

ABSTRACT
Anoxygenic phototrophs represent an environmentally important and phylogenetically diverse group of organisms. They harvest light using bacteriochlorophyll-containing reaction centers. Recently, a novel phototrophic bacterium, Gemmatimonas phototrophica, belonging to a rarely studied phylum, Gemmatimonadetes, was isolated from a freshwater lake in the Gobi Desert. To obtain more information about the environmental distribution of phototrophic Gemmatimonadetes, we collected microbial samples from the water column, upper sediment, and deeper anoxic sediment of Lake Taihu, China. MiSeq sequencing of the 16S rRNA, pufM, and bchY genes was carried out to assess the diversity of local phototrophic communities. In addition, we designed new degenerate primers of aerobic cyclase gene acsF, which serves as a convenient marker for both phototrophic Gemmatimonadetes and phototrophic Proteobacteria. Our results showed that most of the phototrophic species in Lake Taihu belong to Alpha- and Betaproteobacteria. Sequences of green sulfur and green nonsulfur bacteria (phototrophic Chlorobi and Chloroflexi, respectively) were found in the sediment. Using the newly designed primers, we identified a diverse community of phototrophic Gemmatimonadetes forming 30 operational taxonomic units. These species represented 10.5 and 17.3% of the acsF reads in the upper semiaerobic sediment and anoxic sediment, whereas their abundance in the water column was <1%.
IMPORTANCE Photosynthesis is one of the most fundamental biological processes on Earth. Recently, the presence of photosynthetic reaction centers has been reported from a rarely studied bacterial phylum, Gemmatimonadetes, but almost nothing is known about the diversity and environmental distribution of these organisms. The newly designed acsF primers were used to identify phototrophic Gemmatimonadetes from planktonic and sediment samples collected in Lake Taihu, China. The Gemmatimonadetes sequences were found mostly in the upper sediments, documenting the preference of Gemmatimonadetes for semiaerobic conditions. Our results also show that the phototrophic Gemmatimonadetes present in Lake Taihu were relatively diverse, encompassing 30 operational taxonomic units.
INTRODUCTION
Photosynthesis is one of the most important biological processes on Earth. The ability of phototrophic organisms to utilize sunlight provided an unlimited source of energy to sustain life on our planet in all its abundance and complexity. The majority of photosynthetic organisms are formed by oxygenic species: plants, algae, and Cyanobacteria. Aside from these dominant organisms, there exists a large variety of anoxygenic phototrophic (AP) bacteria that harvest light using various forms of bacteriochlorophyll. The absorbed light is transferred into bacterial reaction centers, where it is used for primary photosynthetic reactions. AP species have been found in seven bacterial phyla: Proteobacteria (purple anoxygenic phototrophs), Chlorobi (green sulfur bacteria), Chloroflexi (green nonsulfur bacteria), Firmicutes (heliobacteria), Acidobacteria, and Gemmatimonadetes (1, 2).
The latest-found phylum, Gemmatimonadetes, contains only one phototrophic species, i.e., Gemmatimonas phototrophica, which was isolated from a freshwater lake, Tiān É Hú, in the Gobi desert (2). The new organism contains purple photosynthetic reaction centers but does not fix inorganic carbon (3). The environmental distribution of the novel AP lineage is largely unknown. In general, the members of the Gemmatimonadetes phylum have been identified in polar or alpine environments, soils, and sediments (4), but it is not clear whether the phototrophic members of this group share the same distribution pattern. The performed metagenomics search using the aerobic oxidative cyclase gene acsF as a phylogenetic marker indicated that phototrophic Gemmatimonadetes are distributed in various habitats such as fresh waters, sediments, estuarine waters, biofilms, plant surfaces, intertidal sediment, soil, springs, and wastewater treatment plants, whereas no sequences have been found in seawaters and marine sediments (5). Gemmatimonadetes acsF-like sequences were found in the metagenome of the surface scum community that formed during a cyanobacterial bloom in Lake Taihu, China (5), suggesting the possible existence of phototrophic Gemmatimonadetes in this lake.
Lake Taihu is a subtropical shallow (average depth, 2 m) freshwater lake, located in the Yangtze River delta. With its surface area of 2,250 km2, it is the third largest lake in China. Here, we sought to investigate the diversity patterns of phototrophic Gemmatimonadetes and other phototrophic species in more detail. To determine the preferred habitat of phototrophic Gemmatimonadetes, we collected samples not only from the water column but also from the upper and deeper sediments. The bacterial community was characterized by deep sequencing of the 16S rRNA gene amplicon. To specifically target the phototrophic organisms, we used two commonly used markers: the pufM gene encoding the M subunit of the bacterial reaction centers (6) and the bchY gene encoding the chlorophyllide reductase subunit Y (7). The pufM gene has been applied in a number of environmental studies (8). Unfortunately, the commonly used primers usually only target phototrophic Proteobacteria, whereas the pufM sequences of other AP phyla are not amplified. The newly introduced marker bchY was introduced to cover all AP bacteria (7). Since these two markers are not optimal for identifying phototrophic Gemmatimonadetes, we designed new degenerate primers for the acsF gene to specifically target this unique phototrophic group. The advantage of using the acsF gene is that the sequences originating from phototropic Gemmatimonadetes are clearly separated (<60% sequence identity) from those of phototrophic Proteobacteria (2, 5). We applied these primers to investigate the distribution and diversity of phototrophic Gemmatimonadetes in Taihu water column and sediment.
