

Abstract
CRISPR/Cas9 has been used to genetically modify genomes in a variety of species, including non-human primates. Unfortunately, this new technology does cause mosaic mutations, and we do not yet know whether such mutations can functionally disrupt the targeted gene or cause the pathology seen in human disease. Addressing these issues is necessary if we are to generate large animal models of human diseases using CRISPR/Cas9. Here we used CRISPR/Cas9 to target the monkey dystrophin gene to create mutations that lead to Duchenne muscular dystrophy (DMD), a recessive X-linked form of muscular dystrophy. Examination of the relative targeting rate revealed that Crispr/Cas9 targeting could lead to mosaic mutations in up to 87% of the dystrophin alleles in monkey muscle. Moreover, CRISPR/Cas9 induced mutations in both male and female monkeys, with the markedly depleted dystrophin and muscle degeneration seen in early DMD. Our findings indicate that CRISPR/Cas9 can efficiently generate monkey models of human diseases, regardless of inheritance patterns. The presence of degenerated muscle cells in newborn Cas9-targeted monkeys suggests that therapeutic interventions at the early disease stage may be effective at alleviating the myopathy.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (Cas9) and transcription activator-like effector nucleases (TALENs) have become powerful and versatile tools for genome engineering in a variety of species (1–3). As a result, gene targeting-induced mutations are now possible in large animal embryos for which we lack embryonic stem cell lines for conventional gene-targeting experiments. The recent application of Cas9 and TALEN to primates highlights the potential of using these new technologies to generate non-human primate models of human diseases (4–6). However, there remains a critical concern about using Cas9 and TALEN in large animals and primates: both Cas9 and TALEN reportedly produce mosaic mutations in primates (4–6). Since primates and large animals have much longer breeding times than rodents, it is important to know whether such mosaic mutations can effectively disrupt the function of targeted genes to mimic the loss of function of genetic mutations in human diseases.
Various mutations (missense, non-sense, deletion, insertion, or duplication mutations) in the human dystrophin gene cause Duchenne muscular dystrophy (DMD), a recessive X-linked form of muscular dystrophy that affects ∼1 in 3600 boys (7–9). DMD is characterized by progressive muscle weakness and degeneration, which eventually lead to paralysis and early death by the age of ∼30 as a result of respiratory or cardiac failure (7). Animal models of DMD due to dystrophin deficiency have been investigated extensively to provide important insight into the pathogenesis of DMD (10,11). However, mice lacking the DMD gene develop much milder muscle pathology and phenotypes than humans with DMD (12,13), underscoring the importance of generating large animal models of DMD to better understand its pathogenesis.
Results
In this report, we used Cas9 to disrupt the DMD gene in rhesus monkeys. There are two major hotspots for mutations around exons 3–7 and exons 45–55 in the DMD gene, which consists of a total of 79 exons (14,15) (Fig. 1A). Based on this, we designed Cas9 vectors to target exon 4 and exon 46 of the monkey DMD gene (Fig. 1B). The targeted sequences also show minimal sequence homology to other genes to reduce off-target effects. We used 2–3 targeting regions to design Cas9 vectors and have confirmed the targeted effects of these vectors in transfected HEK293 cells by analyzing sequence mutations in the DMD gene.
Figure 1.

DMD gene mutations generated by Cas9. (A) The location of targeted regions (exon 4 and exon 46) in the human DMD gene. Physical map of the dystrophin gene. Green boxes indicate exons, blue boxes indicate exons specific for each promoter (B1, first brain exon; M1, first muscle exon; P1, first Purkinje exon) and red boxes indicate promoters (BP, brain promoter; G1, general type promoter; MP, muscle promoter; PP, Purkinje promoter; S1, Schwann cell promoter). Not drawn to scale (Adapted from Cohen, N. and Muntoni, F. (2004) Multiple pathogenetic mechanisms in X linked dilated cardiomyopathy. Heart (British Cardiac Society), 90, 835–841). (B) Targeted sequences of the monkey DMD gene by Cas9 vectors.
