

Abstract
(−)-Ambrox is recognised as the prototype of all ambergris odorants. Widely used in perfumery, (−)-Ambrox is an important ingredient due to its unique scent and excellent fixative function. An environmentally friendly and practical preparation of (−)-Ambrox is still unavailable at present although a lot of attention has been paid to this hot research topic for many years. A one-pot synthesis of (−)-Ambrox was studied starting from (−)-sclareol through oxidation with hydrogen peroxide in the presence of a quaternary ammonium phosphomolybdate catalyst {[C5H5NC16H33] [H2PMo12O40]}, which gave the product a 20% overall yield.
Ambergris is a rare product that is produced in the digestive tract of the sperm whale (Physeter macrocephalus) as a waxy substance, presumably to protect it from injuries caused by the sharp beaks of the giant squid (a major dietary staple)1. Ambergris is an indispensable ingredient in the manufacture of perfumes as a fixative, for providing the long-term scent, aiding in mixing ingredients and extending the shelf life of perfumes2. In the 1930 s Firmenich (the world’s largest privately owned company in the fragrance and flavour business) initiated a vast research program into the composition of ambergris due to its limited supply and relatively high cost3. Stoll and his co-workers identified (−)-Ambrox as the most important constituent of the natural ambergris in the 1940 s4. Since then (−)-Ambrox has been recognised as the prototype of all ambergris odorants and used as a valuable ingredient in perfumery because of its unique scent and fixative function5.
Due to its high price and low availability various synthetic alternatives to ambergris have been developed6. Stoll completed the synthesis of Ambrox for the first time by the reduction of sclareolide with LiAlH4, followed by an acid-catalysed cyclization of the resulting diol to produce (−)-Ambrox7. Several other synthetic routes were reported to prepare Ambrox starting with natural terpenoids8,9. For commercial syntheses, the major starting materials investigated included homofarnesic acid, homofarnesol, monocyclohomofarnesic acid, sclareol and monocyclohomofarnesol10,11,12,13. (−)-Sclareol has been extensively used for preparing (−)-Ambrox due to its reasonably priced commercial availability as extract of Salvia sclarea L. All the chemical routes reported to synthesise Ambrox involve several chemical steps having high processing costs, long reaction times, and severe processing conditions such as high pressure and temperature.
The search for alternative cleaner, safer, and environmentally friendly technologies is a priority in the chemistry field14. Traditional “stop-and-go” synthesis comprises reaction, workup, and purification15. Compared with this traditional approach, a one-pot synthesis is a much more efficient method of achieving several transformations and forming several bonds in one step, while at the same time cutting out several purifications, minimising the generation of waste chemicals and saving time. Thus, an environmentally benign one-pot system should be considered when planning a synthesis16. A large range of compounds has been synthesised by one-pot synthesis including Dibenzofurans17, Spiropyrazolones18, 2,3-Difunctionalized 4-Chlorofurans19, (S)-Baclofen20, ABT-34121 and prostaglandin E1 (PGE1) methyl ester22, since this concept was proposed.
In our preliminary research, (−)-Ambrox was detected by accident in 2% yield during the preparation of sclareolide through oxidation of sclareol with hydrogen peroxide catalysed by Na2WO4·2H2O. It became apparent that (−)-Ambrox could be prepared from sclareol using a one-pot reaction if a suitable catalyst was selected. Therefore, in our present work, one-pot synthesis of (−)-Ambrox was investigated starting from (−)-sclareol through oxidation with hydrogen peroxide in the presence of a quaternary ammonium phosphomolybdate catalyst in order to improve the existing preparation methods mentioned above.
Results and Discussion
The results were particularly significant in early research. But chemists were seeking an environmentally friendly and economical method of synthesising (−)-Ambrox. To date, an array of approaches has emerged for the synthesis of (−)-Ambrox with (−)-sclareol as the starting material. The approaches can be divided into three main categories (Fig. 1).
Figure 1. Approaches to (−)-Ambrox.

