

Abstract
Several stimuli induce programmed cell death by increasing Ca2+ transfer from the endoplasmic reticulum (ER) to mitochondria. Perturbation of this process has a special relevance in pathologies as cancer and neurodegenerative disorders. Mitochondrial Ca2+ uptake mainly takes place in correspondence of mitochondria‐associated ER membranes (MAM), specialized contact sites between the two organelles. Here, we show the important role of FATE1, a cancer‐testis antigen, in the regulation of ER–mitochondria distance and Ca2+ uptake by mitochondria. FATE1 is localized at the interface between ER and mitochondria, fractionating into MAM. FATE1 expression in adrenocortical carcinoma (ACC) cells under the control of the transcription factor SF‐1 decreases ER–mitochondria contact and mitochondrial Ca2+ uptake, while its knockdown has an opposite effect. FATE1 also decreases sensitivity to mitochondrial Ca2+‐dependent pro‐apoptotic stimuli and to the chemotherapeutic drug mitotane. In patients with ACC, FATE1 expression in their tumor is inversely correlated with their overall survival. These results show that the ER–mitochondria uncoupling activity of FATE1 is harnessed by cancer cells to escape apoptotic death and resist the action of chemotherapeutic drugs.
Keywords: adrenal cortex, apoptosis, endocrine cancer, MAM, mitochondria
Subject Categories: Autophagy & Cell Death
Introduction
Cancer‐testis antigens (CTAs) are a heterogeneous group of proteins characterized by their predominantly tissue‐restricted expression in testis in physiological conditions and upregulation in one or more cancer types 1, 2. The restricted expression of CTAs in neoplastic tissues, prevalently because of gene promoter demethylation, holds a potential for their use both as cancer biomarkers and as therapeutic targets in cancer treatment. However, even if many different CTAs have been identified, in general the molecular functions of those proteins are poorly understood 2. FATE1 is a gene expressed in fetal and adult testis mapped to Xq28 3 encoding for a protein identified as a CTA in hepatocellular carcinoma and gastric and colon cancer 4. We have previously demonstrated that steroidogenic factor‐1 (SF‐1), a transcription factor playing a key role in adrenal and gonadal development and in adrenocortical tumorigenesis 5, activates FATE1 expression in adrenocortical carcinoma (ACC) cells in a fashion dependent on its dosage 6, 7. The 21‐kDa protein encoded by the FATE1 gene is a member of the Miff protein family, whose founder is Mff (mitochondrial fission factor), a protein involved in the control of mitochondrial and peroxisomal fission 8. FATE1 bears similarity to Mff in its C‐terminal domain, which is provided with a predicted transmembrane segment preceded by a coiled‐coil region. However, it lacks a Mff‐similar N‐terminal domain that is essential for interaction of Mff with the dynamin‐related GTPase Drp1, which operates mitochondrial fission 9. In normal tissues, FATE1 expression is mainly restricted to testis and adrenal gland 3, 6, while it is overexpressed in a variety of cancers 4, 10. Remarkably, FATE1 was identified as one of the genes whose silencing sensitizes a panel of non‐small‐cell lung cancer cell lines to toxicity from the chemotherapeutic drug paclitaxel 11. In the framework of our continuous effort to characterize novel SF‐1 target genes and their role in adrenal tumorigenesis 6, 7, 12, we set out to determine the cellular function of FATE1. Here, we show that FATE1 is localized in mitochondria‐associated ER membranes (MAM) and is implicated in the regulation of Ca2+‐ and drug‐dependent apoptosis in cancer cells by modulating ER–mitochondria distance.
Results
FATE1 localizes at the interface between ER and mitochondria
The H295R/TR SF‐1 ACC cell line we developed overexpresses SF‐1 in a doxycycline (Dox)‐dependent fashion 6. This cell line is a useful cellular model to study the SF‐1‐dependent phenotypes found in ACC 6, 7, 12. Consistent with our previous results 6, FATE1 mRNA and protein expression was very low at the basal level in H295R/TR SF‐1 cells and was strongly induced following Dox treatment (Fig 1A–C). Efficient knockdown of FATE1 was obtained by specific siRNA electroporation (Fig 1B). We also produced H295R‐derived cell lines selectively expressing FATE1 (H295R/TR FATE1) or N‐Flag FATE1 (H295R/TR N‐Flag FATE1) in a Dox‐dependent fashion (Fig 1A). No Dox‐dependent FATE1 expression was detected in the parental H295R/TR cell line (Fig 1A). To define the subcellular localization of the FATE1 protein, we cotransfected Dox‐treated H295R/TR SF‐1 cells with fluorescent markers for ER, mitochondria, and Golgi. Our results show that endogenous FATE1 colocalizes with mitochondria and partially with ER, but not with Golgi (Fig 1D). The same results were obtained in HeLa cells transiently transfected with a FATE1 expression vector (Appendix Fig S1). Consistent with these results, FATE1 colocalizes with the mitochondrial marker HSP60 in H295R/TR SF‐1 cells (Fig 1E). Mitochondrial localization of FATE1 was confirmed by immunoelectron microscopy, which showed that the protein is associated with the mitochondrial surface in H295R/TR N‐Flag FATE1 cells (Fig 1F). Biochemical fractionation of Dox‐treated H295R/TR N‐Flag FATE1 cell extracts confirmed that FATE1 cosediments principally with the heavy membrane fraction (which contains crude mitochondria) and, in smaller amounts, with the light membrane fraction (which contains ER), while it is absent from the soluble cytosolic fraction (Fig 1G). Proteolysis experiments on the intact crude mitochondrial fraction confirmed that FATE1 was accessible to proteolytic digestion, in a manner independent of the presence of the detergent Triton X‐100, similar to the outer mitochondrial membrane (OMM) proteins VDAC1 and TOM20. In contrast, the mitochondrial intermembrane space protein cytochrome c was only digested by protease after Triton X‐100 extraction (Fig 1H). Alkaline extraction showed that FATE1 is membrane‐anchored, similar to the OMM protein VDAC1 (Fig 1I). Altogether, these experiments show that FATE1 is an integral membrane protein facing the cytosol possessing two predicted coiled‐coil domains (CC1 and CC2) and one C‐terminal Mff‐homology domain encompassing a transmembrane region (Fig 2A). Analysis of the FATE1 protein sequence did not reveal a potential mitochondrial targeting sequence. To identify the FATE1 domain(s) that direct(s) its localization, we generated a battery of protein mutants N‐terminally fused with EGFP and assessed their subcellular localization in H295R/TR SF‐1 cells stained with the mitochondrial marker TOM20. The C‐terminal FATE1 domain (aa 125–183) is sufficient to target protein localization to mitochondria, while its isolated N‐terminal domain (aa 1–124) has a prevalent nuclear localization, similar to the aa 1–162 construct (Fig 2B). Stretches of basic residues have been shown to be able to direct protein localization to mitochondria 13. Three amino acid stretches of this type were identified in the FATE1 C‐terminal domain. Reduction of their net positive charge with mutation of basic residues into alanine, either single or in combination, attenuated mitochondrial‐specific targeting of FATE1, with some mutants (RRR146‐147‐149AAA/RHR159‐160‐161AAA and RR138‐139AA/RRR146‐147‐149AAA/RHR159‐160‐161AAA) being distributed throughout intracellular membranes, indicating that basic residues contribute but are not sufficient to address protein localization to mitochondria (Fig EV1 and Appendix Fig S1). Importantly, the L151D mutation, which is predicted to most severely affect the C‐terminal coiled‐coil structure (Appendix Fig S2), impaired mitochondrial localization of EGFP‐FATE1, directing its localization to intracellular membranes (including ER and the nuclear membrane), similar to the EGFP fusion protein harboring only the FATE1 transmembrane domain (TMD) (Fig 2B and Appendix Fig S1). Increased colocalization of the FATE1 TMD and L151D mutants with the ER marker calreticulin and decreased colocalization with the mitochondrial marker HSP60 compared to wild‐type FATE1 in H295R/TR SF‐1 cells are shown in Fig EV2. Similar results were obtained in HeLa cells transfected to express EGFP‐FATE1 deletions and mutant proteins (Fig EV2 and Appendix Fig S1). Subcellular fractionation of transfected HeLa cells showed results consistent with fluorescence analysis (Fig EV3). In particular, this method confirmed the loss of specific mitochondrial (heavy membrane) localization for the FATE1 TMD and L151D mutants. These results show that the C‐terminal domain of the FATE1 protein directs its mitochondrial localization, with an essential role for CC2, and suggest that the FATE1 TMD is inserted in the ER membrane.
Figure 1. FATE1 is associated with mitochondria in human ACC cells.
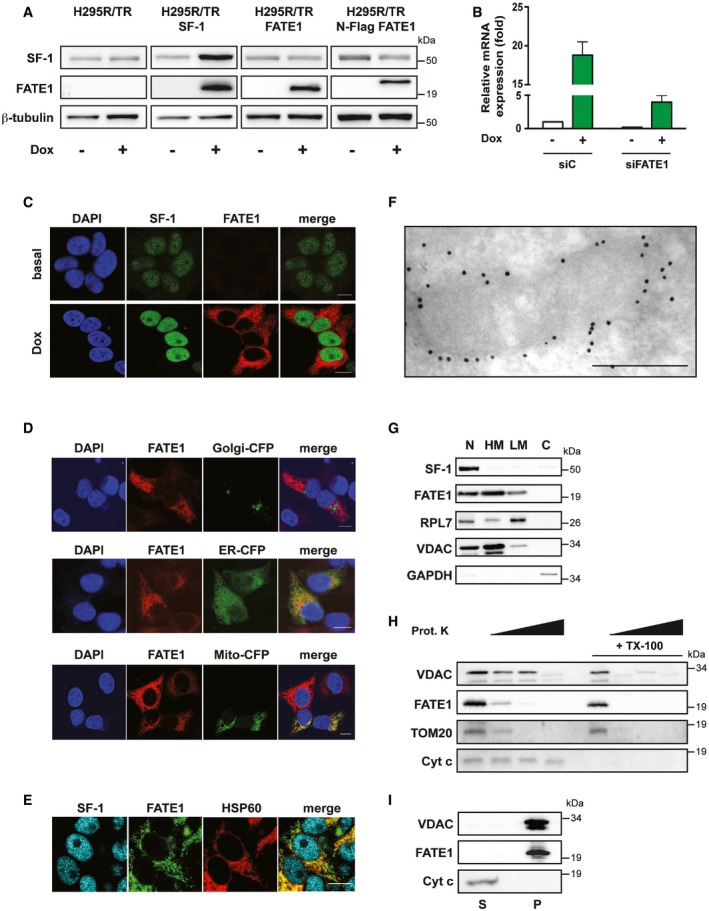
- SF‐1, FATE1 and β‐tubulin protein levels shown in basal condition and after Dox treatment in H295R/TR, H295R/TR SF‐1, H295R/TR FATE1, and H295R/TR N‐Flag FATE1 cells.
- FATE1 mRNA expression is increased in H295R/TR SF‐1 cells by Dox treatment. The efficiency of FATE1 knockdown by specific (siFATE1) vs. control (siC) siRNA nucleofection is shown (mean ± SEM; n = 4 with 3 replicates/experiment).
- Immunofluorescence showing endogenous FATE1 protein (red) induction of expression by Dox treatment of H295R/TR SF‐1 cells. SF‐1 (green), DAPI (blue). Scale bars, 10 μm.
- Subcellular localization of endogenous FATE1 (red) and transfected fluorescent markers for Golgi, ER, and mitochondria, respectively (green) in Dox‐treated H295R/TR SF‐1 cells. DAPI (blue). Scale bars, 10 μm.
- Subcellular localization of the endogenous FATE1 protein (green) in Dox‐treated H295R/TR SF‐1 cells costained with an antibody against the mitochondrial marker HSP60 (red). SF‐1 (blue). Scale bar, 10 μm.
