

To the Editor
The Inhibitor of Kappa Light Polypeptide Gene Enhancer in B-Cells Kinase gene (IKBKG), also known as NF-kappa B Essential Modulator (NEMO) is located on the X chromosome and encodes the regulatory scaffold subunit of the inhibitor of kappa B kinase gamma (IKKγ) of the IKK complex. Upon activation, the IKK complex phosphorylates the inhibitor of kappa B (IKB), leading to its degradation and thereby facilitating nuclear translocation of NF-kappa B and transcription of genes involved in inflammation, immunity and cell survival. Amorphic mutations in IKKγ producing a complete loss of functional protein cause incontinentia pigmenti (IP) in females, while they are embryonically lethal in most affected males[1]. On the other hand, hypomorphic IKBKG/NEMO mutations in males that impair but do not abolish NF-kappa B signaling lead to hypohidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) as well as immunodeficiency without EDA [2]. Multiple transcript variants encoding different isoforms of this gene have been identified [3]. A pseudogene encoding exons 3–10 is located in an adjacent region of the X-chromosome making diagnosis based on sequencing of gDNA challenging [4]. EDA-ID is inherited as an X-linked recessive disorder, although an autosomal dominant form, due to IKBA mutations, has been described [5]. Furthermore, IKBKB mutations cause an autosomal recessive immunodeficiency without signs of ectodermal dysplasia [6].
We report a novel splice site mutation in IKBKG/NEMO leading to exon 9 skipping and a frameshift with premature termination identified in an infant with evidence of EDA and severe immunodeficiency. The patient was born at term to a G1P1 mother with a history of IP. He developed hypoglycemia and hypocalcemia with seizure-like activity shortly after birth. EEG was normal and MRI demonstrated bilateral parietal findings consistent with small areas of ischemia but no structural defects. The infant was discharged home at 72 hours of age. Ten days later he developed poor feeding and body stiffening with eye deviation 6 hours post-circumcision. Upon arrival to the ER he was afebrile but ill appearing and was noted to have sparse hair, absent eyelashes and eyebrows. The glans of his penis was erythematous following circumcision, but without swelling or discharge. He had edema of the scalp, hands, scrotum and lower extremities and appeared sleepy with a poor suck. Initial CBC demonstrated Hb 16.2 g/dL, WBC 6,100/mm3 (ANC 1800/mm3, ALC 3100/mm3) and platelets 66,000/mm3. CRP was 1.6 mg/dL. Chest radiograph revealed multifocal infiltrates consistent with pneumonia. Blood, urine and CSF cultures grew methicillin susceptible Staphylococcus aureus (MSSA). Echocardiography on hospital day 4 revealed a 1 cm mobile, pedunculated mass on the wall of the left ventricle. Repeat MRI of the brain demonstrated reduced diffusivity consistent with microinfarcts, porencephaly, periventricular white matter changes, multiple microabscesses, leptomeningeal and cranial nerve enhancement, and absent frontal incisors.
Over the following 2 weeks the patient’s respiratory status declined; he remained persistently bacteremic despite treatment with oxacillin, rifampin and gentamicin, developed flaccid paralysis and a marked leukocytosis with WBC of 93,000/mm3 with left shift. Immunoglobulin levels were IgG 201mg/dL, IgA<7mg/dL, IgM<5mg/dL, IgE<1IU/mL and he received one dose of IVIG. Total and naïve CD4+ and CD8+ T cells, CD19+ B cells, CD56+ NK cells and TREC levels at newborn screening were normal for age. Chromosomal microarray revealed a normal male without evidence of chromosome imbalances or copy number variation. The patient expired on hospital day eighteen.
The patient’s mother had stigmata consistent with IP manifested by missing incisors and a small hyperpigmented lesion on her abdomen. Three maternal sisters and grandmother also had cutaneous and dental manifestations of IP and their diagnoses were previously made clinically so genetic testing of IKBKG/NEMO was pursued. Coding exons and adjacent splicing sites of the patient’s gDNA (from whole blood) were Sanger sequenced and a donor splice site variant (IKBKG c. 1117+5G>C) was identified (Figure 1E). MutationTaster predicted the loss of the exon 9 splice acceptor leading to IKBKG exon skipping with a frameshift and a premature termination codon. The patient’s cDNA (from PBMCs) yielded only mutant message revealing the predicted IKBKG exon 9 skipping and frameshift/premature termination (Figure 1E). Maternal gDNA (from whole blood) and cDNA (from PBMCs) were examined and demonstrated the same donor splice site variant on gDNA but only wild type allele on cDNA, suggesting nonrandom maternal X-chromosome inactivation. Maternal cDNA was analyzed by cloning and sequencing that revealed only the expression of the wild type allele (Figure 1A). PCR-based human androgen receptor (HUMARA) gene assay showed a 3bp difference in the maternal X-chromosomes but only a single allele was amplified following HpaII digestion supporting nonrandom X-chromosome inactivation (Figure 2E).