After verifying the targeting effects of our vectors, we then performed gene-targeting experiments on rhesus monkeys. MII oocytes were collected from female rhesus monkeys after superovulation and used for intracytoplasmic sperm injection (ICSI). The fertilized eggs were then intracellularly injected with mRNAs of gRNAs/Cas9 (25/200 ng/μl) to target exon 4 alone or exon 4 and exon 46 together. The injected eggs were further cultured for development to the morula or blastocyst stages, which were collected for PCR (Fig. 2A). The PCR products were analyzed via enzymatic digestion of the specific targeted region and T7EN1 cleavage assay. We analyzed a total of 142 embryos for Cas9 targeting, respectively. DNA analysis revealed that Cas9 yielded mutations in 46.47% of embryos (Fig. 2B). Sequence analysis uncovered different indels in the monkey DMD gene by Cas9 targeting, confirming site-specific targeting and mutations via Cas9 (Fig. 2C).
Figure 2.

Monkey embryos injected with Cas9 mRNA and collected for PCR analysis. (A) The normal development of embryos after injection of Cas9 mRNA. (B) Cas9-mediated mutations in exon 4 and exon 46 of the monkey DMD gene. (C) Cas9-mediated DNA mutations in monkey embryos.
Next, we transferred Cas9-injected embryos to surrogate rhesus monkeys. We obtained 14 live monkeys by transferring 179 Cas9-injected embryos to 59 surrogates (Fig. 3A and Table 1). There were eight miscarried fetuses and four full-term stillborn babies resulting from difficult births. These fetuses and stillborn monkeys were found to be both wild-type and mutants in which the targeted DNA regions were confirmed via PCR and sequencing analysis. Thus, the premature deaths of the developing fetuses were unlikely to be due to Cas9 targeting, but rather effects of in vitro fertilization and embryo transfer on early embryo development. Importantly, we detected mutations in the DMD gene in two stillborn and nine live monkeys, for a gene-targeting rate of 61.1% (Fig. 3). In the stillborn monkeys, the targeted exon 4 of the DMD gene contains three types of mutations (−2, +2 and +20), and in live monkeys, five different mutations were found in the targeted exons 4 and 46 (Fig. 3C–E and Table 1). These mutations caused a frame shift of the coding sequences of the DMD gene and terminated full-length protein expression, the same type of mutations seen in patients with DMD. Because Cas9 can tolerate even five mismatches within the target site (16), we identified potentially targeted exon sequences in other genes that differ by five or fewer positions in the targeted region (nucleotides in red within the 20-bp region to which the gRNA hybridizes) (Supplementary Material, Table S1) and amplified these DNA fragments via PCR from the genomic DNAs of a stillborn monkey (Mut-2). We then used a T7EN1 cleavage assay to reveal potentially mutated DNAs, which were also subjected to DNA sequence analysis. The results revealed no authentic mutations (Supplementary Material, Fig. S1), which is consistent with the selectivity of Cas9-mediated targeting in primate genome (4). All Cas9-targeted live monkeys are younger than 6 months and have yet to show any obvious behavioral or movement abnormalities. Since DMD is an age-dependent disorder, whether these Cas9-targeted monkeys show any phenotypes as they develop into adulthood remains to be investigated.
Figure 3.

Generation of live monkeys carrying mutations in the DMD gene. (A) CRISPR/Cas9-mediated targeting in monkeys. (B) Identified sequence mutations in full-term stillborn monkeys. (C) A photo of two monkeys at the age of 76 days, which were found to carry Cas9-mediated mutations. (D) Representative PCR analysis of mutations in exon 4 DMD gene of the DMD gene in full-term stillborn monkeys Mut-1 and Mut-2 and live monkeys (Mut-3, -4, -5 and -6). T7EN1 and enzymatic digestion were performed. WT, wild type; PC, positive control. (E) Identified sequence mutations in live monkeys.
Table 1.
