

Abstract
Frontotemporal dementia (FTD) causes progressive personality, behavior and/or language disturbances and represents the second most common form of dementia under the age of 65. Over half of all FTD cases are classified pathologically as frontotemporal lobar degeneration (FTLD) with TAR DNA-binding protein of 43 kDa (TDP-43) pathology (FTLD-TDP). In FTLD-TDP brains, TDP-43 is phosphorylated, C-terminally cleaved, lost from the nucleus and accumulates in the cytoplasm and processes of neurons and glia. However, the contribution of TDP-43 C-terminal fragments (CTFs) to pathogenesis remains poorly understood. Here, we developed transgenic (Tg) mice with forebrain Camk2a-controlled doxycycline-suppressible expression of a TDP-43 CTF (amino acids 208–414, designated 208 TDP-43 CTF), previously identified in FTLD-TDP brains. In these 208 TDP-43 Tg mice, detergent-insoluble 208 TDP-43 CTF was present in a diffuse punctate pattern in neuronal cytoplasm and dendrites without forming large cytoplasmic inclusions. Remarkably, the hippocampus showed progressive neuron loss and astrogliosis in the dentate gyrus (DG). This was accompanied by phosphorylated TDP-43 in the CA1 subfield, and ubiquitin and mitochondria accumulations in the stratum lacunosum moleculare (SLM) layer, without loss of endogenous nuclear TDP-43. Importantly, 208 TDP-43 CTF and phosphorylated TDP-43 were rapidly cleared when CTF expression was suppressed in aged Tg mice, which ameliorated neuron loss in the DG despite persistence of ubiquitin accumulation in the SLM. Our results demonstrate that Camk2a-directed 208 TDP-43 CTF overexpression is sufficient to cause hippocampal pathology and neurodegeneration in vivo, suggesting an active role for TDP-43 CTFs in the pathogenesis of FTLD-TDP and related TDP-43 proteinopathies.
Introduction
Frontotemporal dementia (FTD) comprises a group of diseases, which manifest with diverse clinical phenotypes including changes in personality, behavior and language (1). Pathologically, over half of all FTD cases are characterized as frontotemporal lobar degeneration (FTLD) with TAR DNA-binding protein of 43 kDa (TDP-43) pathology (FTLD-TDP) (2). Furthermore, over 95% of amyotrophic lateral sclerosis (ALS) and ∼30% of Alzheimer's disease cases also have TDP-43 pathology in the CNS (2,3). Indeed, mutations in the TARDBP gene that encodes TDP-43 account for up to 4% of all familial and ∼1% of sporadic ALS cases, in addition to a small number of FTLD-TDP cases, confirming the pathogenic role of abnormal TDP-43 in disease (2,4).
TDP-43 is a 414 amino acid DNA- and RNA-binding protein located primarily in the nucleus that is involved in a diverse set of essential functions, including transcription, splicing, RNA metabolism and miRNA biogenesis (2,5). In FTLD-TDP, TDP-43 is lost from the nucleus and aberrantly accumulates in the cytoplasm of neurons and glia where it is rendered insoluble, is phosphorylated and is cleaved to generate C-terminal fragments (CTFs), which likewise accumulate in cytoplasmic inclusions (6,7). The contributions of TDP-43 inclusions, CTFs and the loss of nuclear TDP-43 function to neurodegeneration remain poorly understood.
In the cortex and hippocampus of FTLD-TDP patients, TDP-43 CTFs predominate over full-length TDP-43 in the cytoplasmic and dendritic pathology, suggesting a key role for CTFs in neurodegeneration in the brain (8). One of the TDP-43 CTFs detected in FTLD-TDP brains is ∼20-kDa fragment comprising amino acids 208–414 of TDP-43 (referred to here as 208 TDP-43 CTF) (9), which is abnormally phosphorylated similar to full-length TDP-43 in the disease state (7). When exogenously expressed in cell lines, 208 TDP-43 CTF is located primarily in the cytoplasm, where it accumulates in large phosphorylated inclusions and is highly insoluble, reminiscent of the pathology in FTLD-TDP brains (9,10). However, a 202–414 TDP-43 CTF did not cause degeneration in Drosophila (11), and transgenic (Tg) mice expressing 216–414 TDP-43 CTF constitutively in the brain developed cognitive deficits without neuron death, pathology or insoluble TDP-43 accumulation (12). Although Drosophila expressing 219–414 TDP-43 CTF showed a modest rough-eye phenotype indicative of some toxicity (13), adenovirus-mediated expression of 220–414 TDP-43 CTF in the brain and spinal cord of rats resulted in a mild movement disorder, but no detectable neurodegeneration or disease-related TDP-43 pathology (14). Furthermore, in Tg mouse models expressing full-length TDP-43, CTFs are either undetectable or present at only very low levels (15–18). The contribution of any TDP-43 CTFs to disease in these models is also difficult to determine due to the presence of other contributing factors, such as alterations in RNA processing, caused by the overexpression of full-length TDP-43. Therefore, the role that TDP-43 CTFs play in the pathogenesis of TDP-43 proteinopathies, and whether or not TDP-43 CTFs cause neurodegeneration, remains unclear.
Since it has not been possible from previous Tg models to conclusively determine the contribution of FTLD-TDP-associated TDP-43 CTFs to disease in vivo, we generated new Tg mice with doxycycline (Dox)-suppressible expression of 208 TDP-43 CTF under the control of the forebrain-specific Camk2a promoter. In these 208 TDP-43 Tg mice, 208 TDP-43 CTF was present in a diffuse punctate pattern in neuronal cell bodies and dendrites of the brain and was detergent insoluble. Furthermore, although abnormally phosphorylated TDP-43 (pTDP-43) was detected in the CA1 region of the hippocampus, large disease-reminiscent TDP-43 inclusions were not formed and there were no changes in endogenous full-length nuclear TDP-43 in the 208 TDP-43 Tg mice up to 24 months. However, progressive and dramatic loss of granular neurons and astrogliosis occurred in the dentate gyrus (DG) of the hippocampus. Remarkably, when transgene expression was suppressed by reintroduction of mice to Dox, 208 TDP-43 CTF and pTDP-43 were cleared from the brain and DG neuron loss was ameliorated. These findings demonstrate that a TDP-43 CTF found in FTLD-TDP pathology is sufficient to cause neurodegeneration in vivo and suggest that TDP-43 CTFs may play a pathogenic role in TDP-43 proteinopathies.
Results
208 TDP-43 CTF is insoluble and localized to dendrites in 208 TDP-43 Tg mouse brain
Previous results indicated that TDP-43 CTFs are the major components of TDP-43 pathology in the brains of FTLD-TDP patients (8) (Supplementary Material, Fig. S1). We therefore produced bigenic Camk2a-tTA/tetO-208 TDP-43 mice (208 TDP-43 Tg mice) for brain expression of the pathologically relevant human 208 TDP-43 CTF (9), to investigate the role of TDP-43 CTFs in disease. For initial characterization, 208 TDP-43 Tg mice were withdrawn from Dox to induce expression of 208 TDP-43 CTF along with littermate controls and examined by immunoblot (IB) at 6 months off Dox. IB showed expression of 208 TDP-43 CTF at levels ∼3- to 6-fold over endogenous full-length TDP-43 in the cortex, hippocampus and olfactory bulb, with very low levels in the brainstem and cerebellum and no expression in the spinal cord (Fig. 1A). Sequential extraction into RIPA (R)-soluble and urea (U)-soluble protein fractions revealed unaltered solubility of endogenous mouse full-length TDP-43, despite the near exclusive presence of 208 TDP-43 CTF in the U-soluble fraction of all brain regions where it was expressed (Fig. 1A).
Figure 1.

208 TDP-43 CTF is insoluble and located in the cell bodies and dendrites of neurons in 208 TDP-43 Tg mice. (A) IB analysis of RIPA (R)- and urea (U)-soluble protein fractions of various brain regions from littermate control and 208 TDP-43 Tg mice at 6 months off Dox, using C-terminal TDP-43 PAb. Solubility and levels of endogenous full-length TDP-43 are unaltered in 208 TDP-43 Tg mouse, and 208 TDP-43 CTF is expressed in the cortex, hippocampus and olfactory (olf) bulb and is predominantly R-insoluble. Higher intensity images (dark) show the presence of at least four distinct 208 TDP-43 CTF protein bands, and low levels of 208 TDP-43 CTF in the brain stem and cerebellum. GAPDH is shown as a loading control and approximate-molecular-weight markers in kilodaltons are shown in the right. (B) IHC for hTDP-43 in 208 TDP-43 Tg mouse reveals widespread cytoplasmic punctate neuronal 208 TDP-43 CTF in (C) cortex and hippocampus, including the (D) CA1 and (E) DG regions, as well as in (F) olf bulb and (G) striatum at 6 months off Dox. (H) Rare Purkinje cells in the cerebellum are also positive for hTDP-43 with (I) no expression in the spinal cord. Images are representative of n ≥ 3 mice at each of 2 weeks, 6, 10, 16, 19 and 24 months off Dox. (J–O) IF for hTDP-43 shows co-localization of 208 TDP-43 CTF with dendritic marker MAP2 but not axonal marker pNF in 208 TDP-43 Tg mouse at 16 months off Dox. Nuclei (DAPI) are shown in blue in the merged images. Scale bars: B, 500 μm; C–E, 50 μm; F–I, 50 μm; J–O, 20 μm.