MATERIALS AND METHODS
Lake Taihu sampling.
Water and sediment samples were collected at a calm bay located on the southern shore of Lake Taihu (30.95°N, 120.1°E) on 5 May 2015. Temperature, pH, and dissolved oxygen were measure in situ using a portable pH/oxygen meter (SX825; San-Xin, Shanghai, China). Water samples were collected at an ∼50-cm depth, the average water temperature was 18 ± 0.3°C, the pH was 8.6 ± 0.0, and the dissolved oxygen was 13.6 ± 0.1 mg liter−1. The chlorophyll a concentration was determined in acetonic extracts (9) as 4.8 mg liter−1. Sediments were sampled at a shallow area where the surface sediment was well illuminated. The average temperature, pH, and dissolved oxygen of the water immediately above the sediment sampling site were 24°C, 9.5, and 16 mg liter−1, respectively. Sediment cores were collected using a 50-ml Falcon tube with the bottom removed. The upper 3 mm was collected representing the upper sediment layer. The deep (anoxic) sediment was collected at a depth of ∼10 mm. All samples were collected in triplicates.
Genomic DNA extraction from lake samples.
To extract genomic DNA from planktonic samples, 3 liters of lake water was filtered through a 5-μm-pore size Nuclepore track-etched membrane filter (Whatman, United Kingdom) to remove large particles and debris. The bacterial cells were then collected onto 0.22-μm-pore size membrane filters (Xinya, Shanghai, China). For sediment samples, 0.25 g of the sediment was placed in an Eppendorf tube. Genomic DNA was extracted using a PowerSoil DNA isolation kit (MoBio, USA). Purified DNA was dissolved in sterile H2O and kept at −20°C until further use. All DNA samples were extracted in triplicates.
Design of degenerate primers for acsF gene.
The degenerated primers were designed to specifically amplify the acsF gene from anoxygenic phototrophs but not from Cyanobacteria. More than 100 AcsF amino acid sequences from phototrophic Acidobacteria, Proteobacteria, Gemmatimonadetes, Chloroflexi, and Cyanobacteria were downloaded from NCBI GenBank and aligned using BioEdit software (v7.0.5.2). Two sets of degenerate primers were designed in the conserved areas specific for anoxygenic phototrophs but not present in Cyanobacteria (see Fig. S1 in the supplemental material).
Application of the primer to cultured species.
The newly designed primers were first tested with cultured species. The cultures were grown, and the DNA was isolated as described previously (3, 10). Six strains of phototrophic Proteobacteria (Rhodovulum sulfidophilum, Erythrobacter sp. strain NAP1, Congregibacter litoralis, Hoeflea phototrophica, Rubrivivax gelatinosus, and Roseobacter sp. strain COL2P) and Gemmatimonas phototrophica were used as positive-control samples. Heterotrophic Gemmatimonas aurantiaca and the purple bacterium Rhodospirillum rubrum were used as negative controls. The PCR was carried out in 25 μl of reaction mixture containing 20 ng of genomic DNA, 50 nM concentrations of degenerate primers, 1 U of Taq DNA polymerase (TaKaRa, China), 0.25 mM deoxynucleoside triphosphates (dNTPs), and PCR buffer. The PCR program was as follows: 94°C for 5 min, followed by 35 cycles of 94°C for 30 s, using a gradient from 45 to 60°C with six intervals for 45 s and 72°C for 1 min, followed in turn by a final extension at 72°C for 5 min. To verify the effectiveness of the primers, PCR products were gel purified and cloned into a pMD19-T vector (TaKaRa) according to the manufacturer's instructions, followed by Sanger sequencing.
Amplification of acsF, bchY, and pufM from Taihu field samples.
PCR conditions for the acsF gene from field samples were individually optimized. Generally, 25 μl of the reaction mixture contained 14 to 20 ng of genomic DNA, 4 nM degenerate primers, 1.25 U of hot start Taq DNA polymerase (TaKaRa), 0.25 mM dNTPs, and PCR buffer. The PCR program was as follows: 95°C for 10 min, followed by 35 cycles of 95°C for 30 s, 48 or 50°C for 35 s, and 72°C for 25 s, followed in turn by a final extension at 72°C for 10 min. The PCR conditions for bchY and pufM were set according to those set forth by Yutin et al. (6, 7) with expected amplicon lengths of 500 and 250 bp, respectively. After PCR amplification, the PCR products were visualized on agarose or polyacrylamide gels (11). The PCR was run in triplicate for each DNA extract; hence, there were nine replicates PCR runs for each sample. All nine PCR products were combined to run next-generation sequencing (NGS).
NGS and data analysis.
The acsF, bchY, pufM, and 16S rRNA gene products from Taihu samples were sequenced on the Illumina MiSeq platform (Novogen, China). PCR products were prepared according to the manufacturer's instructions. Paired-end reads (250 bp/300 bp) were generated and assigned to each sample according to the unique barcodes. Sequence analyses were performed using the UPARSE software package with the UPARSE-OTU and UPARSE-OTUref algorithms (12). In-house Perl scripts were used to analyze alpha (within-sample) and beta (among-sample) diversity. For 16S rRNA gene analysis, sequences with ≥97% similarity were assigned to the same operational taxonomic units (OTUs). For functional genes, 95% sequence similarity was used to define an OTU.
