

Abstract
Axon injury in the peripheral nervous system (PNS) induces a regeneration-associated gene (RAG) response. Atf3 (activating transcription factor 3) is such a RAG and ATF3's transcriptional activity might induce ‘effector’ RAGs (e.g. small proline rich protein 1a (Sprr1a), Galanin (Gal), growth-associated protein 43 (Gap43)) facilitating peripheral axon regeneration. We provide a first analysis of Atf3 mouse mutants in peripheral nerve regeneration. In Atf3 mutant mice, facial nerve regeneration and neurite outgrowth of adult ATF3-deficient primary dorsal root ganglia neurons was decreased. Using genome-wide transcriptomics, we identified a neuropeptide-encoding RAG cluster (vasoactive intestinal peptide (Vip), Ngf, Grp, Gal, Pacap) regulated by ATF3. Exogenous administration of neuropeptides enhanced neurite growth of Atf3 mutant mice suggesting that these molecules might be effector RAGs of ATF3's pro-regenerative function. In addition to the induction of growth-promoting molecules, we present data that ATF3 suppresses growth-inhibiting molecules such as chemokine (C-C motif) ligand 2. In summary, we show a pro-regenerative ATF3 function during PNS nerve regeneration involving transcriptional activation of a neuropeptide-encoding RAG cluster. ATF3 is a general injury-inducible factor, therefore ATF3-mediated mechanisms identified herein might apply to other cell and injury types.
Keywords: ATF3, axon regeneration, facial nerve, neuropeptide, RAG
1. Background
Upon nerve injury, the axon regeneration success varies in the adult mammalian nervous system. In the central nervous system (CNS), axon regeneration is very limited, whereas severed axons of peripheral nervous system (PNS) neurons regenerate to some extent [1–5]. One factor thought to account for this difference is the potential of PNS neurons to elicit a rapid regeneration-associated gene (RAG) response upon axotomy [5–7]. A typical RAG response encompasses several hundred genes and may vary slightly depending on the injured neuron population [8]. Nevertheless, a prototypical RAG response consists of transcription factor (TF)-encoding and ‘effector’ RAGs [2,4,6,9]. Such RAG-encoded TFs include STAT3 [10], ATF3 [11,12], c-Jun [13], Sox11 [14], Smad1 [15] and p53 [16]. Subsequently, transcriptional activity of these TF RAGs contributes to a second gene transcription wave resulting in expression of effector RAGs. Such effector RAGs encode for genes involved in cell adhesion (e.g. CD44, integrin subunits), neuropeptide signalling (e.g. Galanin, Vip, Npy, Pacap) and cytoskeletal modulation [17]. Subsequently, protein products of these effector RAGs can directly facilitate PNS axon regeneration, e.g. through direct axon growth stimulation [5].
In this study, we analysed ATF3, a RAG-encoded TF of the ATF/cAMP response element-binding protein (CREB) family [18,19]. In brain homeostasis, ATF3 expression is nearly undetectable, whereas pathological stimuli including axotomy, stress and epileptic seizures induce rapid ATF3 expression [18,19]. ATF3 exerts both transcriptional repression and activation of target genes including Hsp27, Sprr1a and c-Jun [18,19]. ATF3 interacts with partner proteins, e.g. c-Jun, to regulate further target genes including the CCL chemokine Ccl2 [20–22]. Chemokine (C-C motif) ligand 2 (CCL2) is involved in neuron–immune cell interactions during axon regeneration and neuronal degeneration [23–29]. Recently, ATF3 was identified as a core hub present in a RAG network after PNS injury [8]. So far, Atf3 mutant mice have not been investigated in axonal regeneration; however, ATF3 overexpression revealed pro-regenerative and neuroprotective functions. This includes stimulation of primary dorsal root ganglia (DRG) neurite growth and regeneration of peripheral and central DRG branches in vivo [8,11,12,30]. In general, ATF3 emerges as a neuroprotective factor. In mouse ALS [31] and epilepsy [32] models, ATF3 overexpression resulted in reduced neurodegeneration. This might be accomplished by ATF3's impact on dendrites, e.g. by providing protection against NMDA-exerted dendrotoxicity [33,34]. In addition, dendritic spine impairments observed in tuberous sclerosis mouse models depend on ATF3 function [35].
Herein, we provide a first analysis of constitutive Atf3 null mouse mutation in PNS axon regeneration employing facial nerve axotomy. The facial nerve connects facial motoneurons (FMN) residing in the facial nucleus (FN) of the brainstem with facial muscles involved in stirring e.g. whisker and eyelid movement [36]. Facial nerve injury triggers a robust RAG response upon injury including ATF3 upregulation [37–42]. We observed reduced facial nerve regeneration in Atf3 mutant mice in vivo and decreased neurite outgrowth on primary DRG neurons. This corresponded with a reduced RAG response in Atf3 mutant mice resulting in blunted expression of a distinct neuropeptide encoding cluster (Vip, Galanin, Grp, Ngf) as well as specific other genes (e.g. Sprr2j, wingless-type MMTV integration site family, member 2B (Wnt2b), Ccl2). The addition of recombinant neuropeptides rescued neurite growth inflicted by ATF3 deficiency. Thus, some of these neuropeptide-encoding genes might operate as effector RAGs transmitting ATF3's pro-regenerative function in vivo.
2. Material and methods
2.1. Atf3 mutant mice
Constitutive Atf3 mutant mice (Atf3−/−) on a C57BL/6 background were a kind gift of Dr T. Hai (Ohio State University, USA). The Atf3 mutant allele lacks exon B, which contains the AUG initiation codon and does not produce any ATF3 protein in the liver [43] or injured FN (figure 1). Genotyping followed a published protocol [43]. As control, offspring harbouring two wild-type (wt) Atf3 alleles (Atf3+/+) were used. Wt and mutant animals were derived from breedings of two Atf3 heterozygous parents (Atf3+/−). In heterozygous Atf3 mice, ATF3 induction after injury was undistinguishable from Atf3+/+ mice, suggesting no dose-dependent effect of reducing one allele (data not shown).
Figure 1.

Temporal sequence ATF3 induction in injured facial motoneurons. (a–f) FN of wt (a–e) and Atf3 mutant (f) animals were stained for ATF3 expression before (a) or several days post-lesion (d.p.l.; b–f). ATF3 is not present in uninjured FMNs (a) or only weakly at 6 h after lesion (b). By contrast, at 3 (c) and 7 (d) d.p.l., ATF3 was localized to FMNs. After three weeks of lesion, there was almost no ATF3 expression (e). In Atf3 mutant mice, no ATF3 expression was visible at 3 d.p.l. (f). (g) Quantification of the number of ATF3 positive FMNs per section at different times after facial nerve injury. (h) Analysis of Atf3, Atf2, Atf4 and Atf6 mRNA abundance in FNs without and 3 d.p.l. in wt and Atf3 mutant mice. Only Atf3, but no other family member, was induced by facial nerve injury in wt neurons. No compensatory expression of Atf2, Atf4 or Atf6 was observed in Atf3 mutant animals. (i,j) Unlesioned (i) and lesioned (j) facial nerves were stained for ATF3 expression. Without injury (i), no ATF3 expression was observed. By contrast, ATF3 was present in Schwann cells of an injured facial nerve (arrows in j). n ≥ 3 animals each bar. Data are presented as mean ± s.d. ***p ≤ 0.001. Scale bar (a–f) = 100 µm; (i,j) = 50 µm.
2.2. Facial nerve transection
Facial nerve transection was performed as described previously [13,44]. Adult mice of either sex (approx. eight weeks old) were anaesthetized, a skin incision was made behind the right ear, and the facial nerve was exposed. Afterwards, the nerve was transected with microscissors 2 mm posterior to the foramen stylomastoideum. The absence of eyelid closure and whisker movement proved successful nerve transection. Regeneration of the facial nerve was quantified by retrograde axonal tracing with fluorogold (FG; Fluorochrome), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; Molecular Probes) or choleratoxin subunit B (Ctx) conjugated with Alexa 488 (choleratoxin Alexa 488 (Ctx488); Molecular Probes). For this, 4 × l µl of FG (4% in H2O), 2 × 1 µl of DiI (2 µg µl−1 in DMSO) or 2 × 1 µl of Ctx488 (1 µg µl−1 in PBS) were injected with a Hamilton syringe at multiple positions in each whisker pad, eyelid or lower jaw, respectively. This injection was performed at 4, 10, 15, 19 or 27 days post-lesion (d.p.l.; figures 2 and 3). The tracers were given for 4 days for retrograde axonal transport, resulting in total regeneration periods of 8, 14, 19, 23 and 31 d.p.l., as indicated in figures 2 and 3. After those 4 days, mice were sacrificed and brains were dissected. FG, DiI or Ctx488 positive neurons of all sections of both FNs per each animal were evaluated before immunohistological staining. All experiments are in accordance with institutional regulations by the local animal ethical committee (Regierungspräsidium Tübingen, Germany).
Figure 2.

Impaired axonal regeneration in Atf3 mutant mice. (a(i–iii)) The unlesioned facial nerve is depicted by a solid black line (a). Unilateral axotomy of the facial nerve was performed at the position marked by an arrow (a(ii)). Axon regeneration is quantified by injection of the fluorescent tracers DiI, FG and Ctx488 in the eyelid, whiskers and lower jaw, respectively (a(iii)). Upon muscle re-innervation by regenerating axons, the tracer is retrogradely transported to FMN cell bodies of the FN (black dotted circle). (b–e) In the unlesioned FN of wt (b) or Atf3−/− (c) mice, FMNs connected to the eyelid were localized in two domains (arrows in b,c). After 23 d.p.l., DiI back-labelled FMNs were dispersed over the FN in both wt (d) and Atf3−/− (e) mice. The number of DiI positive neurons was comparable between the unlesioned and lesioned FN, suggesting rapid re-innervation of the eyelid regardless of genotype (see r). (f–i) In the unlesioned FN of wt (f) or Atf3−/− (g) mice, retrogradely labelled Ctx488 positive FMNs were localized in a medio-dorsal FN quarter (arrows in f,g). Upon lesion, the number of Ctx488 positive FMNs was reduced, although comparably between wt (h) and ATF3-deficient (i) mice. (j–m) FG positive FMNs in unlesioned FN of wt (j) or Atf3−/− (k) mice were restricted to the lateral half of the FN. Upon lesion, FG positive FMNs were spread all-over the entire FN in wt (l) and mutant (m) animals. However, the number of FG positive neurons was reduced in Atf3−/− (m) compared with wt (l) animals (see r). (n–q) Merged images of individual channels presented in (b–m). (r) The average number of DiI, FG or Ctx488 positive FMNs per section in unlesioned or lesioned FN was quantified at 23 d.p.l. in wt (grey circles) and ATF3-deficient (white squares) mice. FG positive neurons were significantly reduced in lesioned FN of mutant compared with wt animals. Each circle or square in (r) represents one mouse. The dotted lines represent the margins of the FN. L, lateral; M, medial; D, dorsal; V, ventral. Data are presented as mean ± s.d. *p ≤ 0.05. Scale bar (b–q) = 100 µm.
Figure 3.