In route A (−)-Ambrox was synthesised through three steps23,24,25, including an oxidative degradation of the (−)-sclareol side chain to give sclareolide, a reduction of sclareolide to ambradiol and a cyclodehydration of ambradiol. It is the most efficient way so far. This system is used in industrial production of (−)-Ambrox in spite of several disadvantages, such as expensive or toxic reagents, elaborate operation, long reaction time and massive pollutant discharge.
In route B alkoxy radicals of several derivatives of sclareol underwent β-fragmentation reaction to provide intermediate 1-(2-iodoethyl)-2,5,5,8a- tetramethyldecahydronaphthalen-2-yl acetate, which was then converted to (−)-Ambrox after three step reactions in 20% overall yield26. Stoichiometric amounts of iodobenzene diacetate and iodine were required, which might result in serious pollution and corrosion of equipment.
In route C (−)-sclareol was transformed into (−)-Ambrox through two steps via β-cleavage of an alkoxy radical intermediate in 11–12% overall yield27. The oxidation was carried out using 70% H2O2 as the oxidant in the presence of stoichiometric amounts of Cu(OAc)2·2H2O, FeSO4·7H2O for 7 d. Obviously this process is not practical due to its low efficacy, possible safety risks and large pollutant discharge.
In our preliminary experiment, (−)-sclareol was oxidized by H2O2 in the presence of Na2WO4·2H2O (10 mol %) and NaH2PO4 (0.1 equiv) as the catalyst, and TBAB as phase transfer catalyst in 1,4-dioxane at 70 °C for 2 h to give 2% yield of (−)-Ambrox. It was interesting to learn that if a suitable catalyst was selected, (−)-Ambrox could be prepared by one-pot reaction. The oxidant is hydrogen peroxide, which is a green oxidant. It is probably the best terminal oxidant after dioxygen with respect to environmental and economic considerations28.
In order to improve the yield of (−)-Ambrox, the reaction mixture was heated to 90 °C after stirring for 2 h at 70 °C. The effect of reaction time at 90 °C on yield was explored and the results are shown in Table 1. The yield of (−)-Ambrox reached the highest value of 3.86% after 60 min (Entry 7, Table 1) and decreased gradually with extended reaction times. Obviously, this was a promising but not ideal yield. It was predicted that various phosphotungstate catalysts might be more effective in view of the real catalytic species produced by the reaction of Na2WO4·2H2O with NaH2PO4.
Table 1. Optimization of the Reaction Times.
| Entry | Reaction time | 4 Yield (%) | Entry | Reaction time | 4 yield (%) | |
|---|---|---|---|---|---|---|
| 1 | 0 min | 2.00 | 11 | 2 h | 2.95 | |
| 2 | 10 min | 2.46 | 12 | 3 h | 3.00 | |
| 3 | 20 min | 2.77 | 13 | 4 h | 3.01 | |
| 4 | 30 min | 2.87 | 14 | 5 h | 2.74 | |
| 5 | 40 min | 3.35 | 15 | 6 h | 2.67 | |
| 6 | 50 min | 3.42 | 16 | 7 h | 2.63 | |
| 7 | 60 min | 3.86 | 17 | 8 h | 2.54 | |
| 8 | 70 min | 3.64 | 18 | 9 h | 2.46 | |
| 9 | 80 min | 3.53 | 19 | 10 h | 2.38 | |
| 10 | 90 min | 3.20 | 20 | 11 h | 2.10 |
*Reaction conditions: (−)-Sclareol 1 (10 mmol), Na2WO4·2H2O (0.1 equiv), NaH2PO4 (0.1 equiv), TBAB (0.1 equiv), 1, 4-dioxane (7 mL), H2O2 (5 mL), 70 °C, 2 h; then rising to 90 °C; TBAB (Tetrabutyl ammonium bromide); GC yield.
Reaction time starts counting when the reaction system temperature rises to 90 °C.