Figure 1E.
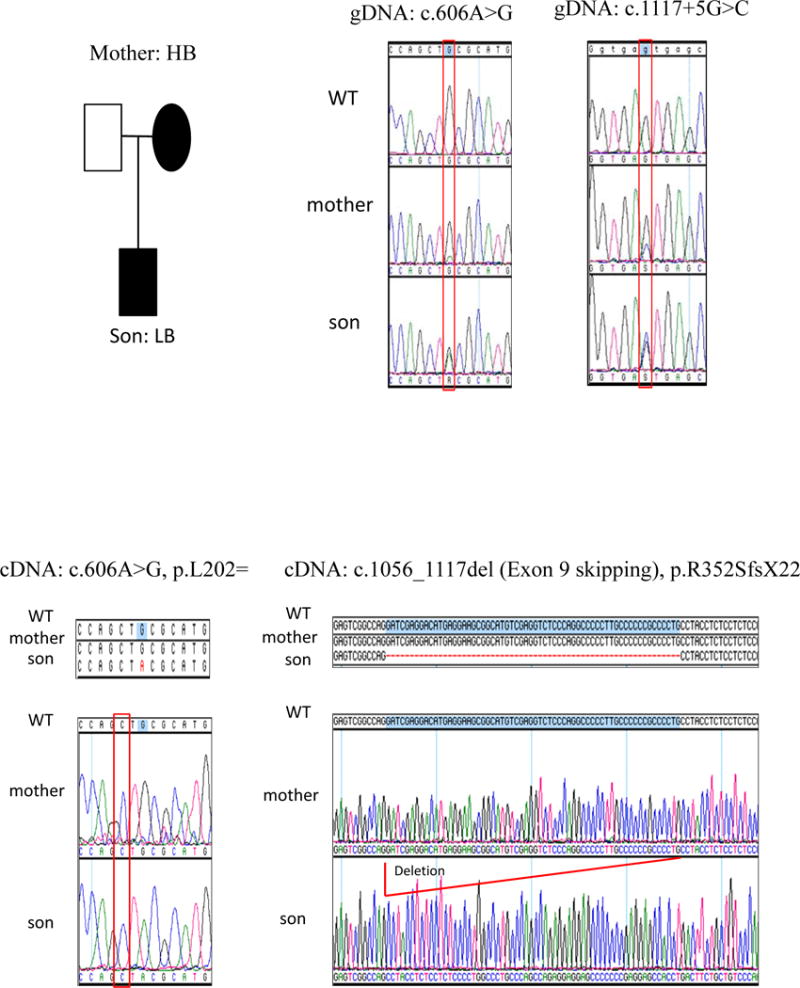
IKBKG/NEMO sequencing
Figure 1A.
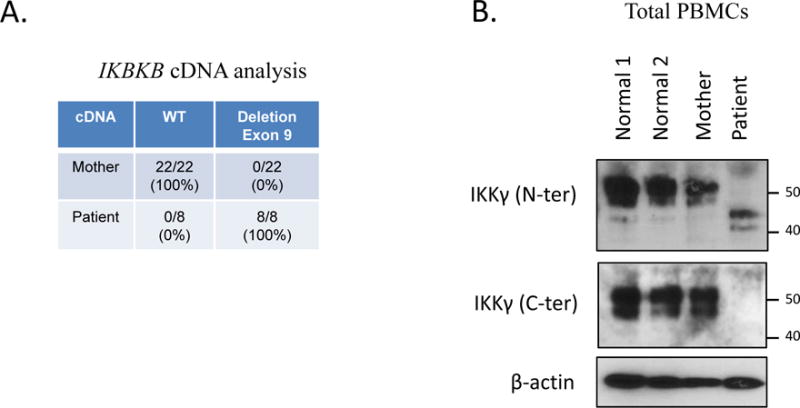
IKBKG/NEMO and IKKg Expression
Figure 2E.
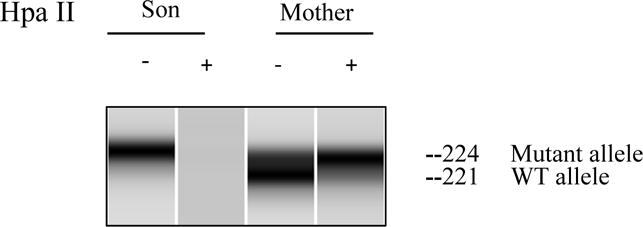
X-chromosome inactivation
While the mother’s PBMCs expressed only wild type protein, western blot data from patient PBMCs showed reduced sized mutant NEMO protein consistent with the cDNA sequencing that predicted exon 9 skipping and frameshift (R352SfsX22) resulting in a premature termination of the C-terminal region (Figure 1A).