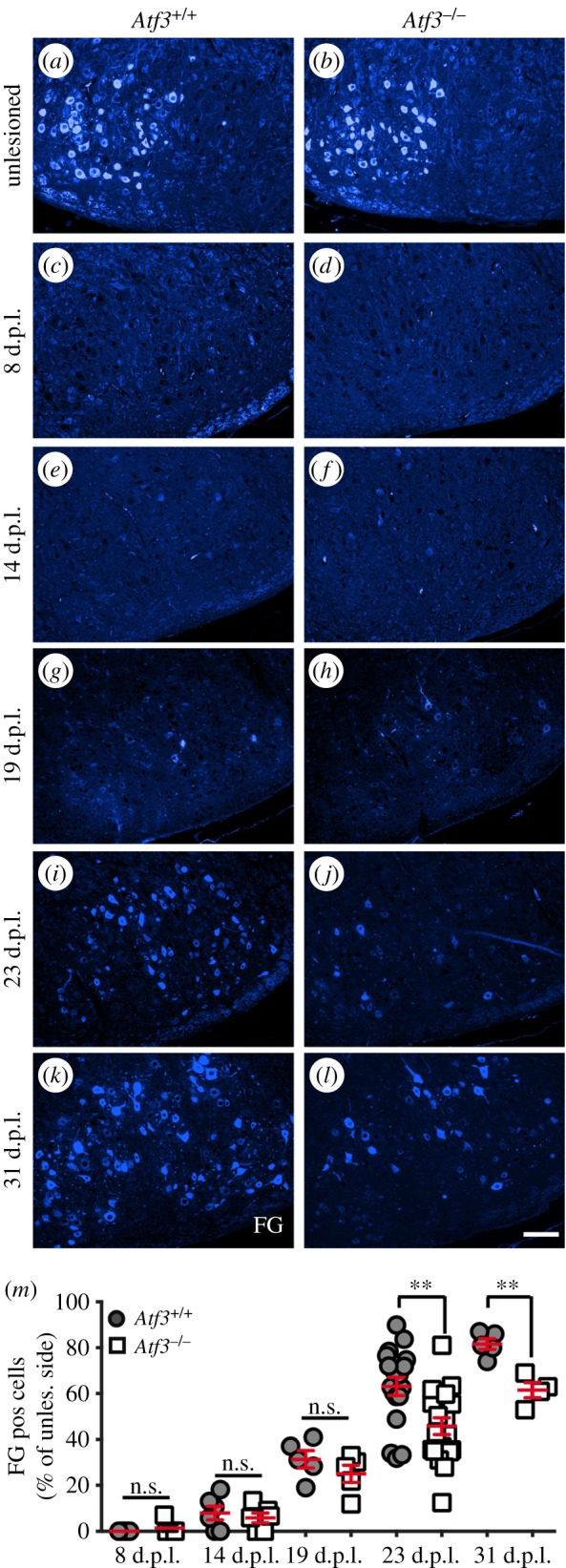
Time course of axonal regeneration in wt and Atf3 mutant mice. (a,b) FG positive FMNs in the unlesioned FN of wt (a) and Atf3−/− (b) mice are restricted to the lateral half of the FN. (c–l) Abundance of FG labelled motoneurons in the FN of wt and Atf3−/− mice is depicted at several timepoints after lesion including 8 (c,d), 14 (e,f), 19 (g,h), 23 (i,j) and 31 (k,l) days post-lesion (d.p.l.). (m) Regeneration of FG positive FMNs was analysed at five different timepoints after injury (8, 14, 19, 23 and 31 days). The extent of regeneration is depicted as a percentage by calculating the ratio between FG positive cells counted on the unlesioned and lesioned FN. At 23 and 31 d.p.l., the percentage of FG positive neurons on the lesioned side was significantly reduced in Atf3−/− animals compared with wt. Each circle or square in (m) represents one mouse. Data are presented as mean ± s.d. **p ≤ 0.01. Scale bar (a–l) = 100 µm.
2.3. Cell biology
Mouse DRG neurons were derived from adult (seven to nine weeks old) wt or Atf3 mutant mice. For neurite growth experiments, 5 × 103 neurons were plated on poly-l-lysine (PLL; 100 µg ml−1) and laminin (5 µg ml−1)-coated 12 mm coverslips. For qPCR experiments, 2.5 × 104 cells were plated on PLL (10 µg ml−1) and laminin (2 µg ml−1)-coated 12-well plates. For chromatin immunoprecipitation (ChIP) experiments, 5 × 106 primary P3–P5 postnatal cerebellar neurons were plated on PLL/laminin-coated 10 cm dishes. The DRG culture medium consisted of neurobasal medium (Gibco) supplemented with B27, glutamine and nerve growth factor (NGF) (50 ng ml−1) if not indicated otherwise. Camptothecin was added at 2 µM for 24 h. Recombinant peptides for mouse VIP (Tocris; no.1911), human gastrin releasing peptide (GRP) (Tocris; no.1789), mouse galanin (Tocris; no.2696) and mouse Wnt2b (R&D systems; no.3900-WN-025) were added at 1 nM (VIP, GRP, galanin) and 1 µg ml−1 (Wn2tb) for 24 h to the cultures. Neurons were cultured for 24 h for neurite growth assays or 3 days for all qPCR/ChIP experiments.
Adenoviral (AV) particles expressing ATF3 (AV-ATF3) or GFP (AV-GFP) were produced in-house at Ulm University. Viral particles (2.5 × 105 for AV-ATF3 and AV-GFP) were added at 2 h after plating and left on cultures throughout the entire duration of the experiment. Electroporation of DRG neurons was performed with 3 µg DNA and 100 µl electroporation solution (Mirus). The CCL2-Cherry expression vector was described previously [45].
2.4. Immunostaining
Cells were fixed for 15 min in 4% PFA/5% sucrose/PBS, permeabilized for 5 min in 0.1% Triton-X-100/PBS and blocked for 30 min in 2% BSA/PBS. Neurons were stained with antibodies directed against βIII tubulin (mouse, 1 : 1000; Covance) incubated overnight at 4°C. The primary antibodies were detected by Alexa488 conjugated secondary antibodies (1 : 1500; Invitrogen). F-actin was stained with Texas Red-X phalloidin (1 : 100; Biotium) added to the secondary antibody solution.
Brains, human nerves or mouse facial nerves were fixed in 4% formaldehyde (FA) for 3 days, followed by preparation of 5 µm paraffin microtome slices. For anti-CD45 and anti-CD4 stainings, 20 µm cryostat sections of unfixed brains were used. Sections were post-fixed with 4% PFA for 10 min and processed for immunofluorescence staining according to standard protocols. Immunohistochemistry was performed using biotin (1 : 500; Vector Laboratories) or Alexa Fluor (1 : 500; Invitrogen) conjugated secondary antibodies and peroxidase-based detection systems using the ABC complex (Vector Laboratories) and DAB as substrate. Primary antibodies included anti-ATF3 (rabbit, 1 : 2000, # HPA001562; Atlas Antibodies), anti-FG (rabbit, 1 : 5000, AB153; Millipore), anti-ionized calcium-binding adapter molecule 1 (IBA1) (rabbit, 1 : 1000, 019-19741; WAKO) anti-GFAP (mouse, 1 : 1000, sc-33673; Santa Cruz Biotechnology), anti-CD45 (mouse, 1 : 100; BD Pharmingen), anti-CD4 (rat, 1 : 100; BD Pharmingen), anti-galanin (rabbit, 1 : 1000, T-4334; Bachem), anti-S100 (mouse, 1 : 200; Abcam) and anti-CCL2 (rabbit, 1 : 100, 500-P113; Peprotech). Informed consent was obtained from all patients included in the study.
2.5. Transmission electron microscopy
Facial nerve parts adjacent to the transection position were harvested 3 days after lesion. In addition, unlesioned control nerves were collected of the same animals. Samples were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) with 1% saccharose overnight and washed with PBS and contrasted with 2% aqueous osmium tetroxide for 1 h. Increasing 1-propanol concentration series (30, 50, 70 and 90%) were used to dehydrate samples, contrasted with saturated alcoholic uranyl acetate solution for 30 min at 37°C and embedded in Epon resin. The sections were cut using the Ultracut UCT ultramicrotome (Leica) using a diamond knife (Diatome, Biel, Switzerland) and mounted on copper grids for transmission electron microscopy (TEM). Samples were analysed at an acceleration voltage of 80 kV in a transmission electron microscope (Zeiss EM 10) after contrasting the sections with lead citrate for 1 min. We analysed a total of 365, 354, 145 and 230 axons in wt unlesioned (n = 3), Atf3 mutant unlesioned (n = 3), wt lesioned (n = 3) and Atf3 mutant lesioned (n = 4) animals, respectively. Sprouts were identified by a bulb-shaped tip morphology with a neck structure clearly separated from the parental axon according to sprout structures described in a previous EM study [46]. To distinguish axonal sprouts from axonal protuberances, axonal sprouts had to show a clear budding off morphology from the parental axon (examples are labelled with arrows in figure 4). The number of axonal sprouts was normalized to the total number of axons present in each frame. For each nerve, at least eight different pictures at 3000× magnification were analysed.
Figure 4.

Analysis of facial nerve sprouting by transmission electron microscopy. (a–d) Three days after injury, unlesioned (a,b) and lesioned (c,d) facial nerves of wt (a,c) and Atf3 mutant (b,d) animals were analysed by EM for the presence of nerve sprouts. In unlesioned nerves, only few axons contained sprout-like structures (a,b). By contrast, lesioned axons contained axonal sprouts (arrows in c,d) with a neck and bulb-like terminal structure (c,d). (e) Quantification of the percentage of axons with sprouts in relation to the total number of axons. In lesioned nerves of wt mice, the number of axons with sprouts was significantly elevated compared with nerves derived from ATF3-deficient animals. Each circle or square represents one mouse. Data are presented as mean ± s.d. *p ≤ 0.05. Scale bar (a–l) = 5 µm.
2.6. Transcriptomics, laser microdissection and quantitative real-time PCR (qPCR)
FN of individual animals were dissected from 300 µm brainstem sections prepared with a tissue chopper using tungsten needles. Total RNA was isolated with the mini RNeasy kit (Qiagen) or peqGOLD MicroSpin Total RNA Kit (Peqlab). For transcriptomics, we subjected a total of four samples (wt lesion, wt no lesion, Atf3 mutant lesion and Atf3 mutant no lesion) to microarray analysis. In each sample, four animals were pooled to obtain sufficient amounts of mRNA (16 animals in total). Thus, although four animals were pooled for one sample, the number of biological replicates is one in figure 6 and no statistical testing was possible. However, four independent biological replicates per condition were used to confirm transcriptomics results in the subsequent qPCR analysis (figure 7). Here, statistical testing was included.
Figure 6.

Transcriptomics analysis of wt and ATF3-deficient facial motor nuclei. (a) Heatmap of genes most strongly (more than sevenfold) altered by facial nerve injury in wt or ATF3-deficient mice. The fold induction in wt and Atf3 mutant mice was calculated in relation to expression values on the uninjured site. Genes depicted in black were not obviously affected by ATF3 deficiency. Expression of genes labelled in red or blue colour was reduced or elevated in Atf3 mutant compared to wt mice, respectively. Green colours in bars depict weakest, whereas red colours indicate strong expression levels. (b) In wt mice, 849 genes out of over 35 000 genes present on the microarray were regulated more than twofold. The number of genes upregulated by injury (594) exceeded those downregulated (255) more than twofold. (c,d) STRING networks of protein clusters identified in wt mice upon facial nerve injury. A first protein cluster (c) revealed known ATF3 interaction partners such as Cdkn1a and Gadd45a and further interactions of ATF3 with for instance components of integrin signalling (Itgam, Itgb2 Itga7). A second protein network (d) consisted of neuropeptide signalling components such as neuropeptides (Pacap, Vip, Ngf, Npy, Grp, Gal) and receptors (Avpr1a, Vipr2). Genes labelled in red were affected by ATF3 deficiency.
Figure 7.