Phosphotungstate catalyst is one of the Polyoxometalates (POMs). POMs are discrete metal-oxide clusters of W, Mo, V, and Nb that have been attracting increasing interest because of their multi-electronic redox activities, photochemical properties, acidic properties and magnetic properties, resulting in potential applications of POMs as catalysts and functional materials29. The Ishii-Venturello system has been used on the epoxidation reaction successfully30,31. As Mo and W belong to the same main group, they display similar characteristics. So our main object is to look for effective phosphotungstate and phosphomolybdate catalysts. A series of quaternary ammonium phosphotungstates and phosphomolybdates were prepared30 and listed in Table 2. All these self-made catalysts were tried as a way of catalysing the oxidation of (−)-sclareol with H2O2 in 1,4-dioxane at 70 °C for 2 h, and then at 90 °C for 1 h. The results are given in Table 3. The catalyst 10 p gave the best yield of 18.20% (entry 20, Table 3), which was almost five times as much as that given by Na2WO4·2H2O. The product was isolated and purified easily. The quaternary ammonium phosphomolybdates usually displayed better catalytic ability (entries 16–21, Table 3) than quaternary ammonium phosphotungstates (entries 4–15, Table 3). All the quaternary ammonium phosphotungstates belong to Keggin-type phosphotungstates30. To explore the catalytic capability of different types of phosphotungstates Dowson-type phosphotungstate HPC (H6P2W18O62.H2O) was also prepared32. The yield given by HPC was 5.47%, which was very close to those values produced by Keggin-type phosphotungstates. The results in Table 3 show that the optimum catalyst for one-pot synthesis for (−)-Ambrox is compound 10 p {[C5H5NC16H33] [H2PMo12O40]}.
Table 2. Synthesis of the catalysts.
| Entry | 8 | 9 | 8:9 | 10 | Chemical compositions of catalyst | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | PTA | HTAC | 1:1 | 10a | {[(CH3) 3C16H33N] [H2PW12O40]} | 80 |
| 2 | PTA | HTAC | 1:2 | 10b | {[(CH3) 3C16H33N]2 [HPW12O40]} | 78 |
| 3 | PTA | HTAC | 1:3 | 10c | {[(CH3) 3C16H33N]3 [PW12O40]} | 81 |
| 4 | PTA | CPC | 1:1 | 10d | {[C5H5NC16H33] [H2PW12O40]} | 76 |
| 5 | PTA | CPC | 1:2 | 10e | {[C5H5NC16H33]2 [HPW12O40]} | 80 |
| 6 | PTA | CPC | 1:3 | 10f | {[C5H5NC16H33]3 [PW12O40]} | 75 |
| 7 | PTA | HMAC | 1:1 | 10g | {[(CH3) 4N] [H2PW12O40]} | 88 |
| 8 | PTA | HMAC | 1:2 | 10h | {[(CH3) 4N]2 [HPW12O40]} | 82 |
| 9 | PTA | HMAC | 1:3 | 10i | {[(CH3) 4N]3 [PW12O40]} | 82 |
| 10 | PTA | TBAB | 1:1 | 10j | {[(CH3CH2 CH2 CH2)4N] [H2PW12O40]} | 83 |
| 11 | PTA | TBAB | 1:2 | 10k | {[(CH3CH2 CH2 CH2)4N]2 [HPW12O40]} | 77 |
| 12 | PTA | TBAB | 1:3 | 10l | {[(CH3CH2 CH2 CH2)4N]3 [PW12O40]} | 79 |
| 13 | PMA | HTAC | 1:1 | 10m | {[(CH3) 3C16H33N] [H2P Mo12O40]} | 72 |
| 14 | PMA | HMAC | 1:1 | 10n | {[(CH3) 4N] [H2P Mo12O40]} | 76 |
| 15 | PMA | TBAB | 1:1 | 10o | {[(CH3CH2 CH2 CH2)4N] [H2P Mo12O40]} | 70 |
| 16 | PMA | CPC | 1:1 | 10p | {[C5H5NC16H33] [H2PMo12O40]} | 82 |
| 17 | PMA | CPC | 1:2 | 10q | {[C5H5NC16H33]2 [HPMo12O40]} | 80 |
| 18 | PMA | CPC | 1:3 | 10r | {[C5H5NC16H33]3 [PMo12O40]} | 81 |
*Reaction conditions: 8 (1 mmol), 9 (relative equiv), deionized water (20 mL), 25 °C, 3 h.
PTA (H3P W12O40), PMA (H3P Mo12O40), HTAC (N-Hexadecyltrimethylammonium Chloride), CPC (Cetylpyridinium chloride), HMAC (Tetramethylammonium chloride), TBAB (Tetrabutyl ammonium bromide).
