

ABSTRACT
DNA damage tolerance (DDT) mechanisms allow cells to synthesize a new DNA strand when the template is damaged. Many mutations resulting from DNA damage in eukaryotes are generated during DDT when cells use the mutagenic translesion polymerases, Rev1 and Polζ, rather than mechanisms with higher fidelity. The coordination among DDT mechanisms is not well understood. We used live-cell imaging to study the function of DDT mechanisms throughout the cell cycle of the fission yeast Schizosaccharomyces pombe. We report that checkpoint-dependent mitotic delay provides a cellular mechanism to ensure the completion of high fidelity DDT, largely by homology-directed repair (HDR). DDT by mutagenic polymerases is suppressed during the checkpoint delay by a mechanism dependent on Rad51 recombinase. When cells pass the G2/M checkpoint and can no longer delay mitosis, they completely lose the capacity for HDR and simultaneously exhibit a requirement for Rev1 and Polζ. Thus, DDT is coordinated with the checkpoint response so that the activity of mutagenic polymerases is confined to a vulnerable period of the cell cycle when checkpoint delay and HDR are not possible.
KEYWORDS: Chk1, DNA damage checkpoint; DNA damage tolerance, homology-directed repair, post-replication repair, Polζ, Polη, Rev1, ubiquitin ligase Rad5
The UV component of sunlight causes base substitution mutations that can give rise to skin cancer in humans.1,2 The molecular analysis of UV-induced mutagenesis has led to numerous insights into the processes by which DNA damage leads to mutations. The vast majority of UV-induced mutations in eukaryotic cells result from the incorporation of incorrect nucleotides opposite damaged bases during translesion DNA synthesis (TLS) by the polymerases Rev1 and Polζ.3,4 These polymerases are unique among the factors required to survive UV radiation in that their elimination reduces the rate of mutation rather than increasing it. For this reason they are termed “mutagenic polymerases.” Cells also have DDT pathways that are not required for UV-induced mutagenesis,5 which we refer to here as “nonmutagenic” pathways. In addition to its role in UV-induced mutagenesis, Polζ is required for the proliferation of mammalian cells6-9 and there is mounting evidence that mutagenic TLS generates a large fraction of the base substitution mutations in the human germline and in cancers formed throughout the body.10-13 Despite the importance of Rev1 and Polζ in maintaining cell viability following DNA damage and in generating mutations that contribute to genetic variability and human disease, the context in which cells employ these enzymes and the exact nature of the structures that they repair have been unclear.
Most mutations resulting from UV exposure are caused by UV-induced cyclobutane pyrimidine dimers (CPDs).14,15 The mutation frequency in proliferating cells exposed to UV increases linearly with the number of CPDs, consistent with a “one-hit” model in which a single CPD leads to a mutation.16,17 The frequency of UV-induced mutagenesis is dramatically higher when cells are irradiated after entering S phase than during G1 phase, suggesting that Rev1 and Polζ may repair structures generated by replication forks.18,19 CPDs have been shown to block the replicative DNA polymerases, generating daughter-strand gaps at sites of damage in the parental strands.20-24 The post-replicative repair (PRR) of these gaps can be observed by using alkaline sucrose gradients to analyze the sizes of nascent DNA strands. Such experiments have revealed that multiple DDT pathways contribute to PRR.
In budding yeast cells, 3 nonmutagenic DDT pathways account for virtually all PRR activity.22,24-26 One pathway, mediated by the HDR machinery, uses redundant information in the sister chromatid for error-free PRR. Two additional pathways require the ubiquitin ligase Rad18, which is thought to function by monoubiquitinating PCNA.27 Rad18 promotes TLS by Polη, a translesion polymerase that reduces the rate of mutation by incorporating the correct nucleotides opposite CPDs,4 and also promotes error-free PRR mediated by the ubiquitin ligase Rad5. It has been postulated that polyubiquitination of PCNA mediated by Rad5 promotes error-free PRR via HDR-independent template-switching to the undamaged sister chromatid.28 The roles of Rad18 and Polη in PRR have been found to be conserved in vertebrate cells,29-32 but less is known about the roles of HDR or Rad5 orthologs in higher eukaryotes.
Polη has a specialized role in tolerating CPDs, but Rev1 and Polζ can use templates containing a wide variety of lesions.4 Despite the diversity of substrates for mutagenic TLS and the dramatic effects of eliminating Rev1 or Polζ on UV-induced mutagenesis, PRR assays generally show little or no repair defect in cells lacking mutagenic TLS.22,25,33 Thus, the role of mutagenic TLS is unclear, especially given the existence of nonmutagenic PRR pathways. One possibility is that Rev1 and Polζ may repair a limited subset of the daughter-strand gaps generated during S phase that is below the level of detection by the usual PRR assays. In budding yeast, Rev1 appears to be involved with a mutagenic process that occurs at a higher rate in late-replicating regions of the genome,12 suggesting that the role of mutagenic TLS may be limited to a subset of daughter-strand gaps produced late in S phase. Consistent with this idea, budding yeast Rev1 expression increases near the end of S phase whereas levels of Polη are relatively constant.34 Expression of human Polζ increases later in the cell cycle as well.9 Consistent with the idea that mutagenic TLS acts late in the cell cycle, the fidelity of DDT appears to be lower in G2 phase than G1 phase in both budding yeast and mammalian cells.35,36
The encounter of replication forks with UV lesions activates a DNA damage checkpoint response that has a role in regulating DDT mechanisms37-39 and delays cell cycle progression to provide additional time for PRR.37,40-44 The checkpoint is activated when sensor proteins detect transitions between double- and single- stranded DNA such as those found in daughter-strand gaps at UV lesions and in double-strand breaks (DSBs) or perhaps other abnormal structures generated at replication forks.2,45 In fission yeast and mammalian cells, checkpoint activation delays cell cycle progression at a point in late G2 phase referred to as the G2/M checkpoint.46-49 After this point in the cell cycle, the induction of DNA damage no longer delays mitosis or cytokinesis.41,50-53 Thus, the cell cycle of most eukaryotes is divided into 2 distinct periods, a “responsive” period from the birth of the cell to the G2/M checkpoint when DNA damage can elicit checkpoint-dependent mitotic delay and a “refractory” period after the G2/M checkpoint.
S. pombe Rev1 and Polζ are required to survive UV radiation but, unlike Polη, these polymerases do not repair daughter-strand gaps during the responsive period of the cell cycle, suggesting that their activity might be limited to the refractory period.42 The functions of the different DDT mechanisms have not been precisely mapped within the cell cycle of any organism, and the influence of checkpoint surveillance on the choice between different mechanisms is unknown.
We report here that the checkpoint response to UV provides a cellular mechanism to ensure that exclusively nonmutagenic DDT mechanisms go to completion before mitosis. Epistasis analysis indicates that 2 nonmutagenic DDT mechanisms operate during the checkpoint response: Rad51-mediated HDR and TLS mediated by the fission yeast homologs of Rad18, Rad5, and Polη. Of the 2 mechanisms, HDR has the more substantial role. We found that cells completely lose the capacity to complete HDR when damage is incurred after the G2/M checkpoint, and that Rev1 and Polζ have a specialized role in DDT at structures generated after this point. The mechanisms by which these structures may be produced are considered in the discussion section. We report evidence that the Rad51 recombinase limits mutagenic TLS during the checkpoint response, thereby favoring the error-free repair of daughter-strand gaps and the preservation of genome integrity.
Results
To study the coordination between cell cycle progression and DDT, we used live-cell imaging to observe the response of individual cells to DNA damage. Asynchronous fission yeast populations were exposed to a short pulse of UV radiation and imaged every 2 minutes over the course of 3 cell cycles. The resulting time-lapse series were analyzed manually to determine the times of cell cleavage (cytokinesis), the lengths of cells in the frame before cleavage, and the terminal phenotypes of cells that failed to divide (Fig. S1). In each experiment we quantified the viability and DNA damage checkpoint-dependent cell cycle delay of 300 cells that incurred DNA damage at different stages of the cell cycle.41 As described in more detail below, our approach allowed us to determine the stages of cells at the time of irradiation without employing a synchronization protocol that could potentially perturb cellular metabolism and cell cycle progression.