Summary of live monkeys with mutations generated by Cas9-mediated targeting
| Monkey | Sex | Age (m) | Targeting region | Mutations |
||
|---|---|---|---|---|---|---|
| TEEN1 diagnosis | Enzyme diagnosis | Identified mutations | ||||
| 1 | F | 6 | exon 4 | Positive | Positive | −5 bp |
| 2 | F | 6 | exon 4 | Positive | Positive | −5 bp |
| exon 46 | Positive | Positive | −7 bp | |||
| 3 | M | 5 | exon 4 | Positive | Positive | −2 bp |
| 4 | F | 5 | exon 4 | Positive | Positive | −2 bp |
| exon 46 | Positive | −7 bp | ||||
| 5 | F | 6 | exon 4 | Negative | Negative | Negative |
| 6 | M | 5 | exon 4 | Negative | Negative | Negative |
| 7 | M | 5 | exon 4 | Positive | Positive | −9 bp |
| 8 | F | 5 | exon 4 | Positive | Positive | −9 bp |
| exon 46 | Positive | Positive | −7 bp | |||
| 9 | M | 5 | exon 4 | Negative | Negative | Negative |
| 10 | M | 5 | exon 4 | Negative | Negative | Negative |
| 11 | M | 5 | exon 4 | Negative | Negative | Negative |
| 12 | F | 4 | exon 4 | Positive | Positive | −2 bp +20 bp |
| 13 | F | 4 | exon 4 | Positive | Positive | −2 bp +20 bp |
| 14 | M | 4 | exon 46 | Positive | Positive | −4 bp |
As reported previously (4), Cas9 targeting caused mosaic mutations in the monkey genome, which might confound potential phenotypes. Such mosaic mutations can result from the prolonged effects of Cas9 after one cell cleavage, though their mRNAs were injected in fertilized eggs at the one-cell stage. Thus, it is important to know how different mutations are distributed in monkey tissues and what the relative rates of these mutations are in the DMD gene in affected tissues, such as muscle. A high rate would indicate that most of the DMD gene has been disrupted to yield the loss of function or to mimic the consequences of DMD mutations in patients.
Of the stillborn babies that developed to full term (>145 days) and died of dystocia or difficult birth, a male monkey (Mut-1), a female monkey (Mut-2) and two wild-type monkeys provided us with different tissues to analyze gene targeting and associated targeting effects. The tissues of Mut-1 and Mut-2 monkeys were found to be positive for DMD mutations via PCR analysis (Fig. 4A). PCR experiments and sequencing of subcloned DNAs showed that Mut-1 and Mut-2 monkeys carried three mutations (−2, +2 and +20) in exon 4 of the DMD gene. We focused on the effect of Cas9 targeting in these monkeys and designed specific primers that can distinguish between wild-type and mutant alleles of the DMD gene via PCR. Under the same PCR conditions, we also used primers that can amplify both mutant and wild-type alleles to obtain the total amount of the DMD alleles (Fig. 4B and C). The ratios of mutant to total PCR products in the same gel were obtained via densitometry, allowing us to estimate the relative amounts of the DMD alleles carrying specific mutations. As a result, this quantitative analysis shows that total DMD mutations occur at 43.5, 39.7, 23.2 and 67.4% in placenta, liver, brain and muscle, respectively, in the Mut-1 monkey (Fig. 4D). The higher mutation rates (124.25, 44.58, 103.33 and 86.99% in placenta, liver, brain and muscle) were seen in the Mut-2 monkey (Fig. 4E). It is possible that some cells in the placenta of the Mut-2 monkey may carry two or more mutations in the same allele, making the total mutation rate slightly over 100%.
Figure 4.

The relative rates of mosaic mutations in stillborn monkey tissues. (A) PCR products of the mutant alleles and the total DMD gene under the same PCR conditions. WT monkey tissues served as controls to show the specific amplification of mutant alleles of the DMD gene in monkey Mut-2. (B and C) Specific primers for each mutation (+20, +2 and −2) were able to amplify the mutant DNA from monkey Mut-1 (B) and Mut-2 (C), but not the WT monkey. Under the same PCR conditions, primers that could amplify both mutant and WT alleles were used to obtain total DMD gene products. (D and E) The levels (%) of each mutant alleles relative to total DMD alleles in different tissues were obtained by densitometry analysis of DNA bands in (B) and (C), respectively. Total mutations represent the sum of different mutations (+20, +2 and −2). The data are mean ± SE (n = 3 independent PCRs).
Given the nature of mosaic mutations created by Cas9, it is important to know whether mosaic mutations can cause dysfunction of the targeted gene. DMD is characterized by muscle degeneration due to the loss of dystrophin (8,9). We first looked at the expression of dystrophin in the muscle of the targeted monkeys and compared with the muscle from a stillborn wild-type monkey. Muscle fibers in the full-term stillborn monkeys are noticeably much smaller than those in an adult monkey (Supplementary Material, Fig. S2A), indicating that the size of muscle cells would continue to grow after birth. Immunohistochemical staining clearly showed that there is a marked reduction of dystrophin in the Mut-1 monkey and an almost complete depletion of dystrophin in the Mut-2 monkey compared with the wild-type monkey (Fig. 5A). High-magnification micrographs demonstrate the staining of dystrophin of the muscle fiber surface in wild-type monkey muscle, which is almost completely eliminated in the Mut-2 monkey muscle (Fig. 5B). Western blotting confirmed a dramatic reduction of dystrophin in the Mut-1 monkey muscle and the loss of dystrophin in the Mut-2 monkey muscle compared with wild type (Fig. 5C). Thus, both immunocytochemical and western blotting analyses provide convincing evidence for the depletion or loss of dystrophin in Cas9-targeted monkey muscle tissues.