Similar to previous reports in cell culture (8), 208 TDP-43 CTF was detected by IB as two major and several minor discrete bands, which were recognized by a human TDP-43 (hTDP-43)-specific MAb (Fig. 1A). This hTDP-43 antibody does not recognize endogenous mouse TDP-43 and binds to an epitope in hTDP-43 between amino acids 261 and 391 (19). The bands corresponded to at least one minor higher molecular mass (Mr) phosphorylated species detected on IB by an antibody against FTLD-TDP/ALS-specific p409/410 TDP-43, which was eliminated by the treatment of the extracts with calf intestinal phosphatase (CIP; Supplementary Material, Fig. S2A). In addition, one minor lower Mr species lacking the 208–220 amino acid region was detected by IB, suggesting post-translational N-terminal cleavage of the 208 TDP-43 CTF (Supplementary Material, Fig. S2B). Furthermore, only one single major band was detected using highly denaturing urea-gel IB, indicating that the several major 208 TDP-43 CTF bands detected by SDS–PAGE IB may represent SDS-resistant conformers that are nonetheless eliminated by urea treatment (Supplementary Material, Fig. S2C). Indeed, different TDP-43 antibodies possessed slightly different affinities for each of the discrete bands, suggesting differences in the exposure of antibody-binding epitopes by SDS–PAGE IB (data not shown). These results indicate that complex post-translational modifications and folding of the 208 TDP-43 CTF occur in vivo and that 208 TDP-43 CTF is predominantly RIPA-insoluble when expressed in mice, which is reminiscent of findings from human FTLD-TDP brain tissue (6).
To define the regional distribution of 208 TDP-43 CTF and the time course of expression, we performed immunohistochemistry (IHC) for hTDP-43 in the brain and spinal cord, for n ≥ 3 mice at each of 1, 2 weeks, 6, 10, 16, 19 and 24 months off Dox. No expression of 208 TDP-43 CTF was detected at 3 days (n = 2), and variable levels were detected at 1 week (data not shown). However, 208 TDP-43 CTF was widely expressed in the brain, including all cortical layers, all subfields of the hippocampus, the olfactory bulb and the striatum and was present in a diffuse and punctate pattern in the perinuclear and dendritic compartments of neurons in all mice examined from 2 weeks of expression until 24 months (Fig. 1B–G). In the cerebellum, only very rare Purkinje cells expressed 208 TDP-43 CTF (Fig. 1H), and no expression was detected in the spinal cord (Fig. 1I), consistent with the IB findings (Fig. 1A). Notably, although the presence of small hTDP-43 puncta and the detergent insolubility suggest that the 208 TDP-43 CTF is present in at least a partially aggregated form in vivo, large cytoplasmic 208 TDP-43 CTF inclusions were not detected in any region at any time point examined. Furthermore, double labeling immunofluorescence (IF) for hTDP-43 and the dendritic marker MAP2 (Fig. 1J–L) or the axonal marker pNF (Fig. 1M–O), along with the lack of hTDP-43 detected in white matter, confirmed that 208 TDP-43 CTF was confined to the somatodendritic compartment of neurons and was not present in axons.
208 TDP-43 CTF is phosphorylated in the CA1 subfield of the hippocampus without the formation of large cytoplasmic inclusions
Both full-length TDP-43 and TDP-43 CTFs are aberrantly phosphorylated at serine residues 403/404 and 409/410 in ALS and FTLD-TDP CNS tissue (6,7,20). To further investigate TDP-43 phosphorylation in the 208 TDP-43 Tg mice, we used specific antibodies for both of these phosphorylation sites for IHC with n ≥ 3 mice at each of 1, 2 weeks, 6, 10, 16, 19 and 24 months off Dox. Despite the widespread expression of 208 TDP-43 CTF in all layers of the cortex and all subfields of the hippocampus (Fig. 1), phosphorylation at serines 409/410 occurred almost exclusively in neurons of the CA1 region of the hippocampus, with only rare p409/410-positive neurons in the cortex (Fig. 2A–C), whereas only weak or negligible p409/410 staining was detected in the CA3 or DG subfields of the hippocampus (Fig. 2D and E). Similarly, no immunoreactivity for p403/404 was detected in controls (Fig. 2F), but intense dendritic CA1 p403/404 immunoreactivity was detected in 208 TDP-43 Tg mice (Fig. 2G–I). Interestingly, p403/404 and p409/410 immunoreactivity was detected at low levels as early as 1–2 weeks and was robustly detected at comparable levels at all later time points examined (Fig. 2G–I, and data not shown). These findings indicate that 208 TDP-43 CTF phosphorylation begins soon after the induction of transgene expression and efficient protein turnover occurs despite the phosphorylation and detergent insolubility, which prevents accumulation over time. Notably, although neurons displayed a punctate distribution of 208 TDP-43 CTF, large cytoplasmic TDP-43-positive inclusions were not detected in any region at any time point with any of the phosphorylation-dependent or -independent TDP-43 antibodies. Consistent with the restricted distribution of pTDP-43 detected by IHC, very low levels of phosphorylated 208 TDP-43 CTF were detected by IB (Supplementary Material, Fig. S2A). However, the specificity of the phosphorylated species was confirmed by enzymatic dephosphorylation of protein extracts followed by analysis by IB (Supplementary Material, Fig. S2A).
Figure 2.

TDP-43 phosphorylation occurs in the CA1 region of the hippocampus of 208 TDP-43 Tg mice. (A and B) IHC for p409/410 TDP-43 is positive predominantly in the CA1 subfield of the hippocampus, with little to none in other regions where 208 CTF TDP-43 is expressed, including (C) the cortex, and (D) the CA3 and (E) DG of the hippocampus. IHC for p403/404 TDP-43 showed no immunoreactivity in (F) littermate controls but was positive in (G–I) 208 TDP-43 Tg mice at all time points examined, including 2 weeks, 6 and 19 months. Scale bars: A, 500 μm; B–I, 50 μm.
208 TDP-43 CTF expression causes progressive neurodegeneration and astrogliosis in the DG of the hippocampus
Using IHC to label neuronal nuclei containing TDP-43, analysis of 208 TDP-43 Tg mouse brains over time did not reveal overt cortical thinning or neuron loss in any region except for a dramatic decrease in DG neurons in aged mice. We therefore analyzed the number of cells in the DG in coronal level-matched regions of the caudal hippocampus in 208 TDP-43 Tg mice and littermate controls over the life span, using N-terminal TDP-43 antibody as a nuclear marker (Fig. 3A–D). Quantitative analysis revealed that the number of DG nuclei did not differ between 208 TDP-43 Tg mice and littermate controls at 5 months (P > 0.05), but there were significantly fewer DG nuclei in 208 TDP-43 Tg mice at 10, 16, and 19 months (Fig. 3E). Notably, DG neuron loss progressed over time with a greater than 80% loss of DG neurons in 208 TDP-43 Tg mice at 19 months. Furthermore, there was a concomitant increase in GFAP immunoreactivity, indicating reactive astrogliosis in response to neurodegeneration, in the DG from 10 months (Fig. 3F–I).
Figure 3.
