

ABSTRACT
Acute myeloid leukemia (AML) is associated with poor natural killer (NK) cell function through aberrant expression of NK-cell-activating receptors and their ligands on tumor cells. These alterations are thought to promote formation of inhibitory NK-target cell synapses, in which killer cell degranulation is attenuated. Allogeneic stem cell transplantation can be effective in treating AML, through restoration of NK cell lytic activity. Similarly, agents that augment NK-cell-activating signals within the immunological synapse may provide some therapeutic benefit. However, the receptor–ligand interactions that critically dictate NK cell function in AML remain undefined. Here, we demonstrate that CD112/CD155 expression is required for DNAM-1-dependent killing of AML cells. Indeed, the low, or absent, expression of CD112/CD155 on multiple AML cell lines resulted in failure to stimulate optimal NK cell function. Importantly, isolated clones with low CD112/155 expression were resistant to NK cell killing while those expressing abundant levels of CD112/155 were highly susceptible. Attenuated NK cell killing in the absence of CD112/CD155 originated from decreased NK–target cell conjugation. Furthermore, we reveal by time-lapse microscopy, a significant increase in NK cell ‘failed killing’ in the absence of DNAM-1 ligands. Consequently, NK cells preferentially lysed ligand-expressing cells within heterogeneous populations, driving clonal selection of CD112/CD155-negative blasts upon NK cell attack. Taken together, we identify reduced CD155 expression as a major NK cell escape mechanism in AML and an opportunity for targeted immunotherapy.
KEYWORDS: AML, cancer, CD155, DNAM-1, immunotherapy, immunological synapse, lymphocytes
Introduction
Acute myeloid leukemia (AML) is a hematological malignancy characterized by the expansion and infiltration of irregularly or poorly differentiated hematopoietic progenitor cells into the bone marrow, peripheral blood and other tissues.1 Accumulation of AML blasts in bone marrow interferes with the normal production of erythrocytes, causing anemia, while immature blasts have poor immune function and promote an immune-compromised state in the patient.1 Intense chemotherapy regimes, typically Cytarabine in combination with an anthyrcycline,2 often invoke remission in AML patients; however, relapse ultimately occurs in the majority of patients.3 In such cases, allogeneic stem cell transplantation (ASCT) can be effective.1 Unfortunately, however, only a minority of patients are deemed suitable for such therapy due to its invasive nature, and graft-versus-host disease (GVHD) limits its efficacy. Thus, there is an urgent need for more target-based immunotherapies against AML.1,2
ASCT efficacy is thought to derive from the graft-vs.-leukemia effect, in which host T cells are re-directed toward a minor mismatch in histocompatibility antigens on leukemic blasts.4 Indeed, T-cell depletion increases the incidence of relapse after ASCT, but does alleviate GVHD.5 Furthermore, poor cytotoxic NK cell activity may be restored through generation of KIR-dependent allo-reactivity, and through re-expression of NK-cell-activating receptors such as DNAM-1 and NKG2D.4,6,7 Importantly, NK cell lytic activity against AML is correlated with disease prognosis.8 AML blasts can escape NK cell-mediated lytic activity through an extensive process of immune-editing, in which multiple NK cell activation receptors and their ligands are manipulated to inhibit NK cell function (conjugation, degranulation and tumor cell lysis), leading to the induction of NK cell tolerance.9 Examples of such immune evasion tactics include a dull NCR profile through downregulation of NKp30 and NKp44,10 loss of NKG2D ligand expression (MICA/B, ULBPs)11 and perturbation of the DNAM-1–CD112/CD155 axis by inducing downregulation of DNAM-1 expression on NK cells.12 Therefore, compounds that can increase the activity of NK cells toward AML are of considerable therapeutic interest. Indeed, cytokines, antibodies and drugs can enhance NK cell activity through a variety of mechanisms, including upregulation of NK-cell-activating receptors and their corresponding ligands on AML cells.9 Thus, identifying the key receptor–ligand interactions that promote this repressed NK cell phenotype is of extreme clinical importance for the generation of targeted therapies.
NK cell receptor–ligand interactions occur within a nanoscale space at the NK cell-tumor contact site, called an immune synapse.13 Regions within the immune synapse can be segregated into domains called supramolecular activation clusters (SMACs).13 Upon synapse formation, signaling molecules that drive NK cell activation are polarized into the central SMAC region (cSMAC), while adhesion receptors such as LFA-1, CD2 and DNAM-1 accumulate around the cSMAC, called the peripheral SMAC (pSMAC).14 Execution of tumor cell killing is dictated by the relative strength of activatory and inhibitory signals, delivered to the NK cell from the tumor cell.15 In the case of activating signal dominance, polarization of the microtubule organizing center (MTOC) occurs, and facilitates the delivery of cytotoxic granules into the synaptic cleft.16 If inhibitory signals prevail, an inhibitory synapse is formed, in which NK cell degranulation fails.15 Because AML is associated with powerful inhibitory NK cell signals, understanding the critical molecular determinants in creating inhibitory NK cell synapses is of great importance for the generation of targeted therapies against AML.
DNAM-1 is a trans-membrane adhesion molecule and member of the immunoglobulin superfamily, which is expressed on T cells and NK cells.17 DNAM-1 drives NK-cell-mediated cytotoxicity and interferon-γ production in response to diverse cancer types and infected cells, by acting as an activation signal within the synaptic pSMAC.18 Upon stimulation, DNAM-1 drives a signaling cascade via a tyrosine- and asparagine-based motif in the cytoplasmic domain.19 This motif is phosphorylated by Src kinases to promote activation of Vav-1, phosphatidylinositol 3 kinase, and phospholipase C-γ1.19 Indeed, DNAM-1 triggers NK cell killing of freshly isolated ovarian carcinoma and neuroblastoma cells20,21 and is required for killing and control of melanoma metastases.22,23 Similarly, mice lacking DNAM-1 exhibit accelerated growth of DNAM-1 ligand-expressing tumors.24 Thus, DNAM-1 appears to be a key regulator of NK-cell-mediated cancer immunosurveilance. Interestingly, the interaction of DNAM-1 with one of its ligands, CD112, is a pathogenic target of herpesvirus, in which a viral gene product (gD) stimulates degradation of CD112 to avoid NK-cell-mediated lysis of infected cells and subsequent viral clearance.25 Similarly, HIV infection drives an exhausted T-cell phenotype, characterized by loss of DNAM-1 expression.26 In line with a central role for DNAM-1 in cancer immunosurveilance, genetic evidence has emerged implicating DNAM-1 mutations as a significant cancer risk factor.27 Taken together, these observations suggest that DNAM-1 is a global regulator of T-cell and NK-cell-mediated cytotoxicity, and perturbations in this pathway are likely to result in dramatically compromised antitumor and pathogen immunity.