ATF3 regulates injury-related expression of neuropeptides and other genes. (a–i) Unlesioned and injured FN of Atf3+/+ and Atf3−/− mice were subjected to qPCR analysis after 3 days of injury to quantify mRNA abundance of primers indicated. In wt mice (grey bars), facial nerve injury resulted in induction of Sprr2j (a), Vip (b), Ngf (c), Wnt2b (d), Galanin (e), Grp (f), Timp1 (g) and the known ATF3 target gene Hsp27 (h). In contrast to wt mice, induction of Sprr2j, Vip, Ngf, Wnt2b, Galanin, Grp and Hsp27 mRNA abundance was reduced upon facial nerve lesion in Atf3 mutant mice (white bars). Timp1 mRNA induction was more pronounced upon ATF3 loss-of-function (g). The Vip2 receptor (Vipr2) was downregulated by facial nerve injury in wt and ATF3-deficient mice (i). Numbers in bars in (b) reflect independent biological replicates for experiments in (a–i). (j–l) Confirmation of reduced galanin expression in ATF3-deficient mice upon facial nerve injury. Deafferented wt (j) and ATF3-deficient (k) FN were stained with anti-galanin directed antibodies. In wt mice (j), galanin localized in secretory vesicle-like structures (see inset) of FMNs (some labelled with an arrow). The number of galanin immunoreactive FMNs was reduced in Atf3 mutant mice (k). (l) Quantification of galanin positive neurons without and 3 and 12 days after lesion. Data are presented as mean ± s.d. **p ≤ 0.01. Scale bar (j,k) = 50 µm; inset = 5 µm.
For microarray analysis, 200 ng total RNA was used as starting material and 5.5 µg ssDNA per hybridization (GeneChip Fluidics Station 450; Affymetrix, Santa Clara, CA, USA). The total RNAs were amplified and labelled following the Whole Transcript (WT) Sense Target Labeling Assay (http://www.affymetrix.com). Labelled ssDNA was hybridized to Mouse Gene 1.0 ST Affymetrix GeneChip arrays (Affymetrix). The chips were scanned with an Affymetrix GeneChip Scanner 3000 and subsequent images analysed using Affymetrix® Expression Console™ Software (Affymetrix). Raw feature data were normalized and intensity expression summary values for each probe set were calculated using robust multiarray average [47].
For laser microdissection, brains were dissected and frozen without fixation. Brain cryosections (20 µm) containing the FN were prepared on membrane slides (Zeiss MembraneSlide-1.0 PEN Cat.#: 415190-9041-000), fixed for 1 min with 70% EtOH and stained with a 1% cresyl-violet solution in 50% ethanol/DEPC treated water. Motoneurons or neighbouring tissue of the FN were collected using the PALM Microbeam Laser capture equipment on a Zeiss Axiovert 200 M microscope frame. Cells from approximately 15 sections per mouse were laser microdissected. RNA was prepared using the RNeasy Plus Micro kit (Qiagen; no.74034). All materials and solutions used were treated to be RNAse free.
For cDNA synthesis, reverse transcription was performed with approximately 150–400 ng of RNA using reverse transcriptase (Promega) and random hexamers. qPCR was performed on a Roche LightCycler® 480 with the SYBR Premix ExTaq (Tli RNase H Plus) PCR Master Mix (TaKaRa). Expression was determined in relation to Gapdh RNA levels. Primer sequences are provided in the electronic supplementary material.
2.7. Chromatin immunoprecipitation
We followed a previously published protocol [48]. Primary P3–P5 postnatal cerebellar cultures were fixed for 15 min at room temperature with 1% FA in PBS. We used 2 µg ml−1 anti-ATF3 (Santa Cruz; SC-335, rabbit) or IgG antibody for each IP. After purification of DNA (PCR purification kit, Qiagen), 2 µl of each input or IP were subjected to qPCR with primers provided in the supplement. Ct values obtained with ATF3 or IgG ChIP were normalized to the respective input values.
2.8. Statistical analysis
Numbers (n) of cell cultures or animals are indicated in figure bars or text. For cell culture, at least three independent experiments were performed and at least 30 neurons were analysed in each experiment. Neurite growth was quantified with the NeuriteTracer plugin of ImageJ [49]. For quantification of immune cells, we used the Keyence BZ Analyzer software with the Hybrid cell count tool. For this, an ellipse of 950 × 750 µm was placed over the FN area. Objects below 5 µm2 were discarded. For IBA-1/CD45 positive or GFAP positive cells, a threshold of 50 or 80, respectively, was used. The software calculated the total area (in square micrometres) of all objects positive for a given marker above these thresholds.
Statistical significance was calculated using Prism6 software with two-way ANOVA multiple comparison tests, with *, **, *** indicating p ≤ 0.05, 0.01 and 0.001, respectively; s.d. is provided if not mentioned otherwise.
3. Results
3.1. Temporal and spatial profile of ATF3 induction upon facial nerve axotomy
So far, ATF3 upregulation after injury was demonstrated at single timepoints after facial nerve lesion [37–42]. We analysed ATF3 expression in FMNs at several timepoints after unilateral axotomy in wild-type (wt) mice (figure 1). In the absence of a lesion (figure 1a) as well as at 6 h post-lesion (h.p.l.; figure 1b), no ATF3 signal was detectable in FMNs (quantified in figure 1g). The maximal number of ATF3 positive FMNs was observed at 3 d.p.l. (figure 1c) and was already slightly weaker at 7 d.p.l. (figure 1d). After 21 d.p.l., ATF3 was no longer present in FMNs (figure 1e,g).
Besides wt mice, we also analysed Atf3 mutant mice (figure 1f,h). At 3 d.p.l., ATF3 protein levels were absent in Atf3 mutant mice (figure 1f). In agreement, qPCR analysis of mRNA levels revealed no Atf3 induction in Atf3 mutant FN at 3 d.p.l. (figure 1h). By contrast, Atf3 mRNA was strongly upregulated upon lesion in wt mice (figure 1h), similar to ATF3 protein (figure 1c). In this injury model, other ATF/CREB family members (Atf2, Atf4, Atf6) were not modulated by nerve injury in wt mice (figure 1h), as opposed to other injury models [50]. Also, the ATF/CREB family member Creb was not altered by ATF3 deficiency (data not shown). Further, no compensatory upregulation of these ATF/CREB members was observed in Atf3 mutant mice (figure 1h).
Besides FMN intrinsic responses, repair programmes are also initiated in Schwann cells [51,52]. Because ATF3 is upregulated upon injury in Schwann cells of the sciatic nerve [53,54], we wondered whether ATF3 expression is also induced in facial nerve Schwann cells. Indeed, we observed an increase in ATF3 in facial nerve Schwann cells in wt mice at 3 d.p.l. (see arrows in figure 1j) compared with the unlesioned facial nerve (figure 1i). However, the number of ATF3 positive cells and signal intensity of ATF3 in the lesioned facial nerve were weaker compared with injured FMNs (figure 1g).
Overall, we observed a strong ATF3 upregulation in FMNs and to a weaker extent also in Schwann cells occurring in the first week after PNS injury.
3.2. Axonal regeneration is reduced in Atf3 mutant mice
We analysed the extent of axonal regeneration in adult wt and Atf3 mutant mice at several timepoints after unilateral facial nerve transection (figures 2 and 3). Responses in the spared contralateral FN served as intra-animal control. Similar to the retino-tectal system [55], FMNs are topographically organized. Here, different FMN populations, e.g. those connected to the eyelid, whisker pad and lower jaw, are confined to specific FN subdomains [56–58].
We analysed the regeneration potential of these different FMN populations and the role of ATF3 in this regeneration process with FMN back-labelling experiments using three different fluorescent tracers (figure 2a(i–iii)). FMNs innervating the eyelid, whiskers and lower jaw were retrogradely labelled through DiI (red), FG (blue) and choleratoxin Alexa 488 (Ctx488, green) injection, respectively (figure 2a(iii)). Injections were performed on the unlesioned and lesioned sides to determine the FMN topographic map before and after lesion. Impaired tracer transport along transected facial nerves to FMNs indicates loss of axonal re-growth to their original postsynaptic muscle targets [13,16]. Of note, this retrograde tracing method allows following of the regeneration process only after facial muscles have been re-innervated by regenerating axons. By contrast, first axon outgrowth responses before reaching muscle targets are not detected by this method.
On the unlesioned side, FMNs were topographically localized as reported before [56–58]. DiI positive FMNs connected with the eyelid occupy two domains (arrows figure 2b,c), whereas FMNs connected to the lower jaw were consistently localized to the dorsomedial FN quarter (arrows in figure 2f,g). Finally, FG positive FMNs innervating the whisker pad were restricted to the lateral half of the FN (figure 2j,k). This FG positive FMN subtype representing the whiskers has the highest numbers in the FN compared with DiI and Ctx488 positive FMNs (figure 2n or o; quantified in r). Thus, eyelid and lower jaw are represented by fewer FMNs, in line with previous reports [56–58]. Comparing FMN localization and numbers between wt (figure 2n) and ATF3-deficient (figure 2o) mice, no obvious differences were discernible on the unlesioned side (quantified in figure 2r).
Next, we inspected the lesioned FN. After 23 d.p.l., a disorganization of the FN topographic map was observed for all three tracers (figure 2p,q). Regenerating FMN axons initially also sprout into topographically wrong target areas [56–58]. Here, they pick up different tracers, resulting in back-labelling of FMNs in topographically aberrant positions. At much later timepoints (not covered in this study), such aberrantly formed sprouts or synapses are eliminated [56–58]. When comparing the localization of individual FMNs within this disorganized topographic map, no obvious differences were observed after lesion between wt (figure 2d,h,l,p) and Atf3 mutant (figure 2e,i,m,q) animals.
Subsequently, we quantified, the number of regenerating FMNs of these different FMN subtypes by counting DiI, FG and Ctx488 positive FMNs on the lesioned side of wt and Atf3 mutant animals (figure 2r). Interestingly, at 23 d.p.l., DiI positive FMNs connected to the eyelid regenerated almost to 100%, however with no difference between genotypes (figure 2b–e; n–q; quantification in r). In contrast with this, only approximately 30% of FMNs innervating the lower jaw regenerated in both wt and mutant animals at the same timepoint (figure 2f–i; quantified in r). Here, overall neuron numbers on the lesioned side were low (figure 2h,i), rendering quantification more difficult (figure 2r). Finally, we inspected the largest FMN population and observed that in wt mice approximately 60% of FG positive FMNs were reconnected to the whisker muscles (figure 2j–m,r). Importantly, for this FMN subtype, we observed a significant difference in axon regeneration between wt and ATF3-deficient animals. Now, only an average 35% of neurons was FG positive in Atf3 mutant animals (figure 2m,q,r). This finding indicates a role of ATF3 in stimulating axon regeneration of lesioned FMNs.
In order to analyse axon regeneration in Atf3 mutant animals more comprehensively, we provide a temporal regeneration profile including a total of five timepoints after lesion (8, 14, 19, 23 and 31 d.p.l.), focusing on FG positive FMNs (figure 3). For quantification, a percentage of regenerated neurons was determined, by calculating the ratio between FG positive FMNs on the lesioned and unlesioned FN (figure 3m). Inspection of wt mice revealed a correlation of the regeneration efficiency of FG positive FMNs with the timespan allowed for regeneration after injury. At 8 d.p.l., no FG positive neurons were observed (figure 3c), whereas 10 and 30% of FMNs were tracer positive at 14 (figure 3e) and 19 (figure 3g) d.p.l. in wt mice, respectively (figure 3m). Thus at injury timepoints before 14 days, axons may have started regeneration but have not yet reached their whisker muscle targets. At later timepoints, more robust re-innervation of facial muscles by regenerating axons was observed in wt mice. Now, 65 and 82% of FMNs were FG positive in the lesioned FN, at 23 (figure 3i) or 31 (figure 3k) d.p.l., respectively (figure 3m).