Figure 5.

Depletion of dystrophin by Cas9 in monkey tissues. (A) Immunohistochemical staining of muscle sections showing the reduced level of dystrophin in Mut-1 monkey muscle and the depletion of dystrophin in Mut-2 monkey muscle compared with wild-type stillborn monkey muscle. (B) High-magnification graphs of immunostained muscle sections from wild-type, Mut-1 and Mut-2 monkeys. (C) Western blot confirming the reduction of dystrophin in Mut-1 monkey muscle and depletion of dystrophin in Mut-2 monkey muscle. Scale bars: (A): 50 μm; (B): 20 μm.
Next, we investigated whether there was any myopathic change mediated by Cas9 targeting. Using hematoxylin and eosin (H&E) staining, we found that both Mut-1 and Mut-2 monkey muscle tissues displayed frequent disruption of muscle structure with groups of hypertrophic fibers, though Mut-2 had more extensive disruption (Fig. 6A, Supplementary Material, Fig. S2B). The hypertrophic muscle cells appear in clusters and, importantly, often contain centralized nuclei, a feature of regenerated muscles after degeneration. In addition, there were large interstitial spaces between these clustered hypertrophic muscles (Fig. 6A), perhaps due to replacement by fat or connective tissues. We examined > 814 cells in each monkey muscle and found that 12.5 and 17.5% of muscle cells showed centralized or multiple nuclei in Mut-1 and Mut-2 monkeys, respectively; these were not found in control muscle from the wild-type stillborn monkey (Fig. 6B). Also, because the stillborn monkeys died of difficult births and their muscle tissues were isolated immediately after death, degenerated muscle cells seen in Cas9-targeted monkeys should already occur before the death of newborn monkeys.
Figure 6.
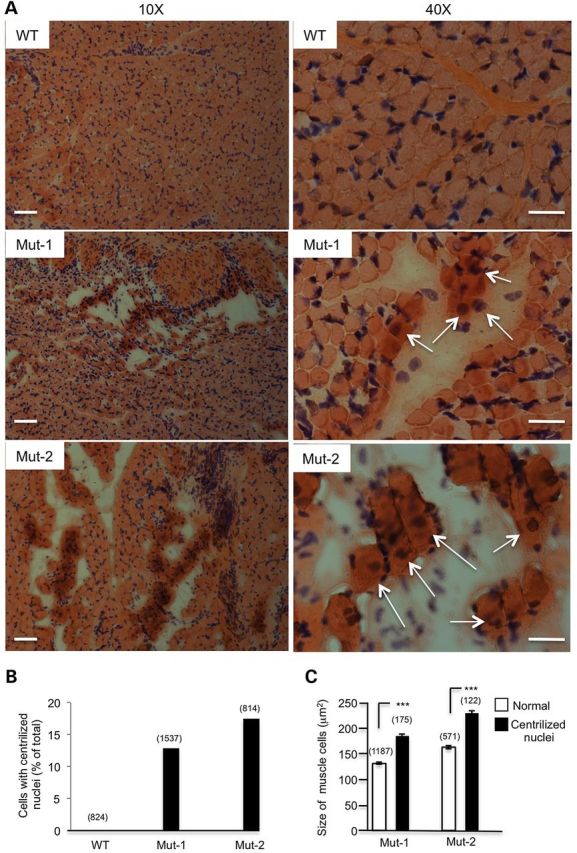
Hypertrophic myopathy caused by Cas9-mediated targeting of the DMD gene. Hematoxylin and eosin (H&E) staining of monkey muscle sections. (A) Low-magnification (×10) micrographs showing the clusters of hypertrophic muscle cells with an increase in the extent of the interstitial space in Mut-1 and Mut-2 monkeys. (B) Higher magnification micrograph of the DMD monkey muscle fibers shows multicentrally nuclei in hypertrophic muscle cells (arrows) compared with WT muscle. (C) Cross-sectional areas of normal muscle cells and hypertrophic muscle fibers containing centralized nuclei. Muscle sections from the stillborn wild-type monkey served as a control. Data are presented as mean ± SE from counting >300 cells in each group. *P < 0.05; **P < 0.01. Scale bars: 50 μm (left panel, ×10); 20 μm (right panel, ×40).