Where possible, we used a dose of 5 J/m2 which introduces 1,455 ± 335 dimeric photoproducts per S. pombe genome42 and is comparable to sunlight exposure.2 This dose is sublethal for most cells (94.8 ± 0.7% viability), indicating that S. pombe has evolved mechanisms to effectively repair and tolerate the level of damage incurred. Irradiation with 5 J/m2 activates a robust checkpoint response that delays the second cell cycle after irradiation.41 Cells only delay during the second cycle because checkpoint activation occurs after lesions are carried into S phase as previously described and as discussed in more detail below. Fission yeast cells continue to elongate during checkpoint-mediated delays, so the length of a cell at cleavage provides a measure of the duration of checkpoint delay that is independent of cycle time. Since there is very little variance in the length of unirradiated cells at the time of cleavage and virtually no checkpoint-independent cell elongation, we have found that increase in cell length at cleavage is the most sensitive and specific metric of the checkpoint response.
Rad51-mediated HDR and the fission yeast homologs of Rad18 and Rad5 are active during the responsive period of the cell cycle
The first question we asked was what DDT pathways are active during the checkpoint response to UV. Elimination of such pathways prolongs the checkpoint response during the second cycle as measured by size of cells at cleavage.42 A point mutation was introduced into the gene encoding Polη (eso1) to eliminate polymerase activity without disrupting an essential sister-chromatid cohesion domain. All other pathways were eliminated using null alleles of DDT genes. We have previously shown that eso1-cs cells lacking Polη activity have a modest prolongation of the checkpoint response after 5 J/m2, while cells lacking Rev1 and Polζ have reduced cell viability but do not have a prolonged checkpoint response even after much higher UV doses.42 Rhp18 and Rad8, the S. pombe homologs of budding yeast Rad18 and Rad5, respectively, are thought to regulate DDT via PCNA ubiquitination.54-56 In the absence of irradiation, cells lacking Rhp18 or Rad8 exhibited cycle times and lengths at cleavage that were similar to those of wild-type cells (Fig. 1A). After exposure to 5 J/m2 of UV, cells lacking either factor progressed through the first cell cycle at the same rate as wild-type cells but exhibited increased lengths at the end of the second cycle (Fig. 1B). The increases in cell length were comparable to those of cells lacking Polη activity.42 These observations indicate that Rad8 and Rhp18 both repair structures during the responsive period of the cell cycle that are recognized by the DNA damage checkpoint system.
Figure 1.
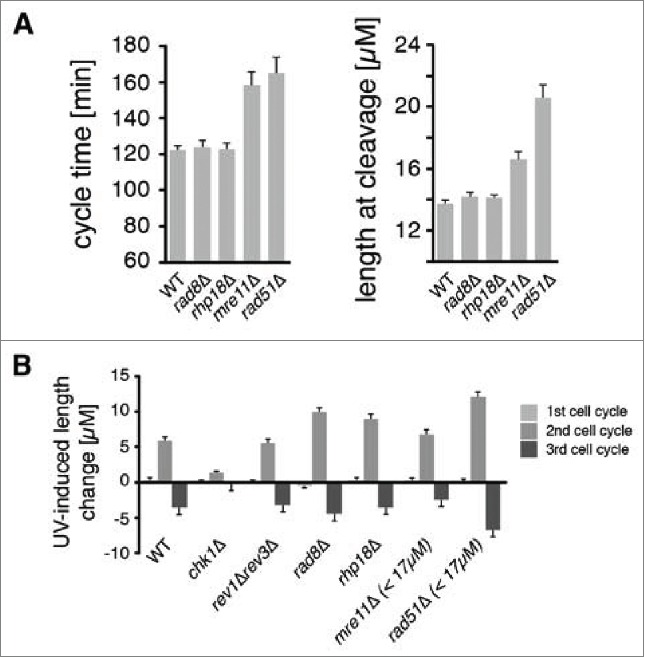
DDT pathways mediated by Rad8, Rhp18, and Rad51 function during the checkpoint response to UV. Asynchronous populations of fission yeast cells were imaged before and after exposure to UV radiation. (A) The average duration of the first cell cycle and the average lengths of cells at the first cleavage are shown for 300 mock-irradiated cells. Error bars denote 95% c.i. (B) Checkpoint responses of mutants in various repair pathways were calculated by subtracting the average length of mock-irradiated cells from the average length of cells exposed to 5 J/m2 of UV (rev3 encodes Polζ). Sample sizes are 300, 600, and 1200 cells for the 1st, 2nd, and 3rd cycles respectively. For mre11Δ and rad51Δ strains, the suppressive effects of spontaneous cell cycle delays were eliminated by restricting analysis to cells < 17 µM at the first cleavage as described in Fig. S2 and in the main text. This procedure reduced the sample size in the mre11Δ and rad51Δ analyses by ∼1/3 and ∼2/3 respectively.
The Rad51 recombinase is required for HDR and contributes to both DDT and the repair of DSBs. The phenotype of cells lacking Rad51 required careful analysis due to the heterogeneous cell cycle progression of mutant strains. In the absence UV, some rad51Δ cells had normal cycle times and underwent cleavage at normal lengths, but a fraction of cells underwent “spontaneous” delays and were elongated at cleavage, presumably due to a checkpoint response to endogenous DNA damage that was not repaired efficiently (Fig. 1A, Fig. S2A). Like mutations in other DDT pathways, deletion of rad51 did not affect the overall kinetics of the first cleavage after irradiation with 5 J/m2 of UV (Fig. S2A). The subset of cells that underwent spontaneous delays during the first cycle exhibited shorter UV-induced checkpoint delays on the second cycle and higher viability than the cells that did not delay on the first cycle (Fig. S2C, E). The likely explanation for these reduced effects of UV is that the cells that underwent first cycle delays had more time for excision repair, so they entered S phase with fewer UV photoproducts and generated fewer abnormal structures requiring Rad51-dependent HDR. We found that if we confined the analysis to cells that did not delay on the first cycle (i.e. divided at a length less than 2 s.d. above the mean, or < 17 µm) we could eliminate the effects of this suppression entirely. The resulting population of rad51Δ cells exhibited exactly the same cleavage kinetics as wild-type cells (Fig. S2G). Elimination of Rad51 resulted in the greatest increase in the UV-induced checkpoint response during the second cycle of any DDT mechanism (Fig. 1B). We conclude that Rad51-dependent HDR operates during the responsive period of the cell cycle and is particularly important in the repair of structures formed after UV lesions are carried into S phase.
Rad51 recombinase has roles in both DDT and DSB repair. To ask whether DSBs were contributing to the checkpoint responses of cells irradiated at 5 J/m2 UV, we studied the effect of deleting mre11, which is known to be required for the repair of DSBs.57 In striking contrast to rad51Δ cells, mre11Δ cells exhibited exactly the same checkpoint response to UV as wild-type cells (Fig. 1B; for a more detailed analysis see Fig. S3). So duration of the UV-induced checkpoint response is not related to the rate at which DSBs can be repaired. We conclude that the checkpoint response is triggered primarily by daughter-strand gaps and perhaps other structures whose repair does not require Mre11.
Live-cell imaging can be used to determine cell cycle stage at the time of irradiation
In addition to simply examining the global checkpoint response to UV irradiation, live-cell imaging allowed us to reconstruct and quantify the responses of cells that incur damage at different stages and to make a much more detailed analysis of the roles of various DDT mechanisms during the cell cycle. The S. pombe cell cycle is similar to that of most eukaryotes, including human cells, except that cleavage occurs in late S phase rather than immediately after mitosis (Fig. 2A). After nuclear division, the 2 daughter nuclei pass through a brief G1 phase and initiate DNA replication. Cleavage then generates 2 daughter cells, each with close to a 2C DNA content, but a low level of DNA synthesis continues in the daughter cells for some time after cleavage.58
Figure 2.

The use of cleavage time to determine cell cycle stage in asynchronous populations. (A) Illustration of the fission yeast cell cycle with sample images from a time-lapse movie of a cell expressing tagged histone H3. C = cleavage/cytokinesis. (B) Determination of the timing of cell cycle landmarks. Confocal microscopy was used to make time-lapse movies of strains expressing GFP fused to α-tubulin (Atb2-GFP) or RPA1 (Rad11-mYFP). Cell cycle landmarks were manually scored as described in Fig. S8. A moving average of the percentage of cells that had passed the indicated landmark was calculated using a 15 min window at increments of 1 min (n = 200 to 300 cells for each experiment). The position of the G2/M checkpoint, determined in panels D and E, is also shown. (C) The percent of cells at the indicated stages were determined from the data in B by calculating the percent of cells that had passed one landmark but had yet to pass the subsequent landmark as described in detail in Fig. S8. (D-F) Cells pass the G2/M checkpoint 1 h before cleavage. Kinetics of the first cleavage event is shown for 300 cells with the indicated genotypes and irradiation conditions (UV or X-rays). (G-H) The checkpoint response to UV occurs prior to mitotic spindle formation. The kinetics of spindle formation and cleavage are shown for 300 cells expressing tagged α-tubulin (Atb2-GFP) imaged after mock irradiation or exposure to 25 J/m2.