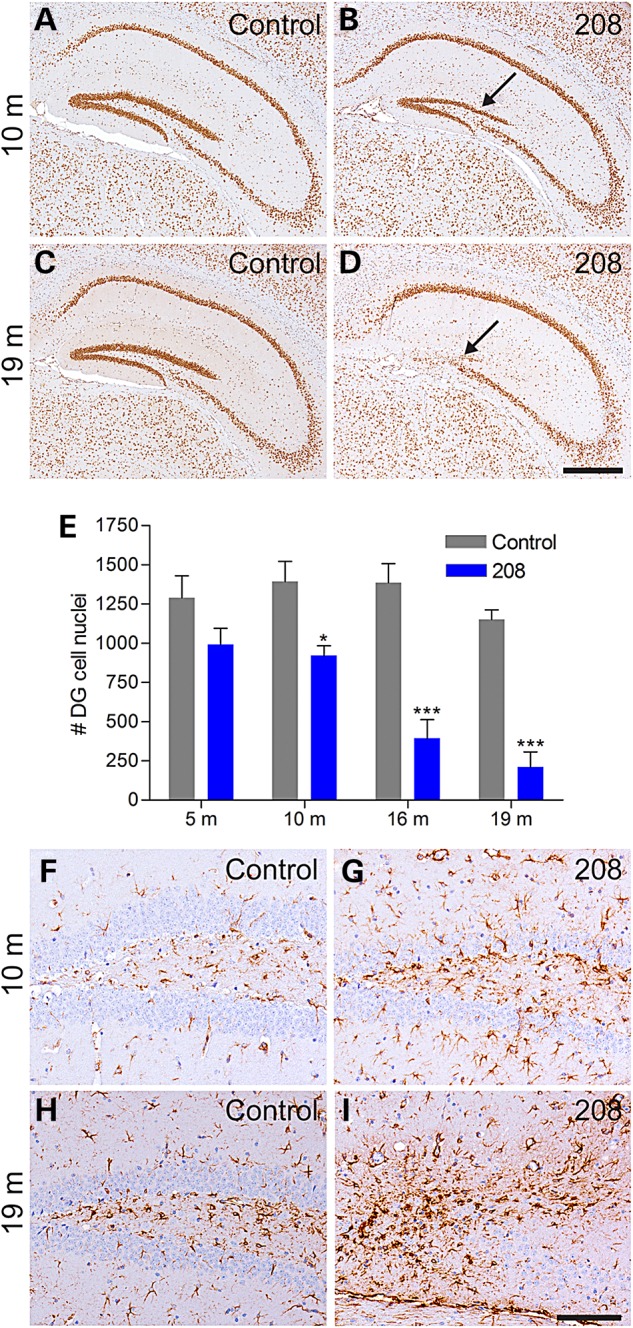
208 TDP-43 CTF expression causes progressive neurodegeneration and astrogliosis. (A–D) IHC of littermate controls and 208 TDP-43 Tg mice for endogenous TDP-43 reveals loss of DG neurons at 10 months, with dramatic loss observed at 19 months (arrows). (E) Quantification of DG cell nuclei based on IHC for endogenous TDP-43 shows significant loss of DG neurons in 208 TDP-43 CTF mice from 10 months by two-way ANOVA followed by Bonferroni's post-test, *P < 0.05 and ***P < 0.001, n = 4 per group per time point. (F–I) 208 TDP-43 Tg mice show astrogliosis concomitant with the neuron loss as shown by IHC for GFAP. Scale bars: A–D, 500 μm; F–I, 100 μm.
208 TDP-43 CTF expression does not affect the level, solubility or subcellular distribution of endogenous TDP-43
In FTLD-TDP brains, TDP-43 is lost from the nuclei of neurons that accumulate cytoplasmic TDP-43 (6). In addition, overexpression of full-length nuclear or cytoplasmic TDP-43 in cell culture and animal models causes downregulation of TDP-43 by auto-regulatory mechanisms (15,21). In order to define the effect of 208 TDP-43 CTF on endogenous TDP-43, we analyzed the brains of 208 TDP-43 Tg mice by IB and IHC over time using an TDP-43 N-terminus-specific antibody, which does not detect the 208 TDP-43 CTF (8). We noted that the levels of 208 TDP-43 CTF observed by IB reached a steady state as early as 2 weeks off Dox and remained unchanged up to 24 months (Fig. 4A), suggestive of a constant process of 208 TDP-43 CTF turnover in the mice. There was no alteration in levels or solubility of 43-kDa endogenous mouse TDP-43 at any time point from 3 days (prior to the detection of 208 TDP-43 CTF expression) until 24 months in 208 TDP-43 Tg mouse cortex (Fig. 4A) or hippocampus (data not shown). Furthermore, IHC showed no change in the abundance or pattern of nuclear immunoreactivity for endogenous TDP-43 in any brain region, including the CA1 subfield (Fig. 4B and C) and the DG (Fig. 4D and E) of the hippocampus, despite significant neuronal loss in the DG (Fig. 3A–E). Indeed, although far fewer neuronal nuclei were present in the DG at later time points, those that did remain were indistinguishable in terms of endogenous nuclear TDP-43 compared with controls. Furthermore, there were no cytoplasmic accumulation of full-length endogenous mouse TDP-43 in any brain region nor were any TDP-43-positive inclusions detected at any time point. Importantly, these results indicate that the insoluble 208 TDP-43 CTF is able to cause neurodegeneration, albeit in a very restricted subset of CNS neurons, in the absence of any change in solubility, levels or distribution of full-length nuclear TDP-43.
Figure 4.
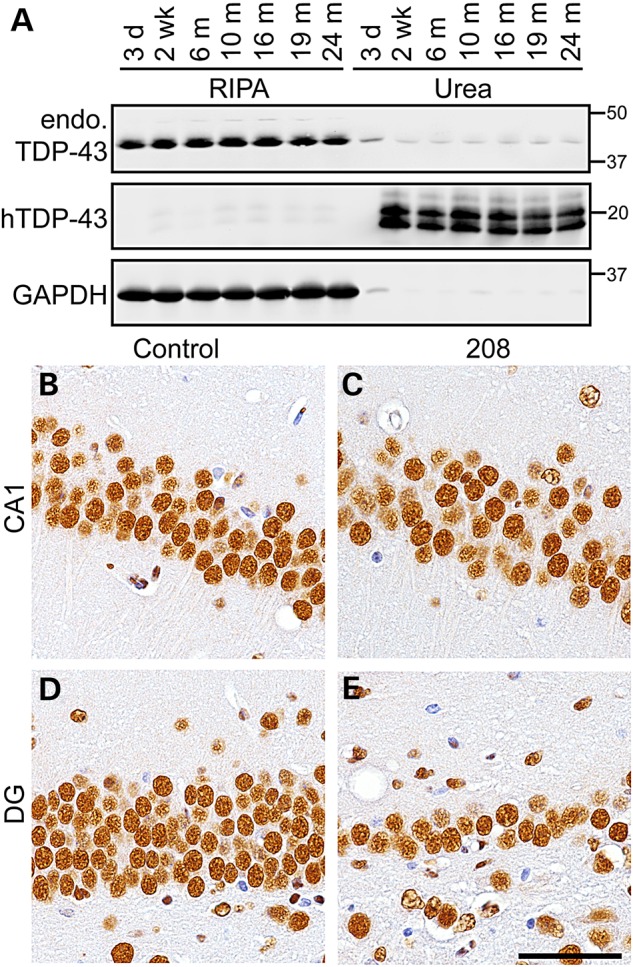
Endogenous full-length TDP-43 levels, solubility and localization are unaltered in 208 TDP-43 Tg mice. (A) IB of RIPA- and urea-soluble protein fractions of 208 TDP-43 Tg mouse cortex at time points as indicated shows levels and solubility of endogenous full-length TDP-43 (endo. TDP-43, detected using N-terminal TDP-43 PAb) are unaltered over time, with detection of 208 TDP-43 CTF (using hTDP-43 MAb) detected from 2 weeks. (B–E) Likewise, IHC for N-terminal TDP-43 in the CA1 and DG subfields of the hippocampus shows no change in endo. TDP-43 in 208 TDP-43 Tg mice compared with littermate controls, despite neuron loss in DG. Images of mice at 19 months are shown. Scale bar: B–E, 50 μm.
208 TDP-43 Tg mice accumulate ubiquitin, p62 and mitochondria in the SLM of the hippocampus
Neurodegenerative diseases, including FTLD-TDP, are characterized by the accumulation of ubiquitinated proteins (22). Likewise, dysfunction of autophagy and subsequent perturbation of mitochondrial homeostasis have also been implicated in neurodegenerative disease pathogenesis (23). In order to investigate the potential development of ubiquitin pathology in 208 TDP-43 Tg mice, we performed IHC at various time points after Dox withdrawal. At 2 week off Dox, there was no difference in ubiquitin immunoreactivity in the brains of 208 TDP-43 Tg mice compared with littermate controls of any time point (Fig. 5A and B). However, although there was no accumulation of ubiquitin in areas with high 208 TDP-43 CTF expression such as the CA1 and DG regions of the hippocampus, we observed intense granular ubiquitin immunoreactivity in the stratum lacunosum moleculare (SLM) layer of the hippocampus in all 208 TDP-43 Tg mice that were off Dox for 5 months or longer, using multiple anti-ubiquitin antibodies (Fig. 5C and D; Supplementary Material, Fig. S3). Under high magnification, the intense ubiquitin immunoreactivity corresponded to a discrete punctate pattern in the SLM (Fig. 5E inset) that was not seen in littermate controls (Fig. 5I).