Table 3. Optimisation of the catalysts.
| Entry | Catalyst | 4 yield (%) | Entry | Catalyst | 4 yield (%) | |
|---|---|---|---|---|---|---|
| 1 | PTA | 6.34 | 12 | 10i | 1.79 | |
| 2 | PMA | 7.05 | 13 | 10j | 2.13 | |
| 3 | HPC | 5.47 | 14 | 10k | 1.47 | |
| 4 | 10a | 7.77 | 15 | 10l | 4.54 | |
| 5 | 10b | 4.78 | 16 | 10m | 10.40 | |
| 6 | 10c | 2.03 | 17 | 10n | 12.30 | |
| 7 | 10d | 8.03 | 18 | 10o | 14.11 | |
| 8 | 10e | 5.07 | 19 | 10p | 18.20 | |
| 9 | 10f | 1.98 | 20 | 10q | 16.78 | |
| 10 | 10g | 5.47 | 21 | 10r | 11.79 | |
| 11 | 10h | 6.13 |
*Reaction conditions: 1 (10 mmol), catalyst (0.1 equiv), 1,4-dioxane (7 mL), H2O2 (5 mL), 70 °C, 2 h; then 90 °C, 1h; GC yield; HPC (Dowson-type H6P2W18O62).
The amount of catalyst 10 p for the reaction was optimised (entries 1–10, Table 4). (−)-Ambrox was produced in the highest yield of 22.79% when 3% equiv. catalyst was used (entry 3, Table 4). The yield decreased from 22.79% to 16.58% when the catalyst loading was lowered from 3% to 1%, whereas the yield didn’t increase with the increment of catalyst loading when it exceeded 3%.
Table 4. Optimisation of the Reaction Conditions when catalysed by 10p.
| Entry | Catalyst amount (mmol)* | 4 Yield (%) | Entry | Reaction time** | 4 Yield (%) | |
|---|---|---|---|---|---|---|
| 1 | 0.01 | 16.58 | 11 | 0 | 3.40 | |
| 2 | 0.02 | 19.76 | 12 | 10 min | 6.87 | |
| 3 | 0.03 | 22.79 | 13 | 20 min | 9.91 | |
| 4 | 0.04 | 20.14 | 14 | 30 min | 13.99 | |
| 5 | 0.05 | 20.17 | 15 | 40 min | 15.38 | |
| 6 | 0.10 | 19.65 | 16 | 50 min | 20.78 | |
| 7 | 0.50 | 19.54 | 17 | 60 min | 22.15 | |
| 8 | 1.00 | 18.34 | 18 | 70 min | 19.98 | |
| 9 | 1.50 | 18.20 | 19 | 80 min | 16.20 | |
| 10 | 2.00 | 17.97 | 20 | 90 min | 14.25 |
*Reaction conditions: (−)-Sclareol 1 (10 mmol), catalyst 10 p (relative equiv), 1,4-dioxane (7 mL), H2O2 (5 mL), 70 °C, 2 h; then 90 °C, 1h; GC yield.
**Reaction conditions: 1 (10 mmol), catalyst 10 p (0.03 equiv), 1, 4-dioxane (7 mL), H2O2 (5 mL), 70 °C, 2 h; then 90 °C. Reaction time starts counting when the reaction system temperature rises to 90 °C.
The reaction time was also optimised when the reaction was catalysed by 10 p. The results are shown in Table 4 (entries 11–20). The results indicate that the yield of (−)-Ambrox increased gradually with extended reaction times from 0 min to 60 min at 90 °C (entries 11–17, Table 4), but decreased from 60 min to 90 min (entries 17–20, Table 4). (−)-Ambrox was obtained in the highest yield of 22.17% after 60 min at 90 °C, which was similar to the case catalysed by Na2WO4·2H2O.
The reaction was repeated under the above optimised conditions and (−)-Ambrox was obtained in an average isolated yield of 20%. The determination of its specific rotation was [α] = −30° (C = 1, toluene), which was similar to that in other literature24.