The position of a cell in the cell cycle is directly related to the age of the cell and is inversely related to the amount of time that elapses before it undergoes cleavage.41,50 Thus, measuring the time of a cell's birth or cleavage can provide information about its stage. We have found that age is a relatively poor predictor of cell cycle stage in fission yeast because of the large variability in the duration of G2 phase (Fig. S4). In contrast, there is relatively little variance in the elapsed time between major cell cycle transitions and the subsequent cleavage event. To determine the relationship between stage and first cleavage time, we used fluorescence microscopy to measure several cell cycle landmarks (Fig. 2B). The median times of these events were: 38 min prior to cleavage for spindle formation, 33 minutes prior to cleavage for nuclear separation, 21 min prior to cleavage for spindle disassembly, and 11 min prior to cleavage for septation. From the timing of cell cycle landmarks, we calculated the average stage of cells as a function of first cleavage time (Fig. 2C). These and other results indicate that first cleavage time can be used to determine the average stage at which cells incur DNA damage during a pulse of radiation without the need to synchronize cells.
A UV dose of 5 J/m2 activates a checkpoint response that delays the second cell cycle following irradiation because most daughter-strand gaps containing UV lesions are generated in binucleated S phase cells that are already committed to cleavage. Thus, the gaps are carried through cytokinesis into daughter cells and trigger a checkpoint response that delays the second cleavage. At higher doses of UV (≥ 10 J/m2), checkpoint-dependent delay occurs on the first cycle as well as the second cycle.41 It is unclear what structures elicit the response during the first cycle, but they may include daughter-strand gaps resulting from a low level of ongoing DNA replication in G2 phase or excision repair gaps generated independently of replication forks.59 A plot of the kinetics of cleavage following irradiation with high doses of UV revealed that cells become refractory to damage-induced checkpoint delay one hour prior to cleavage (Fig. 2D), a point 22 minutes prior to the formation of the mitotic spindle (Fig. 2B). As shown in Figure 2E, entry into the refractory period following exposure to X-rays begins at the same point as for UV (Fig. 2E). As expected, checkpoint delay prior to this point is completely dependent upon Chk1 (Fig. 2F).60 The checkpoint response delays the disappearance of interphase microtubules and the formation of the mitotic spindle (Fig. 2G-H), indicating that fission yeast cells delay in G2 phase after UV exposure as reported for other forms of DNA damage.61 These results indicate that the UV-induced checkpoint response of S. pombe is similar to that of mammalian cells in its requirement for Chk1 and the position of the G2/M checkpoint within the cell cycle (late G2 phase).
The response of S. pombe cells to UV irradiation at different points in the cell cycle is shown in Figure 3. The upper panel in each plot shows the average length of the irradiated cells at the second cleavage, which provides a measure of the DNA damage checkpoint response. The data are plotted as a function of the elapsed time from irradiation to first cleavage, which, as described above, defines the cell cycle position at the time of irradiation (inset). The lower panel shows the percent survival of cells irradiated at different points in the cell cycle. Survival was quantified by scoring cells that continued to divide for 3 generations following irradiation. In wild-type cells, the checkpoint delay and viability curves were both biphasic (Fig. 3A). In the first phase, delay and lethality increased from relatively low values in early G2 phase to maxima in cells irradiated near the beginning of S phase. In the second phase, checkpoint delay and lethality in response to UV irradiation rapidly decreased as cells progressed through S phase.
Figure 3.

The effects of eliminating DDT pathways on UV-induced checkpoint delays and survival as a function of position in the cell cycle. (A-I) Data from time-lapse movies are plotted as a function of the duration between irradiation and first cleavage, a measure of cell cycle stage at the time of irradiation. Cell cycle stage at the time of irradiation is indicated along the x axis, which was inverted so that cell cycle events appear in temporal order. The beginning of S phase was defined as the point of maximal checkpoint delay in WT cells. A moving average of cell length at the 2nd cleavage, a measure of the checkpoint response, is plotted in the top panels for 600 daughter cells. A moving average of the percentage of cells that continued to divide for 3 generations (viability out of 1,200 potential granddaughter cells) is plotted in the bottom panels. A 15 min window was calculated at increments of 1 min. UV doses were chosen so that the mutant strain viabilities and checkpoint delays were in the dynamic range of the assay (rev3 encodes Polζ; the eso1-cs allele is a Polη catalytic site mutant). For a comparison of each strain at 5 J/m2, see Fig. S6. To deduce the stage of rad51Δ cells at the time of UV and to eliminate the suppressive effects of spontaneous cell cycle delays, we restricted our analysis in F to cells that were < 17 µM at the first cleavage as described in the text. This procedure reduced the sample size by ∼2/3.
The first phase of the UV response can be readily explained if the excision repair of UV lesions before S phase reduces checkpoint delay and DNA lethal damage. To investigate this possibility, we eliminated S. pombe pathways known to repair UV lesions. Excision repair of CPDs and other dimeric photoproducts is carried out by 2 mechanisms in fission yeast, nucleotide excision repair (NER) and an additional pathway mediated by the UVDE endonuclease.62 In rhp14Δuve1Δ cells that lack both excision pathways, the cleavage kinetics after 5 J/m2 of UV were similar to those of wild-type cells during the first cycle, but there was a dramatically prolonged checkpoint delay during the second cycle (Fig. S5). Unlike wild-type cells, the duration of this second cycle delay was the same for cells irradiated from early G2 until the onset of S phase (Fig. 3B). This phenotype was not observed in either the uve1Δ or rhp14Δ single mutants. Thus, variation in the UV response during the first phase is due to the repair of UV lesions by NER and UVDE, and the level of checkpoint delay and lethal damage in wild-type cells is correlated with the number of lesions left unrepaired by these processes at the onset of S phase.
The reduction in the UV response during the second phase, which occurred when cells were irradiated during S phase, was the same in wild-type cells and cells lacking excision repair (Fig. 3B). The rapid decline in the UV response during this phase was presumably due to the decreasing number of encounters of replication forks with UV lesions in cells irradiated after an increasing fraction of the genome had completed replication. We conclude that, in both the first and second phases of the UV response, the level of checkpoint delay and lethality is correlated with the number of replication forks expected to encounter UV lesions during S phase.
The observation that the duration of checkpoint delay is not affected by the presence or absence of excision repair when cells incur damage during S phase (Fig. 3B) suggests that the checkpoint response is not due to DSBs generated when replication forks encounter strand breaks created by excision repair of UV lesions. This interpretation is consistent with our conclusion, based on analysis of mre11Δ cells, that UV-induced DSBs do not constitute a major signal for checkpoint activation after 5 J/m2 of UV.
Two nonmutagenic DDT pathways repair daughter-strand gaps generated before the G2/M checkpoint
Cells lacking Polη activity, Rad8, Rhp18, or Rad51, all of which are active in PRR during the responsive period of the cell cycle, showed increased checkpoint responses and cell killing relative to wild-type cells after UV irradiation (Figs. 3C–F, S6). However, the magnitudes of the phenotypes of these mutants at different stages of the cell cycle followed a similar pattern to that of wild-type cells, i.e., the enhanced checkpoint delay and lethality relative to wild-type cells increased from a relatively low value in early G2 to a maximum at the onset of S phase and then decreased rapidly with progression through S phase. Thus, the roles of the DDT pathways that are active during the responsive period of the cell cycle were correlated with the number of replication forks expected to encounter lesions, consistent with the idea that these pathways repair daughter-strand gaps and possibly other structures generated during S phase.
To determine if Polη, Rad8, and Rhp18 function in the same or different DDT pathways, we examined the phenotypes of double mutant cells. Elimination of Rhp18 from rad8Δ cells did not further increase checkpoint delays or reduce viability (Fig. 3G), indicating that Rhp18 functions through Rad8 under the conditions of our experiments. Elimination of Rad8 from eso1-cs cells had little or no further effect on checkpoint delays or cell viability (Fig. 3H), indicating that Rad8 functions primarily through Polη and does not mediate an independent template-switching mechanism. Thus, Rad8, Rhp18, and Polη function in a single pathway that is dependent upon the catalytic activity of Polη. Elimination of Polη activity from rad51Δ cells increased both the duration of checkpoint delays and viability loss (Fig. S7), indicating that Rad51-mediated HDR functions independently from Polη. We conclude that 2 nonmutagenic pathways are operative during the checkpoint response to UV, Rad51-dependent HDR and a second pathway mediated by Rhp18/Rad8/Polη. Of the 2 pathways, HDR has a much more significant role (Figs. 1, S6). The effects of eliminating Rad51 were greater after 2 J/m2 than those observed after 5 J/m2 upon elimination of Rad8, Rad18, or Polη activity (Fig. 3F vs. 3C, D, E - note difference in scale).