Because the size of muscle cells varies during development and in different parts of skeletal muscle, we compared the cross-sectional areas of the muscle cells containing centralized or multiple nuclei with those of normal muscle cells in the same sections. We found that these cells containing centralized nuclei are significantly larger than normal muscle cells [162.93 ± 1.06 (normal) versus 229.59 ± 3.91 (mutant) for Mut-1 and 131.00+ ± 2.03 (normal) versus 184.83 ± 4.56 (mutant) for Mut-2, P < 0.001] (Fig. 6C). Hypertrophic muscle fibers with centralized nuclei are the most abundant histopathological feature of regenerating muscle fibers and represent the cardinal feature of DMD, which is also present in calf muscle in young DMD patients (17), mice (18), dog (19,20) and cat (21,22). Since DMD is an age-dependent disease that results in muscle atrophy following the hypertrophic muscle changes seen in the early disease stages (19), the appearance of large regenerated myofibers with centralized nuclei in stillborn monkeys and the loss of dystrophin indicate that we have generated a monkey model of DMD that shows early muscle atrophy pathology.
Discussion
Although recent studies demonstrated that Cas9 can successfully modify genomes in primates (4), whether this new technology can be used to create non-human primate disease models that show pathology similar to human diseases remains unknown. This issue is critical, because genetic mutations in humans often occur at the one-cell stage before cell division, such that the same mutation is present ubiquitously in every individual cell. The translation of Cas9 mRNA to produce an active enzymatic form is likely delayed until after the first cell division (23), and this delay may play a major role in contributing to genetic mosaicism. Alternatively, differential DNA repair and non-homozygous recombination activities in zygotes and divided embryonic cells can also influence genetic mutation rates and mosaicism. Given the time and cost of breeding non-human primates, it is important to know whether Cas9 can be used to generate primate models that will deplete the expression of the targeted gene and display the expected pathology of a human disease.
Using specific primers to identify particular mutated alleles from monkey genome, we were able to show that Cas9 can induce mosaic mutations that cover 87% of the DMD alleles in muscle, which is also supported by the depletion of dystrophin protein in the targeted monkey muscle and associated muscle pathology. Although more accurate quantification of the mutation rates may be achieved by other methods such as deep sequencing, the loss of dystrophin protein indicates that Cas9 is an efficient tool for disrupting gene function in monkeys via its expression in one-cell embryos.
Another important implication of our study is that it could help us understand the pathogenesis of DMD using large animal models. DMD is a sex-linked muscular dystrophy that affects males only; our findings show that cas9-mediated gene targeting could deplete dystrophin in a female monkey, suggesting that its potency in creating multiple mutations can cause disruption of both alleles of the DMD gene in females. Such a female primate model would be important to reveal the myopathology that is independent of male hormones. Furthermore, the monkeys generated can be investigated as founders without breeding, an advantage for studying large animals, which often require a lengthy time to breed.
Various animal models have led to important advances in our understanding of the pathophysiology of muscular dystrophy and allowed the development of several promising approaches to therapy (19–22,24). However, these animal models have species-specific differences in their clinical courses and phenotypes. Unlike DMD patients, a mouse model (mdx) of DMD does not exhibit extensive muscle fibrosis, severe clinical symptoms or early death (25–27). In all these animal models, hypertrophic muscle is the common pathological feature (21,22,28), which is also seen in young DMD patients (17). Unlike DMD mouse models, our newborn Cas9-targerted monkeys show muscle degeneration at the early stage of disease, suggesting that DMD pathology is species dependent. Since skeletal muscle undergoes differentiation and development during the postnatal period (29), the presence of hypertrophic muscle with centralized nuclei in our newborn Cas9-targeted monkeys also suggests that the loss of dystrophin could affect the development of muscle cells at a very early stage. Thus, therapeutic interventions at the early disease stage may be more effective at alleviating or reducing the myopathy that can otherwise become severe while skeletal muscle tissues are continuously developing after birth.