In Atf3 mutant mice, no FG positive FMNs were observed at 8 d.p.l., similar to wt mice (figure 3d). At 14 d.p.l. injury, no difference in FG positive FMNs was observed between wt and ATF3 lacking mice (figure 3f). At 19 d.p.l., the percentage of regenerated FMNs in Atf3 mutant mice was slightly decreased compared with wt (figure 3h,m). This difference was further pronounced at 23 and 31 days post lesion. At these timepoints, 45 and 60% of FMNs were regenerating at 23 and 31 d.p.l., respectively, resulting in a significant reduction of FG positive FMNs at both timepoints in Atf3 mutant compared with wt mice (figure 3j,l,m).
So far, axonal regeneration responses were only analysed in the FN. Besides the FN, we investigated the presence of axonal sprouts in the facial nerve 3 days after injury (figure 4; n ≥ 3 nerves per condition).
In unlesioned wt (figure 4a) and Atf3 mutant (figure 4b) facial nerves, approximately 10% of all axons had clearly distinguishable sprout-like structures consisting of a bulb and a neck separated from the parental axon (quantified in figure 4e). Three days after facial nerve lesion, the number of nerve sprouts was elevated in wt (arrows in figure 4c) and Atf3 mutant (figure 4d) animals. Now, approximately 30% of all lesioned wt axons protruded sprout-like structures, whereas only 16% of nerves derived from ATF3-deficient animals showed these protrusions (figure 4e). This finding on axonal sprouts in the facial nerve suggests an influence of ATF3 on facial nerve regeneration at an early timepoint after nerve injury.
Taken together, a first analysis of ATF3-deficient mice showed reduced PNS axon regeneration.
3.3. Facial motoneuron number and immune responses are not affected by ATF3 ablation
An obvious factor affecting the outcome of axonal regeneration would be differential FMN loss between wt and Atf3−/− mice. Using Nissl staining to visualize all FMNs present in a FN, we did not observe major differences between genotypes at all injury timepoints analysed (figure 5a–d; quantified in m). Thus, differential FMN loss is not obviously responsible for alterations in axonal regeneration observed in ATF3-deficient mice (figures 2 and 3).
Figure 5.

FMN numbers and immune responses are unaltered in Atf3−/− animals. (a–d) Nissl staining was employed to label all FMNs present in FN. No major FMN loss was observed upon lesion in wt (c) or Atf3−/− (d) mice. (e–l) Microglia (e–h) and astrocytes (i–l) were labelled at 8 d.p.l for IBA-1 and GFAP expression, respectively. In the uninjured FN of wt (e,i) or ATF3-deficient (f,j) animals, no IBA-1 positive microglia (e,f) or GFAP positive astrocyte (i,j) cell infiltration was observed. By contrast, upon injury (g,h and k,l) there was a comparable microglia and astrocyte activation in wt (g,k) and Atf3 mutant (h,l) animals. (m–p) CD4+ cells were found on the unlesioned side of wt (m) and Atf3 mutant (n) animals. CD4+ T cells were slightly upregulated upon lesion in wt (o) as well as Atf3 mutant (p) FNs. (q–t) CD45, a marker for CNS infiltrating monocytes, was nearly absent on the unlesioned FN (q,r). Facial nerve lesion increased CD45 positive cells on the FN of wt (s) and Atf3 mutant (t) animals. Insets in (e–t) show higher magnifications of areas marked with a dashed box. (u–y) The percentage of Nissl positive FMNs on the lesioned FN (control side set to 100%; u), total area (in square micrometres) of GFAP positive astrocytes (v), number of IBA1 positive microglia (w), number of CD4+ cells (x) or total area (in square micrometres) of all CD45+ cells (y) per section were counted and plotted against the different timepoints. Each circle or square in (u–y) represents one mouse. Data are presented as mean ± s.d. Scale bar (a–t) = 100 µm; insets (e–t) = 5 µm.
We also investigated whether ATF3 ablation interfered with induction of a PNS lesion-associated immune response (figure 5e–l,v,w). Upon facial nerve injury, GFAP positive astrocytes and IBA-1 positive microglia are reported to infiltrate the lesioned but not the intact FN [40]. Accordingly, almost no astrocytes or microglia cells were observed in the unlesioned FN of wt (figure 5e,i) or ATF3-deficient (figure 5f,j) mice. By contrast, both brain-resident immune cells were present in the lesioned FN at 8 d.p.l. Astrocyte and microglia numbers were indistinguishable between wt (figure 5g,k) and Atf3 mutant (figure 5h,l) mice (figure 5v,w). A similar finding was observed at other injury timepoints (figure 5v,w).
Besides brain-resident immune cells we inspected CD4 and CD45 positive cells, labelling T-cells and CNS infiltrating monocytes, respectively (figure 5m–t,x,y). In unlesioned FN, CD4+ cells were found in wt (figure 5m) and Atf3 mutant (figure 5n) mice. After three days of lesion, CD4+ cells were slightly but not significantly upregulated on the lesioned side. However, no difference was observed between genotypes (figure 5o,p; quantified in x). CD45 positive cells were nearly absent in the unlesioned FN (figure 5q,r). Upon lesion, CD45 positive cells were upregulated on the lesioned side (figure 5s,t), however in a comparable manner between wt and Atf3 mutant mice (figure 5y).
This suggests no obvious impact of ATF3 deletion on the injury-mediated brain immune response.
3.4. Genome-wide identification of ATF3-dependent genes upon facial nerve injury
The Atf3 gene encodes a RAG-associated TF that may induce expression of further effector RAGs upon injury. So far, no genome-wide data are available on the impact of ATF3 loss-of-function on gene expression. In order to analyse the impact of ATF3 deletion on gene expression, we employed transcriptomics with unlesioned and lesioned FNs of wt and Atf3 mutant mice 3 d.p.l. (figure 6).
In the lesioned FN of wt mice, we observed an induction of a RAG encoding gene regulatory network encompassing several hundreds of genes. The heatmap in figure 6a depicts those genes being up or downregulated in injured wt mice by at least a factor of seven. Notably, the RAG cluster induced by injury in wt mice in this study was almost identical to a previous independent transcriptomics study [40], thereby underscoring specificity and reproducibility of results presented in this study. Prototypical RAGs most strongly induced by nerve injury were Sprr1a, Galanin, Npy, Vip, Atf3, Sprr2j, Cdkn1a (p21) and Itga7 (depicted in figure 6a). In addition, other genes including Gpr151, Wnt2b, Grp, TIMP metallopeptidase Inhibitor (Timp), Gpr133 and Serpinb2, so far not considered as prototypical RAGs, were also induced by facial nerve injury in wt mice (figure 6a).
Out of the more than 35 000 transcripts present on the microarray, the majority of genes (34 602) was largely unchanged or modulated only below a twofold change (less than or equal to twofold) between control and lesioned FN (figure 6b). Among the genes regulated more than twofold, 594 genes were induced, whereas only 255 genes were downregulated upon lesion in wt mice 3 days after injury (figure 6b). The ratio between gene up and downregulation was even more pronounced when focusing on genes with at least sevenfold changed mRNA abundance in wt mice (depicted in figure 6a). Here, 40 genes were upregulated, in contrast with a single gene (Vipr2) being downregulated. This suggests that nerve injury favours gene induction over gene repression.
Employing STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis of the top regulated genes, ATF3 was directly or indirectly connected with other genes induced by facial nerve injury in wt mice (figure 6c). These included integrin-associated signalling members such as Itga7a, Itgb2 and Itgam and the cell cycle regulator p21 (Cdkn1a). STRING analysis and GO (Gene Ontology) annotation uncovered a further module of functionally related genes modulated by facial nerve injury (figure 6d; electronic supplementary material, table S1). This cluster contained members of neuropeptide signalling pathways such as Vip, Ngf, Galanin, Npy, Grp, Pacap (Adcyap1) and Avpr1a all of which, except for the VIP receptor Vipr2, being strongly induced by facial nerve injury in wt mice (figure 6a,d).
Inspection of Atf3 mutant mice revealed that many genes modulated by facial nerve injury in wt mice were in general also induced upon ATF3 deletion' however not to the same extent. For instance, approximately 25% of the strongest regulated genes (at least sevenfold) depicted in figure 6a were at least 1.5-fold altered between lesioned wt and Atf3 mutant animals. In addition, out of the 594 and 255 genes being twofold up or downregulated in wt mice (figure 6b), 135 and 55, respectively were altered at least twofold in Atf3 mutant mice (electronic supplementary material, table S1). Hence, approximately 20–25% of all genes regulated by facial nerve injury in wt mice were to a certain extent ATF3 dependent at 3 days post-injury.
Besides this general impact of ATF3 on the RAG response, specific RAGs were strongly affected by ATF3 deficiency. For instance, Galanin, Sprr2j but also Wnt2b, Grp, Vip, Serpinb2, Itgb2 and Ngf had reduced mRNA abundance in severed Atf3 mutant compared to wt FN (labelled in red in figure 6a). As mentioned above, we noted induction of a cluster of eight neuropeptide signalling encoding genes in injured wt mice (figure 6a,d). Notably, expression of six of these genes (Vipr2, Vip, Ngf, Grp, Gal, Adcyap1) was reduced in Atf3 mutant tissue suggesting a role of ATF3 in wt mice for induction of these RAGs (red in figure 6d). Besides ATF3-mediated gene induction, other genes such as Timp1, nephroblastoma overexpressed gene (Nov) (also called Ccn3) and Ccl2 had elevated mRNA levels in Atf3 mutant mice compared with wt (labelled in blue in figure 6a). This indicates ATF3-dependent gene repression of these genes in wt mice.
In a next step, transcriptomics data were corroborated by qPCR with independent cDNAs derived from different wt and Atf3 mutant animals (figure 7). In agreement with microarray results (figure 6a), mRNA abundance of Sprr2j, Vip, Ngf, Wnt2b, Galanin and Grp was strongly augmented in deafferented FN of wt mice but significantly reduced in lesioned Atf3 mutant FN (figure 7a–f). We also confirmed microarray data for Vipr2 and Timp1 (figure 7i,g).
In principal, we would expect that the identified RAG response occurs predominantly in injured FMNs. However, besides FMNs, other cell types, e.g. immune cells (figure 5), were also present in the lesioned FN and might contribute to the transcriptional changes observed. In order to test, whether FMNs were the main cell type responsible for transcriptional changes after facial nerve injury, we performed laser microdissection experiments (electronic supplementary material, figure S1). Here, FMNs of the unlesioned and lesioned FN were dissected 3 d.p.l. In addition, we included neighbouring tissue on the unlesioned and lesioned sides not containing FMNs (electronic supplementary material, figure S1). Indeed, inspection of eight genes strongly induced by facial nerve transection in our transcriptomics analysis (figure 6) revealed strong mRNA upregulation only in the sample containing FMNs on the lesioned side but not in all other three samples (electronic supplementary material, figure S1). This suggests that the majority of transcriptional changes observed in the microarray analysis (figure 6) took place in FMNs and not surrounding cells.