Given that AML is typified by poor NK cell function, and the role of DNAM-1 and DNAM-1 ligands in this context remains unexplored, we examined the DNAM-1-CD112/CD155 receptor–ligand axis in controlling NK-cell-mediated cytotoxicity toward AML cells.
Results
AML cells promote poor NK-cell-mediated conjugation and killing
To interrogate the mechanisms underpinning poor NK-cell-mediated cytotoxicity in AML, we initially monitored the killing efficiency of allogeneic NK cells against a panel of AML cell lines. The archetypical human NK target cell line, K562, served as a control for efficient NK-cell-mediated lysis. As Fig. 1A demonstrates, all AML cell lines tested exhibited more resistance to NK cell killing over various effector:target ratios (see also Fig. S1). NK-cell-mediated killing was dependent on the cytotoxic granule exocytosis pathway, as the calcium chelator EDTA (which blocks perforin function), suppressed NK-cell-mediated killing entirely (Fig. 1B).
Figure 1.
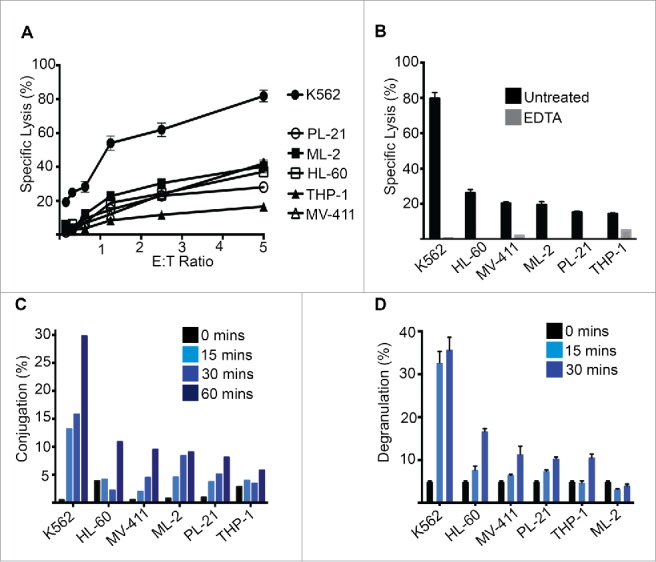
NK cells have poor activity against AML cells. (A) Chromium release assay using donor NK cells as effectors and the indicated AML cell lines as targets. (B) Chromium release assay as in (A) in the presence or absence of EDTA (2 mM). (C) NK cell conjugation assay using the indicated AML cell lines as targets. At the indicated time-points, NK-AML cell conjugates were analyzed by flow cytometry, as described in materials and methods. (D) NK cell degranulation assay in which CD107a exposure was monitored by flow cytometry after incubation with the indicated AML cell lines. Error bars represent the mean ± SEM of triplicate determinations from a representative experiment (n = 3).
We hypothesized that the poor NK-cell-mediated killing of AML cells may be due to sub-optimal conjugation and subsequent failure to form a cytotoxic synapse. Indeed, we found that while K562 cells formed high-frequency NK–tumor cell conjugates, NK cells were impaired in their ability to form stable conjugates with multiple AML cell lines (Fig. 1C). Furthermore, when we monitored NK cell degranulation upon exposure to the AML cell lines, we found that multiple AML cell lines triggered very poor NK cell degranulation (Fig. 1D), consistent with poor target cell lysis (Fig. 1A).
Given that NK cells formed stable cytotoxic synapses with AML cells at a poor frequency (Fig. 1D), this suggested that synapse-associated adhesion/activation interactions were not occurring efficiently in the pSMAC, resulting in a failure to breach the activation threshold necessary to trigger degranulation.28 DNAM-1 is known to be involved in NK cell adhesion, activation and tumor immune surveillance, and is often downregulated on immune cells from AML patients.12 The observed poor NK cell conjugation and killing of AML cells suggested that the DNAM-1-CD112/155 receptor–ligand axis might not be functioning optimally. To investigate this, we initially screened the AML cell lines for expression of the DNAM-1 ligands, CD112 and CD155. While K562 cells expressed DNAM-1 ligands abundantly, all other cell lines expressed poorly, or failed to express, either CD112 or CD155 (Fig. 2A). All cell lines, however, did express the hematopoietic progenitor marker, CD33 (Fig. 2A). Importantly, while low levels of DNAM-1 ligand expression could be detected on PL21, THP-1, MV411 and HL60 cells, the majority of cells within these cultures expressed no CD112 or CD155 (Fig. 2B). Thus, DNAM-1 ligand expression, particularly CD155, is expressed heterogeneously, both among AML cell lines and clonally. We also confirmed by confocal microscopy that particular clones within the AML cell cultures expressed heterogeneous levels of CD155 expression, with only a minority of cells expressing high levels of CD155 (with the majority being negative for CD155 expression) (Fig. 2C). Thus, sub-optimal expression of DNAM-1 ligands was likely responsible for the poor NK cell conjugation and subsequent killing of AML cells we observed (Fig. 1).
Figure 2.