Analysis of prokaryotic diversity using 16S rRNA.
Graphical representation of the relative abundance of bacterial diversity from phylum to species was visualized using a Krona chart. Cluster analysis was preceded by principal-component analysis, which was applied to reduce the dimension of the original variables using the QIIME software package (13). QIIME calculates both weighted and unweighted UniFrac distances, which are phylogenetic parameters of beta diversity (14–16). We used unweighted UniFrac distance for principal coordinate analysis (PCoA) and unweighted pair group method with arithmetic mean (UPGMA) clustering. PCoA helps to get principal coordinates and visualize them from complex, multidimensional data. It takes a transformation from a distance matrix to a new set of orthogonal axes, by which the maximum variation factor is demonstrated by first principal coordinate, and the second maximum one by the second principal coordinate, and so on. UPGMA clustering is a type of hierarchical clustering method using average linkage and can be used to interpret the distance matrix.
Phylogenetic analysis of the acsF, bchY, and pufM genes.
Raw sequences of the three functional genes acsF, bchY, and pufM were grouped into OTUs using 95% nucleotide sequence identity threshold. The OTUs were then identified using the BLASTX tool against the nonredundant (nr) protein sequence database at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/protein). Their validity was manually checked. When necessary, sequences were merged to OTUs using mothur software (17). A phylogenetic tree was constructed using the ML algorithm with MEGA 6.1 (18).
Statistical analysis.
Metastats software was used to confirm differences in the abundances of individual taxonomy between the two groups (19). LEfSe was used for the quantitative analysis of biomarkers within different groups. This method was designed to analyze data in which the number of species is much higher than the number of samples and to provide biological class explanations to establish statistical significance, biological consistency, and effect-size estimation of predicted biomarkers. To identify differences of microbial communities between the two groups, ANOSIM (analysis of similarity) and MRPP (multi-response permutation procedure) were performed based on the Bray-Curtis dissimilarity distance matrices (20).
Accession number(s).
The sequences were submitted to NCBI under accession numbers KX365905 to KX368416, KX368417 to KX368519, KX368520 to KX368619, and KX368620 to KX368720.
RESULTS
Bacterial diversity in Lake Taihu probed by 16S rRNA.
There was a clear difference in the microbial composition of the planktonic and sediment samples (Fig. 1A). The bacterioplankton community in Lake Taihu was composed of members of the phyla Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes, with a smaller proportion of Verrucomicrobia and Cyanobacteria. The compositions of the sediment bacteria were more similar to each other than to the planktonic phase, with the majority of the reads composed of Proteobacteria, Cyanobacteria, Actinobacteria, Bacteroidetes, Acidobacteria, Euryarchaeota, Nitrospirae, Chloroflexi, and Verrucomicrobia. Proteobacteria contributed >50% of the diversity in the sediment. Interestingly, about half of the proteobacterial diversity came from Deltaproteobacteria (495 of 1,040 OTUs).
FIG 1.

(A) UPGMA clustering and relative abundance of top 10 phyla in three samples based on 16S rRNA sequences. “Others” refers to all phyla other than the top ten. (B) Clustering and heat map of the 35 most dominant genera based on 16S rRNA sequences. Phylum abbreviations: bac, Bacteroidetes; pro, Proteobacteria (with a-pro, b-pro, g-pro, and d-pro meaning Alpha-, Beta-, Gamma-, and Deltaproteobacteria, respectively); ver, Verrucomicrobia; fir, Firmicutes; aci, Acidobacteria; eur, Euryarchaeota; nit, Nitrospirae; spi, Spirochaetes. Samples: TH11, water; TH12, 3-mm sediment; TH13, 10-mm sediment.
Figure 1B shows a heat map of the 35 most abundant genera from water and sediments and their double clustering in terms of composition. In the water column the dominant bacteria were aerobic bacteria, such as Limnohabitans, Polynucleobacter, Polaromonas, and Flavobacterium. While in the surface sediment, the dominant taxa belong to semiaerobic or facultative aerobic bacteria. In the 10-mm sediment, the majority of bacteria were anaerobic species such as Desulfococcus, Clostridium, or Thiobacillus.
Diversity of anoxygenic phototrophs revealed by bchY and pufM sequences.
To specifically target the diversity of anoxygenic phototrophs, we first used two commonly used genetic markers: bchY and pufM. We performed NGS sequencing of all the collected samples using both primer sets. Sequencing tag numbers are summarized in Table 1. Overall, there were 801 valid bchY OTUs and 811 valid pufM OTUs. The diversity of AP bacteria, as inferred from both bchY and pufM OTUs, was significantly higher in the sediment than in the water column. The planktonic sample contained 195 bchY OTUs and 314 pufM OTUs, whereas the upper sediment contained 608 bchY and 720 pufM OTUs and the deeper sediment contained 698 bchY and 760 pufM OTUs.
TABLE 1.