To test NF-kappa B function, proliferation to PHA and cytokine production to TLR ligands by PBMCs were measured. The patient’s PBMCs demonstrated diminished proliferation compared with mother and controls, however addition of exogenous IL-2 normalized the patient’s proliferative response (Figure 1B). This finding was consistent with failure of the patient’s T-cells to produce IL-2 in response to CD3/CD28 stimulation (Figure 1B). The patient’s production of TLR-induced IL-6 and TNF-α were also markedly impaired compared with mother and controls (Figure 1B).
Figure 1B.
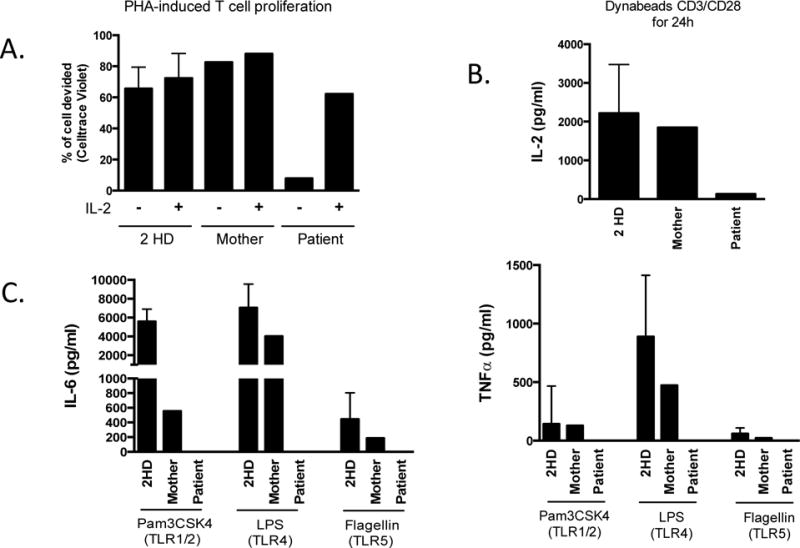
T cell proliferation/IL-2 production and TLR response
This patient’s mutation has not previously been reported and is not present in the LOVD database. Orange et al. reported a patient with an acceptor splice site mutation described to produce IKBKG exon 9 skipping [7]. In contrast to our patient, their patient demonstrated both mutant and WT message and protein, varying at different study time points, suggesting that the splice-site mutation was permissive for residual production of a wild-type transcript.
Our patient’s failure to express wild type IKKγ is consistent with severe immunodeficiency evidenced by overwhelming MSSA sepsis in the first two weeks of life. These findings contrast with the milder clinical presentation of the patient reported by Orange et al. The immunologic findings in their patient suggested a moderate level of immune dysfunction manifesting as reduced specific antibody production, reduced NF-kappa B p65 nuclear translocation, and variable TLR induced TNF production. The relatively low CRP in the face of overwhelming bacterial infection is likely the product of markedly decreased TLR response to multiple agonists with virtual absence of IL-6 production. The clear distinction between these patients both of whom had exon 9 splicing defects is the absence versus presence of wild type IKKγ. This suggests that IKKγ lacking the product of exon 9 and the zinc finger domain may be sufficient for embryogenesis but insufficient for normal ectodermal development and immunity.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH Clinical Center.
References
- 1.Smahi A, et al. Genomic rearrangement in NEMO impairs NF-kappa B activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466–72. doi: 10.1038/35013114. [DOI] [PubMed] [Google Scholar]
- 2.Doffinger R, et al. X-Linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kB signaling. Nature Genetics. 2001;27:277–285. doi: 10.1038/85837. [DOI] [PubMed] [Google Scholar]
- 3.Hanson E, et al. Hypomorphic nuclear factor-k B essential modulator mutation database and reconstitution system identifies phenotypic and immunologic diversity. J Allerg Clin Immunol. 2008;122:1169–1177. doi: 10.1016/j.jaci.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zonana J, et al. A Novel X-Linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO) Am J Hum Genet. 2000;67:1555–1562. doi: 10.1086/316914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortouis G, et al. A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T-cell immunodeficiency. J Clin Invest. 2003;112:1108–1115. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pannicke, et al. Deficiency of innate and acquired immunity caused by an IKBKB Mutation. N Engl J Med. 2013;369:2504–2514. doi: 10.1056/NEJMoa1309199. [DOI] [PubMed] [Google Scholar]
- 7.Orange J, et al. Human Nuclear Factor kB Essential Modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114:650–656. doi: 10.1016/j.jaci.2004.06.052. [DOI] [PubMed] [Google Scholar]