Figure 5.

208 TDP-43 Tg mice develop progressive TDP-43-negative but ubiquitin-, p62- and mitochondrial protein-positive pathology in the SLM of the hippocampus. (A–D) IHC for ubiquitin detects punctate accumulation of ubiquitin (arrows) in the SLM of the hippocampus in 208 TDP-43 Tg mice at later time points, which is not detected at 2 weeks in 208 TDP-43 Tg mice or in littermate controls. IHC for various markers shows that the proteins accumulating in the SLM of 208 TDP-43 Tg mice include (E) ubiquitin, (F) p62 and (G) mitochondrial marker cytochrome c, but not (H) 208 TDP-43 CTF. Boxed regions indicate higher magnification insets, and mice at 19 months are shown. Pathology is confined to the SLM and is not seen in the nearby molecular (Mol) and granular (Gr) layers of the DG, and (I–L) is not detected in littermate controls. IF with antibodies to ubiquitin and another mitochondrial marker, COX IV, reveals colocalization of these proteins in the SLM pathology (M). IF with ubiquitin and (N) the axonal marker pNF or (O) the dendritic marker MAP2 shows that the pathology is present in the distal dendritic compartment of neurons. Nuclei are shown in blue (DAPI). Scale bars: A–D, 500 μm; E–L, 100 μm; M–O, 100 μm.
Since the accumulation of ubiquitinated proteins may reflect a failure of protein degradation or an overload of misfolded proteins, we performed IHC for other protein degradation pathway markers, including the autophagy receptor p62, which is also found in FTLD-TDP pathology, and ubiquilin 2, which has been associated with protein inclusions in a subset of ALS and FTLD cases (24–26). The punctate pattern of p62 distribution mirrored the distribution of ubiquitin in the SLM of 208 TDP-43 Tg mice, albeit in lower abundance than ubiquitin (Fig. 5F and Inset), which was again not seen in littermate controls (Fig. 5J). However, the ubiquitin 2 IHC did not reveal any differences between littermate controls and 208 TDP-43 Tg mice in level or localization in any regions of the brain (data not shown).
Several studies have also implicated mitochondrial dysfunction in TDP-43 proteinopathies, including in TDP-43 cell models and in Tg mice expressing full-length ALS-linked mutant TDP-43 (27–29). Additionally, mutations in the gene encoding the mitochondrial protein CHCHD10 have recently been identified in FTLD and ALS patients (30). In order to investigate mitochondrial integrity in the SLM of 208 TDP-43 Tg mice, we performed IHC and IF for the mitochondrial proteins cytochrome c (Fig. 5G and K) and COX IV (Fig. 5M), which both exhibited a granular staining pattern in 208 TDP-43 Tg mice similar to ubiquitin. Double labeling of COX IV with ubiquitin by IF demonstrated their co-localization in the SLM of 208 TDP-43 Tg mice (Fig. 5M). Remarkably, the SLM accumulations were not immunoreactive for any TDP-43 antibodies (Fig. 5H and L). Finally, double labeling IF for ubiquitin with dendritic marker MAP2 or axonal marker pNF revealed co-localization of the ubiquitin accumulations with dendrites but not axons in the SLM (Fig. 5N and O). Thus, 208 TDP-43 Tg mice develop progressive ubiquitin, p62 and mitochondrial protein accumulations, which do not contain 208 TDP-43 CTF, in distal dendrites of the SLM of the hippocampus.
In order to elucidate if the DG degeneration was due to the expression of 208 TDP-43 CTF in the DG granular cells themselves or was linked with the SLM ubiquitin accumulation, we generated bigenic mice for Dox-suppressible expression under the control of the NEFH promoter, which drives expression broadly in the brain but with only modest levels in the granular cells of the DG (31). Similar to the Camk2a-208 TDP-43 Tg mice, the NEFH-208 TDP-43 Tg mice had high levels of insoluble 208 TDP-43 CTF in the cortex and hippocampus, with accumulation of p409/410 TDP-43 predominantly in the CA1 region and ubiquitin in the SLM region of the hippocampus (Supplementary Material, Fig. S4A–G). However, in the NEFH-208 TDP-43 Tg mice, <5% of DG cells expressed 208 TDP-43 CTF and there was no overt loss of DG neurons up to 19 months (Supplementary Material, Fig. S4H). These findings suggest that expression of 208 TDP-43 CTF within the DG neurons causes neurodegeneration and that the accumulation of pTDP-43 in the CA1 and ubiquitin in the SLM are not the drivers of DG neuron loss in the CAMK-208 TDP-43 Tg mice.
208 TDP-43 CTF is cleared and neurodegeneration is ameliorated when expression is suppressed in 208 TDP-43 Tg mice
We next sought to investigate whether suppression of 208 TDP-43 CTF expression could prevent continued neurodegeneration and allow for clearance of pathology. Thus, we exploited the Dox-suppressible system by returning a cohort (n = 9) of 208 TDP-43 Tg mice to Dox-containing chow after 13 months of transgene expression, which is a time when DG neuron loss has begun (Fig. 3E) and there is accumulation of pTDP-43 (Fig. 2) and ubiquitin (Fig. 5) in the hippocampus. We analyzed these mice at each of 5, 15 days and 6 months back on Dox. By IHC, both hTDP-43 and p409/410 TDP-43 were markedly decreased at 5 days and 15 days back on Dox, with complete clearance at 6 months (Fig. 6A–L). However, immunoreactivity for ubiquitin in the SLM of the hippocampus was unaltered in mice back on Dox, even at 6 months, indicating that this pathology could not be cleared despite clearance of the 208 TDP-43 CTF. Remarkably, after only 5 days back on Dox very little 208 TDP-43 CTF remained by IB, and by 15 days 208 TDP-43 CTF was undetectable (Fig. 6Q).
Figure 6.

208 TDP-43 CTF is cleared and DG neurodegeneration ameliorated when transgene expression is suppressed. (A–H) IHC shows that compared with widespread, robust levels of 208 TDP-43 CTF in mice off Dox (A and E), only low levels of residual 208 TDP-43 CTF are detected in a subset of cortical cells and CA1 dendrites 5 days (B and F) and 15 days (C and G) after return of the 208 TDP-43 Tg mice to Dox, with complete clearance at 6 months back on Dox (D and H). (I–L) IHC for p409/410 TDP-43 shows that pTDP-43 is also cleared over time from the CA1 region. However, (M–P) ubiquitin accumulation persists in the SLM up to 6 months back on Dox. (Q) IB also shows that return of 208 TDP-43 Tg mice to Dox at 13 months off Dox results in rapid clearance of 208 TDP-43 CTF. Darker image shows low levels of 208 TDP-43 CTF are detectible by IB at 5 days back on Dox, and 208 TDP-43 CTF is not detectible after 15 days back on Dox. (R) IHC for endogenous TDP-43 (detected using N-terminal TDP-43 PAb) shows dramatic neuron loss in the DG of 208 TDP-43 Tg mouse at 19 months off Dox, which is ameliorated in 208 TDP-43 Tg mouse at 13 months off Dox + 6 months on Dox. Scale bars: A–P, 50 μm; R, 200 μm.
Importantly, in addition to the rapid clearance of 208 TDP-43 CTF following Dox reintroduction, the progressive loss of DG neurons (Fig. 3) was ameliorated, with mice off Dox for 19 months containing over 70% fewer DG neurons than mice off Dox for 13 months and back on Dox for 6 months (Fig. 6R; P < 0.05 by one-way ANOVA; n = 3–4 per group; Supplementary Material, Fig. S5). These results indicate that the clearance of insoluble 208 TDP-43 CTF prevented continued DG neuron loss (Fig. 7).
Figure 7.

Schematic of pathology development and neurodegeneration as well as the effects of transgene suppression in the hippocampus of 208 TDP-43 Tg mice. (A) Expression of 208 TDP-43 CTF and the presence of pTDP-43 is detected from 2 weeks off Dox. (B) By 10–16 months off Dox, neurodegeneration occurs in the DG, and ubiquitin and mitochondria accumulate in the SLM region. (C) With 6 months back on Dox to suppress transgene expression, 208 TDP-43 CTF and pTDP-43 are cleared and selective DG neurodegeneration is ameliorated, despite continued presence of ubiquitin/mitochondria accumulations in the SLM. (D) However, by 19 months off Dox with continued 208 TDP-43 CTF expression, there is dramatic neurodegeneration in the DG.
