via a disturbance in cilia formation.
Our findings open new avenues for further understanding the autophagic machinery, which is essential for cellular homeostasis, and suggest a novel strategy for treating Joubert syndrome.
Materials and Methods
Reagents and antibodies
The following antibodies were used: rabbit anti‐p62 (MBL; PM045), rabbit anti‐LC3 (MBL; PM036), mouse anti‐LC3 (MBL; M152‐3), mouse anti‐cortactin (p80/85) (Millipore; clone 4F11), mouse anti‐GAPDH (Chemicon; MAb314), mouse anti‐α‐tubulin (Sigma‐Aldrich; B5‐1‐2), rat anti‐LAMP1 (BioLegend; 121601), rabbit anti‐INPP5E (St. John's Laboratory), and mouse anti‐EGFR (MBL; MI‐12‐1). Rabbit anti‐phospho cortactin (Tyr421, Tyr466) antibodies were kindly provided by T. Itoh (Kobe University, Hyogo, Japan). All fluorescent reagents (Alexa Fluor 488, 568, 647‐conjugated goat anti‐rabbit, anti‐mouse, or anti‐rat secondary antibodies) were purchased from Invitrogen. Rhodamine phalloidin (PHDR1) was from Cytoskeleton, Inc. DQ™ Red BSA and LysoSensor Yellow/Blue DND‐160 were from Invitrogen. Recombinant human EGF was from Invitrogen. Wortmannin was from Sigma‐Aldrich. Torin1 was from WAKO. Bafilomycin A1, YM‐201636, and latrunculin A were from Cayman Chemical. Proteinase K was from Novagen. Pepstatin A and E‐64‐d were from Peptide Institute, INC. PI(4,5)P2 and PI(3,5)P2 were from CellSignals. Phosphatidylserine was from Avanti.
Plasmids
The pMRX‐IRES‐puro and pMRX‐IRES‐bsr vectors were kindly provided by S. Yamaoka (Tokyo Medical and Dental University, Tokyo, Japan). Human INPP5E full‐length sequence was amplified with specific primers (5′‐gcggatccggatgccgtccaaggcggag‐3′ and 5′‐gcctcgagtcaagaaacggagcagatgg‐3′) using HeLa cDNA and cloned into pMRX‐mStrawberry (mSt) and pCMV‐Tag2B vectors. Various human INPP5E mutants (∆CAAX, 1–300, 295–644, and D477N) and the Joubert syndrome‐associated mutants (R378C, R435Q, R512/515W, R563H, and K580E) (Bielas et al, 2009) were cloned into the pMRX‐mSt and pCMV‐Tag2B vectors. In addition, pMRX constructs were generated to encode GFP‐Atg5, GFP‐LC3 (Fujita et al, 2013), tandem fluorescent‐tagged LC3 (tfLC3) (Kimura et al, 2007) LAMP1‐mCherry, mCherry‐2xFYVE, OSBP‐PH‐mCherry, mSt‐2xPLC δ1 PH (Hasegawa et al, 2011), GFP‐2xML1N, and mSt‐2xML1N (Li et al, 2013). Recombinant retroviruses were prepared as previously described (Saitoh et al, 2003).
Cell culture and retroviral infections
Plat‐E cells were provided by T. Kitamura (The University of Tokyo, Tokyo, Japan). HeLa cells, MEFs, HEK293A cells, and N1E‐115 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 5 U/ml penicillin, and 50 U/ml streptomycin in a 5% CO2 incubator at 37°C. Stable transformants were selected in growth medium with 1 μg/ml puromycin or 5 μg/ml blasticidin (Invitrogen).
RNA interference
For knockdown of various human phosphoinositide phosphatases, validated siRNAs were purchased from Invitrogen. For knockdown of mouse INPP5E, PIKfyve, Atg2a, Atg2b, and CHMP5, siRNA duplex oligomers were designed (Nippon EGT) as follows:
-
siINPP5E #1: UGAGAUUUGCCUUAGUUUG;
siINPP5E #2: AUAAACAGGGACAUGUACA;
siPIKfyve: UUACAAAAGAACUAUAUGC;
siAtg2a: AGAAACUUGUUGAUCUUGG;
siAtg2b: UUUAAUUCCAUCUGAAUUC;
siCHMP5: CCCAACAGUCCUUUAACAU.