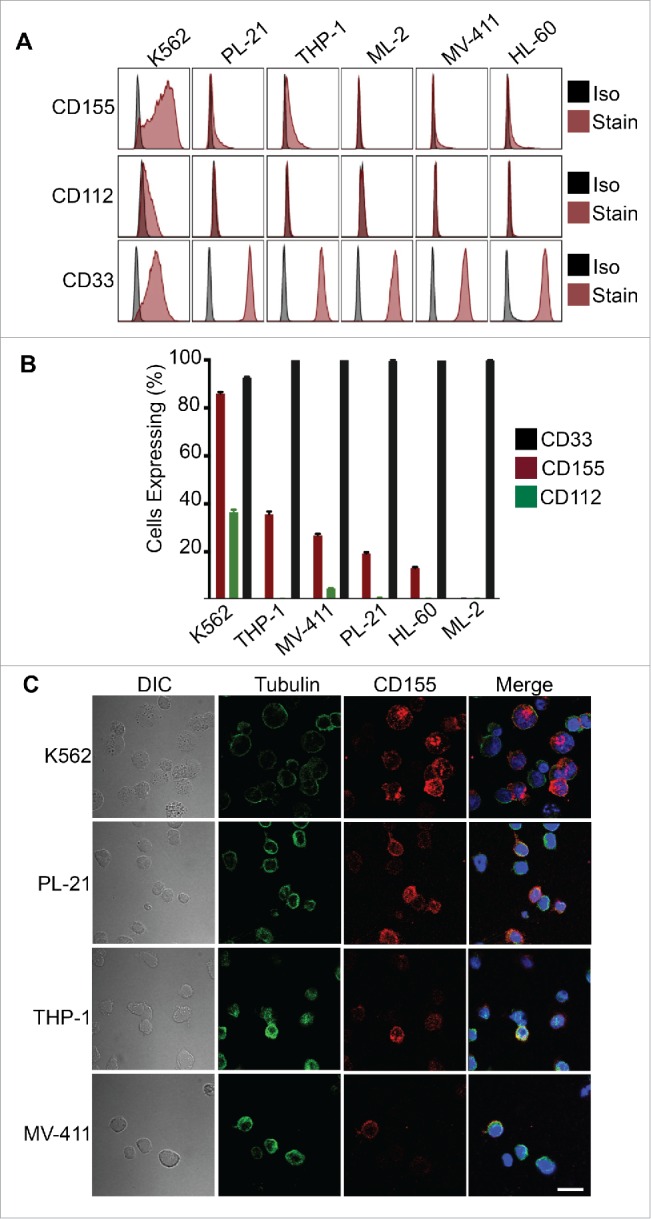
AML cells exhibit no, or heterogeneous, expression of DNAM-1 ligands. (A) The indicated AML cell lines were analyzed for expression of CD112, CD155 and CD33 by flow cytometry. Iso refers to immunoglobulin isotype matched control antibody while stain indicates specific antibody staining. (B) The percentage of cells expressing the indicated ligands within populations of the indicated cell type, as analyzed from (A). (C) CD155 expression on the indicated AML cell lines was visualized by confocal microscopy. Scale bar represents 20 µm.
DNAM-1 ligands promote NK cell degranulation and AML cell lysis
To further explore the heterogeneity of DNAM-1 ligand expression on AML cells, we analyzed the expression of CD112 and CD155 simultaneously on K562 cells. Expression of both ligands ranged from largely negative, to high-expressing clones, suggesting a sub-population of cells may exist that is refractory to DNAM-1 interactions during NK cell attack. To investigate this, we sorted the two populations, now referred to as ‘high’ and ‘low’ AML cell populations based on CD112/CD155 expression (Fig. 3A). We found that cells expressing low levels of DNAM-1 ligands failed to trigger NK cell degranulation to levels observed when the NK cells were exposed to high ligand-expressing AML targets (Fig. 3B). Importantly, this resulted in a dramatic reduction in the ability of NK cells to induce tumor cell lysis (Fig. 3C). Therefore, decreased CD112/155 expression allows AML cells to avoid NK cell-mediated lytic activity. Similarly, CD112/155 expression could be characterized as ‘low’ and ‘high’ on MV-411 cells and separated accordingly by flow cytometry (Fig. 3D). Again, NK cells largely failed to degranulate when exposed to ‘low’ ligand-expressing targets (Fig. 3E), and this translated to dramatically attenuated tumor cell killing (Fig. 3F). Consistent with these data, upregulation of CD155 by the anti-myeloma drug, Bortezomib, restored sensitivity of the ‘low’ ligand-expressing AML targets to NK cell killing (Fig. S2).
Figure 3.
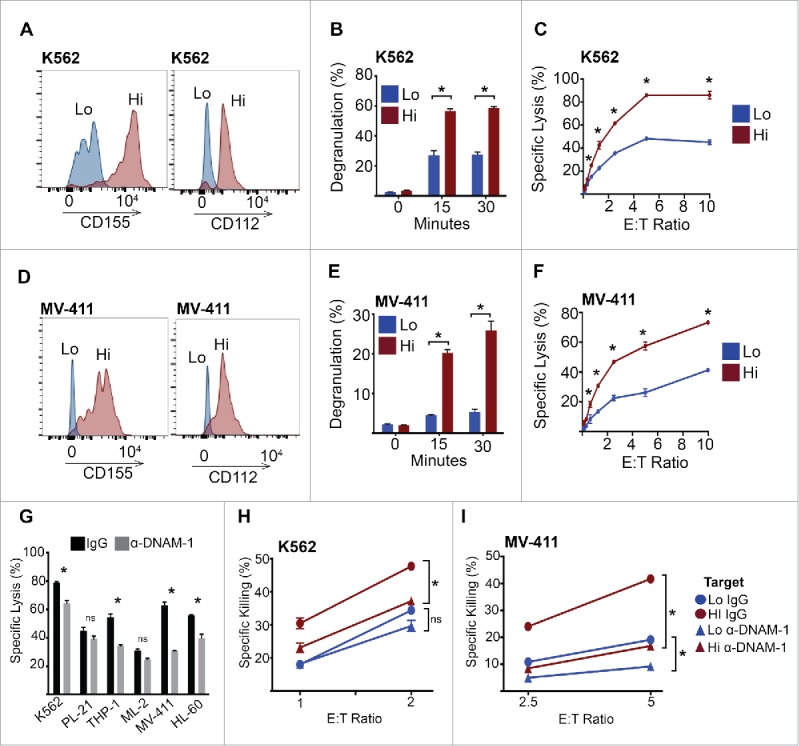
DNAM-1 ligands are required for NK cell activity against AML targets. (A) K562 cells were stained with anti-CD112 and anti-CD155 antibodies, then FACS sorted into high expressing (both CD112 and CD155) and low expressing (both CD112 and CD155) populations. (B) K562 cells were FACS sorted as in (A), then used as targets in an NK cell degranulation assay. (C) K562 cells were FACS sorted as in (A), then used as targets in an NK cell chromium release assay. (D) MV4-11 cells were stained with anti-CD112 and anti-CD155 antibodies, then FACS sorted into high expressing (both CD112 and CD155) and low expressing (both CD112 and CD155) populations. (E) MV4-11 cells were FACS sorted as in (D), then used as targets in an NK cell degranulation assay. (F) MV-411 cells were FACS sorted as in (D), then used as targets in an NK cell chromium release assay. (G) NK cell chromium release assay against the indicated AML targets (5:1 E:T ratio) in the presence or absence of anti-DNAM-1-neutralizing antibody (5 μg/mL). (H–I) NK cell chromium release assay at the indicated E:T ratios using FACS sorted high and low CD112/CD155 expressing K562 and MV-411 cells, in the presence or absence of anti-DNAM-1-neutralizing antibody (5 μg/mL). Error bars represent the mean ± SEM of triplicate determinations from a representative experiment (n = 3). *p < 0.05 by unpaired Student's t test.
