

Abstract
Introduction
Cytosine residues in CpG dinucleotides often undergo various types of modification, such as methylation, deamination, and halogenation. These types of modifications can be pro-mutagenic and can contribute to the formation of mutational hotspots in cells. To analyze mutations induced by DNA modifications in the human genome, we recently developed a system for tracing DNA adducts in targeted mutagenesis (TATAM). In this system, a modified/damaged base is site-specifically introduced into intron 4 of thymidine kinase genes in human lymphoblastoid cells. To further the understanding of the mutagenesis of cytosine modification, we directly introduced different types of altered cytosine residues into the genome and investigated their genomic consequences using the TATAM system.
Findings
In the genome, the pairing of thymine and 5-bromouracil with guanine, resulting from the deamination of 5-methylcytosine and 5-bromocytosine, respectively, was highly pro-mutagenic compared with the pairing of uracil with guanine, resulting from the deamination of cytosine residues.
Conclusions
The deamination of 5-methylcytosine and 5-bromocytosine rather than that of normal cytosine dramatically enhances the mutagenic potential in the human genome.
Keywords: Mutagenesis, Gene targeting, Deamination, Mutagenic potential
Introduction
CpG dinucleotides in the genome are subjected to various types of modification including cytosine methylation. The methylation of cytosine to 5-methylcytosine (5-mC) is a common DNA modification and is important for the epigenetic mechanism of gene regulation in higher eukaryotes. In mammalian cells, 3–6 % of cytosine residues and 70–80 % of cytosine residues in CpG dinucleotides are methylated [1–3]. Such cytosine residues often undergo inappropriate modifications (Fig. 1a), leading to genomic instability.
Fig. 1.
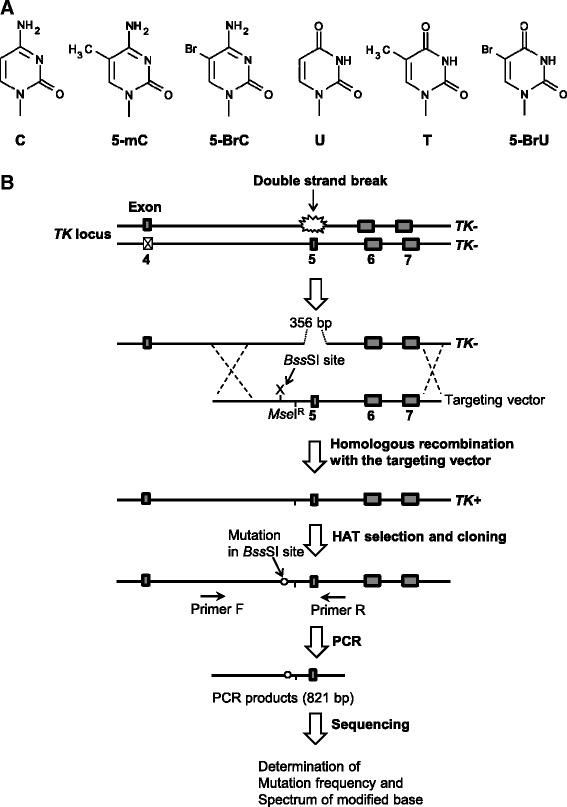
Overview of the TATAM system. Structures of cytosine alteration (a) and the principle of the TATAM system (b). X on the targeting vector indicates the position of cytosine, 5-mC, 5-BrC, U, 5-BrU, or thymine at the BssSI site. The targeting vectors pvINTC:G, pvINT5mC:G, pvINT5BrC:G, pvINTU:G, pvINT5BrU:G, or pvINTT:G and the I-SceI expression plasmid pCBASce were co-transfected into TSCER122 cells. Double-strand break at the I-SceI site enabled gene targeting by inducing site-specific homologous recombination. The targeting vector contained an MseIR site that was resistant to MseI digestion and thereby distinguished targeted and non-targeted revertants of TK. TK revertants were selected by using HAT. Genomic DNA of the revertant colonies was prepared, and part of the TK gene containing the modified DNA integrated site was amplified by PCR. The amplified fragment was sequenced as described in the Materials and Methods section
Cytosine and 5-mC in the genome are often spontaneously deaminated to form U:G and T:G mismatches, respectively [4]. These mismatches are also produced by enzymatic deamination caused by activation-induced deaminase or apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3A (APOBEC3A) [5–7]. The resultant uracil and thymine can pair with adenine during DNA replication, causing C:G to T:A transition mutations. In fact, cytosine residues at CpG dinucleotides in the tumor suppressor gene TP53 is known as a mutational hotspot in carcinoma cells [8]. It has been suggested that in DNA, the hydrolytic deamination of 5-mC occurs more rapidly than that of cytosine [9, 10].