Control siRNA (siControl) was obtained from Nippon EGT. siRNAs (final concentration 20 nM) were transfected into HeLa, MEFs, or N1E‐115 cells with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions, and the expression levels were assessed after 72 h by Western blotting or RT–PCR.
Proteinase K protection assay
The subcellular fractionation (PNS; postnuclear supernatant, LSP; low‐speed pellet, HSP; high‐speed pellet, HSS; high‐speed supernatant) was performed as described previously (Velikkakath et al, 2012). In brief, each fraction of LSP and HSP was treated with 100 μg/ml proteinase K on ice with or without 0.5% Triton X‐100 for 30 min. The fraction samples were precipitated with 10% trichloroacetic acid, washed with ice‐cold acetone three times, resuspended in sample buffer including 3 M urea, and then subjected to SDS–PAGE.
Western blotting
Samples were subjected to SDS–PAGE and transferred to polyvinylidene difluoride membranes (Millipore). The transferred membranes were blocked with TBS‐T (0.1% Tween‐20 and TBS) containing 1% skim milk (Nacalai Tesque) for 30 min and were then incubated overnight at 4°C with primary antibodies. Membranes were washed three times for 10 min with TBS‐T and incubated for 1 h at room temperature (RT) with HRP‐conjugated secondary antibodies (GE Healthcare). The protein bands were visualized by enhanced chemiluminescence using a LAS‐3000 (Fujifilm).
Endocytosis assay
To analyze DQ‐BSA assay, N1E‐115 cells were incubated with 20 μg/ml DQ™ Red BSA in DMEM containing 10% FBS for 12 h at 37°C. After washing with PBS three times, cells were chased in DMEM containing 10% FBS for 2 h, and then fixed and stained with anti‐LAMP1 antibody. To analyze epidermal growth factor (EGF) receptor degradation, N1E‐115 cells were cultured in growth medium, washed with PBS, and incubated in serum‐free medium for 4 h. EGF receptor (EGFR) endocytosis was stimulated by adding 100 ng/ml EGF in serum‐free medium. At 1, 2, and 3 h after EGF stimulation, the cells were lysed and subjected to Western blotting.
Lysosomal enzyme activity
Activities of lysosomal acid hydrolases were measured using 4‐methylumbelliferyl (4‐MU) substrates (Otomo et al, 2011). Cell lysates were prepared by sonication in water and determined protein concentration by Bradford protein assay kit (Bio‐Rad) according to manufacturer's protocol. Small aliquots of cell lysates were incubated with each of the artificial 4‐MU substrates in a reaction volume of 20 μl (Sigma; M1633, M3633, M3567, M8527, M9766, M7633, M9130) in acidic acetate buffer or citrate–phosphate buffer for 1 h at 37°C on 96‐well plates. The reaction was stopped by adding 200 μl glycine–NaOH buffer (0.2 M glycine, 0.2 M NaOH), and fluorescence of Em 450 nm/Ex 365 nm was measured with a microplate reader (TECAN, Infinite 200 PRO). At the same time, 4‐MU standard was also measured in the same buffer, and enzyme activities were calculated as nmol/protein mg/h.
Malachite green assay
FLAG‐tagged INPP5E (WT or D477N) was transfected into HEK293A cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h, FLAG‐INPP5E proteins were precipitated with ANTI‐FLAG M2 agarose gel (Sigma). Hydrolysis of PI(4,5)P2 and PI(3,5)P2 was measured by adding each phosphoinositide (final 25 μM) and phosphatidylserine (final 0.5 mM) in buffer (50 mM HEPES, pH 7.5, 2 mM MgCl2, 5 mM DTT) to the immunoprecipitates at 37°C for 30 min and followed by Biomol Green (Enzo Life Sciences) for additional 10 min. Released inorganic phosphate was measured at 630 nm.
Immunofluorescence microscopy
Cells were fixed with 4% PFA in PBS for 15 min at RT and then were permeabilized with 50 μg/ml digitonin in PBS containing 0.1% gelatin (WAKO) for 5 min at RT, washed three times with PBS, and blocked with PBS containing 0.1% gelatin for 30 min at RT. Cells were incubated with primary antibodies in PBS containing 0.1% gelatin for 60 min. After three washes with PBS, cells were incubated with the appropriate secondary antibodies in PBS containing 0.1% gelatin for 50 min. After a brief wash with PBS, coverslips were mounted onto slides using PermaFluor mountant medium (Thermo Fisher Scientific) and observed under a FluoView 1000‐D confocal microscope equipped with 63x/NA 1.40 oil immersion objective lens (Olympus), or a Delta Vision microscopic system equipped with 60x/NA 1.42 oil immersion objective lens (Applied Precision). Delta Vision SoftWoRx software was used to deconvolve the images.