Rev1 and Polζ are required to survive UV damage incurred between the G2/M checkpoint and anaphase B
To study the effects of eliminating mutagenic TLS, cells were irradiated with 10 J/m2, a UV dose that allowed us to accurately quantify the contributions of Rev1 and Polζ to viability at all stages of the cell cycle. The checkpoint responses of strains lacking mutagenic TLS were identical to wild-type cells after irradiation at all stages, consistent with our previous analysis of the overall population.42 (Fig. 3I, upper panel). Surprisingly, mutant cells completely lacking mutagenic TLS had the same viability as wild-type cells when irradiated at the onset of S phase. This finding is particularly striking because cells irradiated at this point exhibit the greatest dependence on the other DDT mechanisms for survival. Thus, both the viability and checkpoint data indicate that exclusively nonmutagenic DDT mechanisms are employed when damage is incurred during S phase.
In Figure 4A we have overlaid the average UV response data with the median time of occurrence of several cell cycle landmarks. The data indicate that there was no difference in the viabilities of wild-type and mutagenic TLS mutant cells irradiated after late anaphase B through S phase. Cell death resulting from elimination of Rev1/Polζ occurred primarily when the cells were irradiated during a narrow window of the cell cycle from the G2/M checkpoint to late anaphase B. These observations indicate that mutagenic TLS carries out DDT at a subset of DNA damage-induced structures that are formed after the G2/M checkpoint.
Figure 4.
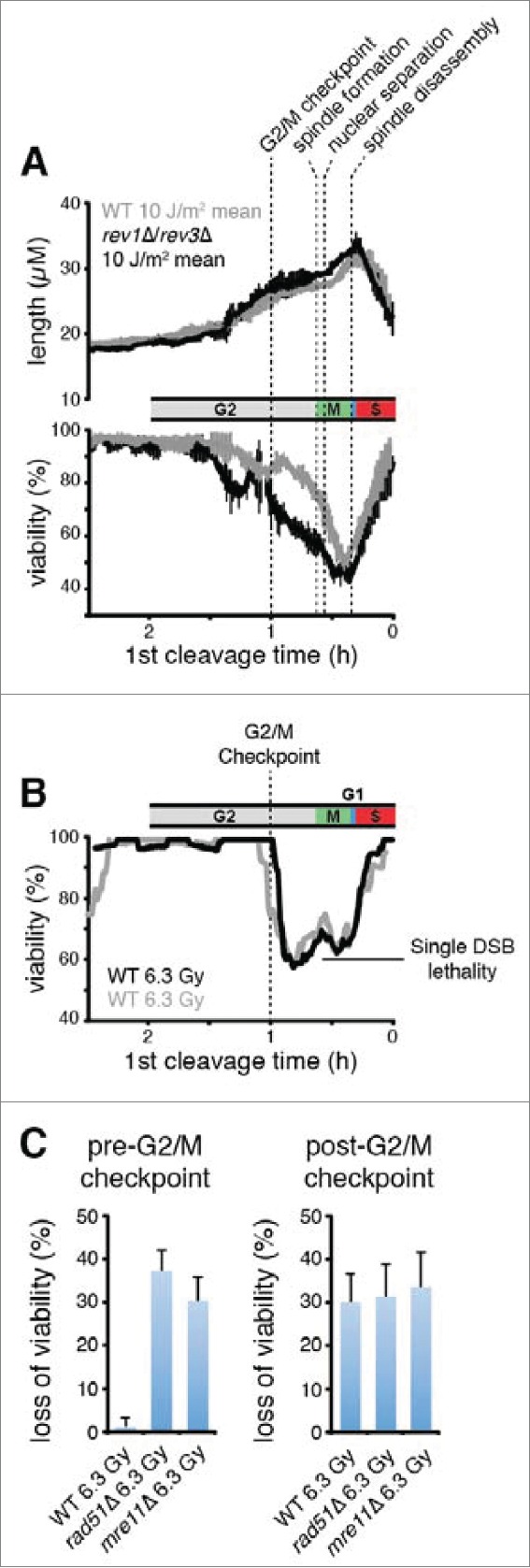
Cells abruptly lose the capacity for HDR of DSBs and begin to exhibit a requirement for mutagenic TLS when they pass the G2/M checkpoint. (A) Rev1 and Polζ are required for survival when damage is incurred between the G2/M checkpoint and mitotic spindle disassembly. The mean and s.d. of the experiments from Figure 3I are plotted. The median times established in Figure 2B are shown for the indicated landmarks. (B) Populations of 300 cells were imaged after exposure to 6.3 Gy of X-rays, a dose chosen to produce 1 DSB/cell on average (0.5 DSB/genome).63 The probability that a genome received one or more DSBs was 0.39 at this level of exposure assuming a Poisson distribution, so 39% loss of viability was expected if each DSB were lethal. Viability of wild-type cells, calculated as in Figure 3, is plotted as a function of cell cycle stage. Two separate experiments are shown. (C) Wild-type cells exhibit the same X-ray sensitivity as cells that lack HDR when irradiated after the G2/M checkpoint. Loss of viability due to X-ray exposure was calculated by subtracting the fraction of viable cells in an X-irradiated population from that of a mock-irradiated population. Error bars show 95% c.i. The pre-G2/M checkpoint subset represents cells that underwent cleavage 1 hour or more after irradiation, and the post-G2/M checkpoint subset is cells that cleaved less than 1 h after irradiation.
The capacity for HDR-mediated repair of DSBs is lost when damage is incurred after the G2/M checkpoint
While our data demonstrated that Rad51-dependent HDR has a particularly critical function prior to the G2/M checkpoint, a remaining question was whether HDR continues to function in parallel with mutagenic TLS after the G2/M checkpoint. The viability of cells irradiated in late G2 and M phase clearly depends upon Rad51 (Fig. 3F). This phenotype is consistent with a role for HDR-mediated DDT during late G2 and M phase, but is also consistent with the hypothesis that HDR functions in the repair of structures generated when lesions introduced in G2 or M phase are carried into the following S phase. To directly examine the capacity of cells for HDR at various stages of the cell cycle, we exposed an asynchronous population of wild-type cells to an X-ray dose expected to produce 1 DSB per cell,63 and followed the fates of individual cells over 3 generations to determine cell viability. DSBs induced by X-rays are potentially lethal, but can be repaired by HDR when an undamaged sister chromatid is available to provide a template for repair synthesis.
We observed that almost all of the cells irradiated before the G2/M checkpoint remained viable, while a significant proportion of those irradiated after the G2/M checkpoint lost viability (Fig. 4B). The percentage of cells that lost viability when irradiated immediately after the G2/M checkpoint (∼40%) was approximately the same as the percentage of genomes expected to incur one or more DSBs (39%), suggesting that each DSB was eventually lethal. So, it appears that exponentially growing fission yeast cells completely lose the capacity to complete HDR when they pass the G2/M checkpoint. Consistent with this interpretation, we observed that that the loss of viability that occurs when wild-type cells are exposed to X-rays after the G2/M checkpoint is similar in magnitude to that observed in rad51Δ and mre11Δ cells that lack HDR altogether (Fig. 4C). Although HDR may still be active after the G2/M checkpoint in wild-type cells, there is likely insufficient time to complete the process before sister-chromatid separation occurs ∼25 minutes later because HDR is a slow process that may take 60 minutes or more to repair a single DSB.64 We conclude from these observations that S. pombe cells simultaneously lose the capacity for HDR and begin to exhibit a requirement for Rev1 and Polζ at the G2/M checkpoint.
Rad51 is required to limit mutagenic TLS prior to the G2/M checkpoint
The temporal correlation between the loss of HDR capacity and the gain of mutagenic TLS function could be explained if the process of HDR were to prevent the action of Rev1/Polζ in some way. We explored this possibility by studying the effects of eliminating mutagenic TLS in the presence and absence of Rad51. Complete elimination of mutagenic TLS from wild-type cells had no effect on the duration of the checkpoint response during the first cycle after UV (Fig. 5A). In contrast, elimination of Polζ from cells lacking Rad51 more than doubled the magnitude of the checkpoint response (Fig. 5A, B). Thus, elimination of Rad51 is sufficient to allow mutagenic TLS function prior to the G2/M checkpoint.