Cytosine modification also occurs during chronic inflammation. At inflammation sites, phagocytic cells generate peroxidases that produce reactive oxidants such as hypobromous acid and hypochlorous acid [11–13]. These oxidants can result in several types of halogenated DNA damages, leading to mutagenesis [14–20]. Among them, the halogenation of cytosine is detrimental to organisms. For example, 5-bromocytosine (5-BrC) and 5-chlorocytosine (5-ClC) in DNA can potentially compromise epigenetic signals by mimicking 5-mC [21]. Moreover, 5-BrC is converted to 5-bromouracil (5-BrU) by APOBEC3A [22], which may result in enhanced mutagenesis in the genome.
The mutagenesis of modified/deaminated cytosine residues has been extensively studied in Escherichia coli and in plasmids introduced into mammalian cells [4, 10, 23–27]. However, the mutagenic consequences of such alterations in the human genome are yet to be completely understood. We recently developed a system for tracing DNA adducts in targeted mutagenesis (TATAM) by directly introducing a DNA modification site specifically into intron 4 of the thymidine kinase (TK) gene in human lymphoblastoid cells (Fig. 1b) [28]. In this study, for better understanding the mutagenesis of cytosine modification in vivo, we introduced cytosine, 5-mC, and 5-BrC paired with guanine and their deamination products U:G, T:G, and 5-BrU:G mismatch at CpG dinucleotides in the genome using the TATAM system.
Materials and methods
Cell culture
The human lymphoblastoid TSCER122 cells used were derivatives of TK6 cells, as reported previously [28]. Cells were maintained in RPMI 1640 medium (Nacalai Tesque) supplemented with 10 % horse serum (JRH Biosciences), 200 μg/ml of sodium pyruvate, 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37 °C under 5 % CO2 and 100 % humidity.
Outline of the TATAM system
TSCER122 cells are compound heterozygous for the TK gene (TK −/−) because of the complete loss of exon 5 in one allele and a point mutation in exon 4 in the other (Fig. 1b). Because there is an I-SceI recognition site in the original exon 5 region, the expression of the I-SceI enzyme in TSCER122 cells generated a double-strand break in the TK gene, allowing for the generation of the wild type TK (TK +/−) by homologous recombination with the targeting vector. TSCER122 cells were co-transfected with the I-SceI expression plasmid and the targeting vector site-specifically containing a synthetic DNA adduct. After 3 days incubation, cells were seeded 96-well plates in the presence of hypoxanthine, aminopterin, and thymidine (HAT) to isolate the DNA adduct-integrated revertant clones. Subsequently, the TK gene loci of the revertant clones were sequenced (Fig. 1b).
Preparation of site-specific modified targeting vector
The targeting vectors pvINTC:G, pvINT5mC:G, pvINT5BrC:G, pvINTU:G, pvINT5BrU:G, and pvINTT:G containing C:G, 5-mC:G, 5-BrC:G, U:G, 5-BrU:G, and T:G base pairs, respectively, in place of the underlined cytosine/guanine at the BssSI site (5′–CTCGTG/5′–CACGAG) were prepared by a polymerase chain reaction (PCR)-based method with the plasmid pTK15, as previously described (Fig. 2) [28, 29]. A 5′–TTCA sequence (MseIR) was labeled near the modified BssSI site. This modified site was resistant to MseI digestion and thus distinguished targeted and non-targeted revertants of TK according to an interallelic recombination (Fig. 1b). The vectors were sequenced to confirm the presence of the modified cytosine at the expected site.