Discussion
The presence of detergent-insoluble TDP-43 CTFs is a hallmark of FTLD-TDP and ALS pathology in the brain (6). However, the contribution of TDP-43 CTFs to neurodegeneration has remained unclear. Here, we describe for the first time a mouse model expressing a highly insoluble TDP-43 CTF identified by N-terminal sequencing in human FTLD-TDP postmortem brain (9). The 208 TDP-43 Tg mice reported here demonstrate that TDP-43 CTFs can cause neurodegeneration in vivo without formation of the large TDP-43 inclusions seen in FTLD-TDP and ALS tissue, and without changes in the level, solubility or subcellular distribution of nuclear full-length TDP-43. Furthermore, neurodegeneration was not dependent on TDP-43 phosphorylation, since the only brain region which showed robust neuron loss, the DG of the hippocampus, showed no or only very little pTDP-43 at any time point. Indeed, the only brain region in which robust TDP-43 phosphorylation was detected, the CA1 of the hippocampus, did not develop overt neuron loss. These results therefore indicate that TDP-43 CTFs are sufficient to cause neurodegeneration without causing the full gamut of human pathology in vivo, suggesting that multiple factors contribute to the formation of TDP-43 pathology in human disease.
The absence of large cytoplasmic TDP-43 inclusions characteristic of human FTLD-TDP and ALS in the 208 TDP-43 Tg mice was surprising, especially since expression of 208 TDP-43 CTF causes the formation of large disease-reminiscent pTDP-43 inclusions in cell culture (9,10). One explanation could be that the level of expression of the transgene in the 208 TDP-43 Tg mice was insufficient to cause TDP-43 inclusion formation, although the 208 TDP-43 CTF was expressed at levels approximately three- to six-fold higher than endogenous TDP-43 in the hippocampus and cortex after accounting for the difference in RIPA-soluble versus RIPA-insoluble partitioning (Fig. 1A). The comparative ratio of 208 TDP-43 CTF to full-length TDP-43 is therefore apparently higher in the 208 TDP-43 Tg mice than occurs in the urea-soluble fractions of FTLD-TDP brain tissue (6,7). Despite the lack of large TDP-43 inclusions, intriguingly the 208 TDP-43 CTF was almost completely RIPA-insoluble in the mice from the earliest time point when expression was consistently detected (2 weeks) up to 24 months (Fig. 4A), mirroring biochemical findings in human patients and cell culture (6,9). Interestingly, our results showed comparable levels of 208 TDP-43 CTF between 2 weeks and 24 months, which suggests that a steady state level is rapidly reached such that cellular degradation mechanisms remain able to maintain a stable level of the protein over time. Our findings also demonstrated that clearance of the 208 TDP-43 CTF occurs rapidly following suppression of the transgene, suggesting that neuronal protein clearance mechanisms remain active even with prolonged expression of the insoluble cytoplasmic 208 TDP-43 CTF. It is therefore possible that additional cellular stress to pathways involved in clearance of TDP-43, including autophagy and proteasomal degradation (32), is required to cause the formation of the TDP-43 inclusions seen in human FTLD-TDP and ALS pathology. Importantly, these findings also indicate that detergent insolubility is not necessarily correlated with the formation of large TDP-43 inclusions, suggesting that multiple post-translational modifications are involved in the aggregation pathway of TDP-43.
Another characteristic of human FTLD-TDP brain tissue, as well as when 208 TDP-43 CTF is expressed in cell culture, is the robust detection of pTDP-43, particularly at serines 403/404 and 409/410 (6,7,9,20). Remarkably, however, pTDP-43 was detected by IHC predominantly in the CA1 region of the hippocampus of the 208 TDP-43 Tg mice, and only at modest levels in a small percentage of neurons in other hippocampus regions and other brain regions, despite widespread expression of the 208 TDP-43 CTF. The reason for this regional selectivity of TDP-43 phosphorylation remains unclear, although a restricted distribution or variation in the cellular complement of kinases and phosphatases that can phosphorylate and dephosphorylate TDP-43 may be one possible explanation. It is also notable that the distribution of pTDP-43, primarily found in the CA1 region, did not correlate with neurodegeneration, which occurred primarily in the DG of the 208 TDP-43 Tg mice. Moreover, large TDP-43-positive inclusions were not formed in the CA1 region of the hippocampus in the 208 TDP-43 Tg mice despite the presence of early and persistent TDP-43 phosphorylation. This provides support for the hypothesis that TDP-43 phosphorylation at amino acids 403/404 and 409/410 is a marker of insoluble TDP-43 species rather than a driver of TDP-43 inclusion formation and neurodegeneration (33). It is also possible that the 403/404 and 409/410 TDP-43 phosphorylation detected in the CA1 neurons may represent a protective mechanism against neurodegeneration. Phosphorylated TDP-43 is regarded as a hallmark of ALS and FTLD-TDP-43 pathology (6), and further investigation of how the regional variation in TDP-43 phosphorylation is mediated, specifically at the disease-relevant 403/404 and 409/410 sites, may reveal information useful for understanding TDP-43 post-translational modifications.
Although insoluble TDP-43 CTFs are readily detected in the brains of FTLD-TDP and ALS patients (6), multiple mechanisms for their generation have been proposed, and the role in disease of the several TDP-43 CTFs of varying size remains unclear. Alternative splicing has been shown to generate an ∼35 kDa TDP-43 CTF (representing amino acids 85–414), which is up-regulated in ALS and co-locates with TDP-43 inclusions in ALS tissue (34,35); however, a wide array of potential cleavage mechanisms have also been implicated in generating TDP-43 CTFs from the full-length protein. TDP-43 CTFs of ∼25 and 35 kDa can be generated by caspase 3 cleavage at Asp89 and Asp219 in cell culture (33,36,37), although similar-sized CTFs are also generated in cells lacking caspase 3 (35). Furthermore, TDP-43 has multiple potential calpain cleavage sites between amino acids 229 and 346, for the generation of ∼33–36-kDa TDP-43 species identified in vitro (38), and asparaginyl endopeptidase can cleave TDP-43 at Asn291 and Asn308 (39). Endoplasmic reticulum (ER)-resident caspase 4 has also been shown to cleave TDP-43, at Asp174, to generate CTFs (40), although it remains uncertain how an ER-resident caspase is able to cleave nuclear or cytoplasmic TDP-43. The mechanism of 208 TDP-43 CTF generation also remains unclear. Given that the presence of TDP-43 CTFs differentiates the brain TDP-43 pathology of FTLD-TDP and ALS patients from the predominantly full-length TDP-43 inclusion pathology seen in the spinal cord of ALS patients (8), regional variation in such cellular TDP-43 cleavage mechanisms may be one determinant of the manifestation of the brain or spinal cord-predominant TDP-43 pathology found in FTLD-TDP or ALS patients. Indeed, recent findings suggest that brain tissues of patients with each of the different subtypes of FTLD-TDP pathology are characterized by predictable and different biochemical patterns of TDP-43 CTFs (41). Importantly, our finding of multiple detectable bands possibly representing different detergent-resistant conformers in the 208 TDP-43 Tg mice suggests that more than one of the multiple TDP-43 CTF bands detected by IB in FTLD-TDP brain tissue could actually be comprised primarily of 208 TDP-43 CTF. Further studies are therefore required to identify the contributions of different mechanisms in generating different-sized TDP-43 CTFs and their relationship to the development of TDP-43 pathology and neurodegeneration in FTLD-TDP and ALS. This could shed light on how dysfunction of the same protein, TDP-43, is able to cause such different but overlapping clinical phenotypes (42).
Our finding of neurodegeneration in the DG of 208 TDP-43 Tg mice, despite the absence of the nuclear TDP-43 clearance found in human pathology (6), indicates that loss of nuclear TDP-43 is not required to cause neurodegeneration. Notably, the 208 TDP-43 CTF lacks the nuclear localization sequence of TDP-43 (at amino acids 82–98) as well as the first RNA recognition motif (RRM1) and part of RRM2 (which approximately spans amino acids 192–265). Therefore, the 208 TDP-43 CTF is located in the cytoplasm and would be predicted to lack RNA binding. Thus, neurodegeneration caused by the 208 TDP-43 CTF is unlikely to be due to altered RNA-binding or splicing functions but is likely caused by some other undetermined gain of toxicity mechanism. Future investigation into how the 208 TDP-43 CTF is able to cause neurodegeneration will allow further differentiation of the effects of TDP-43 CTF compared with cytoplasmic full-length TDP-43. For example, our finding that the 208 TDP-43 CTF is distributed more extensively into the dendrites of neurons than cytoplasmic full-length TDP-43 when expressed in the brains of mice (15) suggests that TDP-43 CTFs are actively transported into the distal processes of neurons and may therefore have more impact on intracellular transport in neurons than full-length TDP-43.