So far, we focused on transcriptional changes after 3 days post-injury (figure 6; electronic supplementary material, figure S1). In order to analyse the time window of injury-regulated gene induction, we inspected lesioned FNs at 7 and 14 days post-injury by qPCR focusing on eight injury-regulated genes (electronic supplementary material, figure S2). After 7 days of injury, mRNA levels of the majority of genes analysed (seven out of eight) were still above control levels and reached significance for Galanin, Ngf, Grp, Sprr1a and Sprr2j (electronic supplementary material, figure S2). One week later, at 14 d.p.l., mRNA abundance was clearly decreased and only selected genes such as Galanin, Grp and Sprr1a were elevated (electronic supplementary material, figure S2). Thus, a facial nerve injury-induced RAG response appears to last for at least 7 days after lesion, with mRNA abundance of selected RAGs persisting up to 14 days post-injury.
In addition, we inspected the reported ATF3 target gene Hsp27 [12,59]. In line with previous reports demonstrating Hsp27 induction by ATF3 overexpression [12], we observed reduced Hsp27 abundance in ATF3-deficient animals upon injury (figure 7h). We also analysed other prototypical RAGs encoding for transcription factors (c-Jun, Smad1, Stat3, Sox11) or effector RAGs (Cap23, Scg10, Gap43, Npy). All these RAGs were induced upon facial nerve lesion, however indistinguishably between wt and Atf3 mutant mice except for Cap23 and Nov (electronic supplementary material, figure S3 and table S1).
In order to confirm mRNA changes observed for the gene encoding the galanin neuropetide at the protein level, lesioned FNs were stained with galanin-directed antibodies at 3 d.p.l. (figure 7j,k). In wt mice, many deafferented FMNs were positive for galanin (arrows figure 7j). Higher magnification suggested the presence of galanin in secretory vesicle-like structures (inset figure 7j). In contrast to this, the number of galanin positive neurons was reduced in ATF3-deficient FN (figure 7k) at two different timepoints (figure 7l).
Overall, this first genome-wide survey in Atf3 mutant mice uncovered regulation of several neuropeptide-encoding genes by ATF3 as well as other potentially novel ATF3 target genes including Wnt2b.
3.5. ATF3 overexpression in peripheral nervous system neurons induces gene expression
Transcriptomics data indicated a stimulatory ATF3 function in transcriptional regulation of neuropeptide expression during PNS axon regeneration (figures 6 and 7). In further experiments, we analysed whether ATF3's function in axonal regeneration might be accomplished through regulation of these neuropeptides, particularly focusing on galanin, Grp, Vip and Ngf. In addition, we included Wnt2b, because several Wnt family members mediate axon growth and regeneration [60,61]. To test whether genes affected by ATF3 loss-of-function were direct target genes, ATF3 overexpression and ATF3-directed ChIP were employed in wt and ATF3-deficient primary neurons (figure 8).
Figure 8.

ATF3 mediates neuropeptide and Wnt2b expression in primary PNS neurons. (a–d) Adult wt or ATF3-deficient mouse DRG neurons were infected with adenoviral (AV) particles resulting in GFP (control) or ATF3 expression. mRNA levels of Atf3 (a), Wnt2b (b), Galanin (c) and Grp (d) were analysed by qPCR. Viral infection strongly enhanced Atf3 mRNA abundance in wt and Atf3 mutant neurons (a). Wnt2b (b), Galanin (c) and Grp (d) mRNA levels were augmented upon viral ATF3 overexpression in wt and Atf3 mutant DRG neurons. (e,f) Primary wt neurons overexpressing GFP or ATF3 were subjected to ChIP analysis with anti-ATF3 or IgG (control) antibodies. ATF3 occupancy at potential ATF3 binding sites of the Galanin (e) and Grp (f) promoter was tested with qPCR. ATF3 promoter occupancy was observed in ATF3-overexpressing samples only in the presence of anti-ATF3 but not IgG antibodies suggesting ATF3 binding at the Galanin (e) and Grp (f) promoter. Numbers in bars reflect independent numbers of experiments. Data are presented as mean ± s.d. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
In the first set of experiments, ATF3 was overexpressed in adult mouse DRG neurons through adenovirus (AV) mediated infection (figure 8a–d). We used adult mouse DRG neurons because adult FMNs did not grow in culture (data not shown). Similar to FMNs, DRG neurons project axons to the periphery and therefore should recapitulate responses of FMNs as closely as possible. After 3 days in culture, mRNA levels of potential ATF3 target genes were quantified by qPCR (figure 8a–d). ATF3 was successfully overexpressed in both wt and Atf3 mutant DRG neurons comparing AV-ATF3 with AV-GFP-infected neurons (figure 8a). Sprr2j, Ngf and Vip mRNA levels were not affected by ATF3 overexpression, suggesting that those genes were not under direct ATF3 control (data not shown). By contrast, Wnt2b (figure 8b), Galanin (figure 8c) and Grp (figure 8d) mRNA levels were induced by ATF3 overexpression in both wt and ATF3-depleted DRG neurons. Therefore, ATF3 overexpression upregulated Wnt2b, Galanin and Grp expression (figure 8), whereas ATF3 deficiency decreased abundance of those genes (figures 6 and 7).
Next, we analysed whether ATF3 may directly bind to promoter regions of Wnt2b, Galanin and Grp. We searched the MotifMap database [62] for ATF3 binding motifs such as an activator protein 1 (AP1), ATF3 or CREB binding motif within 5 kb up and downstream of the transcriptional start site. Galanin and Grp contained a potential AP1 and CREB binding site, respectively. Wnt2b contained no obvious ATF3 recognition site and was not further analysed by ChIP.
ATF3 promoter occupancy was analysed in primary neurons infected with AV-ATF3 or, as control, AV-GFP, followed by ChIP with anti-ATF3 or, as control, anti-IgG directed antibodies (figure 8e,f). Subsequently, qPCR was employed with primers spanning the respective potential TF binding motif. ATF3 bound to the AP1 recognition site of Galanin (figure 8e) and the CREB binding site of Grp (figure 8f) in ATF3 antibody directed ChIP samples of ATF3-expressing neurons. In contrast to this, no specific ATF3 promoter occupancy was observed in GFP-expressing neurons with the anti-ATF3 antibody or in neurons expressing ATF3 subjected to an anti-IgG directed ChIP (figure 8e,f). These findings suggest direct ATF3 binding to Galanin and Grp regulatory elements.
3.6. Impaired neurite growth in Atf3 mutants is rescued by neuropeptide addition
Analysis of facial nerve regeneration in vivo revealed decreased axon regeneration in ATF3-deficient mice (figures 2 and 3). As reduced axon regeneration might be caused by an intrinsic axon growth inhibition induced by ATF3 loss-of-function in PNS neurons, we employed in vitro neurite growth assays with adult wt and ATF3-lacking DRG neurons (figure 9). Besides PNS neurons, the impact of ATF3 deletion was also quantified in CNS neurons. However, in postnatal cerebellar neurons, neurite length in Atf3 mutant compared to wt neurons was only slightly reduced (electronic supplementary material, figure S4), suggesting no strong dependence of CNS neurite growth on ATF3.
Figure 9.

() Neuropeptide treatment elevates neurite growth upon camptothecin inhibition in ATF3-deficient mice. Adult wt or Atf3 mutant DRG neurons were cultured for 24 h followed by labelling for neuron-specific βIII tubulin expression. (a,b) In the absence of NGF, wt DRG neurons (a) elaborated more and longer neurites compared with neurons lacking ATF3 (b). (c,d) In the presence of NGF, Atf3 mutant (d) grew similarly to wt (c) neurons. (e,f) Camptothecin administration for 24 h reduced neurite growth of wt (e) and ATF3-deficient (f) neurons to a similar level. (g,h) Administration of the neuropeptide VIP elevated neurite growth of both wt (g) and ATF3-deficient (h) neurons in the presence of camptothecin and NGF. (i) Quantification of the entire neurite length/neuron for all conditions tested. In Atf3 mutant neurons, GRP and VIP application resulted in statistically significant relief of camptothecin-mediated neurite growth inhibition. Numbers in bars reflect independent cultures analysed. Data are presented as mean ± s.d. *p ≤ 0.05; ***p ≤ 0.001. Scale bar (a–h) = 500 µm.
Wt DRG neurons cultured for 24 h revealed robust neurite growth (figure 9a). On average, the total neurite length of all neurites protruded by a single wt DRG neuron added up to approximately 1500 µm (figure 9i). By contrast, in ATF3-deficient DRG neurons, axon growth was reduced by approximately 40% (figure 9b; quantified in i). Here, the total length of all neurites elaborated by an individual Atf3 mutant neuron accumulated to an average of 950 µm (figure 9i). These data suggest a stimulatory role of ATF3 on PNS axon growth in wt neurons and correspond with enhanced neurite growth reported upon ATF3 overexpression [8,11,12].
In the previous experiment, NGF was omitted, because Ngf mRNA induction was ATF3-dependent in vivo (figures 6 and 7) and Ngf mRNA levels were downregulated by approximately 50% in primary Atf3-deficient DRG neurons in vitro (data not shown). Thus, in the next set of experiments we added recombinant NGF to the culture medium and analysed whether this exogenous NGF application might rescue the neurite growth observed in Atf3 mutant mice in the absence of NGF (figure 9a,b). NGF elevated neurite growth in wt neurons by approximately 30–40% (figure 9c) compared with neurons without NGF administration (figure 9a; quantified in i). Similar to wt neurons, NGF application also elevated axon growth in ATF3-deficient DRG neurons (figure 9d). In fact, neurite growth in Atf3 mutant neurons was (approx. 10%) increased compared with wt neurons (2615 µm versus 2354 µm; figure 9i). This suggests that raising NGF levels in Atf3 mutant neurons by exogenous NGF administration can rescue impaired axon growth associated with ATF3 deficiency.
In a next step, we investigated whether other ATF3-regulated neuropeptides might mediate ATF3's impact on PNS axon growth similar to NGF. Several of the ATF3-regulated neuropeptides, e.g. galanin and VIP, but so far not GRP, promote axon regeneration in vivo and axon growth in vitro [63–68]. This also holds true for several Wnt family members [69–72]; however, Wnt2b has not been analysed so far.
To test if galanin, VIP, GRP or Wnt2b are required for axon growth, recombinant galanin, VIP, GRP or Wnt2b peptides were added to wt and ATF3-lacking DRG neurons. First, we tested whether any of the neuropeptides or Wnt2b administration affected wt DRG neurite growth or rescued neurite growth in Atf3 mutant neurons in NGF-depleted medium. However, under these conditions, none of the recombinant proteins affected wt neurite growth in a significant manner. In Atf3 mutant neurons, only GRP administration improved slightly but not significantly neurite length (electronic supplementary material, figure S5). Thus, under physiological culture conditions, there was no overt effect of neuropeptides or Wnt2b on either wt or Atf3 mutant neurite growth.
In order to investigate whether neuropeptides or Wnt2b modulate neurite growth in an injury-dependent manner, we induced axonal damage in vitro by culturing neurons in the presence of camptothecin for 24 h. Camptothecin is an established neurotoxic agent inducing apoptosis and growth inhibition through DNA topoisomerase I inhibition and subsequent DNA damage accumulation [73]. We added NGF to the cultures to prevent complete neurite growth impairment and overt neuronal cell death.
Camptothecin administration resulted in a robust axon growth reduction in both wt (figure 9e) and ATF3-deficient (figure 9f) neurons. Addition of recombinant GRP enhanced neurite growth in wt neurons in the presence of camptothecin, however not in a statistically significant manner (figure 9i). In Atf3 mutant neurons, GRP elevated neurite growth significantly in the presence of camptothecin. Now, neurite growth reached levels comparable to NGF treatment alone (figure 9i). Similar to GRP, VIP treatment elevated the camptothecin-mediated decrease in neurite growth of both wt (figure 9g) and ATF3-deficient (figure 9h) neurons. Administration of galanin or Wnt2b stimulated neurite growth of wt neurons in the presence of camptothecin. In contrast to this, both recombinant proteins had no effect on Atf3 mutant neurons (figure 9i).