Fig. 2.

Details of the site of modification. The position of a modification is indicated by X in the primer sequence. An unmodified cytosine, 5-mC, 5-BrC, U, 5-BrU, or thymine paired with guanine was inserted at the BssSI site. The MseIR site was placed near the BssSI site
Transfection and cloning of TK revertant cells
DNA transfection was performed as previously described [28]. Briefly, the targeting vector (1 μg) and I-SceI expression plasmid pCBASce (50 μg) were co-transfected into 5 × 106 cells that were suspended in 0.1 ml of Nucleofector Solution V (Lonza) using Nucleofector I, in accordance with the manufacturer’s instructions. After incubation for 72 h, cells were seeded into 96-microwell plates in the presence of HAT (200 μM hypoxanthine, 0.1 μM aminopterin, and 17.5 μM thymidine) for isolating targeting vector-integrated revertant clones. After incubation for 2 weeks, drug-resistant colonies (TK revertants) were analyzed.
Mutation analysis
Genomic DNA templates for PCR were prepared from TK-revertant colonies using alkaline lysis, as previously described [30]. Briefly, cells were treated with 18 μl of 50 mM NaOH at 95 °C for 10 min and neutralized by adding 2 μl of 1 M Tris-HCl (pH 8.0). The cell lysates were then used as templates for PCR to amplify the TK gene fragments containing the modified cytosine integration site. PCR was performed using KOD FX (Toyobo) with the following primers: forward primer 5′–GCT CTT ACG GAA AAG GAA ACA GG–3′ and reverse primer 5′–CTG ATT CAC AAG CAC TGA AG–3′. The resulting DNA fragments were sequenced using an ABI 3730×l DNA analyzer (Applied Biosystems), and clones harboring the MseIR sequence were counted for determining the frequency of modified cytosine integration and numbers of mutations at the BssSI site. The integration frequency of the modified cytosine was calculated by dividing the number of MseIR clones by the total number of revertant clones analyzed. A single point mutation was defined as a single base substitution, insertion, or deletion detected at the modified cytosine. Multiple mutations were multiple base substitutions, deletions, and/or insertions that were detected at sites including the modified cytosine. Base substitutions, deletions, and/or insertions found at sites other than the modified cytosine were defined as non-targeted. Mutant proportions were calculated by dividing the number of mutants by the number of MseIR-bearing clones.
Statistical analysis
Statistical significance was evaluated by Fisher’s exact test. P-values less than 0.01 were considered to be statistically significant.
Results and discussion
To investigate the mutagenic potential of cytosine alterations in the genome, targeting vectors pvINTC:G, pvINT5mC:G, pvINT5BrC:G, pvINTU:G, pvINT5BrU:G, and pvINTT:G were prepared, containing C:G, 5-mC:G, 5-BrC:G, U:G, 5-BrU:G, and T:G base pairs, respectively, as previously reported [28]. The revertant frequencies were comparable between the targeting vectors used (data not shown), indicating that the modified residues on the targeting vector did not influence the efficiency of homologous recombination.
Mutagenic potential of 5-methylcytosine and 5-bromocytosine in the genome
As shown in Table 1, the total proportion of mutants induced by the integration of pvINTC:G, the control vector, was 1.5 %; no C:G to T:A transition mutations were observed (Fig. 3). When pvINT5mC:G was integrated, the proportion of mutants (1.4 %) was comparable to that of pvINTC:G. Some C:G to T:A transition mutations (0.44 %) were detected, followed by one base deletion (0.20 %), one base insertion (0.20 %), and non-targeted mutations, referred to as “others” (0.59 %), indicating that 5-mC itself enhances C:G to T:A transition mutations via its deamination, but the frequency is below that of background mutations in this system. This is in agreement with the finding that the frequency of mutations induced by 5-mC ranges from 10−3 to 10−7 in E. coli with different genetic backgrounds [10, 22, 31].
Table 1.