Figure 5.
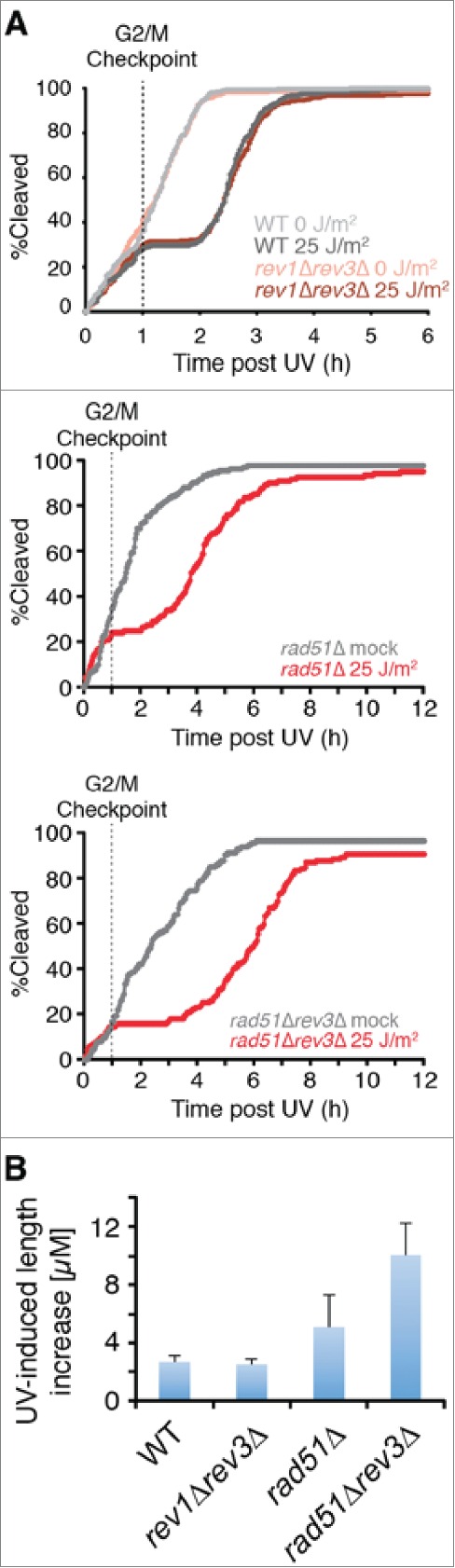
Rad51 is required to prevent Polζ function prior to the G2/M checkpoint. (A) Elimination of Polζ (encoded by rev3) has no effect on the duration of checkpoint delays when UV damage is incurred prior to the G2/M checkpoint in rad51+ cells, but increases delays substantially in rad51Δ cells. Kinetics of the first cleavage are shown for cells of the indicated genotypes exposed to mock irradiation or 25 J/m2 of UV. For strains lacking Rad51, the analysis is restricted to cells with mother cells that were < 17 µM at the cleavage prior to irradiation to reduce the suppressive effects of spontaneous cell cycle delays. (B) UV-induced length increase during the first cycle was calculated for the experiments in A by subtracting the mean length of a mock-irradiated population from the mean length of a population exposed to 25 J/m2 of UV. Error bars denote 95% c.i.
Discussion
All cells take advantage of redundant information in the genome to repair DNA damage without generating mutations. Excision repair processes use redundant information within the double helix to repair base damage and HDR uses redundant information within the sister chromatids to repair DSBs and daughter-strand gaps. Given the apparent selective advantage of high fidelity repair, it has been unclear why cells sometimes employ mutagenic TLS rather than HDR to repair daughter-strand gaps.
To better understand the coordination between the DDT mechanisms that repair daughter-strand gaps, we quantified the effects of eliminating DDT factors on the UV-induced DNA damage response of cells irradiated throughout the cell cycle. The results indicate that 2 nonmutagenic DDT pathways repair structures recognized by the DNA damage checkpoint during the responsive period of the cell cycle. The first pathway requires Rad51-mediated HDR and the second requires Rhp18, Rad8, and the catalytic activity of Polη. Thus, S. pombe cells employ exclusively nonmutagenic DDT mechanisms during the checkpoint response to UV. We found no evidence for a third DDT mechanism involving an independent template-switch. Of the 2 pathways active during the responsive period, HDR had a much more significant role. HDR can, in principal, tolerate any type of lesion whereas Polη function is specialized for CPDs. The ability of HDR to tolerate a broader range of lesions may account for the relatively large effects of eliminating Rad51. Alternatively, the large effects could be explained if cells preferentially use HDR over Polη at all lesions, including CPDs, because of the relatively high error rate of Polη.
DDT sometimes functions to repair structures formed outside of S phase.65-67 However, after a UV dose that might reasonably be expected to occur in nature (5 J/m2), elimination of nonmutagenic DDT factors did not slow the repair of structures that signal to the DNA damage checkpoint until after lesions were carried into S phase. Furthermore, the increase in checkpoint delays and cell killing that resulted from eliminating these pathways were both correlated with the frequency with which replication forks are expected to encounter UV lesions when cells incur damage at different stages of the cell cycle. These observations suggest that HDR and the Rhp18/Rad8/Polη pathway function most frequently to repair replication-dependent structures and that the formation of replication-independent structures requiring DDT is relatively rare under the conditions of our experiments. We observed that the duration of checkpoint delay is not affected by the lack of HDR-mediated DSB repair in mre11Δ cells, suggesting that the most common structures formed after UV lesions are carried into S phase are daughter-strand gaps and perhaps other structures that can be repaired in the absence of Mre11.
Surprisingly, cells lacking mutagenic TLS behaved like wild-type cells when irradiated during S phase. Rev1 and Polζ were required for survival primarily when damage was incurred during a narrow window of the cell cycle between the G2/M checkpoint and anaphase B. Our analysis of cell survival after X-ray exposure revealed that the beginning of this window marks the point at which cells completely lose the capacity to complete HDR. Thus, cells began to exhibit a requirement for mutagenic TLS at the same point at which they lost the ability to carry out HDR. Our findings suggest that HDR and mutagenic TLS function sequentially rather than in parallel, which could potentially explain why mutagenic TLS is required for the proliferation of mammalian cells: there may be no other general mechanism for DDT after the G2/M checkpoint.
We found that elimination of Rad51 was sufficient to allow Polζ to function prior to the G2/M checkpoint. This observation indicates that Rad51 has a critical role in limiting mutagenic TLS before the G2/M checkpoint, and suggests a potential mechanism for the cell cycle regulation of Rev1/Polζ. There is evidence that the ssDNA binding protein RPA is required to stimulate the repair of daughter-strand gaps by TLS.68 One possible explanation for our results is that the formation of Rad51 filaments on ssDNA blocks RPA binding, thereby preventing mutagenic TLS while HDR is active. If this were the case, then the cyclical inactivation of HDR at the metaphase-to-anaphase transition69,70 might relieve a block to mutagenic TLS. Consistent with the idea that mutagenic TLS becomes activated at the metaphase-to-anaphase transition, the levels of Rev1 protein appear to increase at around this point in the S. pombe cell cycle.71 It is unclear how a role for Rad51 in the general inhibition of TLS would allow for the function of Polη that we observed during the responsive period of the cell cycle. It is possible that Polη maintains some activity at Rad51-coated gaps or functions at a subset of daughter-strand gaps that are not coated by Rad51 for some reason.
Although most of the viability loss resulting from elimination of Rev1/Polζ occurred in cells irradiated after the G2/M checkpoint, a small but reproducible loss is evident in cells irradiated just prior to the G2/M checkpoint (Fig. 4A). This population is expected based on our understanding of the UV response of S. pombe. Because UV lesions do not generally elicit a checkpoint response until S phase, lesions introduced prior to the G2/M checkpoint that are not removed by excision repair can be carried into later stages of the cell cycle without inducing delay. Such lesions may be processed after the G2/M checkpoint into structures that require mutagenic TLS for repair.
Because S. pombe cells exhibit a requirement for mutagenic TLS long after S phase is largely completed, the origin of the structures repaired by Rev1 and Polζ is not entirely clear. Daughter-strand gaps could be generated in areas of the genome that have not completed DNA replication by the time of the G2/M checkpoint. In addition, the primary UV lesions could be processed into structures that require mutagenic TLS by mechanisms that are independent of DNA replication. For instance, NER at closely-spaced lesions produces gapped structures with lesions in the single-stranded region that are similar to daughter-strand gaps and can elicit a checkpoint response.66,72 Repair of these gaps by mutagenic TLS generates most of the UV-induced mutations in stationary phase cells, but accounts for only a small fraction of mutations in proliferating cells like those used in this study.16-19,73 Thus, the requirement for Rev1 and Polζ that we observed after the G2/M checkpoint may derive primarily from a role for these polymerases in repairing replication-dependent structures generated after the time of commitment to mitosis (Fig. 6). This interpretation is consistent with the data presented here and can provide a parsimonious explanation for the observation that UV signature mutations in human melanomas occur with an increased frequency in late-replicating regions of the genome.13,74,75
Figure 6.