- Immunogold electron microscopy showing association of FATE1 with the mitochondrial outer surface in Dox‐treated H295R/TR N‐Flag FATE1 cells. Scale bar, 500 nm.
- Dox‐treated H295R/TR N‐Flag FATE1 cells were fractioned into nuclear (N), heavy membranes (HM), light membranes (LM), and cytosolic (C) fractions, and localization of SF‐1, FATE1, ribosomal protein RPL7, VDAC1, and GAPDH was revealed by immunoblot.
- Effect of increasing concentrations (0, 1, 10 and 100 μg/ml) of proteinase K and 0.1% Triton X‐100 (TX‐100) treatment of the mitochondrial fraction from Dox‐treated H295R/TR N‐Flag FATE1 cells on VDAC1, FATE1, TOM20, and cytochrome c.
- FATE1 and VDAC1 are associated with the pellet (membrane; P) fraction after high‐speed centrifugation of the alkaline‐extracted mitochondrial fraction of Dox‐treated H295R/TR SF‐1 cells, while cytochrome c is found in the supernatant (S).
Source data are available online for this figure.
Figure 2. Protein domains directing localization of FATE1 and its association with MAM .
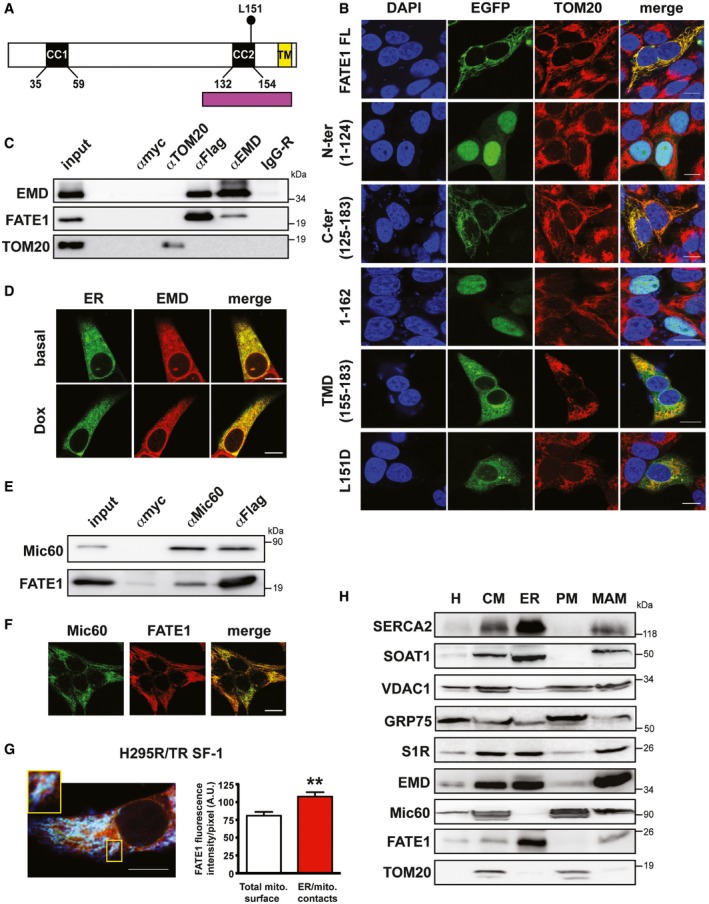
- Structure of the FATE1 protein. Two predicted coiled‐coil domains (CC1 and CC2) and a C‐terminal transmembrane region (TM) are present. Purple, Mff‐homology region. The position of L151 is indicated.
- Subcellular localization of EGFP fusions of full‐length (FL) FATE1 and its mutants: N‐terminal (aa 1–124), C‐terminal (aa 125–183), 1–162, transmembrane domain (aa 155–183), and L151D (green) transfected in H295R/TR SF‐1 cells. Mitochondria were stained by anti‐TOM20 antibody (red) and DNA by DAPI (blue). Scale bars, 10 μm.
- EMD and Flag‐tagged FATE1, but not the other OMM protein TOM20, were coimmunoprecipitated from Dox‐treated H295R/TR N‐Flag FATE1 cells. Control immunoprecipitations were performed with anti‐myc (αmyc) and non‐immune rabbit IgG (IgG‐R) antibodies.
- Confocal fluorescence microscopy showed colocalization of cytoplasmic EMD, as revealed by immunofluorescence (red), with ER, stained by ER‐GFP BacMam (green). Scale bars, 10 μm.
- Mic60/mitofilin and Flag‐tagged FATE1 were coimmunoprecipitated from Dox‐treated H295R/TR N‐Flag FATE1 cells. Control immunoprecipitations were performed with anti‐myc (αmyc) antibody.
- Mitochondrial localization of Mic60/mitofilin (green) and FATE1 (red) in Dox‐treated H295R/TR SF‐1 cells. Scale bar, 10 μm.
- Left, triple‐immunofluorescence labeling of endogenous FATE1 (green), ER labeled by calreticulin (red) and mitochondria labeled by HSP60 staining (blue) in Dox‐treated H295R/TR SF‐1 cells. One area showing close apposition of red, green and blue staining (white signals) is shown at higher magnification. Scale bar, 10 μm. Right, graph showing quantification of FATE1 signal in total mitochondria (white histogram) vs. ER–mitochondria contact sites (red histogram) (mean ± SEM; n = 13). **P < 0.01, Mann–Whitney test.
- H295R/TR SF‐1 cells were fractioned into crude mitochondria (CM), endoplasmic reticulum (ER), purified mitochondria (PM) and mitochondria‐associated membranes (MAM) fractions. H, total homogenate. The localization of SERCA2, SOAT1, VDAC1, GRP75, S1R, EMD, Mic60/mitofilin, FATE1, and TOM20 proteins in the different fractions was revealed by immunoblot.
Source data are available online for this figure.
Figure EV1. Subcellular localization of EGFP‐FATE1 full‐length proteins bearing mutations in C‐terminal domain basic residues in transfected H295R/TR SF‐1 cells.
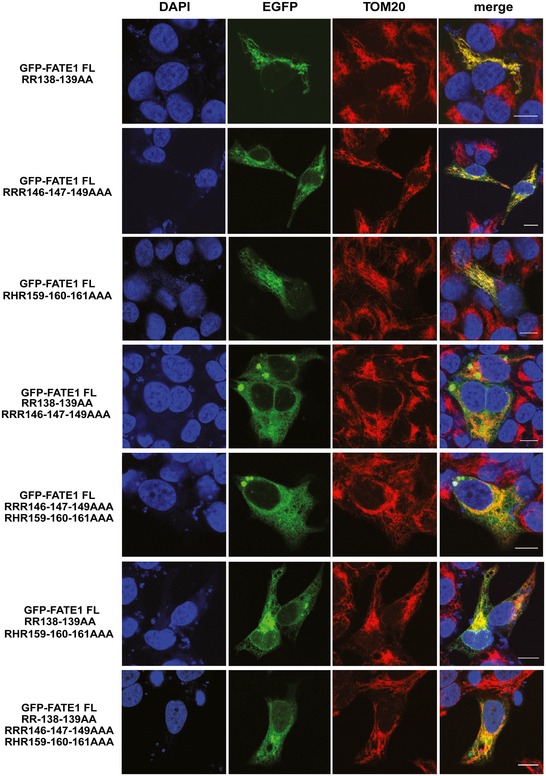
The mitochondrial marker TOM20 is stained in red and DNA in blue with DAPI. Scale bars, 10 μm.
Figure EV2. Subcellular localization of EGFP‐FATE1 L151D and TMD proteins in transfected H295R/TR SF‐1 and HeLa cells.
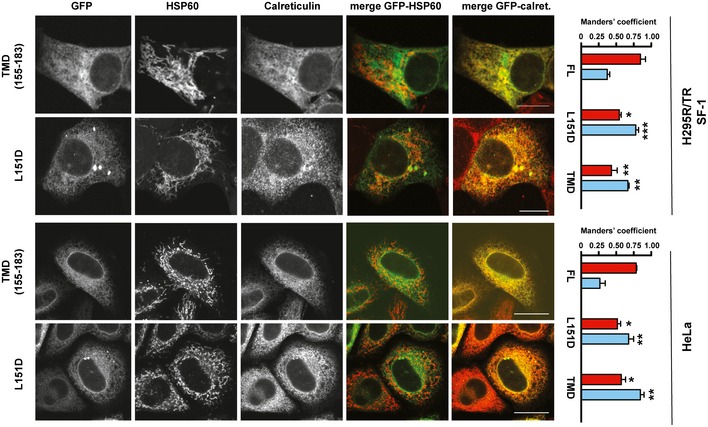
HSP60 was stained as a mitochondrial marker and calreticulin as an ER marker. Manders' colocalization coefficient of both FATE1 mutants with HSP60 (red histograms) is significantly decreased compared to the full‐length wild‐type FATE1, while colocalization with calreticulin (pale blue histograms) is significantly increased (one‐way ANOVA with Bonferroni's correction). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bars, 10 μm.
Figure EV3. Distribution of wild‐type and mutant EGFP‐FATE1 protein in subcellular fractions in transfected HeLa cells.
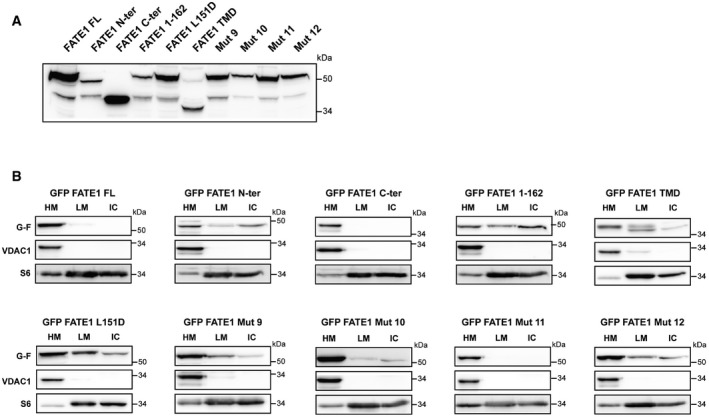
- Immunoblot showing expression of the different EGFP‐FATE1 proteins in whole‐cell lysates. TMD, transmembrane domain (aa 155–183). Mut 9: RR138‐139AA/RRR146‐147‐149AAA. Mut 10: RRR146‐147‐149AAA/RHR159‐160‐161AAA. Mut 11: RR138‐139AA/RHR159‐160‐161AAA. Mut 12: RR138‐139AA/RRR146‐147‐149AAA/RHR159‐160‐161AAA.
- Immunoblot showing the distribution of transfected EGFP‐FATE1 proteins (G‐F) and endogenous VDAC and ribosomal protein S6 in heavy membranes (HM), light membranes (LM), and insoluble cytosolic (IC) fractions.
Source data are available online for this figure.