Mutation spectra induced by integration of the targeting vectors
| Targeting vector | TK revertants analyzed | X:G-integrated revertantsa | No mutation | Single point mutationb | Multiplee | Othersf | Total mutation | NDg | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T | G | A | Delc | Insd | ||||||||
| pvINTC:G | 457 | 410 (100 %) | 403 (98 %) | 0 | 0 | 0 | 2 (0.49 %) | 1 (0.24 %) | 0 | 3 (0.73 %) | 6 (1.5 %) | 1 |
| pvINT5mC:G | 722 | 676 (100 %) | 667 (99 %) | 3 (0.44 %) | 0 | 0 | 1 (0.20 %) | 1 (0.20 %) | 0 | 4 (0.59 %) | 9 (1.4 %) | 0 |
| pvINT5BrC:G | 778 | 705 (100 %) | 700 (99 %) | 0 | 2 (0.28 %) | 0 | 0 | 0 | 0 | 3 (0.43 %) | 5 (0.71 %) | 3 |
| pvINTU:G | 369 | 335 (100 %) | 309 (92 %) | 16 (4.8 %) | 2 (0.60 %) | 0 | 1 (0.30 %) | 2 (0.60 %) | 0 | 3 (0.90 %) | 34 (8.1 %) | 2 |
| pvINTT:G | 187 | 176 (100 %) | 77 (44 %) | 98 (56 %)h | 0 | 0 | 0 | 0 | 0 | 0 | 98 (56 %) | 1 |
| pvINT5BrU:G | 619 | 524 (100 %) | 349 (67 %) | 167 (32 %)h | 4 (0.76 %) | 0 | 1 (0.19 %) | 0 | 2 (0.38 %) | 1 (0.19 %) | 175 (33 %) | 2 |
aX:G indicates C:G, 5-mC:G, 5-BrC:G, U:G, T:G, 5-BrU:G mispair
bA single base substitution, one-base insertion, or one-base deletion detected at the modified base
cOne-base deletion
dOne-base insertion
eMultiple base substitutions, deletions, and/or insertions detected at sites including the modified base in the BssSI site
fMutations found at sites other than the modified base
gNot detectable
h P < 0.01 (significant difference versus pvINTU:G)
Fig. 3.
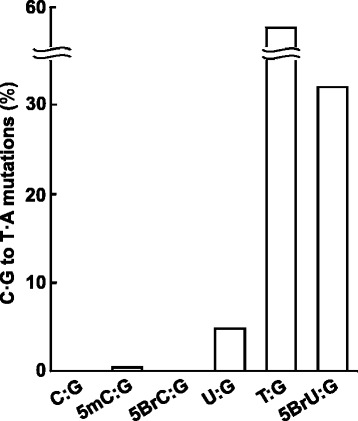
Proportions of C:G to T:A transition mutations induced by the integration of the targeting vector. Proportions of C:G to T:A transition mutations induced by the integration of pvINTC:G, pvINT5mC:G, pvINT5BrC:G, pvINTU:G, pvINT5BrU:G, and pvINTT:G in TSCER122 cells. Data are derived from at least two independent transfections. The results are also tabulated in Table 1
Regarding halogenated cytosine, it has been suggested that 5-ClC causes C:G to T:A transition mutations at rates ranging from 5 to 9 % by mispairing with adenine in E. coli [32]. Based on our results, however, 5-BrC did not induce C:G to T:A transition mutations (0 %, Fig. 3 and Table 1). The total proportion of mutants induced by pvINT5BrC:G (0.71 %) was comparable to that of the control vector. This low pro-mutagenicity of 5-BrC is consistent with an in vitro analysis demonstrating that human DNA polymerases bypass 5-BrC without detectable miscoding [19]. The inconsistency between the previous study on 5-ClC and our results for 5-BrC is probably due to the different atomic radii of the halogens, effects of the specific DNA sequence context, or distinct repair mechanisms between E. coli and human cells.
Mutagenic potential of U:G and 5-BrU:G mismatch in the genome
The integration of pvINTU:G mainly induced C:G to T:A transition mutations (4.8 %), and the total proportion of mutants was 8.1 % (Fig. 3 and Table 1). This mutagenesis caused by the U:G mismatch in the genome is consistent with that in previous reports describing the well-known pro-mutagenicity of the uracil residue [4, 33]. Furthermore, the proportion of mutants was dramatically enhanced when pvINT5BrU:G was integrated (33 %), resulting in an approximately 7-fold higher proportion of C:G to T:A transition mutations than that occurring when pvINTU:G was integrated (4.8 %) (Fisher’s exact test, P < 0.01). This indicates that a bromine atom at the 5′–position of uracil interferes with repair using enzymes such as DNA glycosylases in the genome, thereby resulting in enhanced mutagenesis.