While DNAM-1 is considered the dominant NK-cell-activating receptor that interacts with CD155 and CD112, these ligands also function through CD96/TACTILE and TIGIT (T cell immunoreceptor with Ig and ITIM domains). While CD96 can serve to limit DNAM-1-induced NK cell activation through competitive binding to CD155,29 it may also enhance NK cell killing by promoting NK–tumor cell conjugation.30 Therefore, we investigated if the enhanced NK-cell-mediated killing of CD112/CD155 high ligand-expressing cells was indeed DNAM-1 dependent. To address this question, we neutralized DNAM-1 activity using a blocking monoclonal antibody and found that neutralization of DNAM-1 significantly attenuated NK cell killing of multiple AML cell lines (Fig. 3G). As expected, K562 cells expressing high DNAM-1 ligands were killed rapidly in the presence of a control antibody (Fig. 3H). In the presence of the anti-DNAM-1 antibody, however, this increase was normalized to DNAM-1 ligand ‘low’ levels. Furthermore, anti-DNAM-1 antibody treatment had a negligible effect on the killing of AML cells that expressed minimal DNAM-1 ligands (Fig. 3H). Similar results were obtained with MV-411 cells (Fig. 3I). Thus, CD112/CD115 expression directly impacts on DNAM-1-dependent NK cell killing of AML cells.
The cytotoxic synapse is ‘normal’ in the absence of DNAM-1 ligands, but occurs less frequently
To explore the effect of DNAM-1 ligand expression on NK cell conjugation, we monitored the frequency of NK–tumor cell synapse events by confocal microscopy. K562 and MV-411 cells expressing low levels of DNAM-1 ligands formed very few synapses with NK cells compared to high ligand-expressing cells (Fig. 4A and B). We then interrogated polarization of key molecules to the NK cell synapse interface when exposed to high or low DNAM-1 ligand-expressing cells. We observed efficient polarization of the MTOC, actin, LFA-1, CD2 and perforin to the target contact site in MV-411 ‘high’ cells (Fig. 4C, left panel and D). While these events appeared to also occur in MV-411 ‘low’ cells (Fig. 4C, right panel and D), the frequency in which NK–tumor cell synapses could be detected was severely reduced (Fig. 4A). Similar results were observed for K562 cells (Fig. 4D). Thus, we conclude that the DNAM-1-CD112/155 axis is required to breach a conjugation and subsequent activation threshold,28 which is required for stable synapse formation and delivery of cytotoxic granules. As a result, a large proportion of NK-tumor cell contacts fail to proceed to cytotoxic synapse formation in the absence of sufficient DNAM-1 ligand stimulation.
Figure 4.

DNAM-1 ligands increase the frequency of ‘normal’ NK-target cell synapses. (A–B) FACS sorted (CD112/155 high and low) K562 and MV-411 cells were seeded in chamber slides using serum free media, then overlaid with NK cells 30 min later, followed by fixing. The percentage of targets that had conjugated with an NK cell was then quantitated by confocal microscopy. A minimum of 20 fields of view was analyzed and is representative of two independent experiments. (C) FACS sorted (CD112/155 high and low) MV4-11 cells were seeded in chamber slides using serum free media, then overlaid with NK cells 30 min later. After 1 h, cells were fixed, stained with the antibody combinations indicated, then analyzed by confocal microscopy. Representative images of NK-target cell synapses are presented. Scale bar represents 10 µm. (D) The percentage of NK cells that had polarized LFA-1 and perforin to the synapse was quantified from (C). A minimum of 20 NK-target cell synapses was analyzed and data from two independent experiments was pooled. Error bars represent the mean ± SEM *p < 0.05 by unpaired Student's t test.
Low DNAM-1 ligand expression promotes ‘failed killing’ of AML cells
Having uncovered a diminished frequency of NK cell-AML stable synapse events in the absence DNAM-1 ligand expression (Fig. 4), we used time-lapse microscopy to further interrogate this phenomenon at the single cell level in real time. Upon conjugation of NK cells with target cells, receptor activation triggers calcium influx into the NK cell which is required, but not sufficient, for NK cell degranulation.31 Furthermore, Propidium Iodide (PI) rapidly diffuses through the perforin pore upon delivery of the lethal hit, when added to the assay medium.32 These events can be monitored by imaging the cells in real time using time-lapse microscopy32 and allows accurate quantification of the proportion of NK–tumor cell interactions that trigger calcium influx and result in a successful lethal hit.
We utilized this technique to investigate NK cell interactions with MV-411 AML cells expressing low or high levels of CD112/CD155. Fig. 5A shows a series of still images captured from Movie 1 (supplemental data) in which six AML ‘high’ cells were lysed by the NK cells within 2 h from the time of NK cell addition. The first successful lethal hit can be detected within 45 min and is indicated by sudden PtdIns positivity within the tumor cells (‘PI blush’), which is preceded by robust NK cell calcium fluxing. Interestingly, however, when MV411 cells that express no/low CD112/CD155 were analyzed, NK cells did not kill any of the six available tumor cells within the same time period, despite multiple attempts (Movie 2, supplemental data). Indeed, NK cell calcium flux could be detected to comparable levels as that with high ligand-expressing cells; however, these interactions resulted in ‘failed killings’ (as evident by the absence of PtdIns blush and morphological cell death) (Fig. 5A). Indeed, five AML ‘low’ cells remained alive even after an extended time period of 6.5 h, despite repeated attempts by NK cells to lyse the targets (Fig. S3 and Movie 3, supplemental data). Quantification of these ‘failed killing’ events (Fig. 5B) translated into a poor rate of successful target lysis (Fig. 5C). Consistent with our confocal microscopy, a small number NK cell interactions with low ligand-expressing AML cells did result in successful killing. In these less frequent occurrences, the time from initial NK cell contact to PI blush was not altered by ligand expression (Fig. 5D). We also confirmed these findings using K562 cells as target cells (Fig. 5E–F). Interestingly, when K562 low cells were killed by NK cells, the time from contact to kill was significantly increased, compare to K562 high cells (Fig. 5G). These data suggest that loss of DNAM-1 ligand expression on AML cells promotes a sub-optimal synapse in which an activation threshold, required for cytotoxicity, is rarely breached, resulting in frequent ‘failed killing’ events.
Figure 5.