Tags and OTU numbers of four genes in different samplesa
| Gene | Sample | No. of tags |
OTU |
Gemmatimonadetes |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Taxon | Unclassified | Unique | 95/97% | Valid | OTU (%)b | Tags (%)c | ||
| acsF | TH11 | 30,318 | 28,396 | 1,155 | 767 | 184 | 163 | 11 (6.7) | 204 (0.67) |
| TH12 | 41,278 | 29,863 | 9,384 | 2,031 | 601 | 400 | 26 (6.5) | 4,344 (10.5) | |
| TH13 | 14,846 | 11,058 | 2,307 | 1,481 | 626 | 351 | 28 (8) | 2,571 (17.3) | |
| bchY | TH11 | 36,381 | 33,589 | 5 | 2,787 | 199 | 195 | ||
| TH12 | 36,023 | 32,565 | 464 | 2,994 | 739 | 608 | |||
| TH13 | 42,627 | 35,686 | 1,707 | 5,234 | 1,000 | 698 | |||
| pufM | TH11 | 33,585 | 30,950 | 3 | 2,632 | 316 | 314 | ||
| TH12 | 37,889 | 29,809 | 445 | 7,635 | 784 | 720 | |||
| TH13 | 34,938 | 28,833 | 1,337 | 4,768 | 860 | 760 | |||
| 16S rRNA | TH11 | 26,762 | 25,718 | 0 | 1,044 | 779 | 779 | 12 (1.5) | 99 (0.37) |
| TH12 | 30,529 | 27,671 | 0 | 2,858 | 1,891 | 1,891 | 28 (1.5) | 451 (1.5) | |
| TH13 | 33,457 | 28,649 | 19 | 4,789 | 2,218 | 2,218 | 31 (1.4) | 426 (1.3) | |
Valid OTU refers to the OTU that were annotated as the correct gene. TH11, water; TH12, 3-mm sediment; TH13, 10-mm sediment.
That is, the percentage of Gemmatimonadetes OTUs among the valid OTUs.
That is, the percentage of Gemmatimonadetes tags among the total tags.
Annotation of these bchY OTUs revealed sequences originating from three phototrophic phyla namely Proteobacteria, Chlorobi, and Chloroflexi. All of the Chlorobi and Chloroflexi OTUs were from sediment samples, mostly at a 10-mm depth (Fig. 2A). By the closest matches, it is estimated that around 18 genera of AP bacteria were detected using the bchY primers. Erythrobacter, Rhodobacter, Rhodopseudomonas, Limnohabitans, Sandarakinorhabdus, and Polynucleobacter were the six most abundant phototrophic genera.
FIG 2.

(A) Clustering and heat map of 18 genera of anoxygenic phototrophs detected in Lake Taihu using the bchY gene. (B) Clustering and heat map of 32 genera of anoxygenic phototrophs detected in Lake Taihu using the pufM gene. TH11, water; TH12, 3-mm sediment; TH13, 10-mm sediment. High and low abundances are indicated by range of dark red and dark blue intensities, respectively. The units on the heat map scales were generated by the software HEML.
Meanwhile, pufM sequences originated exclusively from Alpha-, Beta-, and Gammaproteobacteria, which is consistent with the narrow selection of species used for the primer design (6). On the other hand, the pufM gene revealed more AP diversity with 32 identified genera. Sulfitobacter, Loktanella, Limnohabitans, Erythrobacter, Sandarakinorhabdus, and Sphingomonas were identified as the six most abundant genera (Fig. 2B).
Design of degenerate primers for aerobic cyclase gene acsF.
Two forward and two reverse primers were designed (Table 2). Four combinations of these primers were first applied to PCR amplification of phototrophic strain G. phototrophica (positive control) and heterotrophic strain G. aurantiaca (negative control) under various PCR conditions. The primer combinations AcsF F381-396 and AcsF R641-623 produced a PCR product with a correct size 260 bp for G. phototrophica, whereas no product was amplified in the case of G. aurantiaca. This primer combination was also verified with selected phototrophic strains (see Fig. S2 in the supplemental material).
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′) | Source or reference |
|---|---|---|
| AcsF F381-396 | CNGARTWYTCVGGSTG | This study |
| AcsF F479-495 | CGNCAYGCNGGNTTYMT | This study |
| AcsF R624-608 | GCRTAVCCRATYTTYTC | This study |
| AcsF R641-623 | ARATNGTGATRTANCKNGC | This study |
| PufM F | TACGGSAACCTGTWCTAC | 6 |
| PufM R | AYNGCRAACCACCANGCCCA | 6 |
| BchY F | CCNCARACNATGTGYCCNGCNTTYGG | 7 |
| BchY R | GGRTCNRCNGGRAANATYTCNCC | 7 |
The newly designed primers were used for amplification of acsF genes from Taihu DNA samples. PCR products from three environmental samples were further visualized using the polyacrylamide gel electrophoresis (Fig. 3). The results showed that there were two close bands of 230 and 260 bp in the planktonic sample. The efficiency of the acsF primer appeared to be sample specific, since the 260-bp band from planktonic and upper sediment samples were more distinguishable than that from the deep anoxic sediment; the latter showed a smear at 260 bp (Fig. 3). To verify the identity of the obtained product, a small clone library was constructed. Among the five randomly picked clones, three clones carried the 260-bp insertion, and sequences showed confident similarity (>70%) to acsF genes, whereas the other two clones carrying 230 bp were not related to the acsF gene.
FIG 3.