FATE1 is a component of MAM and decreases ER–mitochondria contacts
To identify proteins interacting with FATE1, we immunoaffinity purified FATE1‐interacting proteins from Dox‐treated H295R/TR N‐Flag FATE1 cells and identified them by mass spectrometry (Appendix Table S1). Proteins copurified with FATE1 included proteins from the ER—chaperones and emerin (EMD)—and the mitochondrial protein Mic60/mitofilin. Databases from large‐scale yeast two‐hybrid screening campaigns also report FATE1–EMD interaction 14, 15. FATE1–EMD interaction was confirmed by coimmunoprecipitation, while EMD did not interact with the OMM protein TOM20 (Fig 2C). EMD is known to localize predominantly in the inner nuclear membrane and to have an important role in the organization of nuclear architecture 16, 17. In addition, previous reports showed that a sizeable pool of extranuclear EMD is localized in the ER 18, 19. We confirmed extranuclear EMD localization in the ER in H295R/TR N‐Flag FATE1 cells (Fig 2D). We also confirmed specific interaction between FATE1 and Mic60/mitofilin in H295R/TR N‐Flag FATE1 cells (Fig 2E). Mic60/mitofilin is a mitochondrial protein (Fig 2F) that controls cristae morphology 20 and is part of the conserved mitochondrial contact site (MICOS) complex, which has an essential role in the regulation of mitochondrial architecture, dynamics, and biogenesis 21, 22, 23, 24. The interactions of FATE1 with both ER and mitochondrial proteins prompted us to investigate its implication in ER–mitochondria contacts, which take place at the level of MAM (mitochondria‐associated membranes), sites of close contacts between those two organelles that have a crucial function in lipid metabolism and calcium signaling 25, 26. In crude mitochondrial extracts from Dox‐treated H295R/TR N‐Flag FATE1 cells, FATE1 migrates in a very high molecular weight (about 1 MDa) complex in a blue native gel (Appendix Fig S3). The method published by Wieckowski et al 27 is a robust procedure to fraction cell extracts into crude mitochondria, ER, pure mitochondria, and MAM. Using this method to fraction extracts from Dox‐treated H295R/TR SF‐1 cells, we could detect FATE1 in crude mitochondria, ER, and MAM but not in the pure mitochondrial fraction, consistent with the immunofluorescence experiments that indicate association of the FATE1 TMD with ER (Fig 2H). The efficacy of the fractionation procedure was confirmed by the presence of the known MAM components SERCA2, SOAT1, VDAC1, and Sigma‐1 receptor (S1R) in the MAM fraction, while the OMM protein TOM20 was only found in the crude and pure mitochondrial fractions. Importantly, the FATE1‐interacting proteins EMD and Mic60/mitofilin were also found in the MAM fraction, in addition to their ER and mitochondrial localization, respectively. The chaperone GRP75, which was also found to interact with FATE1 in our mass spectrometry analysis (Appendix Table S1), fractioned prevalently in the pure mitochondrial fraction, even if it was also present in MAM, consistent with previous reports 28.
Consistent with the biochemical fractionation results, in confocal triple‐immunofluorescence microscopy, endogenous FATE1 signal can be detected in close proximity of calreticulin (ER marker) and HSP60 (mitochondrial marker) in discrete regions inside Dox‐treated H295R/TR SF‐1 cells (Fig 2G, left). In these cells, the intensity of FATE1 staining is significantly enriched in correspondence of ER–mitochondria contact sites compared to the total mitochondrial surface (Fig 2G, right). Colocalization profile curves of the FATE1, ER, and mitochondria signals in the image shown in Fig 2G are shown in Appendix Fig S4. Equivalent results were obtained in Dox‐treated H295R/TR N‐Flag FATE1 cells (Appendix Fig S5).
Consistent with its localization associated with ER–mitochondria contact sites, FATE1 expression in H295R/TR N‐Flag FATE1 cells following Dox treatment had the effect of decreasing ER–mitochondria contacts compared to control cells, as shown by a decreased overlap of signals from ER and mitochondria fluorescent probes (Fig 3A) and by decreased signal of a novel split‐GFP‐based probe 29 (Fig 3B). Using transmission electron microscopy (TEM), we could confirm that both the number of ER–mitochondria contacts and the number of mitochondria displaying ER contact sites were reduced in Dox‐treated H295R/TR N‐Flag FATE1 compared to control cells (Fig 3C). Equivalent results were obtained in H295R/TR SF‐1 cells (Fig 3D). In addition, Dox treatment of H295R/TR N‐Flag FATE1 cells decreased mitochondrial fragmentation compared to control cells (Fig 3E and F), shifting the equilibrium of mitochondrial morphology toward a fused state, without altering mitochondrial membrane potential (Fig 3H). Consistent with these data, mitochondrial fragmentation was also decreased by Dox treatment in the H295R/TR SF‐1 cell line (Fig 3G).
Figure 3. Effects of FATE1 expression on ER–mitochondria contacts and mitochondrial shape.
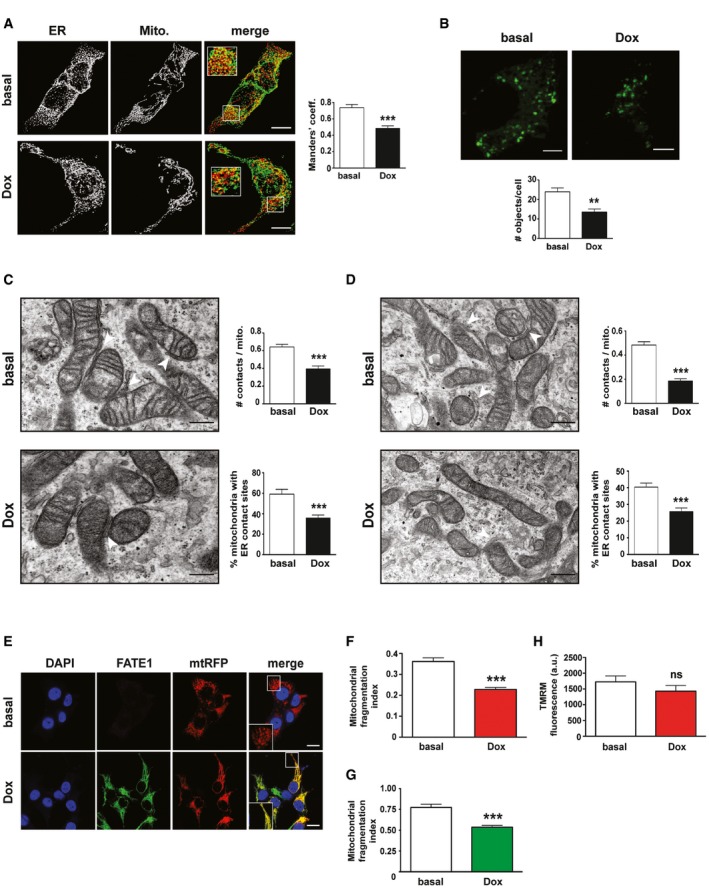
- H295R/TR N‐Flag FATE1 cells were transfected with D1ER marker for ER (green) and mtRFP marker for mitochondria (red). Manders' coefficient for green‐red signals colocalization in basal (white histograms) and Dox‐treated (black histograms) cells is shown on the right (mean ± SEM; n = 12). ***P < 0.001, paired t‐test. Scale bars, 10 μm.
- Effect of FATE1 expression on ER–mitochondria distance in H295R/TR N‐Flag FATE1 cells measured using a split‐GFP probe 29. Quantification is shown in the graph. White histogram, number of fluorescent objects/cell in basal conditions; black histogram, number of fluorescent objects/cell in Dox‐treated cells (mean ± SEM; n = 6 with 90 cells analyzed in total). **P < 0.01, paired t‐test. Scale bars, 5 μm.
- Transmission electron microscopy images of H295R/TR N‐Flag‐FATE1 cells cultured in basal conditions or treated with Dox. ER–mitochondria contacts are indicated by white arrowheads. Scale bars, 200 nm. Right, quantification of the number of contacts normalized by the number of mitochondria and the percentage of mitochondria with ER contact sites is shown in basal conditions (white histograms) and after Dox treatment (black histograms) of cells (mean ± SEM; n = 38 for basal and 47 for Dox‐treated cells). ***P < 0.001, Mann–Whitney test.
- Transmission electron microscopy images of H295R/TR SF‐1 cells cultured in basal conditions or treated with Dox. ER–mitochondria contacts are indicated by white arrowheads. Scale bars, 200 nm. Right, quantification of the number of contacts normalized by the number of mitochondria and the percentage of mitochondria with ER contact sites is shown in basal conditions (white histograms) and after Dox treatment (black histograms) of cells (mean ± SEM; n = 41 for basal and 42 for Dox‐treated cells). ***P < 0.001, Mann–Whitney test.
- Mitochondrial shape (BacMam Mitochondria‐RFP; in red) in H295R/TR N‐Flag FATE1 cells shown by fluorescence confocal microscopy in basal conditions and after Dox treatment. FATE1 (green), DAPI (blue). A higher magnification of merged green and red signal is shown in the insets. Scale bars, 10 μm.
- Mitochondrial fragmentation index in H295R/TR N‐Flag FATE1 cells after Dox treatment (red histogram) compared to cells cultured in basal conditions (white histogram) (mean ± SEM; n = 111 for basal and 101 for Dox‐treated cells). ***P < 0.001, Mann–Whitney test.
- Mitochondrial fragmentation index in H295R/TR SF‐1 cells after Dox treatment (green histogram) compared to cells cultured in basal conditions (white histogram) (mean ± SEM; n = 37 for basal and 49 for Dox‐treated cells). ***P < 0.001, Mann–Whitney test.
- Mitochondrial membrane potential (∆Ψ) measured by TMRM fluorescence after Dox treatment of H295R/TR N‐Flag FATE1 cells (red histogram) compared to cells cultured in basal conditions (white histogram) (mean ± SEM; n = 3 with 12 replicates/experiment). ns, not significant, Mann–Whitney test.