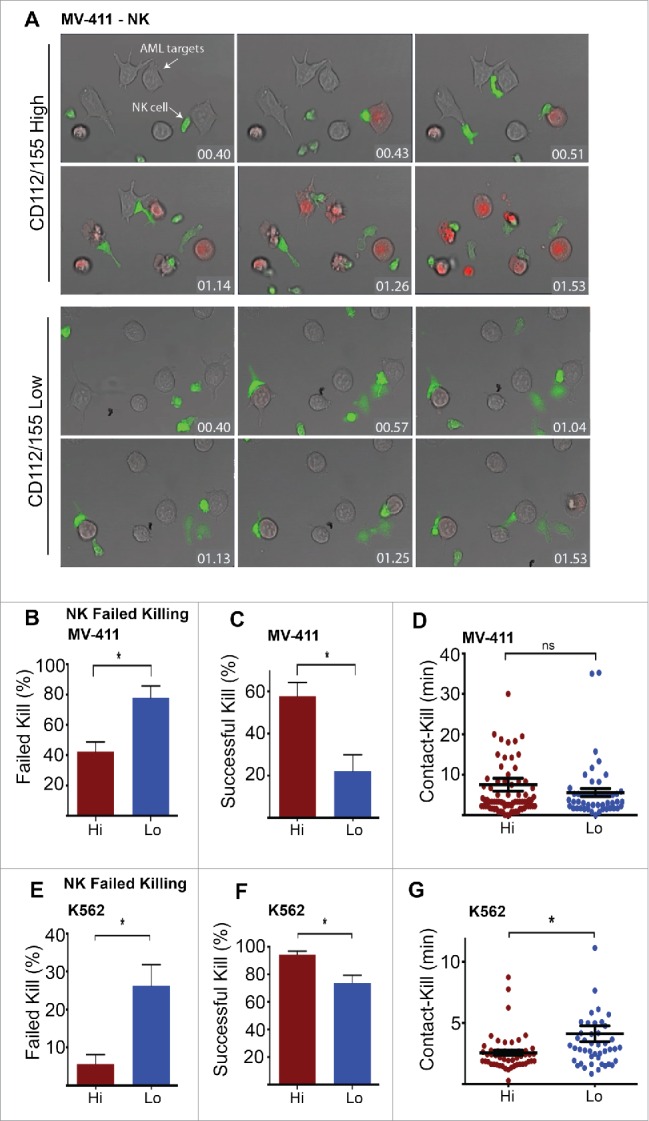
Live imaging reveals that AML cells lacking DNAM-1 ligand expression drive NK cell failed killing. (A) FACS sorted (CD112/155 high and low) MV4-11 cells were seeded in chamber slides using serum free media, then overlaid with NK cells labeled with fluo-4 acetoxymethyl AM (green) to indicate calcium signaling, and analyzed by time-lapse microscopy. PtdIns (red) (100 μg/mL) was added to the medium to indicate perforin-induced target membrane puncture. Representative still images at the indicated time-points are depicted (hr:min). (B–C) Individual NK-MV-411 CD112/CD155 high and low contacts were monitored for events that did (successful kill) or did not (failed kill) result in target killing, as indicated by PI influx and apoptotic morphology. (D) The time interval between initial NK-target cell contact and target cell death (PtdIns influx) was analyzed. (E–G) K562 cells were used as targets in the assays described in (B–D) above. All quantification data is pooled from individual movies (n = 3). Error bars represent the mean ± SEM *p < 0.05 by unpaired Student's t test.
Low DNAM-1 ligand-expressing AML clones are enriched following NK cell attack
AML cells can evade destruction by the immune system through extensive immuno-editing of receptors that promote and inhibit cytotoxic lymphocyte attack to generate a state of immune tolerance.28,33 Given the heterogeneity of CD112 and CD155 expression within AML cultures (Fig. 2), we questioned whether poor NK-cell-mediated lysis of these populations is derived from clones that fail to express these ligands. Initially, we isolated K562 clones expressing high or low CD112/CD155 and labeled these populations with CFSE and CTV dyes, respectively, prior to NK cell exposure. Indeed, specific NK cell killing could be determined through loss of these dyes within the viable cell populations (Fig. 6A). As expected, ‘high’ ligand clones were preferentially eliminated from the target mixture, with ‘low’ ligand-expressing cells evading killing entirely at 2.5 and 1.25 E:T ratios (Fig. 6B). We confirmed our observations using MV411 cells (Fig. 6B). Thus, DNAM-1 ligand negative AML clones, within a heterogeneous population, are resistant to NK cell killing and are enriched following NK cell exposure. To explore this phenomenon during prolonged NK cell attack, we exposed K562 and MV-411 cells to NK cells for 5 d. When the surviving clones were then analyzed, these cells had dramatically reduced expression of DNAM-1 ligands (Fig. 6C and D). Thus, AML cell resistance to NK cell killing is likely to originate through clonal selection, and subsequent outgrowth of DNAM-1 ligand negative blasts.
Figure 6.

NK cells preferentially target DNAM-1 ligand-expressing cells and drive clonal selection of DNAM-1 ligand negativity. (A) FACS-sorted K562 CD112/CD155 high and low cells were labeled with CFSE and CTV, respectively, then exposed to NK cells at the indicated E:T ratios. After 4 h, cells were analyzed by flow cytometry and loss of dye was monitored from viable populations. (B) Extended E:T ratio titration for the assay described in (A), using FACS-sorted K562 and MV-411 CD112/CD155 high and low cells as targets. Error bars represent the mean ± SEM of triplicate determinations from a representative experiment (n = 2). *p < 0.05 by unpaired Student's t test. (C–D) K562 and MV-411 cells were either exposed to NK cells (1:1 E:T Ratio), or not, for 5 d. Viable AML cells (fixable yellow negative, CD33 positive) were then analyzed for CD112 and CD155 expression by flow cytometry, and compared to parental cells (no NK cell exposure). Bar charts represent the number of CD112/CD155 double positive cells after 5 d in the presence or absence of NK cell exposure. Error bars represent the mean ± SEM of triplicate determinations from a representative experiment (n = 3). *p < 0.05 by unpaired Student's t test.
Discussion
NK cell activity (including target cell conjugation and decision to degranulate) is governed by the balance of activatory and inhibitory signals that are delivered to receptors expressed by the NK cells. Thus, the loss of signals that promote conjugation can dramatically affect the propensity of NK cells to degranulate. Furthermore, certain receptors, such as DNAM-1 and LFA-1, can serve to promote conjugation and activation/degranulation simultaneously. Indeed, DNAM-1 is a known adhesion molecule for NK and T-cells, but can also deliver a co-stimulatory/activatory signal through triggering of downstream signaling cascades.34 Therefore, these receptors and their ligands are likely to be deregulated in diseases where NK cell activity is subdued.