Thus, in primary PNS neurons, the neuropeptides GRP and VIP were able to rescue cell damage-associated axon growth in ATF3-lacking neurons.
3.7. ATF3 mediates repression of genes relevant to nerve injury and neurite growth
So far, we analysed genes whose expression was reduced in injured ATF3-deficient compared with wt FN (figure 9). However, in the initial transcriptomics approach (figures 6 and 7), selected genes such as Timp1, Nov or Ccl2 were more upregulated in Atf3 mutants upon nerve injury. Timp1 (figure 7) and Nov (electronic supplementary material, figure S3i) expression profiles obtained in microarrays were confirmed by qPCR.
In further analysis (figure 10), we focused on Ccl2, a gene encoding a secreted chemokine, also known as monocyte chemoattractant protein 1 (MCP-1), previously connected to PNS axon regeneration. PNS injury upregulates Ccl2 in DRGs [27,74,75] and FMNs [76]. So far, CCL2 has been attributed functions during nerve injury-associated immune responses mainly by regulating monocyte rather than neuronal responses [23–29]. In this study, we focus on a potential neuronal response mediated by CCL2 (figure 10). First of all, we confirmed approximately twofold augmented Ccl2 mRNA levels in axotomized FN of Atf3 mutant mice compared with wt (figure 10a; figure 6a). Similarly elevated Ccl2 levels were also observed in Atf3 mutant mice in an ischaemia model [21]. This effect was specific for Ccl2, as other CCL chemokines, such as Ccl3, were unaffected (figure 10b). Ccl2 upregulation upon ATF3 loss-of-function suggests repression of the Ccl2 promoter by wt ATF3. To test whether this might be exerted by direct ATF3 binding to the Ccl2 promoter in neurons, we employed ChIP with ATF3 binding sites reported in other cell types [22]. As seen for neuropeptides (figure 8), ATF3 also occupied potential ATF3 binding sites of the Ccl2 promoter (figure 10c). Furthermore, the CCL2 protein abundance was investigated (figure 10d–f). In unlesioned FN of either genotype, anti-CCL2-directed antibodies did not recognize any positive cells (data not shown), a finding in line with mRNA levels (figure 10a). By contrast, in injured wt FN, several FMNs were CCL2 positive (figure 10d; quantified in f). Inspection of ATF3-deficient injured FN (figure 10e) revealed an increase of CCL2 positive FMNs at several timepoints post-injury (figure 10f).
Figure 10.

CCL2 is upregulated in Atf3 mutants and CCL2 overexpression reduces neurite growth. (a,b) Ccl2 (a) but not Ccl3 (b) mRNA was more strongly induced in injured Atf3 mutant FN compared with wt. (c) In ChIP experiments, ATF3 occupied ATF3 binding sites in the Ccl2 promoter. (d–f) In lesioned wt FN, CCL2-directed antibodies labelled FMNs at 8 d.p.l. (d). The number of CCL2 positive FMNs was elevated in injured ATF3-deficient mice (e). Higher magnification (inset in (e)) suggested CCL2 localization in cytoplasmic vesicles. (f) Quantification of CCL2 positive neurons per section at different times post-injury. (g–l) Wt DRG neurons were electroporated with expression vectors driving Cherry expression alone (g,j) or CCL2-Cherry (h,k). Cherry positive neurons were identified with anti-Cherry (red) and anti-βIII tubulin (green) directed antibodies. CCl2-Cherry positive neurons (arrows in h) and cultures (k) have decreased neurite growth and neuronal network density compared with control condition (g,j). (i,l) Quantification of neurite length of individual Cherry or CCL2-Cherry positive neurons (i) or of all neurites present on the entire coverslip regardless of Cherry expression (l). Numbers in bars indicate numbers of independent experiments or animals analysed. Data are presented as mean ± s.d. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. Scale bar (d,e) = 100 µm; (g,h) = 50 µm; (j,k) = 1 µm.
Next, we analysed functional consequences of a neuronal CCL2 localization such as neurite growth modulation. So far, the impact of CCL2 on neurite growth has not been investigated in great detail. CCL2 induced primary neuronal cell death, suggesting a negative role on neurons [77]. A further study reported that CCL2 administration to the culture medium failed to enhance neurite growth whereas intra-thecal CCL2 delivery primed DRGs for enhanced growth in vitro [27]. To address consequences of elevated intracellular CCL2 levels as observed in ATF3-deficient neurons (figure 10e), we analysed the impact of elevated CCL2 levels in wt DRG neurons (figure 10g–i). For this, CCL2 was overexpressed within DRG neurons by electroporation of a CCL2–Cherry fusion construct rather than applying CCL2 to the medium reported to be ineffective [27].
In control electroporated DRG neurons, expressing Cherry alone, individual Cherry positive DRG neurons elaborated several neurites (arrow, figure 10g). In general, neurons were indistinguishable from Cherry negative neurons on the same coverslip (arrowhead, figure 10g). In contrast to this, overexpression of CCL2-Cherry decreased neurite growth, with most DRG neurons protruding only short neurites (arrows, figure 10h). Thus, elevation of neuron intrinsic CCL2 expression resulted in impaired PNS neurite growth. Following up on this, we wondered whether CCL2 expression might also affect neurite growth of surrounding CCL2 negative DRG neurons in a non-cell-autonomous manner (figure 10j–l). On control coverslips electroporated with Cherry alone (figure 10j), the overall neuronal network density was strongly increased compared with a CCL2-Cherry expressing culture (figure 10k; quantified in l). This finding suggests that CCL2 overexpression decreased neurite extension also by a paracrine mechanism.
In short, electroporation-mediated elevation of CCL2 levels in wt DRG neurons—as observed in Atf3 mutant neurons—decreased primary PNS neurite growth.
3.8. ATF3 is upregulated in human peripheral nervous system injury
So far, to the best of our knowledge, ATF3 expression was not investigated in human peripheral nerve injury. We tested whether ATF3 was also upregulated in human PNS injury, employing neuroma tissue derived from surgical resection of patients with brachial plexus injuries. As control for an intact nerve, we used freshly isolated nervus suralis sections of the same patient (figure 11).
Figure 11.

ATF3 is expressed in human peripheral nerve injury. (a,b,e,f) A non-injured human nervus suralis nerve was stained for ATF3 (a,e) and S100 (b,f). ATF3 was nearly absent from this control nerve sample (a,e). (c,d,g,h) A section of a C5 brachial plexus injury was stained for ATF3 (c,g) and S100 (d,g). ATF3 was expressed in this PNS injury sample and appeared to co-localize with several S100 positive Schwann cells labelled on a neighbouring section (compare (g) and (h)). Scale bar (a–d) = 500 µm; (e–h) = 20 µm.
In the nervus suralis, ATF3 was only weakly expressed (figure 11a,e). Neighbouring nervus suralis sections were stained for the Schwann cell marker S100 to show Schwann cells surrounding peripheral axons (figure 11b,f). By contrast, in a C5 brachial plexus lesion, ATF3 expression was found throughout the entire section (figure 11c,d). To address the cellular identity of ATF3 expressing cells, neighbouring sections were stained for S100 (figure 11d,h). As neuronal cell bodies are not present in these nerve samples, we reasoned that Schwann cells might be upregulating ATF3 upon injury. Indeed, ATF3 and S100 showed similar staining patterns indicating the presence of ATF3 in Schwann cells (compare figure 11g,h). However, not all S100 positive signals appeared to be ATF3 positive suggesting that not all Schwann cells did express ATF3.
Overall this finding points at ATF3 expression in human Schwann cells, similar to PNS injury in rodents (figure 1).
4. Discussion
4.1. The time course and localization of ATF3 expression upon peripheral nervous system nerve injury
ATF3 expression in FMNs peaked in the first week after injury (figure 1) and was nearly absent from 14 d.p.l. onwards, a time period when robust innervation of facial muscles by regenerating motor axons started (figure 3). This expression profile suggests that ATF3-regulated effector RAGs might predominantly influence early processes of FMN regeneration, such as initial outgrowth of motor axons starting two to three days after injury. In accordance with this observation was a reduction in facial nerve sprouting in ATF3-deficient animals at 3 d.p.l. (figure 4). Nevertheless, the mRNA expression profile of several injury-regulated genes points at a function of ATF3 also at later timepoints after injury. For instance, high mRNA levels of several injury-induced genes (e.g. Galanin, Wnt2b. Ngf, Grp, Sprr2j) were observed also at later injury timepoints, i.e. at 7 and sometimes even up to 14 days after facial nerve lesion (electronic supplementary material, figure S2). This suggests that gene products of such ATF3-regulated effector RAGs (figures 6 and 7) might still be present in the second week after injury and later. In accordance, we observed a high protein abundance of the ATF3-regulated RAG galanin in FMNs at 12 days after injury (figure 7l). Thus, at the protein level, ATF3-regulated gene products may influence also later regeneration processes such as long distance axon growth resulting in facial muscle re-innervation, although ATF3 itself is nearly absent. Besides FMNs, ATF3 induction was also observed in rodent (figure 1) and human (figure 11) Schwann cells after peripheral nerve injury. Ablation of the ATF3 partner protein c-Jun uncovered the importance of Schwann cells in PNS regeneration [51,52]. In our study, ATF3 was constitutively deleted from all cells. Thus, besides FMNs, impaired axon regeneration observed in Atf3 mutant mice (figures 2 and 3) might also be a consequence of ATF3 depletion from Schwann cells.
4.2. ATF3's role in peripheral nervous system axon regeneration
In the unlesioned FN, topographic mapping and numbers of FMN subtypes were unchanged between adult wt and Atf3 mutant mice (figure 2n,o;r). In agreement with ATF3's absence in the intact FN (figure 1a), this suggests no major role of ATF3 in topographic map formation and FMN generation during brain development and physiological FMN function. However, ATF3's function became obvious once FMNs were injured and FMN regeneration was analysed. Here, all three FMN subtypes analysed differed with regard to their regeneration potential. Thus, nearly 100% of DiI positive FMNs were re-connected to their eyelid target, whereas only 60% of FG and approximately 30% of Ctx488 positive FMNs reached their targets (figures 2 and 3). As the eyelid is closest to the initial lesion site, this outcome correlates with the distance axons had to bridge to reach their target (figure 2a(ii)). All three FMN subtypes differed also in numbers present in the FN and whiskers were represented by the largest FMN number (figure 2n,o;r).
Inspection of FMN regeneration to the whiskers revealed a significant reduction by approximately 30% in ATF3-deficient mice, indicating a necessary ATF3 contribution to PNS regeneration (figures 2 and 3). Nevertheless, although axon regeneration was slowed down in the absence of ATF3, it was not completely prevented. This points at additional factors contributing to complete nerve regeneration. In a current model of PNS axon regeneration, several TF-encoding RAGs form hubs within several hundred genes encompassing RAG networks. Recently, a detailed analysis of such RAG networks identified and as important TF hubs [8]. Given the presence of several TF hubs, single hub deletion as performed in this study might be compensated for by other hubs [5,6]. In accordance with this model, single deletion of other hub TFs such as c-Jun [13] or p53 [16] reduced FMN regeneration to a similar extent as observed for Atf3 mutant mice in this study. Thus, compound mutagenesis of hub TFs, e.g. Atf3 and c-Jun double mouse mutants, might reveal stronger impairments in PNS regeneration compared with single mutants.