FATE1 negatively affects Ca2+ transfer from the ER to mitochondria and steroid hormone production in ACC cells
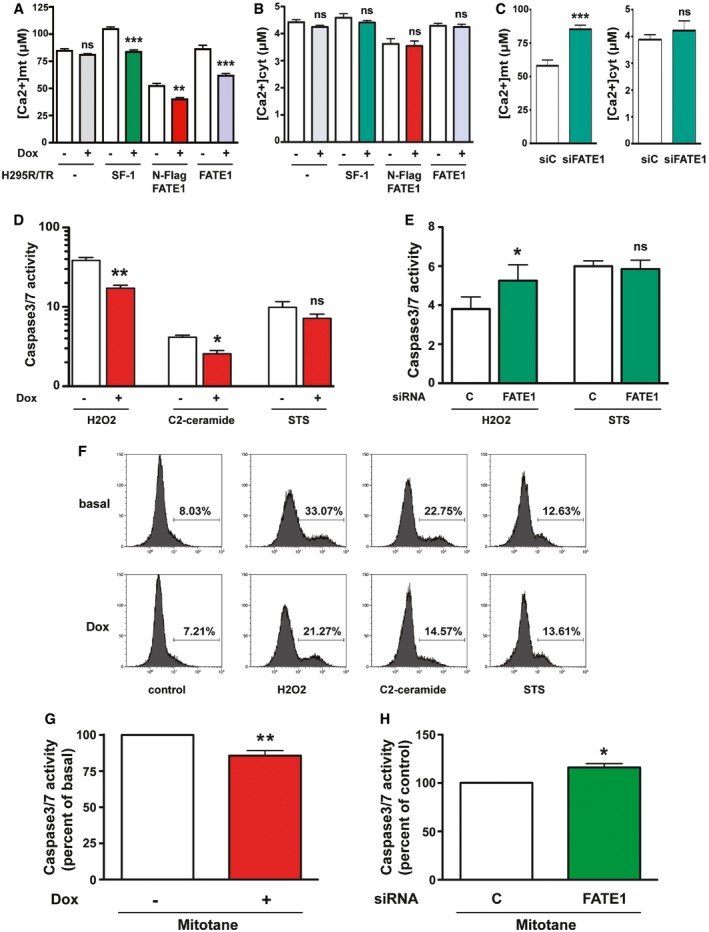
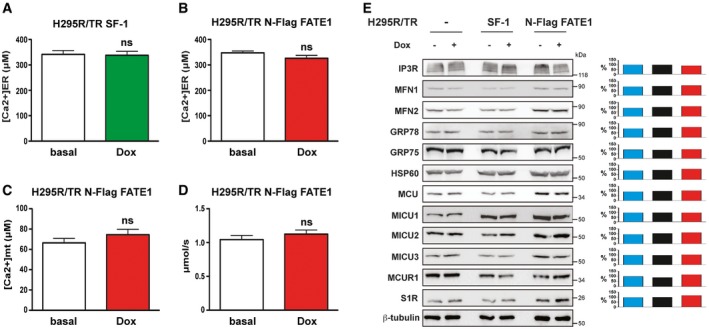
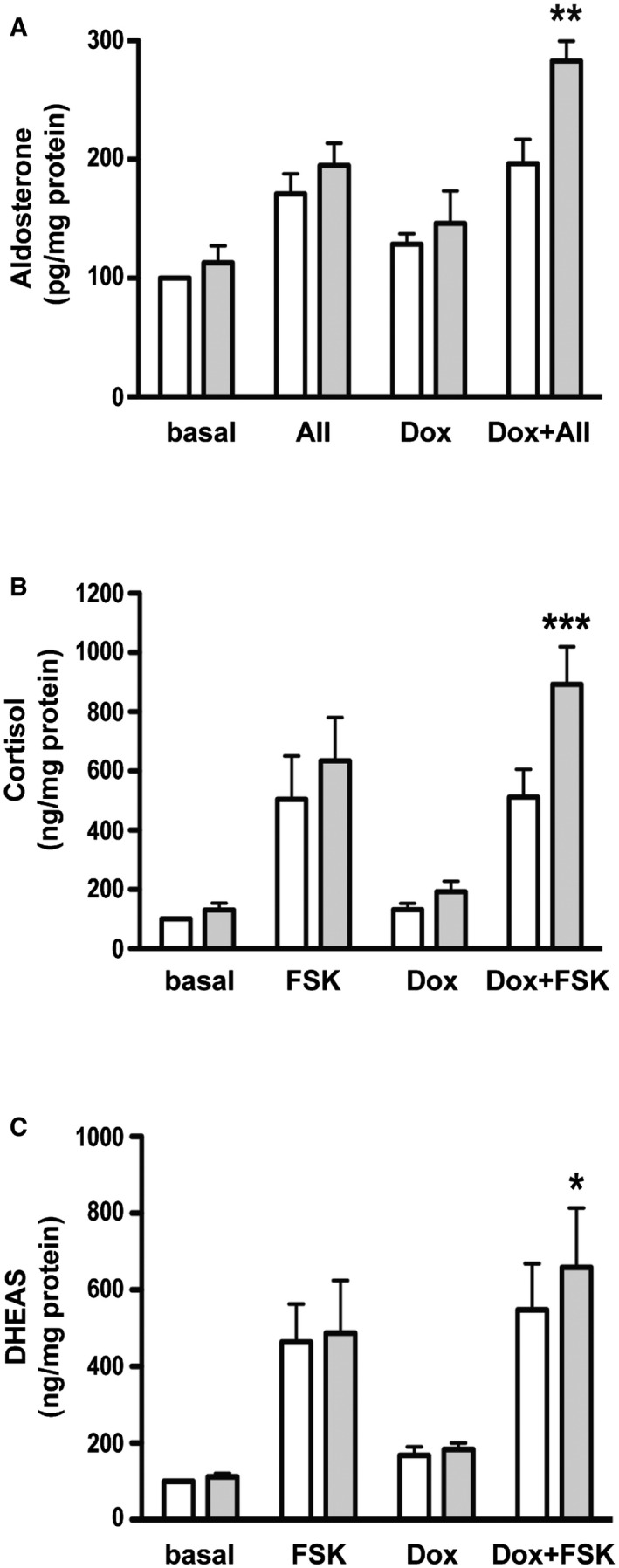
A major function of MAM is the facilitation of Ca2+ transfer from the ER to mitochondria, which, in turn, has a crucial role in the regulation of apoptosis 25, 26. To assess the impact of increased FATE1 expression on mitochondrial and cytosolic Ca2+ uptake, we used aequorin‐based probes targeted to different subcellular compartments 30. In H295R/TR SF‐1 cells, Dox treatment significantly decreased mitochondrial Ca2+ uptake after stimulation with ATP, while it had no effect on the parental cell line (H295R/TR). Mitochondrial Ca2+ concentration was also decreased in both Dox‐treated H295R/TR N‐Flag FATE1 and H295R/TR FATE1 cells compared to control cells (Fig 4A). No effect of SF‐1 or FATE1 overexpression was detected on cytosolic Ca2+ concentration in any of those cell lines (Fig 4B), nor on Ca2+ concentration in the ER (Fig EV4A and B). FATE1 knockdown had the opposite effect on mitochondrial Ca2+ concentration in Dox‐treated H295R/TR SF‐1 cells, significantly increasing it while still having no effect on cytosolic Ca2+ concentration (Fig 4C). The effect of FATE1 expression on mitochondrial Ca2+ uptake is lost after treatment of digitonin‐permeabilized Dox‐treated H295R/TR N‐Flag FATE1 cells with cyclopiazonic acid (CPA), a specific inhibitor of sarco/endoplasmic reticulum Ca2+‐ATPases (Fig EV4C and D). These data show that the effect on Ca2+ transfer into mitochondria triggered by FATE1 expression is MAM‐dependent. On the other hand, the intracellular levels of proteins involved in ER–mitochondria Ca2+ signaling and having a role in the mitochondrial uptake machinery were similar before and after Dox treatment of parental H295R/TR, H295R/TR SF‐1 and H295R/TR N‐Flag FATE1 cells (Fig EV4E). Recent studies implicate ER–mitochondria contacts and MAM in steroid hormone production in steroidogenic cells 31, 32. Consistent with the morphological and Ca2+ uptake data showing uncoupling of ER and mitochondria upon FATE1 expression, FATE1 knockdown in Dox‐treated H295R/TR SF‐1 cells significantly increased angiotensin II‐stimulated aldosterone production and forskolin‐stimulated cortisol and DHEAS production (Fig EV5).
Figure 4. FATE1 expression reduces mitochondrial Ca2+ uptake and apoptosis induced by pharmacological agents and mitotane.
-
A, BMitochondrial Ca2+ uptake (A) and cytosolic Ca2+ concentration (B). H295R/TR parental clone (white/gray histograms), H295R/TR SF‐1 (white/green histograms), H295R/TR N‐Flag FATE1 (white/red histograms), and H295R/TR FATE1 cells (white/violet histograms) (mean ± SEM; n = 3–5 with 12 replicates/condition). **P < 0.01; ***P < 0.001; ns, not significant, one‐way ANOVA with Bonferroni's correction.
-
CMitochondrial (left) and cytosolic (right) Ca2+ concentrations in Dox‐treated H295R/TR SF‐1 cells transfected with control (siC; white histograms) or FATE1‐specific (siFATE1; green histograms) siRNAs (mean ± SEM; n = 3–5 with 12 replicates/condition). ***P < 0.001; ns, not significant, Mann–Whitney test.
-
DCaspase‐3/7 activity in H295R/TR N‐Flag FATE1 cells cultured in basal conditions (white histograms) or in the presence of Dox (red histograms) treated with H2O2 (500 μM), C2‐ceramide (50 μM) or staurosporine (STS) (1 μM) (mean ± SEM; n = 5–8 with 3 replicates/condition). *P < 0.05; **P < 0.01; ns, not significant, one‐way ANOVA with Bonferroni's correction.
-
ECaspase‐3/7 activity in Dox‐treated H295R/TR SF‐1 cells transfected with control (siC; white histograms) or FATE1‐specific (siFATE1; green histograms) siRNAs and treated with H2O2 or STS (mean ± SEM; n = 5–8 with 3 replicates/condition). *P < 0.05; ns, not significant, paired t‐test.
-
FFlow cytometric analysis of TUNEL staining in H295R/TR N‐Flag FATE1 cells cultured in basal conditions or in the presence of Dox and treated with vehicle (control) H2O2 (500 μM), C2‐ceramide (50 μM) or staurosporine (STS) (1 μM).
-
GCaspase‐3/7 activity in H295R/TR N‐Flag FATE1 cells cultured in basal conditions (white histograms) or in the presence of Dox (red histograms) and treated with mitotane (50 μM) (mean ± SEM; n = 5–8 with 3 replicates/condition). **P < 0.01, paired t‐test.
-
HCaspase‐3/7 activity in Dox‐treated H295R/TR SF‐1 cells transfected with control (siC; white histograms) or FATE1‐specific (siFATE1; green histograms) siRNAs and treated with mitotane (mean ± SEM; n = 5–8 with 3 replicates/condition). *P < 0.05, paired t‐test.
Figure EV4. Effect of FATE1 expression on Ca2+ concentration in ER and mitochondria after CPA treatment.
-
A, BH295R/TR SF‐1 cells (A) and H295R/TR N‐Flag FATE1 cells (B) were transduced with adenovirus for expression of ER‐targeted aequorin and then treated with Dox (green or red histograms) or left untreated (white histograms) for 24 h before Ca2+ concentration measurement (mean ± SEM; n = 3–5 with 12 replicates/condition). ns, not significant, Mann–Whitney test.
-
CMitochondrial Ca2+ uptake was measured after permeabilization with digitonin and CPA treatment in H295R/TR N‐Flag FATE1 cells cultured in basal conditions (white histogram) or treated with Dox (red histogram) for 24 h using mitochondrial‐targeted aequorin in cells perfused with 400 nM buffered Ca2+ (mean ± SEM; n = 3 with 12 replicates/condition). ns, not significant, Mann–Whitney test.
-
DSpeed of mitochondrial Ca2+ uptake in permeabilized, CPA‐treated H295R/TR N‐Flag FATE1 cells (mean ± SEM; n = 3 with 12 replicates/condition). ns, not significant, Mann–Whitney test.
-
EImmunoblot showing protein expression in H295R/TR, H295R/TR SF‐1, and H295R/TR N‐Flag FATE1 cells. Cells were cultured in basal conditions or treated with Dox for 24 h. Quantification of the blot using ImageJ is shown on the right side for each protein. Blue histogram, H295R/TR cells. Black histogram, H295R/TR SF‐1 cells. Red histogram, H295R/TR N‐Flag FATE1 cells. Band intensity signals for the expression of each protein in Dox conditions were expressed as percentage of basal signal and normalized by the corresponding β‐tubulin signal using ImageJ software.
Source data are available online for this figure.
Figure EV5. Effect of FATE1 expression on steroid hormone production in H295R/TR SF‐1 cells.
-
A–CAldosterone (A), cortisol (B), and DHEAS (C) levels were measured in H295R/TR SF‐1 cells nucleofected with control (white histograms) or FATE1‐specific (gray histograms) siRNA in basal conditions or after Dox treatment. Cells were either left unstimulated (control) or stimulated with angiotensin II (AII; 10 nM) and forskolin (FSK; 10 μg/ml), respectively (mean ± SEM; n = 5 with 3 replicates/condition). ***P < 0.001; **P < 0.01; *P < 0.05, one‐way ANOVA with Bonferroni's correction.
FATE1 decreases Ca2+‐ and drug‐dependent apoptosis
Oxidative stress induced by H2O2 and C2‐ceramide treatment requires ER–mitochondria Ca2+ transfer to cause apoptotic cell death 33, 34. Consistent with the effect of FATE1 to decrease mitochondrial Ca2+ uptake, FATE1 expression significantly decreased caspase‐3/7 activity after H2O2 and C2‐ceramide treatment in Dox‐treated H295R/TR N‐Flag FATE1 cells, while it had no effect on staurosporine‐induced apoptosis (Fig 4D). Consistent with these data, FATE1 knockdown in Dox‐treated H295R/TR SF‐1 cells significantly increased H2O2‐induced apoptosis without affecting staurosporine‐induced apoptosis (Fig 4E). Flow cytometric TUNEL analysis confirmed the protective effect of FATE1 expression on H2O2‐ and C2‐ceramide‐induced apoptosis and the lack of effect upon staurosporine‐induced cell death in H295R/TR N‐Flag FATE1 cells (Fig 4F).