AML is a disease that can potentially be eradicated by the immune system, as evident by the success of ASCT treatment. However, because this course of action is often not an option, a better understanding of the events that promote a state of immune suppression in AML is critical. AML progression is associated with the failure of NK cells to form cytotoxic synapses with AML blasts.33 Poor NK cell cytotoxicity may be attributed to the deregulation of several critical steps that are required for optimal NK-cell-mediated immunity. Initially, it has been demonstrated that NK cells form low-frequency conjugates with AML cells.35 Downstream of this early event, NK-AML cell conjugation events may not necessarily result in formation of a cytotoxic synapse, in which the tumor cell is successfully lysed.35 Finally, intrinsic resistance to apoptosis may play a role in resistance to NK-cell-mediated killing, downstream of granule entry.36 Here, we demonstrate that poor NK cell function against AML cells originates through an early conjugation defect, which is deregulated through aberrant expression of DNAM-1 ligands. This resulted in a failure of NK cells to degranulate and kill the AML tumor cells. Furthermore, through time-lapse imaging, we reveal for the first time that, in the absence of DNAM-1 ligands, NK-tumor cell contacts fail to proceed to cytotoxic granule delivery, resulting in failed killing.
It was demonstrated that DNAM-1 drives NK cell function through two steps; first, DNAM-1–CD112/CD155 interactions promote NK cell adhesion to target cells, an event that can be uncoupled from signaling.19 Upon stable target cell adhesion/conjugation, DNAM-1 stimulation then triggers an intracellular signaling cascade through its cytoplasmic tyrosine-based tail to drive ERK, AKT and calcium flux.19 Thus, perturbation of the DNAM-1 axis attenuates NK cell function via two distinct mechanisms, which synergize to result in poor cytotoxicity.
Our finding that NK cells attempt, but fail to kill AML targets, has important implications beyond a direct loss in cytotoxicity. It is now appreciated that cytotoxic cells, including CD8+ T cells, are driven into a state of ‘exhaustion’ following prolonged exposure to antigen during cancer or infection.37 This compromised state occurs, at least in part, through upregulation of immune checkpoint inhibitors such as PD-1 and CTLA-4.37 Thus, NK cells that repeatedly fail to lyse target cells due to sub-optimal DNAM-1 stimulation are likely to become exhausted and lose function over time. Furthermore, T cells and NK cells that engage in failed killing hypersecrete pro-inflammatory cytokines and chemokines.38 Importantly, inflammation driven through this process is responsible for the pathogenesis of diseases including hemophagocytic lymphohistiocytosis (FLH)38 and inflammation is a potent driver of tumor initiation, proliferation and metastases.39 Therefore, NK-cell-failed killing through poor DNAM-1 function may actually drive disease progression, independently from a direct loss of cytotoxicity.
Because NK cell function is governed by receptor–ligand interactions, agents that can upregulate NK cell activating receptors and/or their ligands hold great promise therapeutically. Indeed, drugs that have pleiotropic immune-modulatory effects, such as lenalindomide provide some efficacy in hematological malignancies. Lenalidomide is thought to operate, at least in part, through augmenting NK cell synapse formation with tumor cells.40 However, the precise mechanism of action is unknown and problematic side effects limit the use of this agent. Thus, more targeted drugs that directly stimulate upregulation of NK-cell-activating ligands, such as CD112 and CD155, are required. Recent studies demonstrate that DNAM-1 ligands are regulated by ATM and ATR pathways, which are induced by DNA damage, at least in multiple myeloma.41 Consistent with this observation, standard chemotherapeutics such as doxorubicin and melphalan can promote DNAM-1 ligand expression when administered at low concentrations, promoting NK cell cytotoxicity.41 However, as these drugs are intrinsically cytotoxic, further studies are required to understand ways to specifically regulate DNAM-1 ligand expression in AML and other malignancies.
Taken together, we have demonstrated that the DNAM-1 receptor–ligand axis is critical to NK-cell-mediated AML immunity. Perturbation of this axis, through selective pressure of AML clones expressing low levels of these ligands can dramatically attenuate NK cell cytolytic activity against these tumor cells. We therefore identify a critical interaction for targeted immunotherapy.
Materials and methods
Antibodies
Directly conjugated antibodies were used for of the analysis of NK cell ligands and consisted of anti-human CD155-PE (FAB25301, R&D systems), CD112-FITC (FAB2229G, R&D systems), CD33 PECy7 (333946, BD) and anti-mouse CD112-FITC (690912 R&D systems), CD155-PE (690912, R&D systems). For microscopy, the following antibodies were used: rat anti–LFA-1 (55378, BD Biosciences), anti-CD2 (55325, BD Biosciences) and anti-perforin (556434, BD Biosciences); phalloidin-rhodamine (Molecular Probes), and rabbit and rat anti-tubulin (Rockland). Secondary antibodies conjugated to Alexa Fluorophores and ProLong antifade with DAPI were purchased from Molecular Probes.
Cells
The cell lines K562, MV-411, PL-21, ML-2, HL-60, and THP-1 were cultured in RPMI medium (Gibco, Invitrogen) supplemented with 10% FCS (Thermo Scientific) with penicillin/streptomycin (Gibco), and incubated at 37°C in 5% CO2. Primary NK cells were derived from PBMC of healthy volunteer donors and from buffy coat provided by the Australian Red Cross, using negative selection isolation kits (StemCell Technologies, Inc.). NK cell (CD56+, CD16+, CD3−) purity of above 90% purity was considered adequate for assays. NK cells were cultured in RPMI medium supplemented with 10% fetal calf serum (FCS), L-glutamine, penicillin/streptomycin, non-essential amino acids, sodium pyruvate, HEPES, β-2-mercaptoethanol (Calbiochem), and 25 IU/mL recombinant human IL-2 (ROCHE), at 37°C in a 5% CO2 atmosphere.
Flow cytometry
For cell soring by flow cytometry, 10 × 106 AML cells were incubated with anti CD112-FITC and CD155-PE for 20 min at 37°C in PBS, 2% FCS (FACS buffer). Cells were washed three times with FACS buffer prior to capture of ‘high’ and ‘low’ ligand-expressing cells by flow cytometry (Fusion 5, GE Healthcare). Cells were then cultured for at least 24 h under normal conditions prior to use in an assay. Acquisition of all flow cytometry data was performed using a Fortessa analyzer (BD Biosciences). All analysis of flow cytometry data was undertaken using Flowjo (Treestar).