The plausible one-pot synthesis reaction mechanisms were proposed for the current protocol (Fig. 2). (−)-Sclareol was initially epoxidised to compound 11 (Figure S5) or converted to compound 12 (Figure S6) by intramolecular etherification in the presence of the quaternary ammonium phosphomolybdate 10 p. Subsequently, compound 11 or 12 might undergo catalysed substitution with hydrogen peroxide to produce intermediate 13, which fragmented by radical mechanism through radical intermediates 14 and 15 to produce (−)-Ambrox. This one-pot synthesis proceeds through epoxidation30, intramolecular etherification33, free radical substitution27 and free radical fragmentation26.
Figure 2. Proposed one-pot synthesis reaction mechanisms.

A one-pot synthesis of (−)-Ambrox has been accomplished by the use of (−)-sclareol, thus demonstrating the power of catalysis in the synthesis of natural products. This synthesis was made possible by the discovery of a novel route to preparing (−)-Ambrox.
Conclusions
In summary, total syntheses of (−)-Ambrox has been accomplished in 20% total yield by using an inexpensive and simple catalyst and one-pot reactions. This route is not only short and efficient but also has several noteworthy and sustainable features: (1) The total synthesis is performed in only one isolation and chromatographic purification, which reduces the amount of solvent needed and waste formed. (2) The reaction is a highly selective catalytic reaction, involving a quaternary ammonium phosphomolybdates catalyst with 3% catalyst loading and reduces the generation of waste. (3) The oxidant employed in the present synthesis is hydrogen peroxide and water without any pollution is the only theoretical by-product. Thus, the present one-pot synthesis is not only efficient for the synthesis of (−)-Ambrox, but is also environmentally benign. This work is significant in improving the synthesis of (−)-Ambrox based on (−)-sclareol. We are still doing research in our lab in order to further improve the yield of (−)-Ambrox.
Methods
Synthesis of the catalysts
Synthesis of catalysts was illustrated by the synthesis of catalyst 10 p.
PMA (1.82 g, 1 mmol) and deionized water (10 mL) were combined in a 50 mL three-neck flask. The mixture was stirred for 5 min at 25 °C and further CPC (0.36 g, 1 mmol) in deionized water (10 mL) was added after 5 min, then the mixture was stirred for 3 h at 25 °C. When filtered, the filtrate cake was washed with liquid and dried by vacuum to produce H1 (1.76 g, 82%) as a dark green solid.
One-pot synthesis (−)-Ambrox
One-pot synthesis of (−)-Ambrox was illustrated by the reaction of catalysed 10 p.
Sclareol (3.08 g, 10 mmol), 10 p (0.65 g, 0.3 mmol, 3% equiv), 30%H2O2 (5 mL), 1, 4-dioxane (20 mL) were combined in a 50 mL three-neck flask. The mixture was stirred for 2 h at 70 °C and then 1 h at 90 °C. The 1, 4-dioxane was removed under reduced pressure. The residue was extracted with ethyl acetate (20 mL × 3), the organic phases were then washed with a saturated solution of Na2CO3 and brine and then dried over MgSO4. After solvent removal, the residue was purified by flash chromatography on silica gel (petroleum/EtOAc, 30:1) to produce (−)-Ambrox (0.47 g, 20%) as a colourless solid m.p., 74–75 °C, 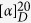 = −30° (C = 1, toluene).
= −30° (C = 1, toluene).
1H NMR (300 MHz, CDCl3) δ: 0.81–0.89 (9H, m, Me-10, 2Me-4), 1.08(3H, s, Me-8), 1.94 (1H, d, J = 11.1 Hz, H-11), 3.82 (1H, q, J = 8.0 Hz, H-12), 3.91 (1H, q, J = 6.8 Hz, H-12) (Figure S2).
13C NMR (75 MHz, CDCl3) δ: 15.0(C-20), 18.4(C-2), 20.6(C-6), 21.1(C-19), 22.6(C-17), 22.6(C-11), 33.0(C-4), 33.6(C-18), 36.1(C-10), 39.7(C-7), 39.9(C-1), 42.4(C-3), 57.2(C-5), 6 0.1(C-9), 64.9(C-12), 79.9(C-8) (Figure S3).