Materials and Methods
Cas9 vectors
Cas9 plasmid (MLM3613, Plasmid #42251) was used to express Cas9 nuclease (Streptococcus pyogenes) under the control of the CMV or T7 promoter. The p-T7-gRNA expression vector used for in vitro transcription of gRNAs was provided by Dr Liangxue Lai at The Guangzhou Institutes of Biomedicine and Health, CAS. gRNAs were designed based on the targeted sequences in the monkey DMD gene and generated using their expression vectors (Cas-msm-3613, PCS2-peas-T, p-T7-gRNA). The targeted regions of the monkey DMD gene were isolated via PCR and subcloned into the p-T7-gRNA, PCS2-peas-T vector to test the effects of gRNA. gRNA GFP-T1 (Addgene, 41819) was used for expression of gRNAs in HEK293 cells.
Template DNAs for in vitro transcription were generated by PCR amplification of the gRNAs plasmids. The PCR products were purified and transcribed by mMESSAGE mMACHINE T7 kit (Ambion, AM1344) in vitro. Cas9-M3613 plasmid was linearized by PmeI and in vitro transcribed using MAXIscript T7 (Ambion, AM1312). The synthesized RNAs were purified using LiCl with an additional ethanol precipitation.
Animals
Healthy female rhesus monkeys (Macaca mulatta), ranging in age from 5 to 8 years with body weights of 4–8 kg, were selected for use in this study. All animals were housed at the Kunming Biomed International (KBI). The KBI is an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. All animal protocols were approved in advance by the Institutional Animal Care and Use Committee of Kunming Biomed International.
Embryo collection
Embryo collection and transfer were performed as previously described (4). In brief, 32 healthy female rhesus monkeys aged 5–8 years with regular menstrual cycles were selected as oocyte donors for superovulation, performed by intramuscular injection with rhFSH (recombinant human follitropin alfa, GONAL-F, Merck Serono) for 8 days, then rhCG (recombinant human chorionic gonadotropin alfa, OVIDREL, Merck Serono) on Day 9. Oocytes were collected by laparoscopic follicular aspiration 32–35 h after rhCG administration. MII (first polar body present) oocytes were used to perform ICSI, and fertilization was confirmed by the presence of two pronuclei.
Cas9/sgRNA injection of one-cell embryos
The zygotes were injected with Cas9 mRNA (200 ng/μl) and gRNAs (25 ng/μl each). Microinjections were performed in the cytoplasm of zygotes using a Nikon microinjection system under standard conditions. The zygotes were cultured in embryo culture medium-10 (HECM-10) containing 10% fetal calf serum (Hyclone Laboratories, SH30088.02) at 37°C in 5% CO2. Cleaved embryos of high quality at the two cell to blastocyst stage were transferred into the oviduct of the matched recipient monkeys. Female monkeys were used as surrogate recipients, and typically, three embryos were transferred into each female. The earliest pregnancy diagnosis was performed by ultrasonography ∼20–30 days after the embryo transfer. Both clinical pregnancy and number of fetuses were confirmed by fetal cardiac activity and presence of a yolk sac as detected by ultrasonography.
PCR and quantitative analysis of targeted alleles
Monkey tissues collected for PCR included: placenta, umbilical cord, liver, brain cortex, muscle, kidney, lung, spleen and heart. Genomic DNA was extracted using a Genomic Extraction Kit (Solarbio, cat NO:D1700, CHN) according to the manufacturer's protocol. The DMD gene sequences including the target sites were determined by PCR. PCR was performed by initial incubation at 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 58°C for 30 s and 72°C for 30 s. The PCR products were purified, and 500 ng of DNA per sample was digested with 0.3 μl of T7 endonuclease (Invitrogen, USA) in a 20-μl reaction volume for 1 h at 37°C. The mixture was then resolved on a 1% agarose gel.
To determine specific mutations, sense primers specific to mutated sequences (5′-tggaaatcataaactgacttg-3′, 5′-tcgtagatggaaatcataaactgactgg-3′ and 5′-taaactgatttatgaaatcataaaccaaa-3′ for −2, +2 and +20 mutated DNAs, respectively) and antisense primer (5′-cactgtggagaagggtttgttattgttag-3′) were used to selectively amplify the mutated DNA via PCR. Under the same PCR conditions, primers (sense 5′-ggaagtactgacatcgtagatggaaatca-3′ and antisense 5′-cactgtggagaagggtttgttattgttag-3′) for the common sequences of mutated and normal alleles of the DMD gene were also used for PCR. The PCR products were resolved in agarose DNA gels, and each band was quantified via densitometry using Gray scale scanning software Image-Pro Plus (IPP). The percentage of each mutated DNA relative to total DMD DNA and the sum of each mutation were obtained from more than three independent PCRs, presented as mean ± SE.