Degranulation assay
NK cells were pre-treated with Golgi Plug (BD Biosciences) for 15 min prior to addition of targets at an E:T ratio of 2:1, in 96-well v bottom plates (Nunc). PE conjugated anti-CD107a (BD Biosciences) was added to the assay medium at a concentration of 1 μg/mL. At the indicated time-points, cells were washed then fixed in 2% PFA, and analyzed by flow cytometry.
Cytotoxicity assay
NK cell cytotoxic activity was measured using a standard chromium release assay as follows: of 1 × 106 targets were labeled with 100 μCi of 51Cr (Perkin Elmer) and added to V bottom plates. NK cells were then added to the targets at the indicated effector to target (E:T) ratios. After 4 h at 37°C in 5% CO2, supernatants were harvested, and the level of 51Cr was quantified by a gamma counter (Wallac Wizard). Percentage specific killing was determined using the formula: (Sample 51Cr release – Spontaneous 51Cr release)/(Total 51Cr release – Spontaneous 51Cr release) × 100.
Immunofluorescent fixed microscopy
Indicated AML cells were adhered onto 8-well chamber slides in serum-free media for 30 min, prior to addition of NK cells for the indicated time periods. Cells were then fixed and permeabilized, then labeled with primary Abs, followed by detection with Alexa Fluor–conjugated secondary Abs and mounted in Prolong anti-fade containing DAPI (Molecular Probes). The slides were imaged using a FV1000 confocal microscope (Olympus, NY). NK cells selected for protein scoring had a single contact site with one tumor cell, indicating a single synapse event. Polarization to the synapse was determined by quantitating the amount of non-nuclear fluorescence in the proximal (P) region (first third of NK cell closest to tumor cell) and distal (D) region (rest of NK cell). A minimum of 20 synapse events were counted per treatment.
Time-lapse microscopy
Target cells were prepared for live cell imaging by seeding 1 × 104 K562 or MV-411 cells into each well of an 8-well chamber slide (Ibidi, Munich, Germany) and incubating overnight at 37°C/10% CO2 in serum-free media to promote adherence to the slide. 2 × 104 NK cells were labeled with Fluo-4 for 20 min (1 μM Fluo-4 and 0.02% [w/v] Pluronic F-127 carrier at 37°C/10% CO2), then added to adherent targets, in media containing 100 μM PI (PtdIns).32 Chamber slides were mounted on a heated stage within a temperature-controlled chamber maintained at 37°C, and constant CO2 concentrations (5 or 7%) were infused using a gas incubation system with active gas mixer (“The Brick;” Ibidi). Optical sections were acquired through sequential scans of Fluo-4 (excitation 488 nm; 5-μm pinhole), PI (excitation 561 nm; 1.38-μm pinhole), or Brightfield/DIC on a TCS SP5 confocal microscope (Leica Microsystems, Deerfield, IL) using a 40× (NA 0.85) air objective and Leica LAS AF software. Images were acquired at ∼6–7 frames/min. Image analysis was performed using MetaMorph Imaging Series 7 software (Universal Imaging, Downingtown, PA).
Statistical analysis
In all cases, data was considered to be statistically significant when p ≤ 0.05, by unpaired students t-test.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We wish to thank Dr Daniel Andrews (Monash University) for critical reading of the manuscript and helpful advice.
Funding
This work was supported by a project grant from the National Health and Medical Research Council of Australia (NHMRC) and the Peter MacCallum Cancer Foundation. PKD and IV are supported by NHMRC Senior Research Fellowships.
References
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015; 373:1136-52; PMID:26376137; http://dx.doi.org/ 10.1056/NEJMra1406184 [DOI] [PubMed] [Google Scholar]
- 2.Dohner H, Estey EH, Amadori S, Appelbaum FR, Buchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA et al.. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115:453-74; PMID:19880497; http://dx.doi.org/ 10.1182/blood-2009-07-235358 [DOI] [PubMed] [Google Scholar]
- 3.Stoiser B, Knobl P, Fonatsch C, Haas OA, Mitterbauer G, Weltermann A, Geissler K, Valent P, Sperr W, Pabinger I et al.. Prognosis of patients with a second relapse of acute myeloid leukemia. Leukemia 2000; 14:2059-63; PMID:11187893; http://dx.doi.org/ 10.1038/sj.leu.2401968 [DOI] [PubMed] [Google Scholar]
- 4.Niewerth D, Creutzig U, Bierings MB, Kaspers GJ. A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood 2010; 116:2205-14; PMID:20538803; http://dx.doi.org/ 10.1182/blood-2010-01-261800 [DOI] [PubMed] [Google Scholar]
- 5.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F et al.. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002; 295:2097-100; PMID:11896281; http://dx.doi.org/ 10.1126/science.1068440 [DOI] [PubMed] [Google Scholar]
- 6.Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, Marsh SG, Geraghty D, Spellman S, Haagenson MD et al.. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 2010; 116:2411-9; PMID:20581313; http://dx.doi.org/ 10.1182/blood-2010-05-283051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruz CR, Bollard CM. T-cell and natural killer cell therapies for hematologic malignancies after hematopoietic stem cell transplantation: enhancing the graft-versus-leukemia effect. Haematologica 2015; 100:709-19; PMID:26034113; http://dx.doi.org/ 10.3324/haematol.2014.113860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tratkiewicz JA, Szer J. Loss of natural killer activity as an indicator of relapse in acute leukaemia. Clin Exp Immunol 1990; 80:241-6; PMID:2357852; http://dx.doi.org/ 10.1111/j.1365-2249.1990.tb05241.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lion E, Willemen Y, Berneman ZN, Van Tendeloo VF, Smits EL. Natural killer cell immune escape in acute myeloid leukemia. Leukemia 2012; 26:2019-26; PMID:22446501; http://dx.doi.org/ 10.1038/leu.2012.87 [DOI] [PubMed] [Google Scholar]
- 10.Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D, Costello RT. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007; 109:323-30; PMID:16940427; http://dx.doi.org/ 10.1182/blood-2005-08-027979 [DOI] [PubMed] [Google Scholar]
- 11.Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D, Gastaut JA, Pende D, Olive D, Moretta A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002; 99:3661-7; PMID:11986221; http://dx.doi.org/ 10.1182/blood.V99.10.3661 [DOI] [PubMed] [Google Scholar]
- 12.Sanchez-Correa B, Gayoso I, Bergua JM, Casado JG, Morgado S, Solana R, Tarazona R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol 2012; 90:109-15; PMID:21383766; http://dx.doi.org/ 10.1038/icb.2011.15 [DOI] [PubMed] [Google Scholar]