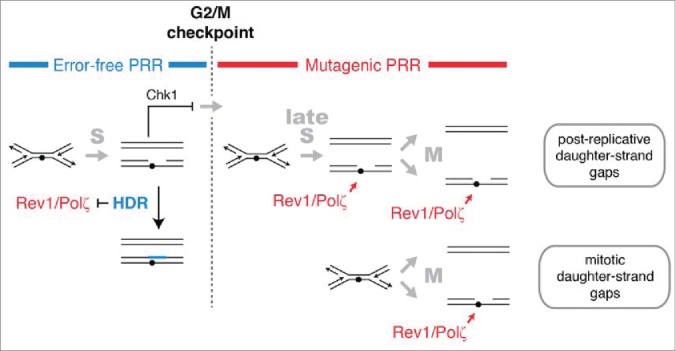
HDR and mutagenic TLS function sequentially and are regulated in concert with checkpoint signaling to maximize the potential for error-free post-replication repair. The DNA damage checkpoint response to UV delays mitosis to allow HDR to complete error-free PRR using redundant information within the sister chromatids. Rad51 recombinase is required to prevent mutagenic TLS by Rev1 and Polζ during the checkpoint delay. A pathway mediated by Rhp18, Rad8, and Polη makes a more limited contribution to PRR at CPDs during the checkpoint delay and may continue to function after the G2/M checkpoint (not shown). After the G2/M checkpoint, cells lose the capacity to complete HDR and rely on Rev1 and Polζ to restore the continuity of the double helix. We suggest that the structures repaired by Rev1 and Polζ are formed most frequently in regions of the genome that remain unreplicated after the G2/M checkpoint. Gaps may form in such regions when replication forks encounter lesions (post-replicative daughter-strand gaps) or when an unreplicated region is unwound during mitosis (mitotic daughter-stand gaps). Rev1 and Polζ may begin to function immediately after cells pass the G2/M checkpoint or, more likely, gain access to DNA when HDR is inactivated at the metaphase-to-anaphase transition.
There are several reasons why DNA replication may not be complete at the time of commitment to mitosis. The mode of initiation of DNA replication in fission yeast (and probably most eukaryotes) is quasi stochastic, so the sizes of replicons have an exponential distribution with a significant number of replicons that are very large and that require a long time to finish replication.76 Failure to complete DNA replication in some regions of the genome by the G2/M checkpoint might also result from chromosomal features that give rise to a low density of origins or create natural impediments to the progress of replication forks.77 For example, it has been shown that common fragile sites in the genome occur at large transcription units that are late replicating because they contain a paucity of replication initiation sites.78 Transcription-associated complexes, G quadruplexes, R-loops and possibly other structures could slow the process of DNA chain elongation in certain regions of the genome and delay their replication until after the G2/M checkpoint.79
When DNA replication has not been completed by the G2/M checkpoint, active replication forks could continue to generate daughter-strand gaps that require mutagenic TLS for repair (Fig. 6, top pathway). Daughter-strand gaps may also be produced when unreplicated DNA containing DNA damage is unwound during the separation of sister chromatids (Fig. 6, bottom pathway). It has been suggested that the ultrafine bridges observed between the separating sister chromatids at anaphase represent, at least in part, regions of DNA that have not completed DNA replication prior to mitosis.78 The resolution of such structures, perhaps by a RecQ helicase and topoisomerase III,80 would be expected to give rise to single-stranded gaps in the daughter chromatids that could require Rev1/Polζ for their repair.
Of the S. pombe DDT pathways operational during the checkpoint response to UV, only HDR is known to provide a general mechanism for error-free DDT and only HDR is inactivated by sister-chromatid separation during mitosis. Given these considerations, it appears that the role of UV-induced checkpoint delay is to maintain cells in a state in which the redundant information within sister chromatids is available for error-free PRR by HDR (Fig. 6). Conversely, the role of Rev1 and Polζ after UV exposure is to restore the continuity of the double helix after cells progress past the G2/M checkpoint and the capacity to use redundant information for repair is irreversibly lost. Thus, regulating the utilization of DDT pathways in concert with checkpoint signaling maximizes the potential for error-free DDT while confining the use of the mutagenic pathway to the interval when survival benefit outweighs the increased mutational burden.
Methods
Time-lapse microscopy
The strains analyzed are listed in Tables S1 and S2. Time-lapse imaging and UV irradiation were carried out as described previously.41 Briefly, a field containing ∼150 cells grown at 30ºC in YE6S rich medium was observed for 3-4 h, irradiated or mock irradiated, then observed until confluent (12 to 24 hours). Images were acquired every 2 minutes. After the movie was completed, 300 cells were marked off at the time of irradiation and both the time of cleavage and length of the cell in the frame before cleavage were manually recorded. These measurements were performed on the cleavage prior to irradiation and 3 cleavages after irradiation. Cycle times were calculated by taking the difference between the cleavage times of a cell and its mother. Loss of viability was recorded when the cell wall appeared to collapse or the cell stopped increasing in length for 1 hour or longer. In all instances, viability was determined from the number of cells that continued to divide for 3 generation after irradiation (e.g. cells that successfully initiated a third cleavage after irradiation).
Irradiation
Cells were UV-irradiated as previously described.41 An X-ray dose of 6.3 Gy was generated using an X-RAD225C irradiator (Precision X-Ray, Inc.., North Branford CT, USA). Cells were irradiated for 55 s at a dose rate of 6.87 Gy/min. This dose was chosen to produce 1 DSB per G2 phase cell based on empirical data reviewed in Prise & Stenerlow.63
Supplementary Material
Abbreviations
- DDT
DNA damage tolerance
- HDR
homology-directed repair
- PRR
post-replication repair
- TLS
translesion DNA synthesis
- DSB
double-strand break
- NER
nucleotide excision repair
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Amanda Pneumann, Emily Clark, and Hiteshkumar Khatnani for their technical work, and John Petrini for comments on the manuscript.
Author contributions
A.C. designed, performed and analyzed experiments. T.K. designed and analyzed experiments, and coordinated the work. A.C. and T.K. wrote the manuscript.
Funding
This work was supported by the National Institutes of Health [5R01GM050806 to T.K.].