The accumulation of mitochondria, ubiquitin and p62 in dendrites of the SLM of the hippocampus, which functionally connects the dendritic tufts of CA1 neurons and projections from the entorhinal cortex (43), also suggests a possible disruption of intracellular transport in neurons of the 208 TDP-43 Tg mice. The lack of TDP-43 within these accumulations likewise indicates that the accumulations are not due to co-aggregation with the 208 TDP-43 CTF itself but form as a result of an upstream dysfunction. Interestingly, p62-positive but TDP-43-negative inclusions are also found in the hippocampus in FTLD-TDP patients with C9orf72 mutations, although these are found to be perinuclear in pyramidal cells rather than in the SLM (44). Likewise, phospho-tau accumulates in dilated structures in the dendritic compartment of the SLM in the hippocampus in early-stage Alzheimer's disease brain (45), and ubiquitin-positive spheroids are also detected in the SLM of patients with Lewy body dementia (46). However, since neurodegeneration was halted in the 208 TDP-43 Tg mice upon suppression of transgene expression despite the continued presence of the SLM ubiquitin accumulations, it is likely that this pathology is epiphenomenal rather than causative of neurodegeneration. Another explanation is that the constant expression of 208 TDP-43 CTF leads to an overload of cellular degradation machinery, such that the degradation of other proteins and organelles is slowed, resulting in the SLM pathology. Further studies will be required to identify the molecular causes and consequences of these changes.
The physiological relevance of mitochondrial accumulation in the distal dendrites of CA1 neurons of the hippocampus in the 208 TDP-43 Tg mice also remains unclear. It is possible that this mitochondrial accumulation is a result of potential defective axonal transport caused by the insoluble 208 TDP-43 CTF in the more proximal dendrites of these neurons. However, our finding of persistent mitochondrial accumulation in the SLM of the hippocampus despite clearance of the 208 TDP-43 CTF and amelioration of neuron loss upon return of the 208 TDP-43 Tg mice to Dox for 6 months suggests that this mitochondrial accumulation is not primarily responsible for the neuron loss detected in the hippocampus. Any negative consequences resulting from these aberrant mitochondrial accumulations, such as perturbation in energy metabolism or production of potentially detrimental free radicals, are interesting areas for future study.
The functional outcome of the neurodegeneration caused by 208 TDP-43 CTF remains unclear, since we did not detect a consistent difference between 208 TDP-43 Tg mice and littermate controls in motor and learning tasks, including rotarod, wirehang and y-maze test, at 10–12 months, due to large variation from animal to animal (data not shown). However, given our relatively small group size (n ≤ 10) for these tests and the mixed C57BL/6JxC3HeJ background of the mice in this cohort, these studies were likely underpowered to detect any changes. Although expression of 208 TDP-43 CTF and the formation of pathology was consistent in the 208 TDP-43 Tg mice, behavior tests are particularly sensitive to animal-to-animal variation, and further experiments with a larger group size and mice back-crossed to a pure background will likely lead to more definitive results on the behavioral outcome of 208 TDP-43 CTF expression. That said, the 208 TDP-43 Tg mice lived a normal life span without overt behavioral abnormalities. Regardless, our results show that the expression of 208 TDP-43 CTF causes highly selective DG neuronal degeneration in vivo, which may have direct relevance to the neuron loss seen in human FTLD-TDP patients. Indeed, our dramatic finding of amelioration of DG neuron loss upon 208 TDP-43 transgene suppression further strengthens the hypothesis that prevention of further TDP-43 CTF accumulation via modulation of TDP-43 cleavage or clearance mechanisms could be beneficial for future disease treatments.
Our results are the first in vivo demonstration of robust neurodegeneration caused by a disease-relevant TDP-43 CTF. Importantly, the highly selective DG neuron loss was not dependent on TDP-43 phosphorylation, loss of nuclear full-length TDP-43, or accumulation of the large cytoplasmic TDP-43-positive inclusions seen in human TDP-43 proteinopathies, despite the disease-reminiscent detergent insolubility of the 208 TDP-43 CTF. Further understanding of the contribution of different full-length and cleaved TDP-43 proteins will be crucial for the development of appropriate therapeutic strategies for FTLD-TDP, ALS and other neurodegenerative diseases characterized by TDP-43 pathology.
Materials and Methods
Tg mouse generation and maintenance
Camk2a-tTA Tg mice (Jackson Laboratories #3010) (47) were maintained on a pure C57BL/6J background by crossing of monogenic male mice with C57BL/6 females (Jackson Laboratory). tetO-208 TDP-43 Tg mice were generated by injection of a NotI-linearized modified moPrP vector (48) containing the coding sequence for amino acids 208–414 of human TDP-43 (TARDBP, RefSeq: NP_031401.1) under the control of the tetracycline-responsive element with Tet operator (tetO) and a minimal sequence for human cytomegalovirus (CMV2) immediate-early promoter, into the pronucleus of fertilized eggs from C57BL/6JxC3HeJ F1 matings. The tetO-208 TDP-43 Tg mice were maintained by breeding with wild-type C57BL/6JxC3HeJ F1 mice. Three founders were generated, and the line with the highest inducible expression of 208 TDP-43 CTF (line #2) was studied in detail as reported here, although the other two lines showed the same 208 TDP-43 CTF distribution and biochemical properties (data not shown). Bigenic 208 TDP-43 Tg mice were produced by crossing tetO-208 TDP-43 Tg mice with Camk2a-tTA Tg mice, resulting in 75% C57BL/6J background and 25% C3HeJ background contribution. Control groups included non-Tg and monogenic littermates. NEFH-tTA Tg mice (31) were similarly crossed to tetO-208 TDP-43 Tg mice to generate a cohort of NEFH-208 TDP-43 Tg mice and littermate controls. Breeding mice were provided with chow containing 200 mg/kg doxycycline (Dox) to prevent expression during pre- and post-natal development (Dox Diet #3888, Bio-Serv). Expression of 208 TDP-43 CTF was induced in mice at ∼1 month of age by switching to standard chow lacking Dox (Rodent Diet 20 #5053, PicoLab). To suppress the expression of 208 TDP-43 CTF, mice were returned to Dox Diet for the indicated periods of time. Procedures were performed in accordance with the NIH Guide for the Care and Use of Experimental Animals. Studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Mouse tissue
Mice were anesthetized with ketamine and xylazine and perfused intracardially with phosphate-buffered saline. The brain and spinal cord were removed, and the brain was bisected through the midline. The left half of the brain was dissected into regions and these and part of the spinal cord were immediately frozen on dry ice and stored at −80°C until processing. The right half of the brain and part of the spinal cord were immediately placed in 10% buffered formalin and allowed to post-fix for ∼16 h at room temperature. Fixed tissues were washed with Tris-buffered saline (50 mM Tris–HCl and 150 mm NaCl), pH 7.4, and embedded in paraffin prior to sectioning at a thickness of 6 μm.
Human FTLD-TDP tissue
Six-micrometer-thick sections of formalin-fixed, paraffin-embedded frontal cortex tissues from a 62-year-old female with the primary neuropathological diagnosis of FTLD-TDP type C were used for IHC analysis. Tissue was obtained from the Center for Neurodegenerative Disease Research (CNDR) Brain Bank, and autopsy and sample processing was performed as described previously (49). Informed consent was obtained in accordance with the University of Pennsylvania Institutional Review Board guidelines.
Preparation of mouse brain lysates
CNS tissues were thawed on ice and sonicated to homogenize in 5× v/w of radio-immunoprecipitation (RIPA) buffer (50 mm Tris, 150 mm NaCl, 1% NP-40, 5 mm EDTA, 0.5% sodium deoxycholate, 0.1% SDS, pH 8.0 with 1 mm PMSF and phosphatase and protease inhibitor cocktails). Homogenates were centrifuged at ∼100 000g at 4°C for 30 min and the supernatant collected as the RIPA-soluble fraction. The pellet was washed with RIPA buffer and centrifuged as above and the resultant pellet was sonicated with 2× v/w of urea buffer (7 M urea, 2 M thiourea, 4% CHAPS, 30 mm Tris, pH 8.5). Homogenates were centrifuged at ∼100 000g at 22°C for 30 min and the supernatant collected as the urea (U)-soluble fraction.