Mutagenic potential of T:G mismatch in the genome
Unexpectedly, the integration of the T:G mismatch (pvINTT:G) accounted for the highest proportion of mutants (56 %) (Table 1). Notably, all these mutants harbored C:G to T:A transition mutations, and the proportion of such mutations was 12-fold higher than that associated with the integration of a U:G mismatch (4.8 %) (Fisher’s exact test, P < 0.01) (Fig. 3). This high pro-mutagenicity of T:G mispairing is in contrasts with a previous report describing that T:G mismatches in episomal DNA are preferentially repaired to C:G at an approximate efficiency of 90 % by mismatch repair in mammalian cells [27]. Although our cell lines are mismatch repair proficient [34], the integrated T:G mismatch in the TK locus did not seem to have been corrected. Therefore, the repair efficiency of the T:G mismatch by the specific mismatch repair might depend on the genomic loci where the mismatch has been integrated. Our in vivo results are in agreement with those in a previous in vitro study demonstrating that the repair of mismatched T:G is far less efficient than that of mismatched U:G at a mutational hotspot sequence in the TP53 gene [35].
On the basis of our results, T:G and 5-BrU:G mismatches, resulting from the deamination of 5-mC:G and 5-BrC:G, respectively, markedly enhanced the mutagenic potential compared with that of the U:G mismatch. Although it has been suggested that human thymine DNA glycosylase and methyl-CpG binding protein 4 excise thymine and 5-BrU paired with guanine at CpG dinucleotides [21, 36–38], they might play minor roles in repair in cells. Thus, once deamination of the modified cytosine occurs, the deaminated residues could steadily induce mutations. Because the frequencies of C:G to T:A transition mutations induced by 5-mC and 5-BrC were 0.44 % (4.4 × 10−3) and 0 % (<10−3), respectively (Table 1), the frequencies of deamination of them might be equal to or less than the order of 10−3 in TSCER122 cells. Taking these findings together, we emphasize that those deaminated bases contribute to the mutagenesis and formation of mutational hotspots at specific loci, for example, CpG dinucleotides, in the genome.
Conclusion
Overall, we revealed the mutagenic potential of modified/deaminated cytosine residues in the human genome. Because T:G and 5-BrU:G mismatches can be highly pro-mutagenic, the rate-limiting step in the formation of mutational hotspots might be the deamination of modified cytosine residues. Our results are also useful to further study the mechanisms by which genomic integrity is maintained.
Acknowledgements
We thank Dr. Kenichi Masumura (National Institute of Health Sciences) for helpful discussions and suggestions. We also thank Enago (www.enago.jp) for the English-language review. This research was supported by Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology and for Health Science Foundation (H27-food-general-002) from the Ministry of Health, Labor and Welfare in Japan.
Abbreviations
- 5-BrC
5-bromocytosine
- 5-BrU
5-bromouracil
- 5-ClC
5-chlorocytosine
- 5-mC
5-methylcytosine
- PCR
polymerase chain reaction
- TATAM
tracing DNA adducts in targeted mutagenesis
- TK
thymidine kinase
Footnotes
Akira Sassa and Yuki Kanemaru contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AS, YK, MH, and MY designed the research and discussed the study. AS, YK, NK, and MY performed the experiments and analyzed the data. AS and MY wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Akira Sassa, Email: a-sassa@nihs.go.jp.
Manabu Yasui, Email: m-yasui@nihs.go.jp.