References
- 1.Brash DE. UV signature mutations. Photochem Photobiol 2015; 91:15–26; PMID:25354245; http://dx.doi.org/ 10.1111/php.12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callegari AJ, Kelly TJ. Shedding light on the DNA damage checkpoint. Cell Cycle 2007; 6:660–6; PMID:17387276; http://dx.doi.org/ 10.4161/cc.6.6.3984 [DOI] [PubMed] [Google Scholar]
- 3.Lemontt JF. Genetic and physiological factors affecting repair and mutagenesis in yeast. Basic Life Sci 1980; 15:85–120; PMID:7011312 [DOI] [PubMed] [Google Scholar]
- 4.Waters LS, Minesinger BK, Wiltrout ME, D'Souza S, Woodruff RV, Walker GC. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 2009; 73:134–54; PMID:19258535; http://dx.doi.org/ 10.1128/MMBR.00034-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sale JE. Competition, collaboration and coordination–determining how cells bypass DNA damage. J Cell Sci 2012; 125:1633–43; PMID:22499669; http://dx.doi.org/ 10.1242/jcs.094748 [DOI] [PubMed] [Google Scholar]
- 6.Wittschieben J, Shivji MK, Lalani E, Jacobs MA, Marini F, Gearhart PJ, Rosewell I, Stamp G, Wood RD. Disruption of the developmentally regulated Rev3l gene causes embryonic lethality. Curr Biol 2000; 10:1217–20; PMID:11050392; http://dx.doi.org/ 10.1016/S0960-9822(00)00725-9 [DOI] [PubMed] [Google Scholar]
- 7.Bemark M, Khamlichi AA, Davies SL, Neuberger MS. Disruption of mouse polymerase zeta (Rev3) leads to embryonic lethality and impairs blastocyst development in vitro. Curr Biol 2000; 10:1213–6; PMID:11050391; http://dx.doi.org/ 10.1016/S0960-9822(00)00724-7 [DOI] [PubMed] [Google Scholar]
- 8.Esposito G, Godindagger I, Klein U, Yaspo ML, Cumano A, Rajewsky K. Disruption of the Rev3l-encoded catalytic subunit of polymerase zeta in mice results in early embryonic lethality. Curr Biol 2000; 10:1221–4; PMID:11050393; http://dx.doi.org/ 10.1016/S0960-9822(00)00726-0 [DOI] [PubMed] [Google Scholar]
- 9.Bhat A, Andersen PL, Qin Z, Xiao W. Rev3, the catalytic subunit of Polzeta, is required for maintaining fragile site stability in human cells. Nucleic Acids Res 2013; 41:2328–39; PMID:23303771; http://dx.doi.org/ 10.1093/nar/gks1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR. Human mutation rate associated with DNA replication timing. Nat Genet 2009; 41:393–5; PMID:19287383; http://dx.doi.org/ 10.1038/ng.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen CL, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d'Aubenton-Carafa Y, Arneodo A, Hyrien O, et al.. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res 2010; 20:447–57; PMID:20103589; http://dx.doi.org/ 10.1101/gr.098947.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang GI, Murray AW. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evolution 2011; 3:799–811; PMID:21666225; http://dx.doi.org/ 10.1093/gbe/evr054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499:214–8; PMID:23770567; http://dx.doi.org/ 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito T, Yamasaki T, Domon M, Ishizaka S, Matsudai Y.. Ultraviolet-Induced Photoreversible Genetic Change Observed in Heterozygous Diploid System of Yeast - a Study of Uv Action Spectrum for Induction and of Photoreversion. Japanese J Genet 1964; 39:136; http://dx.doi.org/ 10.1266/jjg.39.136 [DOI] [Google Scholar]
- 15.You YH, Lee DH, Yoon JH, Nakajima S, Yasui A, Pfeifer GP. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J Biol Chem 2001; 276:44688–94; PMID:11572873; http://dx.doi.org/ 10.1074/jbc.M107696200 [DOI] [PubMed] [Google Scholar]
- 16.Yamasaki T, Ito T, Matsudaira Y. Studies on the Genetic Multiplicity of a Gene in Yeast Cells. 2. Effects of Ultraviolet Light. J Radiat Res 1963; 4:85–90; PMID:14193265; http://dx.doi.org/ 10.1269/jrr.4.85 [DOI] [PubMed] [Google Scholar]
- 17.Van Zeeland AA, Simons JW. Linear dose–response relationships after prolonged expression times in V-79 Chinese hamster cells. Mutat Res 1976; 35:129–37; PMID:178997; http://dx.doi.org/ 10.1016/0027-5107(76)90175-5 [DOI] [PubMed] [Google Scholar]
- 18.Enninga IC, Groenendijk RT, van Zeeland AA, Simons JW. Differential response of human fibroblasts to the cytotoxic and mutagenic effects of UV radiation in different phases of the cell cycle. Mutat Res 1985; 148:119–28; PMID:3969076; http://dx.doi.org/ 10.1016/0027-5107(85)90215-5 [DOI] [PubMed] [Google Scholar]
- 19.Ostroff RM, Sclafani RA. Cell cycle regulation of induced mutagenesis in yeast. Mutat Res 1995; 329:143–52; PMID:7603496; http://dx.doi.org/ 10.1016/0027-5107(95)00030-M [DOI] [PubMed] [Google Scholar]
- 20.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol 1968; 31:291–304; PMID:4865486; http://dx.doi.org/ 10.1016/0022-2836(68)90445-2 [DOI] [PubMed] [Google Scholar]
- 21.Lehmann AR. Postreplication repair of DNA in ultraviolet-irradiated mammalian cells. J Mol Biol 1972; 66:319–37; PMID:5037019; http://dx.doi.org/ 10.1016/0022-2836(72)90418-4 [DOI] [PubMed] [Google Scholar]
- 22.Prakash L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol Gen Genet 1981; 184:471–8; PMID:7038396; http://dx.doi.org/ 10.1007/BF00352525 [DOI] [PubMed] [Google Scholar]
- 23.di Caprio L, Cox BS. DNA synthesis in UV-irradiated yeast. Mutat Res 1981; 82:69–85; PMID:7022172; http://dx.doi.org/ 10.1016/0027-5107(81)90139-1 [DOI] [PubMed] [Google Scholar]
- 24.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 2006; 21:15–27; PMID:16387650; http://dx.doi.org/ 10.1016/j.molcel.2005.11.015 [DOI] [PubMed] [Google Scholar]
- 25.Torres-Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 2002; 22:2419–26; PMID:11884624; http://dx.doi.org/ 10.1128/MCB.22.7.2419-2426.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gangavarapu V, Prakash S, Prakash L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 2007; 27:7758–64; PMID:17785441; http://dx.doi.org/ 10.1128/MCB.01331-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res 2008; 18:162–73; PMID:18157158; http://dx.doi.org/ 10.1038/cr.2007.114 [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci U S A 2005; 102:15954–9; PMID:16247017; http://dx.doi.org/ 10.1073/pnas.0504586102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann AR, Kirk-Bell S, Arlett CF, Paterson MC, Lohman PH, de Weerd-Kastelein EA, Bootsma D. Xeroderma pigmentosum cells with normal levels of excision repair have a defect in DNA synthesis after UV-irradiation. Proc Natl Acad Sci U S A 1975; 72:219–23; PMID:1054497; http://dx.doi.org/ 10.1073/pnas.72.1.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita YM, Okada T, Matsusaka T, Sonoda E, Zhao GY, Araki K, Tateishi S, Yamaizumi M, Takeda S. RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. Embo J 2002; 21:5558–66; PMID:12374756; http://dx.doi.org/ 10.1093/emboj/cdf534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tateishi S, Niwa H, Miyazaki J, Fujimoto S, Inoue H, Yamaizumi M. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol Cell Biol 2003; 23:474–81; PMID:12509447; http://dx.doi.org/ 10.1128/MCB.23.2.474-481.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 2008; 30:519–29; PMID:18498753; http://dx.doi.org/ 10.1016/j.molcel.2008.03.024 [DOI] [PubMed] [Google Scholar]
- 33.Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M, et al.. Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J 2003; 22:3188–97; PMID:12805232; http://dx.doi.org/ 10.1093/emboj/cdg308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci U S A 2006; 103:8971–6; PMID:16751278; http://dx.doi.org/ 10.1073/pnas.0510167103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang D, Piening BD, Paulovich AG. The preference for error-free or error-prone postreplication repair in Saccharomyces cerevisiae exposed to low-dose methyl methanesulfonate is cell cycle dependent. Mol Cell Biol 2013; 33:1515–27; PMID:23382077; http://dx.doi.org/ 10.1128/MCB.01392-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, Livneh Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 2012; 40:170–80; PMID:21908406; http://dx.doi.org/ 10.1093/nar/gkr596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulovich AG, Armour CD, Hartwell LH. The Saccharomyces cerevisiae RAD9, RAD17, RAD24 and MEC3 genes are required for tolerating irreparable, ultraviolet-induced DNA damage. Genetics 1998; 150:75–93; PMID:9725831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kai M, Wang TS. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev 2003; 17:64–76; PMID:12514100; http://dx.doi.org/ 10.1101/gad.1043203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gangavarapu V, Santa Maria SR, Prakash S, Prakash L. Requirement of replication checkpoint protein kinases Mec1/Rad53 for postreplication repair in yeast. Mbio 2011; 2:e00079-11; PMID:21586645; http://dx.doi.org/ 10.1128/mBio.00079-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neecke H, Lucchini G, Longhese MP. Cell cycle progression in the presence of irreparable DNA damage is controlled by a Mec1- and Rad53-dependent checkpoint in budding yeast. Embo J 1999; 18:4485–97; PMID:10449414; http://dx.doi.org/ 10.1093/emboj/18.16.4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callegari AJ, Kelly TJ. UV irradiation induces a postreplication DNA damage checkpoint. Proc Natl Acad Sci U S A 2006; 103:15877–82; PMID:17043220; http://dx.doi.org/ 10.1073/pnas.0607343103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callegari AJ, Clark E, Pneuman A, Kelly TJ. Postreplication gaps at UV lesions are signals for checkpoint activation. Proc Natl Acad Sci U S A 2010; 107:8219–24; PMID:20404181; http://dx.doi.org/ 10.1073/pnas.1003449107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010; 141:255–67; PMID:20403322; http://dx.doi.org/ 10.1016/j.cell.2010.02.028 [DOI] [PubMed] [Google Scholar]