4.3. Identification of ATF3 target genes associated with peripheral nervous system axon regeneration
In this study, we provide a first genome-wide survey of ATF3 target genes employing ATF3 loss-of-function. Approximately 20–25% of the FMN RAG programme was to some extent ATF3-dependent (figure 6; electronic supplementary material, table S1), suggesting overall a modest impact of ATF3 on the total RAG response. However, individual genes or gene sets identified in this study showed a stronger ATF3 dependency. Specific RAGs including Sprr2j and Wnt2b and, particularly, a neuropeptide (Pacap, Grp, Vip, Galanin, Ngf, Npy) or neuropeptide receptor (Vipr2, Avpr1a) encoding gene cluster were strongly affected by ATF3 deficiency upon injury (figures 6–8). So far, transcriptional regulation of this gene cluster by ATF3 was not reported. Further analysis suggested indirect regulation of Vip, Ngf, Pacap and Vipr2 by ATF3, whereas Galanin and Grp might be direct ATF3 target genes (figure 8). Laser microdissection experiments suggest that the RAG response primarily takes place in FMNs and not in other cell types (electronic supplementary material, figure S1).
Besides transcriptional activation, individual genes appeared to be under negative ATF3 transcriptional control. So far, gene repression by ATF3 is known in non-neuronal cells [18,78,79]. In this study, we identified genes whose expression is potentially repressed by ATF3 in neurons. For instance, Timp1, Nov and Ccl2 were more upregulated in lesioned Atf3 mutants suggesting gene repression by wild-type ATF3 (figures 6 and 10; electronic supplementary material, figure S3). Ccl2 was also elevated in Atf3 mutants during cerebral ischaemia [21] indicating a more general mechanism of Ccl2 inhibition by ATF3.
4.4. ATF3-associated mechanisms of peripheral nervous system axon regeneration
In this report, impaired facial nerve regeneration in Atf3 mutants was associated with reduced induction of several RAGs including the neuropeptides Galanin, Grp, Vip, Ngf and Wnt2b as well as Sprr2j. Besides Grp, all the aforementioned genes or closely related family members can stimulate neurite growth and axon regeneration [63–68,80,81]. In addition, many neuropeptides affect neuronal survival, pain perception and neurotransmitter release [82,83]. We directly tested whether any of these ATF3-regulated gene products can rescue neurite growth reduced upon ATF3 loss-of-function in vitro. Indeed, addition of recombinant GRP, VIP and, most pronounced, NGF were able to rescue neurite growth impairment induced by ATF3 deficiency (figure 9). Although confirmation of such a rescue effect is missing upon facial nerve injury in vivo, these in vitro data highlight NGF, GRP and VIP as potential candidates responsible for ATF3's pro-regenerative function (see summary figure 12).
Figure 12.

Summary of ATF3's function in facial nerve regeneration. (a) In injured wild-type mice, ATF3 is involved in transcriptional activation of the RAGs Gal, Vip, Wnt2, Grp and Ngf, whereas Ccl2 is repressed. Neuropeptides would stimulate axonal growth and thereby increase axonal regeneration. By contrast, CCL2 levels in FMNs are suppressed by ATF3 and therefore CCL2's potential to decrease axon growth is reduced. (b) In lesioned Atf3 mutant mice, induction of Gal, Vip, Wnt2, Grp and Ngf is reduced, resulting in weaker stimulation of axonal growth and also reduced regeneration potential of FMNs. In addition, Ccl2 is not repressed by ATF3 anymore and enhanced CCL2 levels in injured FMNs decrease axonal growth and regeneration.
Data above suggest positive regulation of direct growth-promoting molecules by ATF3 upon nerve injury (see summary figure 12). In addition, we also observed a potential ATF3-mediated repression of genes such as Ccl2 and Nov (figures 6 and 10; electronic supplementary material, table S1 and figure S3). Both Ccl2 (figure 10) and Nov (electronic supplementary material, figure S3) were more strongly upregulated in injured Atf3 mutant FN. In axon regeneration, CCL2 has beneficial functions on immune cells, e.g. during removal of myelin debris. However, CCL2 also triggers axonal damage in mouse models of motoneuron disease [23] and multiple sclerosis [25]. The latter finding is in agreement with our in vitro data, demonstrating neurite growth inhibition by CCL2 overexpression (figure 10). Similar to CCL2, Nov (CCN3) has also been attributed growth inhibitory functions [84]. Thus, a further mechanism by which ATF3 enhances axon regeneration might involve repression of axon growth-inhibiting molecules such as CCL2 and CCN3 (see summary figure 12). At first glance, this appears contradictory, because Ccl2 and Nov were also induced in wild-type mice upon injury (figure 6 and electronic supplementary material, table S1). As CCL2 mediates an immune cell response after injury, certain CCL2 levels might be beneficial for axon regeneration. However, excess CCL2 levels, as observed in ATF3-deficient neurons or upon CCL2 overexpression in wt neurons (figure 10), might counteract the neuron intrinsic axon growth potential. Thus, ATF3 might play a role in balancing CCL2 levels for an optimal regeneration outcome (see summary, figure 12).
5. Conclusion
In this study, we provide a first description of an ATF3 pro-regenerative function employing Atf3 mouse mutants. For this, ATF3 appears to induce axon growth-stimulating molecules such as neuropeptides (e.g. galanin, VIP, GRP and NGF) and downregulate growth-inhibiting molecules such as CCL2. ATF3 loss-of-function data in this study fit well with ATF3 gain-of-function results, showing enhanced axon growth and PNS regeneration in neurons or mice overexpressing ATF3 [8,11,12,30]. In general, ATF3's stimulatory role in regeneration is in accordance with other ATF3 functions such as providing neuroprotection in mouse epilepsy [32,85], ischaemia [34], neurotoxicity [33] or ALS [31] models. As ATF3 is induced by numerous types of neuronal injury in many cell types, our findings on ATF3-mediated mechanisms in PNS injury might also hold true in other neuronal injury conditions.
Supplementary Material
Supplementary Material
Acknowledgements
Atf3 mutant mice were thankfully received from Dr Tsonwin Hai (Ohoi State University). We thank Paul Walther and colleagues of the Central Electron Microscopy Facility at Ulm University for their support. We appreciate the help of Ruth Simon for the laser microdissection experiments.
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
M.G., R.W., C.S. and D.S. performed and analysed all experiments. M.T.P. provided human material. B.K. conceived the project, supervised all research and has written the manuscript.
Competing interests
We declare we have no competing interests.
Funding
B.K. is supported by the DFG (Deutsche Forschungsgemeinschaft) through SFB1149, an Ulm University and Bundeswehrkrankenhaus research initiative and the Schram, Gottschalk and Gemeinnützige Hertie foundation.
References
- 1.Kiryu-Seo S, Kiyama H. 2011. The nuclear events guiding successful nerve regeneration. Front. Mol. Neurosci. 4, 53 (doi:10.3389/fnmol.2011.00053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore DL, Goldberg JL. 2011. Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 71, 1186–1211. (doi:10.1002/dneu.20934) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patodia S, Raivich G. 2012. Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 5, 8 (doi:10.3389/fnmol.2012.00008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tedeschi A. 2011. Tuning the orchestra: transcriptional pathways controlling axon regeneration. Front. Mol. Neurosci. 4, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Kesteren RE, Mason MR, Macgillavry HD, Smit AB, Verhaagen J. 2011. A gene network perspective on axonal regeneration. Front. Mol. Neurosci. 4, 46 (doi:10.3389/fnmol.2011.00046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma TC, Willis DE. 2015. What makes a RAG regeneration associated? Front. Mol. Neurosci. 8, 43 (doi:10.3389/fnmol.2015.00043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makwana M, Raivich G. 2005. Molecular mechanisms in successful peripheral regeneration. FEBS J. 272, 2628–2638. (doi:10.1111/j.1742-4658.2005.04699.x) [DOI] [PubMed] [Google Scholar]
- 8.Chandran V, et al. 2016. A systems-level analysis of the peripheral nerve intrinsic axonal growth program. Neuron 89, 956–970. (doi:10.1016/j.neuron.2016.01.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patodia S, Raivich G. 2012. Downstream effector molecules in successful peripheral nerve regeneration. Cell Tissue Res. 349, 15–26. (doi:10.1007/s00441-012-1416-6) [DOI] [PubMed] [Google Scholar]
- 10.Bareyre FM, Garzorz N, Lang C, Misgeld T, Buning H, Kerschensteiner M. 2011. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl Acad. Sci. USA 108, 6282–6287. (doi:10.1073/pnas.1015239108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seijffers R, Allchorne AJ, Woolf CJ. 2006. The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell. Neurosci. 32, 143–154. (doi:10.1016/j.mcn.2006.03.005) [DOI] [PubMed] [Google Scholar]
- 12.Seijffers R, Mills CD, Woolf CJ. 2007. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920. (doi:10.1523/JNEUROSCI.5313-06.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raivich G, et al. 2004. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67. (doi:10.1016/j.neuron.2004.06.005) [DOI] [PubMed] [Google Scholar]
- 14.Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, Koerber HR, Albers KM. 2009. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 1256, 43–54. (doi:10.1016/j.brainres.2008.12.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parikh P, Hao Y, Hosseinkhani M, Patil SB, Huntley GW, Tessier-Lavigne M, Zou H. 2011. Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl Acad. Sci. USA 108, E99–E107. (doi:10.1073/pnas.1100426108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Giovanni S, Knights CD, Rao M, Yakovlev A, Beers J, Catania J, Avantaggiati ML, Faden AI. 2006. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. Embo J. 25, 4084–4096. (doi:10.1038/sj.emboj.7601292) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tetzlaff W, Alexander SW, Miller FD, Bisby MA. 1991. Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J. Neurosci. 11, 2528–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hai T, Wolford CC, Chang YS. 2010. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: is modulation of inflammation a unifying component? Gene Expr. 15, 1–11. (doi:10.3727/105221610X12819686555015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunt D, Raivich G, Anderson PN. 2012. Activating transcription factor 3 and the nervous system. Front. Mol. Neurosci. 5, 7 (doi:10.3389/fnmol.2012.00007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Luan S, OuYang H, Huang Z, Wu S, Ma C, Wei J, Xin W. 2015. Upregulation of CCL2 via ATF3/c-Jun interaction mediated the Bortezomib-induced peripheral neuropathy. Brain Behav. Immun. 53, 96–104. (doi:10.1016/j.bbi.2015.11.004) [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Deng S, Lu Y, Zhang Y, Yang L, Guan Y, Jiang H, Li H. 2012. Increased inflammation and brain injury after transient focal cerebral ischemia in activating transcription factor 3 knockout mice. Neuroscience 220, 100–108. (doi:10.1016/j.neuroscience.2012.06.010) [DOI] [PubMed] [Google Scholar]