References
- 1.Vanyushin BF, Tkacheva SG, Belozersky AN. Rare bases in animal DNA. Nature. 1970;225:948–9. doi: 10.1038/225948a0. [DOI] [PubMed] [Google Scholar]
- 2.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A. 1993;90:11995–9. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird AP. Gene number, noise reduction and biological complexity. Trends Genet. 1995;11:94–100. doi: 10.1016/S0168-9525(00)89009-5. [DOI] [PubMed] [Google Scholar]
- 4.Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–1. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 5.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–7. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–60. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 7.Wijesinghe P, Bhagwat AS. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012;40:9206–17. doi: 10.1093/nar/gks685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich M, Norris KF, Wang RY, Kuo KC, Gehrke CW. DNA cytosine methylation and heat-induced deamination. Biosci Rep. 1986;6:387–93. doi: 10.1007/BF01116426. [DOI] [PubMed] [Google Scholar]
- 10.Shen JC, Rideout WM, 3rd, Jones PA. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 1994;22(6):972–6. doi: 10.1093/nar/22.6.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss SJ, Test ST, Eckmann CM, Roos D, Regiani S. Brominating oxidants generated by human eosinophils. Science. 1986;234:200–3. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 12.Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976;251:1371–4. [PubMed] [Google Scholar]
- 13.Foote CS, Goyne TE, Lehrer RI. Assessment of chlorination by human neutrophils. Nature. 1983;301:715–6. doi: 10.1038/301715a0. [DOI] [PubMed] [Google Scholar]
- 14.Henderson JP, Byun J, Takeshita J, Heinecke JW. Phagocytes produce 5-chlorouracil and 5-bromouracil, two mutagenic products of myeloperoxidase, in human inflammatory tissue. J Biol Chem. 2003;278:23522–8. doi: 10.1074/jbc.M303928200. [DOI] [PubMed] [Google Scholar]
- 15.Henderson JP, Byun J, Williams MV, McCormick ML, Parks WC, Ridnour LA, Heinecke JW. Bromination of deoxycytidine by eosinophil peroxidase: a mechanism for mutagenesis by oxidative damage of nucleotide precursors. Proc Natl Acad Sci U S A. 2001;98:1631–6. doi: 10.1073/pnas.98.4.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asahi T, Kondo H, Masuda M, Nishino H, Aratani Y, Naito Y, Yoshikawa T, Hisaka S, Kato Y, Osawa T. Chemical and immunochemical detection of 8-halogenated deoxyguanosines at early stage inflammation. J Biol Chem. 2010;285:9282–91. doi: 10.1074/jbc.M109.054213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen Z, Mitra SN, Wu W, Chen Y, Yang Y, Qin J, Hazen SL. Eosinophil peroxidase catalyzes bromination of free nucleosides and double-stranded DNA. Biochemistry. 2001;40:2041–51. doi: 10.1021/bi001961t. [DOI] [PubMed] [Google Scholar]
- 18.Masuda M, Suzuki T, Friesen MD, Ravanat JL, Cadet J, Pignatelli B, Nishino H, Ohshima H. Chlorination of guanosine and other nucleosides by hypochlorous acid and myeloperoxidase of activated human neutrophils. catalysis by nicotine and trimethylamine. J Biol Chem. 2001;276:40486–96. doi: 10.1074/jbc.M102700200. [DOI] [PubMed] [Google Scholar]
- 19.Sassa A, Ohta T, Nohmi T, Honma M, Yasui M. Mutational specificities of brominated DNA adducts catalyzed by human DNA polymerases. J Mol Biol. 2011;406:679–86. doi: 10.1016/j.jmb.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Sassa A, Kamoshita N, Matsuda T, Ishii Y, Kuraoka I, Nohmi T, Ohta T, Honma M, Yasui M. Miscoding properties of 8-chloro-2′-deoxyguanosine, a hypochlorous acid-induced DNA adduct, catalysed by human DNA polymerases. Mutagenesis. 2013;28:81–8. doi: 10.1093/mutage/ges056. [DOI] [PubMed] [Google Scholar]