Electron microscopy
N1E‐115 cells were grown on carbon‐coated sapphire disks in 35‐mm dishes to 60–80% confluency. Cells were cryo‐immobilization by HPF with a high pressure freezing machine (HPM 100; Leica). Samples were further processed by freeze substitution (FS) and embedding in a temperature‐controlling device (model AFS2; Leica). FS occurred at −90°C for 5 h with 0.1% (wt/vol) uranyl acetate in glass‐distilled acetone. The temperature was then raised to −50°C (15°C/h), and samples were washed with acetone and infiltrated with increasing concentrations (25, 50, and 75%; 2 h each) of Lowicryl in acetone. About 100% Lowicryl was exchanged three times in 3‐h steps and samples were UV polymerized at −50°C for 48 h, after which the temperature was raised to 20°C (5°C/h) and UV polymerization continued for 48 h. Seventy‐nanometer sections were cut with a microtome (Ultracut UCT; Leica) and a diamond knife and picked up on carbon‐coated 200‐mesh copper grids. Sections were observed using JEOL (JEM‐1011).
RNA extraction and quantitative RT–PCR
Total RNA was extracted from cells using RNeasy Plus Mini Kit (QIAGEN) according to the manufacturer's protocol. Total RNA was reverse‐transcribed using Transcriptor First Strand cDNA Synthesis Kit (Roche). Semi‐quantitative RT–PCR analysis was performed using following primers:
-
mouse and human INPP5E: forward (fw) 5′‐acgcatcgtgtctcagatcaagac‐3′ and reverse (rv) 5′‐ggacagatgtcacccttgtg‐3′;
mouse and human β‐actin: fw 5′‐gctccggcatgtgcaa‐3′ and rv 5′‐aggatcttcatgaggtagt‐3′;
mouse PIKfyve: fw 5′‐aaggctcatgtcttcctctgtg‐3′ and rv 5′‐gggattgtagctgtttcctgga‐3′;
mouse Atg2a: fw 5′‐gtctccaaagctctgaatccca‐3′ and rv 5′‐caccagtgtggagaaggtactc‐3′;
mouse Atg2b: fw 5′‐atattgaccaggacgccttgtt‐3′ and rv 5′‐cataccaagtccttcaggcctt‐3′;
mouse CHMP5: fw 5′‐ggatagcagggcagaatccatt‐3′ and rv 5′‐gtccaaattcatccaccagcac‐3′.
Quantitative real‐time PCRs were performed using a Power SYBR Green PCR Master Mix (ABI) and a QuantStudio 7 Flex Real‐Time PCR system (ABI). The primer sequences used in this method are as follows:
-
mouse Atg7: fw 5′‐tccgttgaagtcctctgctt‐3′ and rv 5′‐ccactgaggttcaccatcct‐3′;
mouse LC3b: fw 5′‐acaaagagtggaagatgtccggct‐3′ and rv 5′‐tgcaagcgccgtctgattatcttg‐3′;
mouse SNAP‐29: fw 5′‐cccattgacaggcagcagta‐3′ and rv 5′‐gttctaggactcctcgctgc‐3′;
mouse VAMP‐8: fw 5′‐gacttggaagccacgtctga‐3′ and rv 5′‐gggatggtaccagtggcaaa‐3′;
mouse syntaxin 17: fw 5′‐actgcgctccaatatccgag‐3′ and rv 5′‐ggaactctgccgtagctgtt‐3′;
mouse β‐actin: fw 5′‐ggctgtattcccctccatcg‐3′ and rv 5′‐ccagttggtaacaatgccatgt‐3′.
Differences between samples were calculated using the ∆∆C t method.
Statistical analysis
Statistically significant differences were determined using the Student's t‐test. Differences were considered significant if P < 0.05.