To further evaluate potential therapeutic consequences of FATE1 overexpression in ACC, we analyzed the effect of its expression on chemotherapy‐induced apoptosis. Mitotane (o,p'‐DDD), a derivative of the insecticide DDT, is a cornerstone of current medical therapy in advanced ACC 35. Of note, FATE1 expression also significantly decreased apoptosis induced by 50 μM mitotane in Dox‐treated H295R/TR N‐Flag FATE1 cells (Fig 4G). This dose of mitotane falls inside the therapeutic window (14–20 mg/l) for patients with ACC 36. Conversely, FATE1 knockdown in Dox‐treated H295R/TR SF‐1 cells significantly increased their sensitivity to mitotane (Fig 4H). Altogether, these data show that FATE1 increases resistance of ACC cells to mitotane.
FATE1 expression levels correlate with prognosis in ACC patients
Immunohistochemical analysis showed low FATE1 expression in the normal adrenal cortex and in its benign neoplastic states (hyperplasia and adenoma), while it was expressed at high levels in about 40% of a cohort of 141 ACCs studied, similar to testicular seminiferous tubuli, as previously shown 10 (Fig 5A and B). Conversely, FATE1 was not expressed in a series of 77 non‐steroidogenic tumors analyzed (Fig 5B). As previously reported, elevated tumor stage 37 and high SF‐1 expression 38 were negative prognostic indicators in this cohort of ACC (Fig 5C). Remarkably, high FATE1 and combined high SF‐1/high FATE1 expression were also inversely correlated with patients' overall survival both in univariate and in multivariate (adjusted for age, sex, and ENSAT tumor stage) analysis (Fig 5C and D). These data show that high FATE1 expression in the tumor is a poor prognosis indicator in ACC patients.
Figure 5. Expression of FATE1 in tissues and correlation of its expression with ACC patients' survival.
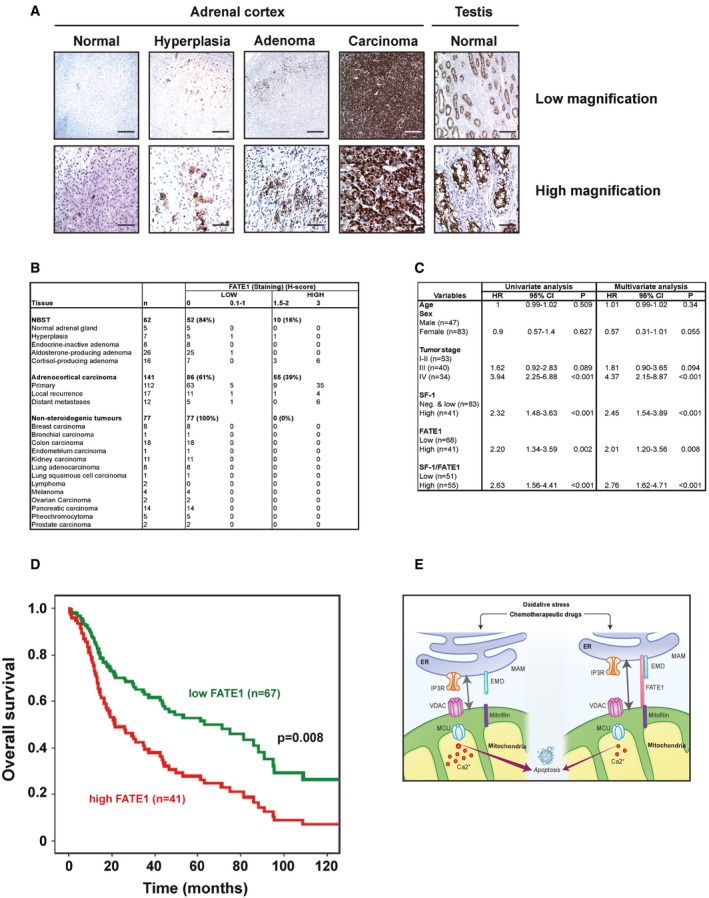
- Immunohistochemical analysis of FATE1 expression in normal adrenal cortex and adrenal hyperplasia, adenoma, and carcinoma (ACC). Testis tissue was used as a positive control. Scale bars, 200 μm (low magnification), 50 μm (high magnification).
- Table summarizing the relative intensity of FATE1 IHC staining (H‐score) in groups of normal, benign and malignant tissues. The highest FATE1 expression levels were found in ACC. n, number of samples studied for each category of tissues. NBST, normal and benign steroidogenic tissues.
- Prognostic parameters in ACC patients. Results are presented as hazard ratios (HR) including 95% confidence interval (CI). The significance level was set at α = 5% for all comparisons. The results for SF‐1, FATE1 and combined SF‐1/FATE1 expression levels are shown both in univariate and multivariate (adjusted for age, sex and ENSAT tumor stage) analysis.
- Overall survival of ACC patients according to FATE1 expression in their tumors, as measured by quantitative immunohistochemistry (low, green curve; high, red curve) in multivariate analysis. P = 0.008, Mantel–Cox test.
- Diagram illustrating FATE1 effect on ER–mitochondria distance and Ca2+ influx into mitochondria. In the presence of FATE1 (right) at the level of MAM, coupling between ER and mitochondria and Ca2+ influx into mitochondria decrease, with consequent resistance to apoptosis triggered by oxidative stress and chemotherapeutic drugs.
Discussion
Tight regulation of ER–mitochondria coupling is critical for the control of functional interactions between the two organelles, which range from Ca2+ transfer to lipid biosynthesis and metabolism 25, 26. Dysfunctions in this process are important in the pathogenesis of neurodegenerative 39, cardiovascular 40, and metabolic 41 diseases. The molecular mechanisms implicated in the regulation of ER–mitochondria connections in mammalian cells are only in their initial phase of characterization 26. A breakthrough was made with the identification of the role of the dynamin‐related GTPase mitofusin 2 (Mfn2) in this process. It was initially shown that Mfn2 functions as a tether between ER and mitochondria 42, but two recent studies challenged this model and suggested that Mfn2 rather acts as a negative regulator of ER–mitochondria apposition 43, 44. Other proteins were described to have an anti‐tethering function between ER and mitochondria, as trichoplein/mitostatin (TCHP), which localizes at the ER–mitochondria interface and negatively regulates ER–mitochondria tethering through interaction with Mfn2 45. Furthermore, presenilin‐1 (PS1) and presenilin‐2 (PS2), the catalytic components of γ‐secretase, an enzyme involved in the pathogenesis of Alzheimer's disease, have been shown to be highly enriched in MAM and inhibit, probably in an indirect manner, ER–mitochondria contacts 39. In our study, using different approaches, we have shown that upregulation of the cancer‐testis antigen FATE1 by transcription factor SF‐1 represents a novel mechanism exploited by cancer cells to increase the distance between ER and mitochondria and functionally uncouple the two organelles, with the consequence to induce resistance to apoptotic cell death caused by pharmacological agents and mitotane (Fig 5E). Remarkably, a previous study showed that FATE1 knockdown also sensitized a panel of human non‐small‐cell lung cancer cell lines to toxicity by paclitaxel, another chemotherapeutic drug 11. While FATE1 is associated with mitochondria in intact cells, biochemical purification of cell extracts shows that it localizes in the crude mitochondria, ER, and MAM fractions but not in pure mitochondria. We believe that these apparently conflicting results may be reconciled by the hypothesis that FATE1 inserts in ER membranes through its C‐terminal transmembrane domain and interacts with mitochondrial partner(s) through its C‐terminal coiled‐coil domain (CC2 in Fig 2A). This hypothesis is consistent with the experiments showing that GFP‐fusion proteins containing either full‐length FATE1 bearing the L151D mutation (predicted to disrupt CC2) or the isolated FATE1 TMD are colocalized with ER and lose mitochondrial localization (Figs 2B and EV2, and Appendix Fig S1). One FATE1‐interacting protein that may play a role to direct its mitochondrial localization is Mic60/mitofilin. This protein harbors coiled‐coil domains 46 and, similar to its yeast ortholog Fcj, plays a pivotal role in the organization of the MICOS complex, which regulates mitochondrial morphology and function [21, 22, 23, 24; reviewed in 47]. While the role of Mic60/mitofilin in the organization of the MICOS complex inside mitochondria is well defined, several studies have shown that it associates with the periphery of the organelle 20, 46, 48. Here, we have shown that Mic60/mitofilin can also be found in the MAM fraction (Fig 2H). It is then possible to hypothesize that this protein may have additional structural roles in the organization of multiprotein complexes connecting ER and mitochondria termed ER–mitochondria organizing network (ERMIONE) in 47. Since FATE1 resides in a high molecular weight complex, as shown in Appendix Fig S3, further studies are required to establish whether it directly interacts with mitofilin, possibly through its CC2, or whether interaction is mediated by other partners. Our results show that FATE1 is significantly enriched in correspondence of ER–mitochondria contact sites (Fig 2G), but clearly a pool of the protein is also found associated with the outer surface of mitochondria at sites that are not in close contact with the ER. We can speculate that for this protein pool, tight interactions with mitochondrial partners may be sufficient to mediate association with that organelle in situations where the FATE1 C‐terminal TMD may not be functional due to misfolding or shielding interactions with other partners.
Another FATE1 interactor identified in our study is EMD, the protein mutated in Emery–Dreifuss muscular dystrophy (EDMD) 16. This finding is confirmed by data present in databases from high‐throughput yeast two‐hybrid screening campaigns, which also report FATE1–EMD interaction 14, 15. Most of the studies on EMD have focused on its nuclear function of regulator of chromatin architecture and gene expression 17. However, a pool of extranuclear EMD is associated with the ER 18, 19 and is present in MAM (Fig 2H). Further studies are needed to assess the role of EMD in ER–mitochondria communication and its potential impact on the pathogenesis of EDMD.
Consistent with its ER–mitochondria anti‐tethering function, FATE1 expression is associated with a decrease in mitochondrial Ca2+ uptake. Mitochondrial fragmentation, which is usually linked to cell death and correlates with increased mitochondrial Ca2+ uptake 49, is also decreased consequently to FATE1 expression (Fig 3E–G). The function of FATE1 as an ER–mitochondria anti‐tethering factor is also consistent with its negative effect on steroid hormone production in ACC cells (Fig EV5). Recent studies have shown in fact that steroidogenic stimulation induces an increase in the number of ER–mitochondria contact sites in mouse Leydig MA‐10 cells, with the ATAD3 protein playing an important role in this process 31. Furthermore, steroidogenic acute regulatory protein (StAR) interaction with MAM proteins is an essential step for its translocation into mitochondria and activation of steroidogenesis 32. FATE1 localization at MAM may also account for its effect on mitotane‐induced apoptosis, since this drug has been shown to inhibit the SOAT1 enzyme, which is localized in MAM, inducing accumulation of toxic cholesterol esters inside the cell and ER stress 50. It is then possible that in the presence of FATE1, SOAT1 inhibition by mitotane may be less efficient.