- 13.Dustin ML. The immunological synapse. Cancer Immunol Res 2014; 2:1023-33; PMID:25367977; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dustin ML. Modular design of immunological synapses and kinapses. Cold Spring Harb Perspect Biol 2009; 1:a002873; PMID:20066081; http://dx.doi.org/ 10.1101/cshperspect.a002873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495-502; PMID:18425106; http://dx.doi.org/ 10.1038/ni1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orange JS, Harris KE, Andzelm MM, Valter MM, Geha RS, Strominger JL. The mature activating natural killer cell immunologic synapse is formed in distinct stages. Proc Natl Acad Sci U S A 2003; 100:14151-6; PMID:14612578; http://dx.doi.org/ 10.1073/pnas.1835830100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, Kitamura T, Nicholl J, Sutherland GR, Lanier LL et al.. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996; 4:573-81; PMID:8673704; http://dx.doi.org/ 10.1016/S1074-7613(00)70060-4 [DOI] [PubMed] [Google Scholar]
- 18.de Andrade LF, Smyth MJ, Martinet L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol Cell Biol 2014; 92:237-44; PMID:24343663; http://dx.doi.org/ 10.1038/icb.2013.95 [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM-1 controls NK cell activation via an ITT-like motif. J Exp Med 2015; 212:2165-82; PMID:26552706; http://dx.doi.org/ 10.1084/jem.20150792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlsten M, Bjorkstrom NK, Norell H, Bryceson Y, van Hall T, Baumann BC, Hanson M, Schedvins K, Kiessling R, Ljunggren HG et al.. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res 2007; 67:1317-25; PMID:17283169; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2264 [DOI] [PubMed] [Google Scholar]
- 21.Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, Bottino C, Moretta A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res 2004; 64:9180-4; PMID:15604290; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2682 [DOI] [PubMed] [Google Scholar]
- 22.Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol 2010; 184:902-11; PMID:20008292; http://dx.doi.org/ 10.4049/jimmunol.0903225 [DOI] [PubMed] [Google Scholar]
- 23.Lakshmikanth T, Burke S, Ali TH, Kimpfler S, Ursini F, Ruggeri L, Capanni M, Umansky V, Paschen A, Sucker A et al.. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J Clin Invest 2009; 119:1251-63; PMID:19349689; http://dx.doi.org/ 10.1172/JCI36022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya K, Shibuya A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med 2008; 205:2959-64; PMID:19029379; http://dx.doi.org/ 10.1084/jem.20081611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grauwet K, Cantoni C, Parodi M, De Maria A, Devriendt B, Pende D, Moretta L, Vitale M, Favoreel HW. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc Natl Acad Sci U S A 2014; 111:16118-23; PMID:25352670; http://dx.doi.org/ 10.1073/pnas.1409485111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cella M, Presti R, Vermi W, Lavender K, Turnbull E, Ochsenbauer-Jambor C, Kappes JC, Ferrari G, Kessels L, Williams I et al.. Loss of DNAM-1 contributes to CD8+ T-cell exhaustion in chronic HIV-1 infection. Eur J Immunol 2010; 40:949-54; PMID:20201043; http://dx.doi.org/ 10.1002/eji.200940234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu ZX, Peng Y, Li WM. CD226 gene polymorphisms are associated with non-small-cell lung cancer in the Chinese Han population. Ther Clin Risk Manag 2015; 11:1259-64; PMID:26346602; http://dx.doi.org/ 10.2147/TCRM.S90365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmes TD, El-Sherbiny YM, Davison A, Clough SL, Blair GE, Cook GP. A human NK cell activation/inhibition threshold allows small changes in the target cell surface phenotype to dramatically alter susceptibility to NK cells. J Immunol 2011; 186:1538-45; PMID:21191066; http://dx.doi.org/ 10.4049/jimmunol.1000951 [DOI] [PubMed] [Google Scholar]
- 29.Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol 2014; 15:431-8; PMID:24658051; http://dx.doi.org/ 10.1038/ni.2850 [DOI] [PubMed] [Google Scholar]
- 30.Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J Immunol 2004; 172:3994-8; PMID:15034010; http://dx.doi.org/ 10.4049/jimmunol.172.7.3994 [DOI] [PubMed] [Google Scholar]
- 31.Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, Sjöqvist S, Hufnagel M, Schulze I, Bass T et al.. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A 2011; 108:3324-9; PMID:21300876; http://dx.doi.org/ 10.1073/pnas.1013285108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RH, Johnston A, Bird CH, Bird PI, Whisstock JC et al.. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood 2013; 121:2659-68; PMID:23377437; http://dx.doi.org/ 10.1182/blood-2012-07-446146 [DOI] [PubMed] [Google Scholar]
- 33.Teague RM, Kline J. Immune evasion in acute myeloid leukemia: current concepts and future directions. J Immunother Cancer 2013; 1; PMID:24353898; http://dx.doi.org/ 10.1186/2051-1426-1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibuya K, Lanier LL, Phillips JH, Ochs HD, Shimizu K, Nakayama E, Nakauchi H, Shibuya A. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity 1999; 11:615-23; PMID:10591186; http://dx.doi.org/ 10.1016/S1074-7613(00)80136-3 [DOI] [PubMed] [Google Scholar]
- 35.Khaznadar Z, Henry G, Setterblad N, Agaugue S, Raffoux E, Boissel N, Dombret H, Toubert A, Dulphy N. Acute myeloid leukemia impairs natural killer cells through the formation of a deficient cytotoxic immunological synapse. Eur J Immunol 2014; 44:3068-80; PMID:25041786; http://dx.doi.org/ 10.1002/eji.201444500 [DOI] [PubMed] [Google Scholar]
- 36.Testa U, Riccioni R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica 2007; 92:81-94; PMID:17229639; http://dx.doi.org/ 10.3324/haematol.10279 [DOI] [PubMed] [Google Scholar]
- 37.Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492-9; PMID:21739672; http://dx.doi.org/ 10.1038/ni.2035 [DOI] [PubMed] [Google Scholar]
- 38.Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, Voskoboinik I, Trapani JA. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med 2015; 212:307-17; PMID:25732304; http://dx.doi.org/ 10.1084/jem.20140964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/ 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lagrue K, Carisey A, Morgan DJ, Chopra R, Davis DM. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood 2015; 126:50-60; PMID:26002964; http://dx.doi.org/ 10.1182/blood-2015-01-625004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A et al.. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009; 113:3503-11; PMID:19098271; http://dx.doi.org/ 10.1182/blood-2008-08-173914 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.