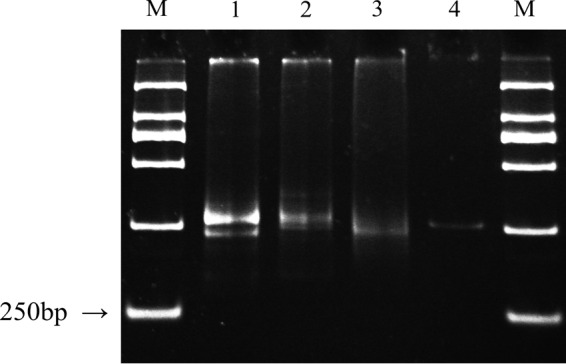
Polyacrylamide gel of acsF gene PCR products from Taihu samples. Lanes: M, DNA ladder; 1, water sample; 2, 3-mm sediment sample; 3, 10-mm sediment sample; 4, G. phototrophica AP64 (positive control).
Diversity of anoxygenic phototrophs detected by acsF degenerate primers.
The 260-bp products were used for NGS. The sequencing results are summarized in Table 1. With 95% similarity, there were 163, 400, and 351 valid OTUs in the planktonic, upper-sediment, and deep-sediment samples, respectively. Overall, there were 452 OTUs, and they were 69 to 100% similar to known acsF gene sequences. More than half of them (240 OTUs) share more than 90% similarity to known sequences. The diversity of AP bacteria containing the acsF gene was the highest in the upper sediment compared to the planktonic or the deep-sediment sample.
Annotation of these 452 OTUs revealed that 285 of them were similar to acsF genes from Alphaproteobacteria, 109 were similar to acsF genes from Betaproteobacteria, and 28 were similar to acsF genes from Gammaproteobacteria. Thirty OTUs were found to be similar to the sequence of G. phototrophica, which indicated that they represented phototrophic Gemmatimonadetes (similarity range, 84 to 100%). By the closest matches, it was estimated that 31 genera of anoxygenic phototrophs were detected in Lake Taihu by the newly designed acsF primers. No cyanobacterial acsF sequences were amplified, which documents the good specificity of our primers in excluding Cyanobacteria.
A heat map of the 31 genera was plotted for all three collected samples (Fig. 4). The 10 most abundant genera were Nevskia, Rubrivivax, Rhodobacter, Gemmatimonas, Sandarakinorhabdus, Limnohabitans, Porphyrobacter, Erythrobacter, Methylobacterium, and Afifella. Nevskia, Rhodobacter, and Limnohabitans were the dominant genera in the planktonic sample, whereas Rubrivivax, Porphyrobacter, Sandarakinorhabdus, and Gemmatimonas were dominant in the upper sediment, and Nevskia, Gemmatimonas, and Rhodobacter were dominant in the deeper sediment.
FIG 4.
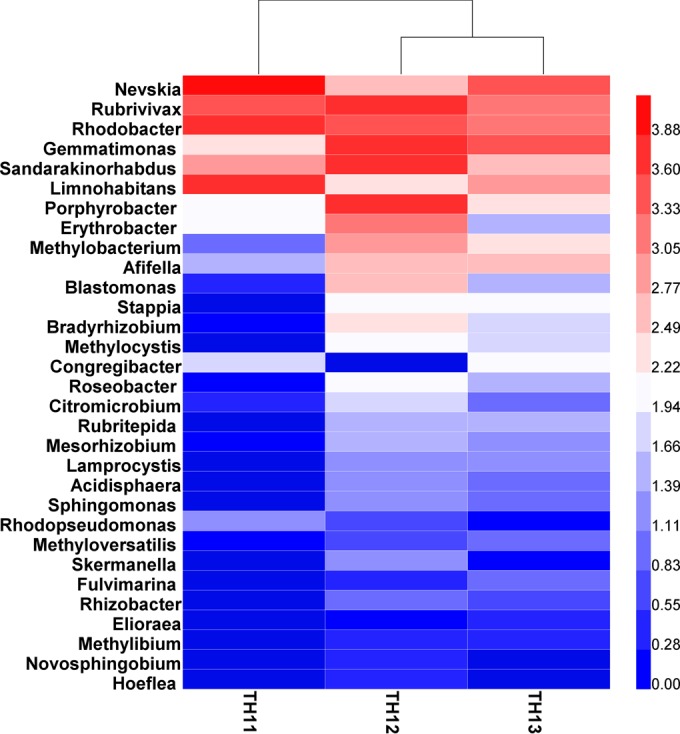
Clustering and heat map of 31 genera of anoxygenic phototrophs detected in Lake Taihu by the newly designed acsF primers. TH11, water; TH12, 3-mm sediment; TH13, 10-mm sediment.
Distribution of Gemmatimonadetes in Lake Taihu.
To better understand the environmental distribution of Gemmatimonadetes species, we compared the results of 16S rRNA gene of NGS, which documented all Gemmatimonadetes species, to the acsF gene, which specifically identified the phototrophic Gemmatimonadetes (Table 1).
Using the 16S rRNA gene, there were 32 OTUs assigned to the phylum Gemmatimonadetes. Among these, 12 OTUs were found in the water column, whereas 28 and 31 OTUs were found in the upper and deeper sediments. All OTUs from the water column could be found in sediments, whereas most OTUs from sediments were absent from the water column.