- 44.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2011; 465:951–5; http://dx.doi.org/ 10.1038/nature09097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179–204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tobey RA. Different drugs arrest cells at a number of distinct stages in G2. Nature 1975; 254:245–7; PMID:46594; http://dx.doi.org/ 10.1038/254245a0 [DOI] [PubMed] [Google Scholar]
- 47.al-Khodairy F, Carr AM. DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. Embo J 1992; 11:1343–50; PMID:1563350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep 2003; 4:671–7; PMID:12835754; http://dx.doi.org/ 10.1038/sj.embor.embor887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukas J, Lukas C, Bartek J. Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 2004; 3:997–1007; PMID:15279786; http://dx.doi.org/ 10.1016/j.dnarep.2004.03.006 [DOI] [PubMed] [Google Scholar]
- 50.Puck TT, Steffen J. Life Cycle Analysis of Mammalian Cells. I. A Method for Localizing Metabolic Events within the Life Cycle, and Its Application to the Action of Colcemide and Sublethal Doses of X-Irradiation. Biophys J 1963; 3:379–97; PMID:14062457; http://dx.doi.org/ 10.1016/S0006-3495(63)86828-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walters RA, Petersen DF. Radiosensitivity of mammalian cells. II. Timing and dose-dependence of radiation-induced division delay. Biophys J 1968; 8:1475–86; PMID:5753223; http://dx.doi.org/ 10.1016/S0006-3495(68)86567-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han A, Sinclair WK, Yu CK. Ultraviolet light-induced division delayed in synchronized Chinese hamster cells. Biophys J 1971; 11:540–9; http://dx.doi.org/ 10.1016/S0006-3495(71)86233-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whitson GL. Concepts in radiation cell biology. New York,: Academic Press, 1972 [Google Scholar]
- 54.Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, Furuya K, Watts FZ, Carr AM, Lehmann AR. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell 2006; 17:2976–85; PMID:16641370; http://dx.doi.org/ 10.1091/mbc.E05-11-1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coulon S, Ramasubramanyan S, Alies C, Philippin G, Lehmann A, Fuchs RP. Rad8Rad5/Mms2-Ubc13 ubiquitin ligase complex controls translesion synthesis in fission yeast. EMBO J 2010; 29:2048–58; PMID:20453833; http://dx.doi.org/ 10.1038/emboj.2010.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding L, Forsburg SL. Essential domains of Schizosaccharomyces pombe Rad8 required for DNA damage response. G3 2014; 4:1373–84; PMID:24875629; http://dx.doi.org/full_text [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tavassoli M, Shayeghi M, Nasim A, Watts FZ. Cloning and characterisation of the Schizosaccharomyces pombe rad32 gene: a gene required for repair of double strand breaks and recombination. Nucleic Acids Res 1995; 23:383–8; PMID:7885834; http://dx.doi.org/ 10.1093/nar/23.3.383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nasmyth K, Nurse P, Fraser RS. The effect of cell mass on the cell cycle timing and duration of S-phase in fission yeast. J Cell Sci 1979; 39:215–33; PMID:528581 [DOI] [PubMed] [Google Scholar]
- 59.Sertic S, Pizzi S, Lazzaro F, Plevani P, Muzi-Falconi M. NER and DDR: classical music with new instruments. Cell Cycle 2012; 11:668–74; PMID:22373527; http://dx.doi.org/ 10.4161/cc.11.4.19117 [DOI] [PubMed] [Google Scholar]
- 60.al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Lehmann AR, Carr AM. Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell 1994; 5:147–60; PMID:8019001; http://dx.doi.org/ 10.1091/mbc.5.2.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carr AM, Moudjou M, Bentley NJ, Hagan IM. The chk1 pathway is required to prevent mitosis following cell-cycle arrest at ‘start’. Curr Biol 1995; 5:1179–90; PMID:8548290; http://dx.doi.org/ 10.1016/S0960-9822(95)00234-X [DOI] [PubMed] [Google Scholar]
- 62.Yonemasu R, McCready SJ, Murray JM, Osman F, Takao M, Yamamoto K, Lehmann AR, Yasui A. Characterization of the alternative excision repair pathway of UV-damaged DNA in Schizosaccharomyces pombe. Nucleic Acids Res 1997; 25:1553–8; PMID:9092661; http://dx.doi.org/ 10.1093/nar/25.8.1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prise KM, Ahnstrom G, Belli M, Carlsson J, Frankenberg D, Kiefer J, Löbrich M, Michael BD, Nygren J, Simone G, et al.. A review of dsb induction data for varying quality radiations. Int J Radiat Biol 1998; 74:173–84; PMID:9712547; http://dx.doi.org/ 10.1080/095530098141564 [DOI] [PubMed] [Google Scholar]
- 64.Connolly B, White CI, Haber JE. Physical monitoring of mating type switching in Saccharomyces cerevisiae. Mol Cell Biol 1988; 8:2342–9; PMID:2841579; http://dx.doi.org/ 10.1128/MCB.8.6.2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, Canman CE. REV1 and polymerase zeta facilitate homologous recombination repair. Nucleic Acids Res 2012; 40:682–91; PMID:21926160; http://dx.doi.org/ 10.1093/nar/gkr769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kozmin SG, Jinks-Robertson S. The mechanism of nucleotide excision repair-mediated UV-induced mutagenesis in nonproliferating cells. Genetics 2013; 193:803–17; PMID:23307894; http://dx.doi.org/ 10.1534/genetics.112.147421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chan K, Resnick MA, Gordenin DA. The choice of nucleotide inserted opposite abasic sites formed within chromosomal DNA reveals the polymerase activities participating in translesion DNA synthesis. DNA Repair (Amst) 2013; 12:878–89; PMID:23988736; http://dx.doi.org/ 10.1016/j.dnarep.2013.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol Cell 2008; 29:625–36; PMID:18342608; http://dx.doi.org/ 10.1016/j.molcel.2007.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al.. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011–7; PMID:15496928; http://dx.doi.org/ 10.1038/nature02964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trickey M, Grimaldi M, Yamano H. The anaphase-promoting complex/cyclosome controls repair and recombination by ubiquitylating Rhp54 in fission yeast. Mol Cell Biol 2008; 28:3905–16; PMID:18426916; http://dx.doi.org/ 10.1128/MCB.02116-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uchiyama M, Terunuma J, Hanaoka F. The Protein Level of Rev1, a TLS Polymerase in Fission Yeast, Is Strictly Regulated during the Cell Cycle and after DNA Damage. PLoS One 2015; 10:e0130000; PMID:26147350; http://dx.doi.org/ 10.1371/journal.pone.0130000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, Muzi-Falconi M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 2010; 40:50–62; PMID:20932474; http://dx.doi.org/ 10.1016/j.molcel.2010.09.004 [DOI] [PubMed] [Google Scholar]
- 73.Maher VM, Dorney DJ, Mendrala AL, Konze-Thomas B, McCormick JJ. DNA excision-repair processes in human cells can eliminate the cytotoxic and mutagenic consequences of ultraviolet irradiation. Mutat Res 1979; 62:311–23; PMID:503098; http://dx.doi.org/ 10.1016/0027-5107(79)90087-3 [DOI] [PubMed] [Google Scholar]
- 74.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, Ye K, et al.. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010; 463:191–6; PMID:20016485; http://dx.doi.org/ 10.1038/nature08658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu L, De S, Michor F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat Commun 2013; 4:1502; PMID:23422670; http://dx.doi.org/ 10.1038/ncomms2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 2006; 17:308–16; PMID:16251353; http://dx.doi.org/ 10.1091/mbc.E05-07-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aparicio OM. Location, location, location: it's all in the timing for replication origins. Genes Dev 2013; 27:117–28; PMID:23348837; http://dx.doi.org/ 10.1101/gad.209999.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mankouri HW, Huttner D, Hickson ID. How unfinished business from S-phase affects mitosis and beyond. EMBO J 2013; 32:2661–71; PMID:24065128; http://dx.doi.org/ 10.1038/emboj.2013.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wickramasinghe CM, Arzouk H, Frey A, Maiter A, Sale JE. Contributions of the specialised DNA polymerases to replication of structured DNA. DNA Repair (Amst) 2015; 29:83–90; PMID:25704659 http://dx.doi.org/ 10.1016/j.dnarep.2015.01.004 [DOI] [PubMed] [Google Scholar]
- 80.Suski C, Marians KJ. Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 2008; 30:779–89; PMID:18570879; http://dx.doi.org/ 10.1016/j.molcel.2008.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.