- 21.Valinluck V, Liu P, Kang JI, Jr, Burdzy A, Sowers LC. 5-halogenated pyrimidine lesions within a CpG sequence context mimic 5-methylcytosine by enhancing the binding of the methyl-CpG-binding domain of methyl-CpG-binding protein 2 (MeCP2) Nucleic Acids Res. 2005;33:3057–64. doi: 10.1093/nar/gki612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suspene R, Aynaud MM, Vartanian JP, Wain-Hobson S. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PLoS One. 2013;8:e63461. doi: 10.1371/journal.pone.0063461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–80. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 24.Choi JH, Pfeifer GP. DNA damage and mutations produced by chloroacetaldehyde in a CpG-methylated target gene. Mutat Res. 2004;568:245–56. doi: 10.1016/j.mrfmmm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Lee DH, Pfeifer GP. Deamination of 5-methylcytosines within cyclobutane pyrimidine dimers is an important component of UVB mutagenesis. J Biol Chem. 2003;278:10314–21. doi: 10.1074/jbc.M212696200. [DOI] [PubMed] [Google Scholar]
- 26.Kamiya H, Tsuchiya H, Karino N, Ueno Y, Matsuda A, Harashima H. Mutagenicity of 5-formylcytosine, an oxidation product of 5-methylcytosine, in DNA in mammalian cells. J Biochem. 2002;132:551–5. doi: 10.1093/oxfordjournals.jbchem.a003256. [DOI] [PubMed] [Google Scholar]
- 27.Brown TC, Jiricny J. A specific mismatch repair event protects mammalian cells from loss of 5-methylcytosine. Cell. 1987;50:945–50. doi: 10.1016/0092-8674(87)90521-6. [DOI] [PubMed] [Google Scholar]
- 28.Yasui M, Kanemaru Y, Kamoshita N, Suzuki T, Arakawa T, Honma M. Tracing the fates of site-specifically introduced DNA adducts in the human genome. DNA Repair (Amst) 2014;15:11–20. doi: 10.1016/j.dnarep.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Arakawa T, Ohta T, Abiko Y, Okayama M, Mizoguchi I, Takuma T. A polymerase chain reaction-based method for constructing a linear vector with site-specific DNA methylation. Anal Biochem. 2011;416:211–7. doi: 10.1016/j.ab.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 30.Sassa A, Kamoshita N, Kanemaru Y, Honma M, Yasui M. Xeroderma pigmentosum group A suppresses mutagenesis caused by clustered oxidative DNA adducts in the human genome. PLoS One. 2015;10:e0142218. doi: 10.1371/journal.pone.0142218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandaru B, Wyszynski M, Bhagwat AS. HpaII methyltransferase is mutagenic in Escherichia coli. J Bacteriol. 1995;177:2950–2. doi: 10.1128/jb.177.10.2950-2952.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fedeles BI, Freudenthal BD, Yau E, Singh V, Chang SC, Li D, Delaney JC, Wilson SH, Essigmann JM. Intrinsic mutagenic properties of 5-chlorocytosine: a mechanistic connection between chronic inflammation and cancer. Proc Natl Acad Sci U S A. 2015;112:E4571–80. doi: 10.1073/pnas.1507709112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabral-Neto JB, Gentil A, Cabral RE, Sarasin A. Implication of uracil in spontaneous mutagenesis on a single-stranded shuttle vector replicated in mammalian cells. Mutat Res. 1993;288:249–55. doi: 10.1016/0027-5107(93)90091-S. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto A, Sakamoto Y, Masumura K, Honma M, Nohmi T. Involvement of mismatch repair proteins in adaptive responses induced by N-methyl-N′-nitro-N-nitrosoguanidine against gamma-induced genotoxicity in human cells. Mutat Res. 2011;713:56–63. doi: 10.1016/j.mrfmmm.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Schmutte C, Yang AS, Beart RW, Jones PA. Base excision repair of U:G mismatches at a mutational hotspot in the p53 gene is more efficient than base excision repair of T:G mismatches in extracts of human colon tumors. Cancer Res. 1995;55:3742–6. [PubMed] [Google Scholar]
- 36.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–4. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 37.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J Am Chem Soc. 2006;128:12510–9. doi: 10.1021/ja0634829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neddermann P, Gallinari P, Lettieri T, Schmid D, Truong O, Hsuan JJ, Wiebauer K, Jiricny J. Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem. 1996;271:12767–74. doi: 10.1074/jbc.271.48.30986. [DOI] [PubMed] [Google Scholar]