In terms of abundance, the total reads of Gemmatimonadetes were 99, 451, and 426 in the water column, upper sediments, and deeper sediments, respectively. Gemmatimonadetes-related sequences made up 1.3 to 1.5% of the 16S rRNA tags in the sediments, while in the water column they only represented 0.37% of the tags (Fig. 5). In terms of diversity (by OTUs), the Gemmatimonadetes represented ∼1.5% of the identified OTUs in both the planktonic and the sediment samples.
FIG 5.
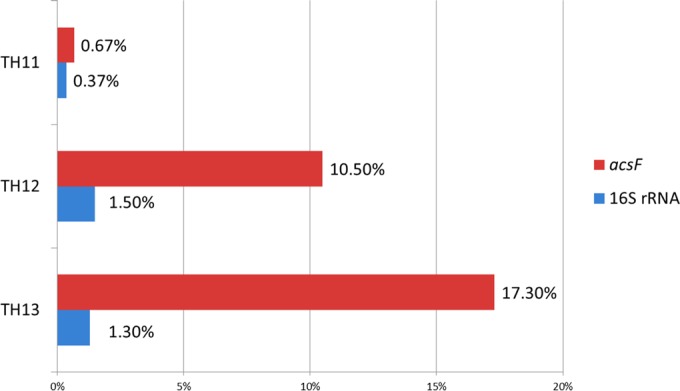
Abundance of Gemmatimonadetes in three samples detected by acsF and 16S rRNA gene, respectively. TH11, water; TH12, 3-mm sediment; TH13, 10-mm sediment.
A similar distribution was observed for phototrophic Gemmatimonadetes based on acsF gene sequences. Overall, there were 30 OTUs assigned to the phylum Gemmatimonadetes, which indicates a high diversity of these organisms (Fig. 6). Among these, 11 OTUs were found in the water column, whereas 26 and 28 OTUs were found in upper and deeper sediments, respectively. All OTUs from the water column could be found in sediments, whereas most OTUs from sediments were absent in the water column. Regarding the diversity, phototrophic Gemmatimonadetes represented from 6.5 to 8% of all the identified OTUs. In terms of abundance, Gemmatimonadetes made up 0.67% of the acsF tags in the planktonic phase, whereas in the upper and deeper sediments they formed 10 and 17% of the acsF tags, respectively (Fig. 5).
FIG 6.
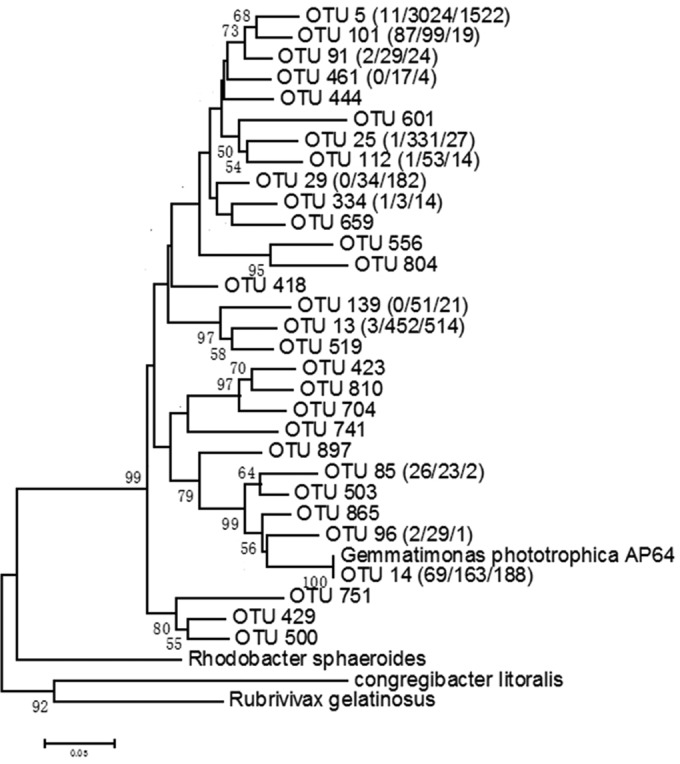
Phylogenetic tree of 30 OTUs belonging to the phylum Gemmatimonadetes and the reference acsF genes from G. phototrophica AP64 and others. The phylogenetic tree was constructed using the ML algorithm with MEGA 6.1. Bootstrap values are based on 1,000 replicates, and only values of >50% are shown. The numbers in parentheses after the OTUs are the OTU reads in water column, upper- and deep-sediment samples, separated by slashes (“/”). OTUs without a number in parentheses means the overall reads for this OTU were <10.
DISCUSSION
The recent introduction of NGS techniques represented a major breakthrough in the study of natural microbial communities. NGS offers both an economical and highly effective tool to study microbial diversity. Most often, 16S rRNA is used as a general marker to assess the microbial diversity. However, the relationship between the bacterial taxonomy and environmental function is not straightforward. Therefore, functional genes could offer more in-depth insights into ecological roles of specific functional groups in the natural environments.
Our 16S rRNA analysis of microbial community in Lake Taihu revealed that the water column sample contained representatives of Proteobacteria, Actinobacteria, Bacteriodetes, and Firmicutes. This result is very similar to the recent pyrosequencing analysis of Lake Taihu which identified Proteobacteria, Actinobacteria, and Bacteroidetes as the dominant groups (21). Also, the composition of the sediment samples was consistent with the previous investigation (22). The main genera in the sediments are clearly different from those found in the water column, which documents that our samples are representative for the selected environments and that they are free of any potential cross-contamination.