In ACC cells, FATE1 induction is part of the transcriptional program orchestrated by an increased SF‐1 dosage linked to adrenocortical tumorigenesis 5, 6. This program involves negative modulation of the expression of antiapoptotic factors 7, 12 and increased expression of genes involved in cell cycle transition, response to oxidative stress, adhesion to extracellular matrix, and cell invasion 6, 7, 51, 52. However, since SF‐1 is a tissue‐specific transcription factor 5, it is likely that other mechanisms are responsible for FATE1 aberrant expression in other tumors, one of them probably being promoter demethylation, similar to other CTAs 1, 2. On the basis of our results in ACC cells, we suggest that FATE1 expression may confer resistance to pro‐apoptotic stimuli and chemotherapeutic drugs in other cancer types 4, 10, 11. This is in agreement with a recently published study showing that FATE1 inhibits pro‐apoptotic signaling in a variety of cancer cell lines by destabilizing the pro‐apoptotic BIK protein 53. However, BIK expression was not modulated by FATE1 in any of the H295R‐derived cell lines used in this study (Appendix Fig S6). These data suggest that modulation of ER–mitochondria distance is the main mechanism by which FATE1 counteracts apoptosis in ACC cells. Because of the restricted expression pattern of FATE1 in cancer, new therapeutic strategies targeted at antagonizing its expression and/or function may represent a valuable new tool to specifically sensitize cancer cells to the effect of chemotherapy.
Materials and Methods
Antibodies
Primary antibodies used in this study are listed in Appendix Table S2.
DNA constructs, cloning, and site‐directed mutagenesis
Full‐length FATE1 cDNA was amplified by PCR using human adrenal cDNA as a template and cloned into the pcDNA4/TO (Invitrogen) and pSG5 vectors for expression of N‐terminally Flag‐tagged FATE1. Wild‐type and mutant FATE1 were also subcloned into pEGFP‐C2 (Invitrogen) using standard PCR methods. Mutations (R138A, R139A, R146A, R147A, R149A, R159A, H160A, R161A, L151D) were introduced in various combinations in the FATE1 cDNA by site‐directed mutagenesis using the QuikChange kit (Agilent). In Fig EV3, FATE1 mutants are indicated as follows:
Mut 9: RR138‐139AA/RRR146‐147‐149AAA
Mut 10: RRR146‐147‐149AAA/RHR159‐160‐161AAA
Mut 11: RR138‐139AA/RHR159‐160‐161AAA
Mut 12: RR138‐139AA/RRR146‐147‐149AAA/RHR159‐160‐161AAA.
All constructs were verified by sequencing.
Cell culture and transient transfection
H295R/TR and H295R/TR SF‐1 cells were cultured as described 6. H295R/TR FATE1 and H295R/TR N‐Flag FATE1 stable clones were established as described 6 and expressed FATE1 and N‐terminally flagged FATE1, respectively, in a Dox‐inducible manner. HeLa cells were cultured in DMEM (4.5 g/l glucose; Invitrogen) supplemented with penicillin–streptomycin and 10% fetal calf serum (Invitrogen). H295R/TR SF‐1 cells were transfected with Lipofectamine 2000 (Invitrogen) and HeLa cells with JetPEI (Polyplus Transfection) to express Golgi‐targeted (Golgi‐CFP), ER‐targeted (ER‐CFP), or mitochondrial‐targeted (Mito‐CFP) fluorescent proteins (Clontech). For mRNA knockdown experiments, Dox‐treated H295R/TR SF‐1 cells were transfected with FATE1‐specific siRNA (UGCGAUCAUAAUGGAAACGUAUGCC), and control (medium GC) Stealth siRNA oligos (Invitrogen) using the Amaxa nucleofection method (Lonza). Cells were electroporated with 80 pmol siRNA/106 cells using solution R with T‐020 program and then plated in 24‐well plates. Infections with adenovirus for expression of aequorin probes were performed the day after nucleofection, as described previously 30.
RNA extraction and RT–qPCR
Total RNA was isolated from cultured cells using the RNeasy Mini Kit (Qiagen). Reverse transcription was realized using SuperScript III reverse transcriptase (Invitrogen), and quantitative polymerase chain reaction for transcript quantification was performed as described 6 using the SYBR Green dye assay on a LC480 (Roche) instrument using human TBP mRNA as a reference transcript. Results are expressed using the 2−ΔΔCt method 54.
Immunofluorescence
Immunofluorescence was performed as described 6, using primary antibodies listed in Appendix Table S2 and Alexa Fluor‐conjugated secondary antibodies (Invitrogen). Cell nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI). In some experiments, mitochondria were stained with HSP60 antibody, MitoTracker, or BacMam reagents (Life Technologies) following the manufacturer's protocols. Images were collected on a LSM780 laser scanning confocal microscope (Carl Zeiss) equipped with a PlanApochromat 63X/1.4 NA objective and acquired with the ZEN software. The ImageJ software (http://imagej.nih.gov/ij/) was used for image treatment. For colocalization analysis of fluorescence signals, the overlapping RGB intensity profiles on three different images were obtained using an in‐house developed ImageJ macro software. Mitochondrial FATE1 fluorescence intensity measurements were made using another in‐house developed ImageJ macro software. For each image, the mitochondrial and ER images were segmented to extract two different masks, which were combined by logical operators to obtain the fraction of the mitochondria intersecting the ER. The different mitochondrial masks were applied on the FATE1 fluorescence images to obtain the corresponding intensity measurements in those areas per area surface. All intensities were averaged for each image considered.
Electron microscopy
For ultrastructural analysis, cells were fixed in 1.6% glutaraldehyde in 0.1 M phosphate buffer, rinsed in 0.1 M cacodylate buffer, and post‐fixed for 1 h in 1% osmium tetroxide and 1% potassium ferrocyanide in 0.1 M cacodylate buffer to enhance the staining of membranes. Cells were rinsed in distilled water, dehydrated in alcohols, and lastly embedded in epoxy resin. Contrasted ultrathin sections (70 nm) were analyzed under a JEOL 1400 transmission electron microscope mounted with a Morada Olympus CCD camera. For quantification of ER–mitochondria contacts, both the number of contacts (defined as ER–mitochondria appositions at distances lower than 50 nm corrected by the number of mitochondria present in each image) and the percentage of mitochondria displaying contacts with the ER were taken into consideration. Analysis was performed on 41 and 42 EM images for basal and Dox‐treated H295R/TR SF‐1 cells, respectively, and on 38 and 47 EM images for basal and Dox‐treated H295R/TR N‐Flag FATE1 cells, respectively.
For immunogold stainings, cells were fixed with 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer (PB) (pH 7.4) for 2 h and were processed for ultracryomicrotomy according to a slightly modified Tokuyasu method. In brief, cell suspension was spun down in 10% gelatin. After immersion in 2.3 M sucrose in PB overnight at 4°C, the samples were rapidly frozen in liquid nitrogen. Ultrathin (70 nm thick) cryosections were prepared with an ultracryomicrotome (Leica EMFCS) and mounted on formvar‐coated nickel grids (Electron Microscopy Sciences). Immunostainings were processed with an automated immunogold labeling system (Leica EM IGL) as follows: The grids were incubated successively in PBS containing 50 mM NH4Cl (2×, 5 min each), PBS containing 1% BSA (2×, 5 min each), PBS containing the anti‐FATE1 (Abcam) primary antibody in 1% BSA for 1 h, PBS containing 0.1% BSA (3×, 5 min each), PBS containing 1% BSA and 15 nm colloidal gold‐conjugated protein A/G (CMC), PBS containing 0.1% BSA for 5 min, and PBS for 5 min twice. Last, the samples were fixed for 10 min with 1% glutaraldehyde, rinsed in distilled water, and contrasted with a mixture of methylcellulose/sucrose and 0.3% uranyl acetate on ice. After having been dried in air, sections were examined under a JEOL 1400 transmission electron microscope 55.
Cell viability and apoptosis assay
For H295R/TR N‐Flag FATE1 cells, FATE1 expression was induced with Dox for 24 h and a treatment with apoptotic stimuli was applied during the last 4 h of treatment [500 μM H2O2 (Sigma‐Aldrich), 50 μM C2‐ceramide (Sigma‐Aldrich), or 1 μM staurosporine (Sigma‐Aldrich)]. For H295R/TR SF‐1 cells, SF‐1 expression was induced by Dox (1 μg/ml; Sigma‐Aldrich) treatment for 24 h before nucleofection with the indicated siRNAs. The day after, cells were treated with Dox or medium (ethanol) for 24 h before a 4‐h apoptotic stimulus (500 μM H2O2 or 500 nM staurosporine). For mitotane treatment experiments, a 6‐h Dox pretreatment followed by a 16‐h treatment with mitotane (50 μM; Sigma‐Aldrich) was performed. Caspase‐3/7 activity was assessed with Caspase‐3/7‐Glo assay (Promega) and normalized to cell viability measured with CellTiter‐Glo assay (Promega). Data are reported from 5 to 8 independent experiments each one performed in triplicate. The TUNEL assay was performed using the fluorescein In situ cell death detection kit (Roche), and the percentages of TUNEL‐positive cells were measured by flow cytometry.
Cell fractionation
Cell fractionation was performed at 4°C, as described 56. Each fraction was resuspended in Laemmli buffer–4 M urea, the protein concentration was measured by the Bradford assay, and an equal amount of protein (20 μg) was subjected to SDS–PAGE for Western blot analysis as previously described 6. To define FATE1 topology and localization, proteinase K digestion and alkaline treatment of intact mitochondria were performed. Briefly, cells were resuspended in a hypotonic buffer (0.25 M sucrose, 50 mM Tris–HCl pH 7.5, 25 mM KCl, 5 mM MgCl2). 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitors were added just before fractionation, and cells were disrupted with a Dounce homogenizer. The lysate was centrifuged at 500 g for 5 min to pellet the crude nuclear fraction, while the supernatant was centrifuged at 10,000 g for 5 min to obtain an enriched mitochondrial pellet fraction. Crude mitochondria were washed twice with hypotonic buffer without protease inhibitors and then incubated in homogenizing buffer (without protease inhibitors) alone or in the presence of proteinase K (1, 10 or 100 μg/ml; Sigma‐Aldrich) in the absence or in the presence of Triton X‐100 (0.1% final concentration; Sigma‐Aldrich). After 30‐min incubation on ice, 1 mM PMSF (Sigma‐Aldrich) was added to inactivate proteolysis. Samples were centrifuged for 10 min at 15,000 g at 4°C. Pellet and supernatant were then boiled in Laemmli loading buffer for 5 min and analyzed by immunoblotting. For alkaline treatment, pellets were resuspended in 0.1 M Na2CO3 (pH 12.5) and incubated on ice for 20 min. The soluble supernatant and insoluble membrane fractions were separated by ultracentrifugation at 100,000 g for 30 min, and samples were resuspended in Laemmli buffer–4 M urea. Protein concentration was measured by the BCA protein assay (Thermo), and an equal amount of protein (20 μg) was subjected to SDS–PAGE for Western blot.
Coimmunoprecipitation analysis and mass spectrometry
Crude mitochondria prepared as described before were lysed by gentle agitation for 30 min at 4°C in lysis buffer (50 mM Tris–HCl pH 7.4, 1% NP‐40, 150 mM NaCl, 50 mM NaF, 1 mM PMSF, and protease inhibitors cocktail). The extract was cleared by centrifugation for 15 min at 12,000 g at 4°C. The supernatant extract was used for coimmunoprecipitation with the anti‐Flag M2 antibody (Sigma‐Aldrich) overnight at 4°C. Twenty‐five microliters of Protein A or G Dynabeads (Invitrogen) was then added and incubated for 1 h at 4°C. The antibody–antigen complexes on beads were washed four times with lysis buffer and eluted in Laemmli loading buffer at 70°C, and proteins were electrophoresed in SDS–PAGE. The gel was stained with colloidal Coomassie blue and protein bands excised from the gel. Molecular masses and peptides sequence were determined by matrix‐assisted laser desorption/ionization–time of flight (MALDI‐TOF/TOF; AB SCIEX 4800 Plus). For analysis, MS spectra were recorded manually in a mass range of 700–4,000 Da resulting from 200 laser shots of constant intensity fixed at 3600. Data were collected using 4000 Series Explorer (AB SCIEX) experiments. Protein identification was performed using Mascot database search (http://www.matrixscience.com) and using peptide mass fingerprint mode. Mass tolerance was fixed at 50 ppm against the UniProtKB/Swiss‐Prot/NCBI database.