IR (KBr) ν:3751, 2920, 2869, 1457, 1382, 1273, 1200, 1125, 1005, 914, 839, 715, 478, 416 cm−1 (Figure S4).
Additional Information
How to cite this article: Yang, S. et al. One-pot synthesis of (−)-Ambrox. Sci. Rep. 6, 32650; doi: 10.1038/srep32650 (2016).
Supplementary Material
Acknowledgments
This work was supported by General funding project of Beijing Education Commission Research Project (SQKM201610011003), the Importation and Development of High-Calibre Talents Project of Beijing Municipal Institutions (CIT&TCD20140306), the National Key Technology R&D Program (2014BAD04B06).
Footnotes
Author Contributions S.X.Y., H.Y.T. and B.G.S. onceived of the project and designed the experiments. S.X.Y. wrote most of the paper. Y.G.L., Y.F.H. and Y.Y.L.carried out the experiments, analysed the data. All authors discussed the results and commented on the manuscript.
References
- Leffingwell J. & Leffingwell D. Chiral chemistry in flavours & fragrances. Speciality Chemicals Magazine. 30–33 (2011). [Google Scholar]
- Fráter G., Bajgrowicz J. A. & Kraft P. Fragrance chemistry. Tetrahedron 54, 7633–7703 (1998). [Google Scholar]
- Ruzicka L. & Janot M. M. Höhere Terpenverbindungen L. Zur Kenntnis des Sclareols. Helv. Chim. Acta. 14, 645–650 (1931). [Google Scholar]
- Lederer E., Marx F., Mercier D. & Perot G. Sur les constituants de l’ambre gris II. Ambreine et coprostanone. Helv. Chim. Acta. 29, 1354–1365 (1946). [Google Scholar]
- Chauffat C. & Morris A. From ambergris to Cetalox laevo tradition, innovation and creation. Perfum. flavor. 29, 34–41 (2004). [Google Scholar]
- Martínez-Guido S. I. et al. A multiobjective optimization approach for the development of a sustainable supply chain of a new fixative in the perfume industry. ACS Sustain. Chem. Eng. 2, 2380–2390 (2014). [Google Scholar]
- Stoll M. & Hinder M. Odeur et constitution III. Les substances bicyclohomofarnésiques. Helv. Chim. Acta. 33, 1251–1260 (1950). [Google Scholar]
- Suwancharoen S., Pornpakakul S. & Muangsin N. Synthesis of ent-ambrox® from (−)-nidorellol. Tetrahedron Lett. 53, 5418–5421 (2012). [Google Scholar]
- Fehr C., Magpantay I., Saudan L. & Sommer H. trans-Tetrahydrofurans by OH-Assisted Ru-Catalyzed Isomerization of 2-Butene-1, 4-diols. Eur. J. Org. Chem. 32, 6153–6156 (2010). [Google Scholar]
- Rüdinger C., Eberle H. J., Zeitler N., Wollin M. & Wittman G. Consortium für elektrochemische Industrie GmbH. DE 19649426 (1998).