Immunohistochemistry and immunofluorescence
For IHC, sections were deparaffinized in 100% xylene and hydrated through a descending series of ethanol, and endogenous peroxidases were blocked by incubation in 5% H2O2 in methanol for 30 min. For IF, sections were similarly processed but without blocking of endogenous peroxidases. Sections were washed and microwave antigen retrieval was performed at 95°C for 15 min using Antigen Unmasking Solution (Vector) for all sections except when using anti-ubiquitin antibodies. Sections were washed and non-specific antibody binding was blocked with 2% fetal bovine serum in 0.1 m Tris, pH 8.0. Primary antibodies were applied in blocking solution overnight at 4°C. Primary antibodies used for IHC were as follows: mouse anti-human TDP-43 (hTDP-43) monoclonal antibodies (MAbs; 0.06 µg/ml, clones 241, 5095, 5104, 5123) (19), a rat anti-p409/410 TDP-43 MAb (1:200, TAR5P-1D3, Ascenion, Munich, Germany) (7), a rabbit anti-p403/404 TDP-43 polyclonal antibody (PAb; 1:2000, CosmoBio TIP-PTD-P05), a rabbit anti-N-terminal TDP-43 PAb (0.22 μg/ml, CNDR N1065) (8), a rabbit anti-C-terminal TDP-43 PAb (0.15 μg/ml, CNDR C1039) (8), a rat anti-glial fibrillary acidic protein (GFAP) MAb (1:2000, clone 2.2B10, CNDR) (50), a mouse anti-ubiquitin MAb (1:10 000, clone Ubi-1, Millipore MAB1510), a rabbit anti-ubiquitin PAb (1:1000, Dako z0458), a mouse anti-ubiquitin MAb (1:1000, clone Ub1B4, CNDR) (6), a mouse anti-p62 MAb (1.0 μg/ml, clone 2C11, Abnova H00008878-M01) and a mouse anti-cytochrome c MAb (0.8 μg/ml, clone A-8, Santa Cruz sc-13156). Primary antibodies used for IF were as follows: a rat anti-phospho-neurofilament medium and heavy-chain (pNF) MAb (1:200, clone TA51, CNDR) (51), a rabbit anti-COX IV PAb (1.0 μg/ml, Abcam ab16056), Ubi-1 (1:2500) and a rabbit anti-microtubule-associated protein 2 (MAP2) PAb (1:1000, CNDR 17028) (52). For IHC, Vectastain ABC kit was used for detection using 0.05% 3,3′-diaminobenzidine tetrahydrochloride hydrate (Sigma D5637) with 0.003% hydrogen peroxide in 0.1 m phosphate buffer, pH 7.4. Sections were counterstained with haemotoxylin and cover-slipped with Cytoseal 60 toluene mounting medium (Thermo Scientific). For IF, AlexaFluor-488 or -594-labeled goat anti-mouse, anti-rabbit or anti-rat secondary antibodies were used (1:1000, Molecular Probes), lipofuscin autofluorescence was quenched with 0.3% Sudan Black B in 70% ethanol (Sigma) and slides were mounted using Fluoromount Gold plus DAPI (Southern Biotech). Images were acquired with a Nikon Eclipse Ni inverted microscope, using a DS-Fi2 camera (brightfield) or DS-Qi1Mc camera (fluorescence).
Immunoblotting
RIPA-soluble and urea-soluble proteins were analyzed by SDS–PAGE for IB with Tris-glycine buffer system, nitrocellulose membranes and blocking buffer for fluorescent western blotting (Rockland). Protein concentrations of the RIPA-soluble fraction were determined using the bicinchoninic acid assay (Pierce). The amounts of U-soluble fractions used for immunoblotting were calculated based on volume from the corresponding RIPA-soluble protein concentration. Primary antibodies used were as follows: N1065, 5095, 5104, C1039 (all at the above dilutions) and a mouse anti-GAPDH MAb (1:10 000, clone 6C5, Advanced Immunochemical). Goat anti-mouse or anti-rabbit IRDye-680 or IRDye-800-conjugated secondary PAbs (Li-Cor or Rockland) were used and images acquired with a Li-Cor Odyssey imaging system. Band infrared fluorescent signals were quantified using Li-Cor Image Studio Version 2.0. For separation of 208–220-immunoreactive bands by IB, urea-soluble protein fractions from 208 TDP-43 Tg mouse cortex were electrophoresed with 12% Bis–Tris gels (Life Technologies) using MES buffer. Primary antibodies used were as follows: 5104 and a rabbit anti-TDP-43 PAb (raised against amino acids 208–220, 1:500, CNDR 2052). For urea-gel IB, urea-soluble protein fractions from 208 TDP-43 Tg mouse cortex were electrophoresed through 6 M urea-containing 12% acrylamide SDS–PAGE at 70 V for ∼15 h. For protein dephosphorylation, urea-soluble protein fractions from 208 TDP-43 Tg mouse cortex were dialyzed at 4°C overnight with gentle mixing through 10 000 MW cutoff membranes into 50 mm Tris, pH 8.0 with 0.5 mm PMSF. Samples were treated with or without calf intestinal phosphatase (CIP; NEB #M0290) at 37°C for 1 h and analyzed by IB.
Neuron quantification
N1065-immunostained and haemotoxylin-counterstained coronal sections of matched levels of the caudal hippocampus from age-matched littermate control and 208 TDP-43 Tg mice were imaged. Nuclei in the DG of the hippocampus of the right hemisphere were counted by two independent blinded observers using ImageJ software.
Statistics
Statistical analysis of DG neuron number was performed using Prism 4 (GraphPad). Two-way analysis of variance (ANOVA) was used to examine the effect of genotype and time, and Bonferonni's post-test was used when significant main effects and interaction of genotype and time were detected. All results are presented as mean ± standard error of the mean (SEM), and P < 0.05 was considered statistically significant.
Supplementary Material
Funding
This work was supported by National Institutes of Health/National Institute on Aging (AG032953, AG17586); and an Australian National Health & Medical Research Council C.J. Martin Biomedical Early Career Fellowship (1036835 to A.W.).
Supplementary Material
Acknowledgements
We thank Drs Sílvia Porta, Todd Cohen, Edward B. Lee and Krista Spiller for helpful discussion, Chi Li for technical assistance, Drs Manuela Neumann and Elizabeth Kremmer for providing the phosphorylation-specific TDP-43 rat MAb TAR5P-1D3 and Dr Jean Richa of the University of Pennsylvania Transgenic and Chimeric Mouse Facility for Tg mouse production.
Conflict of Interest statement. None declared.