To analyze AP diversity, we applied the two commonly used markers: bchY and pufM genes. Unfortunately, these markers did not clearly distinguish phototrophic Gemmatimonadetes, probably because of their high similarity to the sequences of phototrophic Proteobacteria. To avoid this ambiguity, we recently introduced a novel marker, the acsF gene, which in our previous studies proved to be a reliable phylogenetic marker for a large range of anoxygenic bacteria (10). The novel primer successfully amplified acsF genes from Lake Taihu samples and showed high efficiency and specificity. In our experiments, the ratios of valid OTUs to total OTUs of the three gene markers in the water column were always higher than those in sediments; this is probably due to higher purity of gDNA from water samples and higher fidelity in PCR amplification.
In comparison, the diversity of AP probed by acsF, bchY, and pufM was varied. It is estimated that 18, 32, and 31 genera of AP bacteria were detected in Lake Taihu by bchY, pufM, and acsF primers, respectively. bchY sequences originated from three phototrophic phyla: Proteobacteria, Chlorobi, and Chloroflexi. pufM sequences originated exclusively from Proteobacteria, and acsF sequences originated from Proteobacteria and Gemmatimonadetes. Despite the different affinity of these three gene markers to the AP community, they all detected some common dominant genera, such as Sandarakinorhabdus, Limnohabitans, Rhodobacter, and Erythrobacter (Fig. 2A, 2B, and 4). Members of the genera Limnohabitans and Rhodobacter were already identified earlier as dominant AP species in the Delaware River (23) and in German freshwater lakes (24). Interestingly, the genus Rhodobacter, which was recognized by 16S rRNA, bchY, and acsF sequences, was not identified in the pufM amplicon. The likely reason is that the automatic affiliation routine incorrectly classified these sequences as Sulfitobacter and Loktanella, which are marine species with pufM sequences closely related to those of freshwater Rhodobacter spp. (25). This problem illustrates another weakness of using the pufM gene as a marker, which is its complex phylogeny, which frequently hampers determining a clear phylogenetic affiliation (25, 26). Another interesting finding is the presence of Nevskia-related sequences in the acsF amplicon, which was not noted in pufM or bchY amplicons. The gammaproteobacterium Nevskiaramosa is a common member of the freshwater epineuston (27). The presented data suggest that members of this genus may form a significant part of the limnic AP community.
Another interesting finding is a striking difference in the distribution of main prokaryotic genera and the distribution of the main AP groups. Although the composition of total prokaryotes was fundamentally different between water column and sediment samples (see Fig. 1B), many AP genera (Rhodobacter, Rubrivivax, Sandarakinorhabdus, and Limnohabitans) were found in both limnic and sediment samples without any strict preference (Fig. 2A, 2B, and 4). It is noteworthy that species belonging to the Erythrobacter-Erythromicrobium cluster, which encompasses typical aerobic anoxygenic phototrophs, had more reads in the sediment than in the limnic phase. This lack of clear partitioning indicates that the dominant phototrophic Proteobacteria are very flexible in terms of substrate, oxygen, and light preferences.
The main advantage of the acsF gene is its capacity to discriminate the presence of phototrophic Gemmatimonadetes. The advantage of our approach is that our focused sampling allowed for better characterization of the distribution of phototrophic Gemmatimonadetes in the environment than in previous investigations of unrelated metagenome studies. The phototrophic Gemmatimonadetes represented 17.3 and 10.5% of the reads in the deep- and shallow-sediment samples, respectively. In the water column they represented only 0.67% of the acsF reads. This finding corresponds relatively well to the recent metagenomics study by Zeng et al. (5), which documented that phototrophic Gemmatimonadetes made up 0 to 11.9% of the acsF-containing prokaryotic phototrophs in various environments. The high abundance of phototrophic Gemmatimonadetes in sediment was consistent with its microaerophilic character (3). Furthermore, our data documented high diversity among phototrophic Gemmatimonadetes, which may indicate their longer evolution and greater speciation in the studied environment.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported financially by the National Natural Science Foundation of China (21137003) and the 973 Program (2014CB441103). This research was also supported by Czech Science Foundation project 13-11281S and project Algatech Plus (LO1416).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01063-16.