Author contributions
JH and TY designed the experiments and analyzed the data. JH, RI, TO, AN, and MH performed the experiments. JH and TY wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 7
Acknowledgements
We thank S. Yamaoka for providing pMRX‐IRES‐puro and pMRX‐IRES‐bsr; T. Kitamura for providing the Plat‐E cells; R. Muramatsu for providing the N1E‐115 cells and help with real‐time PCR; T. Takenawa and T. Umemoto for the siRNAs; S. Ohnishi for help with plasmid construction. This study was supported in part by the Ministry of Education, Culture, Sports, Science and Technology (MEXT, to J. Hasegawa and T. Yoshimori); and the Takeda Science Foundation (to J. Hasegawa); and the ONO Medical Research Foundation (to J. Hasegawa).
The EMBO Journal (2016) 35: 1853–1867
See also: L Li & Q Zhong (September 2016)
References
- Backer JM (2008) The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J 410: 1–17 [DOI] [PubMed] [Google Scholar]
- Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93: 1019–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al‐Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel‐Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M et al (2009) Mutations in INPP5E, encoding inositol polyphosphate‐5‐phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41: 1032–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB, Reggiori F (2012) Phosphatidylinositol‐3‐phosphate clearance plays a key role in autophagosome completion. Curr Biol 22: 1545–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez M, Ena S, Van Sande J, de Kerchove d'Exaerde A, Schurmans S, Schiffmann SN (2015) Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev Cell 34: 338–350 [DOI] [PubMed] [Google Scholar]
- Dall'Armi C, Devereaux KA, Di Paolo G (2013) The role of lipids in the control of autophagy. Curr Biol 23: R33–R45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443: 651–657 [DOI] [PubMed] [Google Scholar]
- Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson CJ, Lenk GM, Meisler MH (2009) Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 18: 4868–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, Haraguchi T, Guan J‐L, Iwai K, Tokunaga F, Saito K, Ishibashi K, Akira S, Fukuda M, Noda T, Yoshimori T (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Gonzalo FR, Phua SC, Roberson EC, Garcia G III, Abedin M, Schurmans S, Inoue T, Reiter JF (2015) Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev Cell 34: 400–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa J, Tokuda E, Tenno T, Tsujita K, Sawai H, Hiroaki H, Takenawa T, Itoh T (2011) SH3YL1 regulates dorsal ruffle formation by a novel phosphoinositide‐binding domain. J Cell Biol 193: 901–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong NH, Qi A, Weaver AM (2015) PI(3,5)P2 controls endosomal branched actin dynamics by regulating cortactin‐actin interactions. J Cell Biol 210: 753–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The Hairpin‐type Tail‐Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, Gergely F, Riley JH, Pérez‐Morga D, Woods CG, Schurmans S (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41: 1027–1031 [DOI] [PubMed] [Google Scholar]
- Jefferies HBJ, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, Parker PJ (2008) A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep 9: 164–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Gomez TS, Metlagel Z, Umetani J, Otwinowski Z, Rosen MK, Billadeau DD (2010) WASH and WAVE actin regulators of the Wiskott‐Aldrich syndrome protein (WASP) family are controlled by analogous structurally related complexes. Proc Natl Acad Sci USA 107: 10442–10447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T (2001) Beclin‐phosphatidylinositol 3‐kinase complex functions at the trans‐Golgi network. EMBO Rep 2: 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 3: 452–460 [DOI] [PubMed] [Google Scholar]
- Kisseleva MV, Wilson MP, Majerus PW (2000) The isolation and characterization of a cDNA encoding phospholipid‐specific inositol polyphosphate 5‐phosphatase. J Biol Chem 275: 20110–20116 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard‐Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT et al (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4: 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T (2005) Impairment of starvation‐induced and constitutive autophagy in Atg7‐deficient mice. J Cell Biol 169: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong AM, Speed CJ, O'Malley CJ, Layton MJ, Meehan T, Loveland KL, Cheema S, Ooms LM, Mitchell CA (2000) Cloning and characterization of a 72‐kDa inositol‐polyphosphate 5‐phosphatase localized to the Golgi network. J Biol Chem 275: 24052–24064 [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2004) The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036 [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14: 759–774 [DOI] [PubMed] [Google Scholar]