- Snowden R. L. Cetalox® and Analogues: Synthesis via Acid‐Mediated Polyene Cyclizations. Chem. Biodivers. 5, 958–969 (2008). [DOI] [PubMed] [Google Scholar]
- Kawanobe T., Kogami K. & Matsui M. New Syntheses of (±)-Ambrox, (+)-Ambra Oxide and Their Stereoisomers. Agric. Biol. Chem. 50, 1475–1480 (1986). [Google Scholar]
- Snowden R. L. et al. Internal nucleophilic termination in biomimetic acid mediated polyene cyclizations: stereochemical and mechanistic implications. Synthesis of (±)-Ambrox and its diastereoisomers. J. Org. Chem. 57, 955–960 (1992). [Google Scholar]
- Grondal C., Jeanty M. & Enders D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. chem. 2, 167–178 (2010). [DOI] [PubMed] [Google Scholar]
- Hayashi Y. Pot economy and one-pot synthesis. Chem. Sci. 7, 866–880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Climent M. J., Corma A. & Iborra S. Heterogeneous catalysts for the one-pot synthesis of chemicals and fine chemicals. Chem. Rev. 111, 1072–1133 (2010). [DOI] [PubMed] [Google Scholar]
- Zhao H. et al. A One-Pot Synthesis of Dibenzofurans from 6-Diazo-2-cyclohexenones. Org. Lett. 17, 5744–5747 (2015). [DOI] [PubMed] [Google Scholar]
- Hack D. et al. Asymmetric Synthesis of Spiropyrazolones by Sequential Organo‐and Silver Catalysis. Angew. Chem. Int. Ed. 54, 1797–1800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram R. N., Gupta D. K. & Soni V. K. Copper (I)/Ligand-Catalyzed 5-endo Radical Cyclization-Aromatization of 2, 2, 2-Trichloroethyl Vinyl Ethers: Synthesis of 2, 3-Difunctionalized 4-Chlorofurans. J. Org. Chem. 81, 1665–1674 (2016). [DOI] [PubMed] [Google Scholar]
- Hayashi Y., Sakamoto D. & Okamura D. One-Pot Synthesis of (S)-Baclofen via Aldol Condensation of Acetaldehyde with Diphenylprolinol Silyl Ether Mediated Asymmetric Michael Reaction as a Key Step. Org. Lett. 18, 4–7 (2015). [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Honma M. & Hayashi Y. One-Pot High-Yielding Synthesis of the DPP4‐Selective Inhibitor ABT-341 by a Four-Component Coupling Mediated by a Diphenylprolinol Silyl Ether. Angew. Chem. Int. Ed. 50, 2824–2827 (2011). [DOI] [PubMed] [Google Scholar]
- Hayashi Y. & Umemiya S. Pot economy in the synthesis of prostaglandin A1 and E1 methyl esters. Angew. Chem. Int. Ed. 52, 3450–3452 (2013). [DOI] [PubMed] [Google Scholar]
- Moulines J., Lamidey A. M. & Desvergnes-Breuil V. A practical synthesis of Ambrox® from sclareol using no metallic oxidant. Synthetic Commun. 31, 749–758 (2001). [Google Scholar]
- Barrero A. F., Alvarez-Manzaneda E. J., Chahboun R. & Arteaga A. F. Degradation of the Side Chain of (−)-Sclareol: A Very Short Synthesis of nor-Ambreinolide and Ambrox. Synthetic Commun. 34, 3631–3643 (2004). [Google Scholar]
- Chapuis C. Enantioselective Access to (−)-Ambrox® Starting from β-Farnesene. Helv. Chim. Acta. 97, 197–214 (2014). [Google Scholar]
- Christenson P. A. Synthesis of dodecahydro-3a, 6, 6, 9a-tetramethyl naphtho [2, 1-b] furan via alkoxy radical fragmentation. Tetrahedron 44, 1925–1932 (1988). [Google Scholar]
- Decorzant R., Vial C., Näf F. & Whitesides G. M. A short synthesis of ambrox® from sclareol. Tetrahedron 43, 1871–1879 (1987). [Google Scholar]
- Lane B. S. & Burgess K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 103, 2457–2474 (2003). [DOI] [PubMed] [Google Scholar]
- Cronin L. & Müller A. Special thematic issue on polyoxometalates. Chem. Soc. Rev. 41, 7325–7648 (2012). [Google Scholar]
- Zuwei X., Ning Z., Yu S. & Kunlan L. Reaction-controlled phase-transfer catalysis for propylene epoxidation to propylene oxide. Science 292, 1139–1141 (2001). [DOI] [PubMed] [Google Scholar]
- Wang C. & Yamamoto H. Tungsten-Catalyzed Asymmetric Epoxidation of Allylic and Homoallylic Alcohols with Hydrogen Peroxide. J. Am. Chem. Soc. 136, 1222–1225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baronetti G., Briand L., Sedran U. & Thomas H. Heteropolyacid-based catalysis. Dawson acid for MTBE synthesis in gas phase. Appl. Catal. A-Gen. 172, 265–272 (1998). [Google Scholar]
- Singh S. & Patel A. Oxidative Esterification of Aldehydes to Esters over Anchored Phosphotungstates. Catal. Lett. 144, 1557–1567 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.