Mitochondrial sample preparation and blue native PAGE
Extracts for BN‐PAGE were prepared as described previously 57. Briefly, mitochondrial pellet was resuspended in a solubilization buffer containing 50 mM imidazole/HCl pH 7, NaCl 50 mM, EDTA 1 mM, and different detergents: 1% (v/v) n‐dodecyl‐β‐D‐maltoside, 1% digitonin or 1% SDS (all from Sigma‐Aldrich). The suspension was kept on ice for 1 h with occasional vortexing and was then clarified by centrifugation at 20,000 g for 30 min. Following centrifugation, the supernatant was mixed with glycerol (2%) and with 1 μl 5% Coomassie blue G‐250 dye stock to give a detergent:dye ratio equal to 6 (w/w). Mitochondrial protein complexes were thereafter separated by BN‐PAGE using gradient gels (3–20%, w/v acrylamide; Expedeon) and transferred to a PVDF membrane as described previously 57. Immunoblotting was performed as described before using an anti‐Flag antibody (Sigma‐Aldrich).
Ca2+ measurements
Cells were seeded on 96‐well plates and infected with adenovirus coding for cytosolic (cytAEQ) or mitochondrial‐targeted (mutated mtAEQ) aequorin 58 and treated with Dox where indicated. For measurement of mitochondrial Ca2+ uptake after permeabilization, cells were seeded on 13‐mm glass coverslips and infected with adenovirus coding mitochondrial‐targeted (mutated mtAEQ) aequorin, and treated with Dox where indicated. After 24 h, cells were processed for [Ca2+] measurements as described 30. Cells were permeabilized by 1 min perfusion with 50 μM digitonin during luminescence measurements. Where indicated cells were treated with CPA (Sigma‐Aldrich), a specific SERCA (sarco/endoplasmic reticulum Ca2+‐ATPase) inhibitor (20 μM). For ER Ca2+ uptake and release measurements, cells were seeded on 13‐mm glass coverslips and infected with adenovirus encoding ER‐targeted aequorin 59 and treated with Dox where indicated. After 24 h, cells were processed for [Ca2+] measurements. At the end of each experiment, the remaining aequorin pool was estimated by perfusing a hypotonic Ca2+‐rich solution (10 mM CaCl2 in H2O) containing 100 μM digitonin. The light signal was collected and calibrated into [Ca2+] values by an algorithm based on the Ca2+ response curve of aequorin at physiological conditions of pH, [Mg2+], and ionic strength. Data presented derived from at least 3 independent experiments with twelve replicates per condition.
Measurement of mitochondrial membrane potential
Basal mitochondrial membrane potential (∆Ψ) was measured by loading cells with 20 nM tetramethyl rhodamine methyl ester (TMRM; Invitrogen) for 30 min at 37°C. Images were taken on an inverted microscope (Zeiss Axiovert 200) equipped with a Plan‐Fluor 40x/1.3 NA objective, a Photometrics Evolve Delta EMCCD and a 75 W Xenon arc lamp coupled to a monochromator (PTI Deltaram V). TMRM excitation was performed at 560 nm, and emission was collected through a 590–650 nm bandpass filter. Images were taken every 3 s with a fixed 300‐ms exposure time. A total of 10 μM CCCP (carbonyl cyanide p‐tricloromethoxyphenylhydrazone), an uncoupler of oxidative phosphorylation, was added after 20 acquisitions to completely collapse the electrical gradient established by the respiratory chain. After background correction, the fluorescence value after CCCP addition was subtracted for each cell. Data presented derived from twelve cells per experiment from three independent experiments.
MAM preparation
Mitochondria‐associated membranes (MAM) were prepared from H295R/TR SF‐1 cells after Dox treatment. All samples were maintained in ice and centrifugations were performed at 4°C. MAM and pure mitochondria were isolated as previously described 27. Briefly, 2 × 108 cells were scraped with PBS, resuspended in isolation buffer (30 mM Tris pH 7.4, 75 mM sucrose, and 225 mM mannitol) and homogenized in a glass Dounce homogenizer with a Teflon pestle on ice. The homogenate was spun at 600 g for 5 min to remove intact cells and nuclei, and this step was repeated once. The supernatant was recovered and further centrifuged for 10 min at 10,000 g. The resulting pellet (crude mitochondria; CM) was collected, while the supernatant was centrifuged at 20,000 g for 30 min at 4°C. Further centrifugation of the supernatant obtained (100,000 g for 1 h) results in isolation of ER (pellet) and cytosolic fraction (supernatant). The CM pellet was resuspended in buffer 1 (250 mM mannitol, 5 mM HEPES pH 7.4, 0.5 mM EGTA) and carefully homogenized. Then, a centrifugation at 95,000 g for 30 min on a 30% Percoll density gradient in buffer 2 (225 mM mannitol, 25 mM HEPES, 1 mM EGTA, and PMSF 1 mM) was performed. The low‐density band (MAM fraction) and the high‐density band (pure mitochondria fraction) were collected. The mitochondrial layer obtained was washed by two centrifugation steps at 10,000 g for 10 min. The MAM fraction was washed in buffer 2 at 6,300 g for 10 min, and the supernatant was centrifuged at 100,000 g for 90 min to obtain the MAM fraction.
Analysis of ER–mitochondria contact sites using fluorescence microscopy
Cells plated on 14‐mm‐diameter coverslips were transfected with vectors for expression of D1ER 60 to label ER and mtRFP 61 to label mitochondria, while FATE1 expression was induced by Dox treatment for 24 h. Fluorescence was analyzed in living cells by a Leica SP5 confocal microscope. Cells were excited separately at 488 or 547 nm, and single images were recorded. Confocal stacks were acquired every 0.2 μm along the z‐axis (for a total of 40 images) with a 100× objective. Cells were maintained in KRB containing 1 mM CaCl2 and 5.5 mM glucose during the acquisition of images. For mitochondria–ER interaction analysis, stacks were automatically thresholded using ImageJ, deconvoluted, 3D‐reconstructed, and surface‐rendered by using the VolumeJ plugin in ImageJ. The colocalization of green and red signals was analyzed in 12 cells per condition. Interactions were quantified by Manders' colocalization coefficient, as described 62. We used a complementary methodology based on the split‐GFP‐based technology 63. The GFP1‐10 fragment targeted to the OMM and the ER‐targeted β‐11 strand were kindly provided by T. Calì 29. Image analysis was performed with the “3D Object counter” function, an image‐processing package based on ImageJ. The threshold was set manually, and objects with a volume below 5 voxels or above 500 voxels were removed in order to eliminate noise and aggregation artifacts, respectively. On each image, the number of puncta was divided by the total number of cells analyzed. At least 90 cells per condition were analyzed. Data are presented as mean objects per cell ± standard error and derived from six independent experiments. Representative images and relative histograms are shown in Fig 3B.
Analysis of mitochondrial network
To quantify the impact of FATE1 expression on structural mitochondrial network, H295R/TR N‐Flag FATE1 and H295R/TR SF‐1 cells were treated with or without Dox (1 μg/ml) for 24 h in the presence of mitochondria‐targeted red fluorescent protein (mito‐RFP) BacMam reagent (Life Technologies). Images obtained by fluorescence confocal microscopy (TCS SP5; Leica Microsystems) were analyzed to quantify mitochondrial fragmentation by using an in‐house developed ImageJ macro program. The mitochondrial network was quantified by measuring for each cell its length and the number of objects it is composed of. In total, 111 and 101 H295R/TR N‐Flag FATE1 cells were analyzed in basal and Dox‐treated conditions, respectively, while 37 and 49 H295R/TR SF‐1 cells were analyzed in basal and Dox‐treated conditions, respectively. After filtering, segmentation, and skeletization, the length and the number of subunits of the skeleton were determined. A fragmentation index was then calculated as the ratio of number of subunits to the total length of mitochondrial network.
Steroid hormone assays
Aldosterone, cortisol, and DHEAS concentrations were measured in the supernatant of H295R/TR SF‐1 cells nucleofected with siC or siFATE1 siRNAs, treated or not with Dox, in basal conditions or after stimulation with angiotensin II (AII; 10 nM) or forskolin (FSK; 10 μg/ml) using specific EIA Kits (Diagnostics Biochem Canada). Steroid concentrations were normalized by cell protein levels measured with a Bradford kit (Bio‐Rad).
Immunohistochemistry
All patients donating adrenocortical tissues gave written informed consent for collecting tissue and clinical data, and the study was approved by the ethics committee of the University of Würzburg. Tissue samples from 141 adrenocortical carcinomas, 57 adenomas and hyperplasias, five normal adrenal glands, and 77 non‐adrenocortical malignancies included in tissue microarrays (TMA) were processed and analyzed as described 38. The FATE1 protein was detected in IHC by the monoclonal mouse anti‐FATE1 antibody (Santa Cruz). Staining intensity was graded as negative (0), low (1), medium (2), or strong (3). The percentage of positive tumor cells was calculated for each specimen and scored 0 if 0% were positive, 0.1 if 1–9%, 0.5 if 10–49%, and 1 if 50% or more. A quantitative H‐score was then calculated by multiplying the staining intensity grading score by the proportion score 38.
Statistical analysis
Statistical analysis was performed on GraphPad Prism 5.0 software using the Mann–Whitney test, paired t‐test or one‐way ANOVA with Bonferroni's correction for multiple testing. A P‐value < 0.05 was considered to be statistically significant. Survival analysis for ACC patients was performed as described 38 using the Kaplan–Meier method, and differences between groups were assessed with log‐rank and Cox proportional hazards statistics, using the SPSS software package (version 15.0.0) after adjustment for sex, age, and tumor stage. Overall survival was defined as time elapsed from primary resection of ACC to death or last follow‐up visit.
Author contributions
MD‐B, VG, SS, IS, and SL‐G performed experiments. FB contributed to microscopy data acquisition and image analysis. MF and RR supervised experiments. EL performed experiments, supervised the whole project, and wrote the manuscript together with MD‐B, receiving inputs from all other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgements
We thank J. Cazareth for flow cytometry, S. Scarzello for mass spectrometry, G. Drin and R. Gautier for discussion, and B. Bardoni, T. Calì, M. Chami, S. Feliciangeli, M. Kroiss, and D. Toniolo for gift of reagents. This work was supported by the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 259735, by Institut National Du Cancer grant no. 2008‐045, and by the French National Research Agency (ANR) through the “Investments for the Future” LABEX SIGNALIFE (ANR‐11‐LABX‐0028‐01).