- de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbé S, Clague MJ (2009) PIKfyve regulation of endosome‐linked pathways. Traffic 10: 883–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J‐Y, Koga H, Kawaguchi Y, Tang W, Wong E, Gao Y‐S, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao T‐P (2010) HDAC6 controls autophagosome maturation essential for ubiquitin‐selective quality‐control autophagy. EMBO J 29: 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang X, Zhang X, Zhao M, Tsang WL, Zhang Y, Yau RGW, Weisman LS, Xu H (2013) Genetically encoded fluorescent probe to visualize intracellular phosphatidylinositol 3,5‐bisphosphate localization and dynamics. Proc Natl Acad Sci USA 110: 21165–21170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Harper CB, May LM, Coulson EJ, Meunier FA, Osborne SL (2013) Inhibition of PIKfyve by YM‐201636 dysregulates autophagy and leads to apoptosis‐independent neuronal cell death. PLoS ONE 8: e60152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney AJ, Zhang Y, Weisman LS (2014) Phosphatidylinositol 3,5‐bisphosphate: low abundance, high significance. BioEssays 36: 52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi N, Chen Y, Wang S, Chen M, Zhao M, Yang G, Ma M, Su Q, Luo S, Shi J, Xu J, Guo Q, Gao N, Sun Y, Chen Z, Yu L (2015) CapZ regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat Cell Biol 17: 1112–1123 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol 12: 823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Mochizuki Y, Ohashi R, Kawamura T, Iwanari H, Kodama T, Naito M, Hamakubo T (2013) Phosphatidylinositol 3‐phosphatase myotubularin‐related protein 6 (MTMR6) is regulated by small GTPase Rab1B in the early secretory and autophagic pathways. J Biol Chem 288: 1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oser M, Mader CC, Gil‐Henn H, Magalhaes M, Bravo‐Cordero JJ, Koleske AJ, Condeelis J (2010) Specific tyrosine phosphorylation sites on cortactin regulate Nck1‐dependent actin polymerization in invadopodia. J Cell Sci 123: 3662–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo T, Higaki K, Nanba E, Ozono K, Sakai N (2011) Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J Biol Chem 286: 35283–35290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Orhon I, Patel B, Sridhar S, Díaz‐Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM (2013) Functional interaction between autophagy and ciliogenesis. Nature 502: 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova OV, Seo S, Cottle DL, Conduit S, Hakim S, Dyson JM, Mitchell CA, Smyth IM (2015) INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci 128: 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani M, Micalizzi A, Valente EM (2013) Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 12: 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S (2003) TWEAK induces NF‐kappaB2 p100 processing and long lasting NF‐kappaB activation. J Biol Chem 278: 36005–36012 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, Suzuki A (2009) Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 48: 307–343 [DOI] [PubMed] [Google Scholar]
- Schneider JL, Cuervo AM (2014) Autophagy and human disease: emerging themes. Curr Opin Genet Dev 26C: 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3‐kinase complexes. J Cell Biol 186: 773–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM (2011) Autophagy in the cellular energetic balance. Cell Metab 13: 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi‐Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, Noda T (2010) Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic 11: 468–478 [DOI] [PubMed] [Google Scholar]
- Takáts S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, Kovács AL, Hegedűs K, Juhász G (2013) Autophagosomal Syntaxin17‐dependent lysosomal degradation maintains neuronal function in Drosophila . J Cell Biol 201: 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q (2013) Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502: 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikkakath AKG, Nishimura T, Oita E, Ishihara N, Mizushima N (2012) Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell 23: 896–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas‐Cezanne T, Laporte J, Deretic V (2009) Control of autophagy initiation by phosphoinositide 3‐phosphatase Jumpy. EMBO J 28: 2244–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicinanza M, D'Angelo G, Di Campli A, De Matteis MA (2008) Function and dysfunction of the PI system in membrane trafficking. EMBO J 27: 2457–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DM, Vaughn MB, Shiflett SL, White PL, Pollock AL, Hill J, Schnegelberger R, Sundquist WI, Kaplan J (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV‐1 budding in mammalian cells. J Biol Chem 280: 10548–10555 [DOI] [PubMed] [Google Scholar]
- Zhuo C, Ji Y, Chen Z, Kitazato K, Xiang Y, Zhong M, Wang Q, Pei Y, Ju H, Wang Y (2013) Proteomics analysis of autophagy‐deficient Atg7‐/‐ MEFs reveals a close relationship between F‐actin and autophagy. Biochem Biophys Res Commun 437: 482–488 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 7