References
- 1.Warren J.D., Rohrer J.D., Rossor M.N. (2013) Clinical review. Frontotemporal dementia. B.M.J., 347, f4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ling S.C., Polymenidou M., Cleveland D.W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uryu K., Nakashima-Yasuda H., Forman M.S., Kwong L.K., Clark C.M., Grossman M., Miller B.L., Kretzschmar H.A., Lee V.M., Trojanowski J.Q. et al. (2008) Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol., 67, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton A.E., Chio A., Traynor B.J. (2014) State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci., 17, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winton M.J., Igaz L.M., Wong M.M., Kwong L.K., Trojanowski J.Q., Lee V.M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem., 283, 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M. et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 7.Neumann M., Kwong L.K., Lee E.B., Kremmer E., Flatley A., Xu Y., Forman M.S., Troost D., Kretzschmar H.A., Trojanowski J.Q. et al. (2009) Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol., 117, 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Igaz L.M., Kwong L.K., Xu Y., Truax A.C., Uryu K., Neumann M., Clark C.M., Elman L.B., Miller B.L., Grossman M. et al. (2008) Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol., 173, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Igaz L.M., Kwong L.K., Chen-Plotkin A., Winton M.J., Unger T.L., Xu Y., Neumann M., Trojanowski J.Q., Lee V.M. (2009) Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J. Biol. Chem., 284, 8516–8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y.T., Kuo P.H., Chiang C.H., Liang J.R., Chen Y.R., Wang S., Shen J.C., Yuan H.S. (2013) The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem., 288, 9049–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y., Ray P., Rao E.J., Shi C., Guo W., Chen X., Woodruff E.A. III, Fushimi K., Wu J.Y. (2010) A Drosophila model for TDP-43 proteinopathy. Proc. Natl Acad. Sci. USA, 107, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caccamo A., Majumder S., Oddo S. (2012) Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am. J. Pathol., 180, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregory J.M., Barros T.P., Meehan S., Dobson C.M., Luheshi L.M. (2012) The aggregation and neurotoxicity of TDP-43 and its ALS-associated 25 kDa fragment are differentially affected by molecular chaperones in Drosophila. PLoS One, 7, e31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dayton R.D., Gitcho M.A., Orchard E.A., Wilson J.D., Wang D.B., Cain C.D., Johnson J.A., Zhang Y.J., Petrucelli L., Mathis J.M. et al. (2013) Selective forelimb impairment in rats expressing a pathological TDP-43 25 kDa C-terminal fragment to mimic amyotrophic lateral sclerosis. Mol. Ther., 21, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Igaz L.M., Kwong L.K., Lee E.B., Chen-Plotkin A., Swanson E., Unger T., Malunda J., Xu Y., Winton M.J., Trojanowski J.Q. et al. (2011) Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest., 121, 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wils H., Kleinberger G., Janssens J., Pereson S., Joris G., Cuijt I., Smits V., Ceuterick-de Groote C., Van Broeckhoven C., Kumar-Singh S. (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl Acad. Sci. USA, 107, 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wegorzewska I., Bell S., Cairns N.J., Miller T.M., Baloh R.H. (2009) TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl Acad. Sci. USA, 106, 18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Y.F., Gendron T.F., Zhang Y.J., Lin W.L., D'Alton S., Sheng H., Casey M.C., Tong J., Knight J., Yu X. et al. (2010) Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci., 30, 10851–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwong L.K., Irwin D.J., Walker A.K., Xu Y., Riddle D.M., Trojanowski J.Q., Lee V.M. (2014) Novel monoclonal antibodies to normal and pathologically altered human TDP-43 proteins. Acta Neuropathol. Commun., 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T.G., Buratti E., Baralle F., Morita M. et al. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol., 64, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayala Y.M., De Conti L., Avendano-Vazquez S.E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E. et al. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J., 30, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dantuma N.P., Bott L.C. (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci., 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menzies F.M., Fleming A., Rubinsztein D.C. (2015) Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci., 16, 345–357. [DOI] [PubMed] [Google Scholar]
- 24.Brettschneider J., Van Deerlin V.M., Robinson J.L., Kwong L., Lee E.B., Ali Y.O., Safren N., Monteiro M.J., Toledo J.B., Elman L. et al. (2012) Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol., 123, 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fecto F., Siddique T. (2012) UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve, 45, 157–162. [DOI] [PubMed] [Google Scholar]
- 26.Porta S., Kwong L.K., Trojanowski J.Q., Lee V.M. (2015) Drosha inclusions are new components of dipeptide-repeat protein aggregates in FTLD-TDP and ALS C9orf72 expansion cases. J. Neuropathol. Exp. Neurol., 74, 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stribl C., Samara A., Truembach D., Augustin R., Neumann M., Fuchs H., Gailus-Durner V., Hrabe de Angelis M., Rathkolb B., Wolf E. et al. (2014) Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem., 289, 10769–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssens J., Wils H., Kleinberger G., Joris G., Cuijt I., Ceuterick-de Groote C., Van Broeckhoven C., Kumar-Singh S. (2013) Overexpression of ALS-associated p.M337V human TDP-43 in mice worsens disease features compared to wild-type human TDP-43 mice. Mol. Neurobiol., 48, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong K., Li Y., Duan W., Guo Y., Jiang H., Li W., Li C. (2012) Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett., 530, 144–149. [DOI] [PubMed] [Google Scholar]
- 30.Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E.C., Lacas-Gervais S., Fragaki K., Berg-Alonso L., Kageyama Y., Serre V., Moore D.G. et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain, 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker A.K., Spiller K.J., Ge G., Zheng A., Xu Y., Zhou M., Tripathy K., Kwong L.K., Trojanowski J.Q., Lee V.M. (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol., doi:10.1007/s00401-015-1460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scotter E.L., Vance C., Nishimura A.L., Lee Y.B., Chen H.J., Urwin H., Sardone V., Mitchell J.C., Rogelj B., Rubinsztein D.C. et al. (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci., 127, 1263–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dormann D., Capell A., Carlson A.M., Shankaran S.S., Rodde R., Neumann M., Kremmer E., Matsuwaki T., Yamanouchi K., Nishihara M. et al. (2009) Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J. Neurochem., 110, 1082–1094. [DOI] [PubMed] [Google Scholar]
- 34.Xiao S., Sanelli T., Chiang H., Sun Y., Chakrabartty A., Keith J., Rogaeva E., Zinman L., Robertson J. (2015) Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol., 130, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishimoto Y., Ito D., Yagi T., Nihei Y., Tsunoda Y., Suzuki N. (2010) Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J. Biol. Chem., 285, 608–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y.J., Xu Y.F., Dickey C.A., Buratti E., Baralle F., Bailey R., Pickering-Brown S., Dickson D., Petrucelli L. (2007) Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci., 27, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y.J., Xu Y.F., Cook C., Gendron T.F., Roettges P., Link C.D., Lin W.L., Tong J., Castanedes-Casey M., Ash P. et al. (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl Acad. Sci. USA, 106, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamashita T., Hideyama T., Hachiga K., Teramoto S., Takano J., Iwata N., Saido T.C., Kwak S. (2012) A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun., 3, 1307. [DOI] [PubMed] [Google Scholar]
- 39.Herskowitz J.H., Gozal Y.M., Duong D.M., Dammer E.B., Gearing M., Ye K., Lah J.J., Peng J., Levey A.I., Seyfried N.T. (2012) Asparaginyl endopeptidase cleaves TDP-43 in brain. Proteomics, 12, 2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Q., Yokoshi M., Okada H., Kawahara Y. (2015) The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat. Commun., 6, 6183. [DOI] [PubMed] [Google Scholar]
- 41.Tsuji H., Arai T., Kametani F., Nonaka T., Yamashita M., Suzukake M., Hosokawa M., Yoshida M., Hatsuta H., Takao M. et al. (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain, 135, 3380–3391. [DOI] [PubMed] [Google Scholar]
- 42.Bennion Callister J., Pickering-Brown S.M. (2014) Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp. Neurol., 262, 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capogna M. (2011) Neurogliaform cells and other interneurons of stratum lacunosum-moleculare gate entorhinal-hippocampal dialogue. J. Physiol., 589, 1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Sarraj S., King A., Troakes C., Smith B., Maekawa S., Bodi I., Rogelj B., Al-Chalabi A., Hortobagyi T., Shaw C.E. (2011) p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol., 122, 691–702. [DOI] [PubMed] [Google Scholar]
- 45.Braak E., Braak H. (1997) Alzheimer's disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon's horn. Acta Neuropathol., 93, 323–325. [DOI] [PubMed] [Google Scholar]
- 46.Iseki E., Li F., Odawara T., Kosaka K. (1997) Hippocampal pathology in diffuse Lewy body disease using ubiquitin immunohistochemistry. J. Neurol. Sci., 149, 165–169. [DOI] [PubMed] [Google Scholar]
- 47.Mayford M., Bach M.E., Huang Y.Y., Wang L., Hawkins R.D., Kandel E.R. (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science, 274, 1678–1683. [DOI] [PubMed] [Google Scholar]
- 48.Jankowsky J.L., Slunt H.H., Gonzales V., Savonenko A.V., Wen J.C., Jenkins N.A., Copeland N.G., Younkin L.H., Lester H.A., Younkin S.G. et al. (2005) Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med., 2, e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toledo J.B., Van Deerlin V.M., Lee E.B., Suh E., Baek Y., Robinson J.L., Xie S.X., McBride J., Wood E.M., Schuck T. et al. (2014) A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement., 10, 477–484 e471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee V.M., Page C.D., Wu H.L., Schlaepfer W.W. (1984) Monoclonal antibodies to gel-excised glial filament protein and their reactivities with other intermediate filament proteins. J. Neurochem., 42, 25–32. [DOI] [PubMed] [Google Scholar]
- 51.Sobue G., Hashizume Y., Yasuda T., Mukai E., Kumagai T., Mitsuma T., Trojanowski J.Q. (1990) Phosphorylated high molecular weight neurofilament protein in lower motor neurons in amyotrophic lateral sclerosis and other neurodegenerative diseases involving ventral horn cells. Acta Neuropathol., 79, 402–408. [DOI] [PubMed] [Google Scholar]
- 52.Volpicelli-Daley L.A., Luk K.C., Patel T.P., Tanik S.A., Riddle D.M., Stieber A., Meaney D.F., Trojanowski J.Q., Lee V.M. (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.