Western blot analysis
Tissues from the dead monkeys were isolated and used for analysis. Muscle tissues were homogenized in RIPA buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.1% SDS, 0.5% deoxycholate, 1% Triton X-100, 1:1000 protease inhibitor cocktail) at 4°C using a Teflon-glass homogenizer. Total proteins were then extracted, and their concentrations were determined using the Bradford assay (Pierce). Equal amounts (50–60 mg) of protein extracts with loading dye were boiled before loading into 7% polyacrylamide gels (Bio-Rad). After electrophoresis, proteins were transferred onto a PVDF membrane (Bio-Rad) using Bio-Rad's Trans-Blot. The blots were blocked in 5% skimmed milk for 1 h, incubated with the primary antibodies rabbit monoclonal anti-dystrophin (abcam15277, 1:5000 dilution) and GAPDH (Sigma; 1:10 000 dilution), followed by secondary antibodies for detecting proteins.
Off-target analysis
Possible Cas9 off-target sites (OTS) for exon 4 of the monkey dystrophin gene were predicted by screening the M. mulatta genome. Ten putative OTS were found, allowing a maximum of 3 to 4 bases of mismatches. The OTS were PCR amplified from the genomic DNA in muscle tissues of the Cas9-targeted monkey (Mut-2). The isolated DNA fragments from the potential targeted regions were subjected to T7EN1 cleavage assay. DNA fragments that showed some cleaved products were also subjected to DNA sequence analysis. Our study demonstrated no detectable off-target mutation induced by Cas9 and gRNA in the monkey.
Quantitative analysis of hypertrophic muscle cells
The size of muscle cells was analyzed using Microscope Software AxioVision Rel 4.8. Five to 6 random images (×10) per section and 8–10 sections from each animal were analyzed. The edge of each cell was used to determine the area of the cell using the measurement tool of Microscope Software AxioVision Rel 4.8, and data were exported as Microsoft Excel files. The number and size of the normal and hypertrophic muscle cells were then calculated using Microsoft Excel quantitation tools. For all quantifications, the mean value of muscle cell size from the same genotype was calculated and compared with a Student t-test.
Supplementary Material
Funding
This work was supported by the National Key Basic Research Program of China (2012CBA01304, 2012CBA01300, 2014CB560701), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13000000), the National High Technology Research and Development Program of China (2012AA020701), the National Natural Science Foundation of China (91332206, U1302227, 31271599), Yunnan Innovation Talents of Science and Technology (No. 2012HA013, 2013HB133), the State Key Laboratory of Molecular Developmental Biology, China, Yunnan Academic Leaders and Reserve Personnel, and the Taiwan youth visiting scholar program in the Chinese Academy of Sciences.
Supplementary Material
Acknowledgements
We thank Peng Yin, Fengyang Wang, Lianghe Zhang, Qiang Sun, Jinquan Gao, Xudong Liu, Li Yan and Jiaoni Cheng for technical assistance and Cheryl Strauss for critical reading of this manuscript.
Conflict of Interest statement. None declared.