REFERENCES
- 1.Bryant DA, Costas AMG, Maresca JA, Chew AGM, Klatt CG, Bateson MM, Ward DM. 2012. Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic acidobacterium. Science 317:523–526. doi: 10.1126/science.1143236. [DOI] [PubMed] [Google Scholar]
- 2.Zeng Y, Feng F, Medová H, Dean J, Koblížek M. 2014. Functional type 2 photosynthetic reaction centers found in the rare bacterial phylum Gemmatimonadetes. Proc Natl Acad Sci U S A 111:7795–7800. doi: 10.1073/pnas.1400295111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeng Y, Selyanin V, Lukeš M, Dean J, Kaftan D, Feng F, Koblížek M. 2015. Characterization of the microaerophilic, bacteriochlorophyll a-containing bacterium Gemmatimonas phototrophica sp. nov., and emended descriptions of the genus Gemmatimonas and Gemmatimonas aurantiaca. Int J Syst Evol Microbiol 65:2410–2419. doi: 10.1099/ijs.0.000272. [DOI] [PubMed] [Google Scholar]
- 4.DeBruyn JM, Nixon LT, Fawaz MN, Johnson AM, Radosevich M. 2011. Global biogeography and quantitative seasonal dynamics of Gemmatimonadetes in soil. Appl Environ Microbiol 77:6295–6300. doi: 10.1128/AEM.05005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng Y, Baumbach J, Barbosa EG, Azevedo V, Zhang C, Koblížek M. 2015. Metagenomic evidence for the presence of phototrophic Gemmatimonadetes bacteria in diverse environments. Environ Microbiol Rep doi: 10.1111/1758-2229.12363. [DOI] [PubMed] [Google Scholar]
- 6.Yutin N, Suzuki MT, Béjà O. 2005. Novel primers reveal wider diversity among marine aerobic anoxygenic phototrophs. Appl Environ Microbiol 71:8958–8962. doi: 10.1128/AEM.71.12.8958-8962.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yutin N, Suzuki MT, Rosenberg M, Rotem D, Madigan MT, Süling J, Imhoff JF, Béjà O. 2009. BchY-based degenerate primers target all types of anoxygenic photosynthetic bacteria in a single PCR. Appl Environ Microbiol 75:7556–7559. doi: 10.1128/AEM.01014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koblížek M. 2015. Ecology of aerobic anoxygenic phototrophsin aquatic environments. FEMS Microbiol Rev 39:854–870. doi: 10.1093/femsre/fuv032. [DOI] [PubMed] [Google Scholar]
- 9.Čuperová Z, Holzer E, Salka I, Sommaruga R, Koblížek M. 2013. Temporal changes and altitudinal distribution of aerobic anoxygenic phototrophs in mountain lakes. Appl Environ Microbiol 79:6439–6446. doi: 10.1128/AEM.01526-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boldareva-Nuianzina EN, Bláhová Z, Sobotka R, Koblížek M. 2013. Distribution and origin of oxygen-dependent and oxygen-independent forms of Mg-protoporphyrin monomethylester cyclase among phototrophic proteobacteria. Appl Environ Microbiol 79:2596–2604. doi: 10.1128/AEM.00104-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 12.Edgar RC. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 13.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lozupone CA, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lozupone CA, Lladser ME, Knights D, Stombaugh J, Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J 5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lozupone CA, Hamady M, Kelley ST, Knight R. 2007. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulson JN, Pop M, Bravo HC. 2011. Metastats: an improved statistical method for analysis of metagenomic data. Genome Biol 12:17. doi: 10.1186/gb-2011-12-s1-p17. [DOI] [Google Scholar]
- 20.Clarke KR. 1993. Nonparametric multivariate analysis of changes in community structure. Aust J Ecol 18:117–143. [Google Scholar]
- 21.Tang X, Li L, Shao K, Wang B, Cai X, Zhang L, Chao J, Gao G. 2015. Pyrosequencing analysis of free-living and attached bacterial communities in Meiliang Bay, Lake Taihu, a large eutrophic shallow lake in China. Can J Microbiol 61:22–31. doi: 10.1139/cjm-2014-0503. [DOI] [PubMed] [Google Scholar]
- 22.Chen N, Yang JS, Qu JH, Li HF, Liu WJ, Li BZ, Wang ET, Yuan HL. 2015. Sediment prokaryote communities in different sites of eutrophic Lake Taihu and their interactions with environmental factors. World J Microbiol Biotechnol 31:883–896. doi: 10.1007/s11274-015-1842-1. [DOI] [PubMed] [Google Scholar]
- 23.Waidner LA, Kirchman DL. 2005. Aerobic anoxygenic photosynthesis genes and operons in uncultured bacteria in the Delaware River. Environ Microbiol 7:1896–1908. doi: 10.1111/j.1462-2920.2005.00883.x. [DOI] [PubMed] [Google Scholar]
- 24.Salka I, Čuperová Z, Mašín M, Koblížek M. 2011. Rhodoferax-related pufM gene cluster dominates the aerobic anoxygenic phototrophic communities in German freshwater lakes. Environ Microbiol 13:2865–2875. doi: 10.1111/j.1462-2920.2011.02562.x. [DOI] [PubMed] [Google Scholar]
- 25.Koblížek M, Moulisová V, Muroňová M, Oborník M. 2015. Horizontal transfers of two types of puf operons among phototrophic members of the Roseobacter clade. Folia Microbiol 60:37–43. doi: 10.1007/s12223-014-0337-z. [DOI] [PubMed] [Google Scholar]
- 26.Zeng YH, Shen W, Jiao NZ. 2009. Genetic diversity of aerobic anoxygenic photosynthetic bacteria in open ocean surface waters and upper twilight zones. Mar Biol 156:425–437. doi: 10.1007/s00227-008-1095-8. [DOI] [Google Scholar]
- 27.Cypionka H, Babenzien H-D, Glöckner FO, Amann R. 2006. The genus Nevskia, p 1152–1155. In Falkow S, Dworkin M (ed), The prokaryotes, 3rd ed, vol 6 Proteobacteria: gamma subclass. Springer, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.