EMBO Reports (2016) 17: 1264–1280
References
- 1. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ (2005) Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer 5: 615–625 [DOI] [PubMed] [Google Scholar]
- 2. Whitehurst AW (2014) Cause and consequence of cancer/testis antigen activation in cancer. Annu Rev Pharmacol Toxicol 54: 251–272 [DOI] [PubMed] [Google Scholar]
- 3. Olesen C, Larsen NJ, Byskov AG, Harboe TL, Tommerup N (2000) Human FATE is a novel X‐linked gene expressed in fetal and adult testis. Mol Cell Endocrinol 184: 25–32 [DOI] [PubMed] [Google Scholar]
- 4. Dong XY, Su YR, Qian XP, Yang XA, Pang XW, Wu HY, Chen WF (2003) Identification of two novel CT antigens and their capacity to elicit antibody response in hepatocellular carcinoma patients. Br J Cancer 89: 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lalli E (2010) Adrenocortical development and cancer: focus on SF‐1. J Mol Endocrinol 44: 301–307 [DOI] [PubMed] [Google Scholar]
- 6. Doghman M, Karpova T, Rodrigues GA, Arhatte M, De Moura J, Cavalli LR, Virolle V, Barbry P, Zambetti GP, Figueiredo BC et al (2007) Increased Steroidogenic Factor‐1 dosage triggers adrenocortical cell proliferation and cancer. Mol Endocrinol 21: 2968–2987 [DOI] [PubMed] [Google Scholar]
- 7. Doghman M, Figueiredo BC, Volante M, Papotti M, Lalli E (2013) Integrative analysis of SF‐1 transcription factor dosage impact on genome‐wide binding and gene expression regulation. Nucl Acids Res 41: 8896–8907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gandre‐Babbe S, van der Bliek AM (2008) The novel tail‐anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191: 1141–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang XA, Dong XY, Qiao H, Wang YD, Peng JR, Li Y, Pang XW, Tian C, Chen WF (2005) Immunohistochemical analysis of the expression of FATE/BJ‐HCC‐2 antigen in normal and malignant tissues. Lab Invest 85: 205–213 [DOI] [PubMed] [Google Scholar]
- 11. Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, Minna JD, Michnoff C, Hao W, Roth MG et al (2007) Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446: 815–819 [DOI] [PubMed] [Google Scholar]
- 12. Doghman M, Arhatte M, Thibout H, Rodrigues G, De Moura J, Grosso S, West AN, Laurent M, Mas JC, Bongain A et al (2007) Nephroblastoma overexpressed/cysteine‐rich protein 61/connective tissue growth factor/nephroblastoma overexpressed gene‐3 (NOV/CCN3), a selective adrenocortical cell proapoptotic factor, is down‐regulated in childhood adrenocortical tumors. J Clin Endocrinol Metab 92: 3253–3260 [DOI] [PubMed] [Google Scholar]
- 13. Horie C, Suzuki H, Sakaguchi M, Mihara K (2002) Characterization of signal that directs C‐tail–anchored proteins to mammalian mitochondrial outer membrane. Mol Biol Cell 13: 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rual JF, Venkatesan K, Hao T, Hirozane‐Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi‐Guedehoussou N et al (2005) Towards a proteome‐scale map of the human protein–protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- 15. Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R et al (2014) A proteome‐scale map of the human interactome network. Cell 159: 1212–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D (1994) Identification of a novel X‐linked gene responsible for Emery‐Dreifuss muscular dystrophy. Nat Genet 8: 323–327 [DOI] [PubMed] [Google Scholar]
- 17. Berk JM, Tifft KE, Wilson KL (2013) The nuclear envelope LEM‐domain protein emerin. Nucleus 4: 298–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Östlund C, Ellenberg J, Hallberg E, Lippincott‐Schwartz J, Worman HJ (1999) Intracellular trafficking of emerin, the Emery‐Dreifuss muscular dystrophy protein. J Cell Sci 112: 1709–1719 [DOI] [PubMed] [Google Scholar]
- 19. Salpingdou G, Smerlenko A, Hausmanowa‐Petrucewicz I, Hussey PJ, Hutchison CJ (2007) A novel role for the nuclear membrane protein emerin in association of the centrosome to the outer nuclear membrane. J Cell Biol 178: 897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JML, Rangell L, Bennett MJ, Zha J (2005) The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell 16: 1543–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. von der Malsburg K, Müller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska‐Lwowska A, Wiese S, Rao S, Milenkovic D et al (2011) Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell 21: 694–707 [DOI] [PubMed] [Google Scholar]
- 22. Harner M, Körner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U, Griffith J, Mann M, Reggiori F, Neupert W (2011) The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J 30: 4356–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoppins S, Collins SR, Cassidy‐Stone A, Hummel E, Devay RM, Lackner LL, Westermann B, Schuldiner M, Weissman JS, Nunnari J (2011) A mitochondrial‐focused genetic interaction map reveals a scaffold‐like complex required for inner membrane organization in mitochondria. J Cell Biol 195: 323–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alkhaja AK, Jans DC, Nikolov M, Vukotic M, Lytovchenko O, Ludewig F, Schliebs W, Riedel D, Urlaub H, Jakobs S et al (2012) MINOS1 is a conserved component of mitofilin complexes and required for mitochondrial function and cristae organization. Mol Biol Cell 23: 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Naon D, Scorrano L (2014) At the right distance: ER‐mitochondria juxtaposition in cell life and death. Bioch Biophys Acta 1843: 2184–2194 [DOI] [PubMed] [Google Scholar]
- 27. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: 1582–1590 [DOI] [PubMed] [Google Scholar]
- 28. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski M, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone‐mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175: 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Granatiero V, Giorgio V, Calì T, Patron M, Brini M, Bernardi P, Tiranti V, Zeviani M, Pallafacchina G, De Stefani D et al (2016) Reduced mitochondrial Ca2+ transients stimulate autophagy in human fibroblasts carrying the 13514A>G mutation of the ND5 subunit of NADH dehydrogenase. Cell Death Diff 23: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Granatiero V, Patron M, Tosatto A, Merli G, Rizzuto R (2014) Using targeted variants of aequorin to measure Ca2+ levels in intracellular organelles. Cold Spring Harb Protoc 2014: 86–93 [DOI] [PubMed] [Google Scholar]
- 31. Issop L, Fan J, Lee S, Rone MB, Basu K, Mui J, Papadopoulos V (2015) Mitochondria‐associated membrane formation in hormone‐stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 156: 334–345 [DOI] [PubMed] [Google Scholar]
- 32. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS (2015) Mitochondria‐associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)‐voltage‐dependent anion channel 2 (VDAC2) interaction. J Biol Chem 290: 2604–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide‐induced apoptosis: significance for the molecular mechanism of Bcl‐2 action. EMBO J 20: 2690–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- 35. Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade‐Brittinger C, Lacroix A, Jarzab B et al (2012) Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 366: 2189–2197 [DOI] [PubMed] [Google Scholar]
- 36. Hermsen IG, Fassnacht M, Terzolo M, Houterman S, den Hartigh J, Leboulleux S, Daffara F, Berruti A, Chadarevian R, Schlumberger M et al (2011) Plasma concentrations of o, p'DDD, o, p'DDA, and o, p'DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab 96: 1844–1851 [DOI] [PubMed] [Google Scholar]
- 37. Fassnacht M, Libé R, Kroiss M, Allolio B (2011) Adrenocortical carcinoma: a clinician's update. Nat Rev Endocrinol 7: 323–335 [DOI] [PubMed] [Google Scholar]
- 38. Sbiera S, Schmull S, Assie G, Voelker HU, Kraus L, Beyer M, Ragazzon B, Beuschlein F, Willenberg HS, Hahner S et al (2010) High diagnostic and prognostic value of Steroidogenic Factor‐1 expression in adrenal tumors. J Clin Endocrinol Metab 95: E161–171 [DOI] [PubMed] [Google Scholar]
- 39. Area‐Gomez E, Lara Castillo MDC, Tambini MD, Guardia‐Laguarta C, de Groof AJ, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL et al (2012) Upregulated function of mitochondria‐associated ER membranes in Alzheimer disease. EMBO J 31: 4106–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z, McMurtry MS, Michalak M, Vance JE, Sessa WC et al (2011) The role of Nogo and the mitochondria‐endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med 3: 88ra55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nasrallah CM, Horvath TL (2014) Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol 10: 650–658 [DOI] [PubMed] [Google Scholar]
- 42. de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum and mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- 43. Cosson P, Marchetti A, Ravazzola M, Orci L (2012) Mitofusin‐2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS ONE 7: e46293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum‐mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, Baffa R, Dimmer KS, Scorrano L (2010) Trichoplein/mitostatin regulates endoplasmic reticulum‐mitochondria juxtaposition. EMBO Rep 11: 854–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Odgren P, Toukatly G, Bangs PL, Gilmore R, Fey EG (1996) Molecular characterization of mitofilin (HMP), a mitochondria‐associated protein with predicted coiled coil and intermembrane space targeting domains. J Cell Sci 109: 2253–2264 [DOI] [PubMed] [Google Scholar]
- 47. van der Laan Bohnert M, Wiedemann N, Pfanner N (2012) Role of MINOS in mitochondrial membrane architecture and biogenesis. Trends Cell Biol 22: 185–192 [DOI] [PubMed] [Google Scholar]
- 48. Jans DC, Wurm CA, Riedel D, Wenzel D, Stagge F, Deckers M, Rehling P, Jakobs S (2013) STED super‐resolution microscopy reveals an array of MINOS clusters along human mitochondria. Proc Natl Acad Sci USA 110: 8936–8941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R (2006) Mitochondrial dynamics and Ca2+ signalling. Bioch Biophys Acta 1763: 442–449 [DOI] [PubMed] [Google Scholar]
- 50. Sbiera S, Leich E, Liebisch G, Sbiera I, Schirbel A, Wiemer L, Matysik S, Eckhardt C, Gardill F, Gehl A et al (2015) Mitotane inhibits Sterol‐O‐Acyl Transferase 1 triggering lipid‐mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells. Endocrinology 156: 3895–3908 [DOI] [PubMed] [Google Scholar]
- 51. Lalli E, Doghman M, Latre de Late P, El Wakil A, Mus‐Veteau I (2013) Beyond steroidogenesis: novel target genes for SF‐1 discovered by genomics. Mol Cell Endocrinol 371: 154–159 [DOI] [PubMed] [Google Scholar]
- 52. Latre de Late P, El Wakil A, Jarjat M, de Krijger RR, Heckert LL, Naquet P, Lalli E (2014) Vanin‐1 inactivation antagonizes the development of adrenocortical neoplasia in Sf‐1 transgenic mice. Endocrinology 155: 2349–2354 [DOI] [PubMed] [Google Scholar]
- 53. Maxfield KE, Taus PJ, Corcoran K, Wooten J, Macion J, Zhou W, Borromeo M, Kollipara RK, Yan J, Xie Y et al (2015) Comprehensive functional characterization of cancer‐testis antigens defines obligate participation in multiple hallmarks of cancer. Nat Commun 6: 8840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCt method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 55. Brahimi‐Horn MC, Lacas‐Gervais S, Adaixo R, Ilc K, Rouleau M, Notte A, Dieu M, Michiels C, Voeltzel T, Maguer‐Satta V et al (2015) Local mitochondrial‐endolysosomal microfusion cleaves voltage‐dependent anion channel 1 to promote survival in hypoxia. Mol Cell Biol 35: 1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lalli E, Ohe K, Hindelang C, Sassone‐Corsi P (2000) Orphan receptor DAX‐1 is a shuttling RNA binding protein associated to polyribosomes via messenger RNA. Mol Cell Biol 20: 4910–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wittig I, Braun HP, Schägger H (2006) Blue native PAGE. Nat Protoc 1: 418–428 [DOI] [PubMed] [Google Scholar]
- 58. Porcelli AM, Pinton P, Ainscow EK, Chiesa A, Rugolo M, Rutter GA, Rizzuto R (2001) Targeting of reporter molecules to mitochondria to measure calcium, ATP, and pH. Methods Cell Biol 65: 353–380 [DOI] [PubMed] [Google Scholar]
- 59. Mitchell KJ, Pinton P, Varadi A, Tacchetti C, Ainscow EK, Pozzan T, Rizzuto R, Rutter GA (2001) Dense core secretory vesicles revealed as a dynamic Ca(2+) store in neuroendocrine cells with a vesicle‐associated membrane protein aequorin chimaera. J Cell Biol 155: 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palmer AE, Jin C, Reed JC, Tsien RY (2004) Bcl‐2‐mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA 101: 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Legros F, Lombès A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13: 4343–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Manders EMM, Verbeek FJ, Aten JA (1993) Measurement of co‐localisation of objects in dual‐colour confocal images. J Microsc 169: 375–382 [DOI] [PubMed] [Google Scholar]
- 63. Cabantous S, Terwilliger TC, Waldo GS (2005) Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nat Biotechnol 23: 102–107 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2