References
- 1.Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C., et al. (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol., 29, 731–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsu P.D., Lander E.S., Zhang F. (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niu Y., Shen B., Cui Y., Chen Y., Wang J., Wang L., Kang Y., Zhao X., Si W., Li W., et al. (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell, 156, 836–843. [DOI] [PubMed] [Google Scholar]
- 5.Liu H., Chen Y., Niu Y., Zhang K., Kang Y., Ge W., Liu X., Zhao E., Wang C., Lin S., et al. (2014) TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell, 14, 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Z., Zhou X., Zhu Y., Chen Z.F., Yu B., Wang Y., Zhang C.C., Nie Y.H., Sang X., Cai Y.J., et al. (2014) Generation of a monkey with MECP2 mutations by TALEN-based gene targeting. Neurosci. Bull., 30, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moser H. (1984) Duchenne muscular dystrophy: pathogenetic aspects and genetic prevention. Hum. Genet., 66, 17–40. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman E.P., Brown R.H., Jr, Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 9.Emery A.E. (2002) The muscular dystrophies. Lancet, 359, 687–695. [DOI] [PubMed] [Google Scholar]
- 10.Banks G.B., Chamberlain J.S. (2008) The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top Dev. Biol., 84, 431–453. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura A., Takeda S. (2011) Mammalian models of Duchenne Muscular Dystrophy: pathological characteristics and therapeutic applications. J. Biomed. Biotechnol., 2011, doi:10.1155/2011/184393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dangain J., Vrbova G. (1984) Muscle development in mdx mutant mice. Muscle Nerve, 7, 700–704. [DOI] [PubMed] [Google Scholar]
- 13.Tanabe Y., Esaki K., Nomura T. (1986) Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol., 69, 91–95. [DOI] [PubMed] [Google Scholar]
- 14.Muntoni F., Torelli S., Ferlini A. (2003) Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol., 2, 731–740. [DOI] [PubMed] [Google Scholar]
- 15.Ferlini A., Neri M., Gualandi F. (2013) The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscul. Disord., 23, 4–14. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol., 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cros D., Harnden P., Pellissier J.F., Serratrice G. (1989) Muscle hypertrophy in Duchenne muscular dystrophy. A pathological and morphometric study. J. Neurol., 236, 43–47. [DOI] [PubMed] [Google Scholar]
- 18.Coulton G.R., Morgan J.E., Partridge T.A., Sloper J.C. (1988) The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol., 14, 53–70. [DOI] [PubMed] [Google Scholar]
- 19.Kornegay J.N., Childers M.K., Bogan D.J., Bogan J.R., Nghiem P., Wang J., Fan Z., Howard J.F., Jr., Schatzberg S.J., Dow J.L., et al. (2012) The paradox of muscle hypertrophy in muscular dystrophy. Phys. Med. Rehabil. Clin. N. Am., 23, 149–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharp N.J., Kornegay J.N., Van Camp S.D., Herbstreith M.H., Secore S.L., Kettle S., Hung W.Y., Constantinou C.D., Dykstra M.J., Roses A.D., et al. (1992) An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics, 13, 115–121. [DOI] [PubMed] [Google Scholar]
- 21.Carpenter J.L., Hoffman E.P., Romanul F.C., Kunkel L.M., Rosales R.K., Ma N.S., Dasbach J.J., Rae J.F., Moore F.M., McAfee M.B., et al. (1989) Feline muscular dystrophy with dystrophin deficiency. Am. J. Pathol., 135, 909–919. [PMC free article] [PubMed] [Google Scholar]
- 22.Gaschen F.P., Hoffman E.P., Gorospe J.R., Uhl E.W., Senior D.F., Cardinet G.H., 3rd, Pearce L.K. (1992) Dystrophin deficiency causes lethal muscle hypertrophy in cats. J. Neurol. Sci., 110, 149–159. [DOI] [PubMed] [Google Scholar]
- 23.Oh B., Hwang S., McLaughlin J., Solter D., Knowles B.B. (2000) Timely translation during the mouse oocyte-to-embryo transition. Development, 127, 3795–3803. [DOI] [PubMed] [Google Scholar]
- 24.Cooper B.J., Winand N.J., Stedman H., Valentine B.A., Hoffman E.P., Kunkel L.M., Scott M.O., Fischbeck K.H., Kornegay J.N., Avery R.J., et al. (1988) The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature, 334, 154–156. [DOI] [PubMed] [Google Scholar]
- 25.Bulfield G., Siller W.G., Wight P.A., Moore K.J. (1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA, 81, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. (1991) The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature, 352, 536–539. [DOI] [PubMed] [Google Scholar]
- 27.Chamberlain J.S., Metzger J., Reyes M., Townsend D., Faulkner J.A. (2007) Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J., 21, 2195–2204. [DOI] [PubMed] [Google Scholar]
- 28.Vos J.H., van der Linde-Sipman J.S., Goedegebuure S.A. (1986) Dystrophy-like myopathy in the cat. J. Comp. Pathol., 96, 335–341. [DOI] [PubMed] [Google Scholar]
- 29.Buckingham M., Bajard L., Chang T., Daubas P., Hadchouel J., Meilhac S., Montarras D., Rocancourt D., Relaix F. (1986) The formation of skeletal muscle: from somite to limb. J. Anat., 202, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.