- 22.Zmuda EJ, Viapiano M, Grey ST, Hadley G, Garcia-Ocana A, Hai T. 2010. Deficiency of Atf3, an adaptive-response gene, protects islets and ameliorates inflammation in a syngeneic mouse transplantation model. Diabetologia 53, 1438–1450. (doi:10.1007/s00125-010-1696-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S, Martini R. 2010. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Human Mol. Genet. 19, 3530–3543. (doi:10.1093/hmg/ddq269) [DOI] [PubMed] [Google Scholar]
- 24.Kwon MJ, Shin HY, Cui Y, Kim H, Thi AH, Choi JY, Kim EY, Hwang DH, Kim BG. 2015. CCL2 mediates neuron–macrophage interactions to drive proregenerative macrophage activation following preconditioning injury. J. Neurosci. 35, 15 934–15 947. (doi:10.1523/JNEUROSCI.1924-15.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno M, Bannerman P, Ma J, Guo F, Miers L, Soulika AM, Pleasure D. 2014. Conditional ablation of astroglial CCL2 suppresses CNS accumulation of M1 macrophages and preserves axons in mice with MOG peptide EAE. J. Neurosci. 34, 8175–8185. (doi:10.1523/JNEUROSCI.1137-14.2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muessel MJ, Klein RM, Wilson AM, Berman NE. 2002. Ablation of the chemokine monocyte chemoattractant protein-1 delays retrograde neuronal degeneration, attenuates microglial activation, and alters expression of cell death molecules. Brain Res. 103, 12–27. (doi:10.1016/S0169-328X(02)00158-4) [DOI] [PubMed] [Google Scholar]
- 27.Niemi JP, DeFrancesco-Lisowitz A, Cregg JM, Howarth M, Zigmond RE. 2015. Overexpression of the monocyte chemokine CCL2 in dorsal root ganglion neurons causes a conditioning-like increase in neurite outgrowth and does so via a STAT3 dependent mechanism. Exp. Neurol. 275, 25–37. (doi:10.1016/j.expneurol.2015.09.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ousman SS, David S. 2001. MIP-1alpha, MCP-1, GM-CSF, and TNF-α control the immune cell response that mediates rapid phagocytosis of myelin from the adult mouse spinal cord. J. Neurosci. 21, 4649–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perrin FE, Lacroix S, Aviles-Trigueros M, David S. 2005. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 128, 854–866. (doi:10.1093/brain/awh407) [DOI] [PubMed] [Google Scholar]
- 30.Fagoe ND, Attwell CL, Kouwenhoven D, Verhaagen J, Mason MR. 2015. Over-expression of ATF3 or the combination of ATF3, c-Jun, STAT3 and Smad1 promotes regeneration of the central axon branch of sensory neurons but without synergistic effects. Hum. Mol. Genet. 18, 6788–6800. (doi:10.1093/hmg/ddv383) [DOI] [PubMed] [Google Scholar]
- 31.Seijffers R, et al. 2014. ATF3 expression improves motor function in the ALS mouse model by promoting motor neuron survival and retaining muscle innervation. Proc. Natl Acad. Sci. USA 111, 1622–1627. (doi:10.1073/pnas.1314826111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. 2009. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 5, e1000604 (doi:10.1371/journal.pgen.1000604) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahlgren H, Bas-Orth C, Freitag HE, Hellwig A, Ottersen OP, Bading H. 2014. The nuclear calcium signaling target, activating transcription factor 3 (ATF3), protects against dendrotoxicity and facilitates the recovery of synaptic transmission after an excitotoxic insult. J. Biol. Chem. 289, 9970–9982. (doi:10.1074/jbc.M113.502914) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang SJ, et al. 2011. A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J. Neurosci. 31, 4978–4990. (doi:10.1523/JNEUROSCI.2672-10.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nie D, et al. 2015. The stress-induced Atf3–gelsolin cascade underlies dendritic spine deficits in neuronal models of tuberous sclerosis complex. J. Neurosci. 35, 10 762–10 772. (doi:10.1523/JNEUROSCI.4796-14.2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moran LB, Graeber MB. 2004. The facial nerve axotomy model. Brain Res. Rev. 44, 154–178. (doi:10.1016/j.brainresrev.2003.11.004) [DOI] [PubMed] [Google Scholar]
- 37.Park BG, Lee JS, Lee JY, Song DY, Jeong SW, Cho BP. 2011. Co-localization of activating transcription factor 3 and phosphorylated c-Jun in axotomized facial motoneurons. Anat. Cell Biol. 44, 226–237. (doi:10.5115/acb.2011.44.3.226) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruff CA, et al. 2012. Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J. Neurochem. 121, 607–618. (doi:10.1111/j.1471-4159.2012.07706.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt AB, et al. 2003. Identification of regeneration-associated genes after central and peripheral nerve injury in the adult rat. BMC Neurosci. 4, 8 (doi:10.1186/1471-2202-4-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stern S, Sinske D, Knoll B. 2012. Serum response factor modulates neuron survival during peripheral axon injury. J. Neuroinflamm. 9, 78 (doi:10.1186/1742-2094-9-78) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tetzlaff W, Bisby MA, Kreutzberg GW. 1988. Changes in cytoskeletal proteins in the rat facial nucleus following axotomy. J. Neurosci. 8, 3181–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zujovic V, Luo D, Baker HV, Lopez MC, Miller KR, Streit WJ, Harrison JK. 2005. The facial motor nucleus transcriptional program in response to peripheral nerve injury identifies Hn1 as a regeneration-associated gene. J. Neurosci. Res. 82, 581–591. (doi:10.1002/jnr.20676) [DOI] [PubMed] [Google Scholar]
- 43.Hartman MG, et al. 2004. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell. Biol. 24, 5721–5732. (doi:10.1128/MCB.24.13.5721-5732.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stern S, Haverkamp S, Sinske D, Tedeschi A, Naumann U, Di Giovanni S, Kochanek S, Nordheim A, Knoll B. 2013. The transcription factor serum response factor stimulates axon regeneration through cytoplasmic localization and cofilin interaction. J. Neurosci. 33, 18 836–18 848. (doi:10.1523/JNEUROSCI.3029-13.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anastasiadou S, Liebenehm S, Sinske D, Meyer zu Reckendorf C, Moepps B, Nordheim A, Knöll B. 2015. Neuronal expression of the transcription factor serum response factor modulates myelination in a mouse multiple sclerosis model. Glia 63, 958–976. (doi:10.1002/glia.22794) [DOI] [PubMed] [Google Scholar]
- 46.Duce IR, Keen P. 1976. A light and electron microscope study of changes occurring at the cut ends following section of the dorsal roots of rat spinal nerves. Cell Tissue Res. 170, 491–505. (doi:10.1007/BF00361707) [DOI] [PubMed] [Google Scholar]
- 47.Irizarry RA, et al. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. (doi:10.1093/biostatistics/4.2.249) [DOI] [PubMed] [Google Scholar]
- 48.Nelson JD, Denisenko O, Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1, 179–185. (doi:10.1038/nprot.2006.27) [DOI] [PubMed] [Google Scholar]
- 49.Pool M, Thiemann J, Bar-Or A, Fournier AE. 2008. NeuriteTracer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J. Neurosci. Methods 168, 134–139. (doi:10.1016/j.jneumeth.2007.08.029) [DOI] [PubMed] [Google Scholar]
- 50.Martin-Villalba A, Winter C, Brecht S, Buschmann T, Zimmermann M, Herdegen T. 1998. Rapid and long-lasting suppression of the ATF-2 transcription factor is a common response to neuronal injury. Brain Res. Mol. Brain Res. 62, 158–166. (doi:10.1016/S0169-328X(98)00239-3) [DOI] [PubMed] [Google Scholar]
- 51.Arthur-Farraj PJ, et al. 2012. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647. (doi:10.1016/j.neuron.2012.06.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fontana X, et al. 2012. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 198, 127–141. (doi:10.1083/jcb.201205025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blom CL, Martensson LB, Dahlin LB. 2014. Nerve injury-induced c-Jun activation in Schwann cells is JNK independent. BioMed Res. Int. 2014, 83–93. (doi:10.1155/2014/392971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stenberg L, Kanje M, Dolezal K, Dahlin LB. 2012. Expression of activating transcription factor 3 (ATF 3) and caspase 3 in Schwann cells and axonal outgrowth after sciatic nerve repair in diabetic BB rats. Neurosci. Lett. 515, 34–38. (doi:10.1016/j.neulet.2012.03.011) [DOI] [PubMed] [Google Scholar]
- 55.Knoll B, Drescher U. 2002. Ephrin-As as receptors in topographic projections. Trends Neurosci. 25, 145–149. (doi:10.1016/S0166-2236(00)02093-2) [DOI] [PubMed] [Google Scholar]
- 56.Grosheva M, et al. 2008. Local stabilization of microtubule assembly improves recovery of facial nerve function after repair. Exp. Neurol. 209, 131–144. (doi:10.1016/j.expneurol.2007.09.016) [DOI] [PubMed] [Google Scholar]
- 57.Guntinas-Lichius O, et al. 2007. Manual stimulation of facial muscles improves functional recovery after hypoglossal–facial anastomosis and interpositional nerve grafting of the facial nerve in adult rats. Neurobiol. Dis. 28, 101–112. (doi:10.1016/j.nbd.2007.07.006) [DOI] [PubMed] [Google Scholar]
- 58.Guntinas-Lichius O, Irintchev A, Streppel M, Lenzen M, Grosheva M, Wewetzer K, Neiss WF, Angelov DN. 2005. Factors limiting motor recovery after facial nerve transection in the rat: combined structural and functional analyses. Eur. J. Neurosci. 21, 391–402. (doi:10.1111/j.1460-9568.2005.03877.x) [DOI] [PubMed] [Google Scholar]
- 59.Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H. 2003. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J. Neurosci. 23, 5187–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clark CE, Liu Y, Cooper HM. 2014. The Yin and Yang of Wnt/Ryk axon guidance in development and regeneration. Sci. China Life Sci. 57, 366–371. (doi:10.1007/s11427-014-4640-3) [DOI] [PubMed] [Google Scholar]
- 61.Ono S. 2010. Dynamic regulation of sarcomeric actin filaments in striated muscle. Cytoskeleton (Hoboken) 67, 677–692. (doi:10.1002/cm.20476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Daily K, Patel VR, Rigor P, Xie X, Baldi P. 2011. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinform. 12, 495 (doi:10.1186/1471-2105-12-495) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hobson SA, Bacon A, Elliot-Hunt CR, Holmes FE, Kerr NC, Pope R, Vanderplank P, Wynick D. 2010. Galanin acts as a trophic factor to the central and peripheral nervous systems. In Galanin, Experientia Supplementum, vol. 102 (ed. T Hökfelt), pp. 25–28. Basel, Switzerland: Springer Basel. [DOI] [PubMed] [Google Scholar]
- 64.Hobson SA, Vanderplank PA, Pope RJ, Kerr NC, Wynick D. 2013. Galanin stimulates neurite outgrowth from sensory neurons by inhibition of Cdc42 and Rho GTPases and activation of cofilin. J. Neurochem. 127, 199–208. (doi:10.1111/jnc.12379) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holmes FE, Mahoney S, King VR, Bacon A, Kerr NC, Pachnis V, Curtis R, Priestley JV, Wynick D. 2000. Targeted disruption of the galanin gene reduces the number of sensory neurons and their regenerative capacity. Proc. Natl Acad. Sci. USA 97, 11 563–11 568. (doi:10.1073/pnas.210221897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Passemard S, Sokolowska P, Schwendimann L, Gressens P. 2011. VIP-induced neuroprotection of the developing brain. Curr. Pharm. Design 17, 1036–1039. (doi:10.2174/138161211795589409) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sachs HH, Wynick D, Zigmond RE. 2007. Galanin plays a role in the conditioning lesion effect in sensory neurons. Neuroreport 18, 1729–1733. (doi:10.1097/WNR.0b013e3282f0d3f4) [DOI] [PubMed] [Google Scholar]
- 68.Suarez V, Guntinas-Lichius O, Streppel M, Ingorokva S, Grosheva M, Neiss WF, Angelov DN, Klimaschewski L. 2006. The axotomy-induced neuropeptides galanin and pituitary adenylate cyclase-activating peptide promote axonal sprouting of primary afferent and cranial motor neurones. Eur. J. Neurosci. 24, 1555–1564. (doi:10.1111/j.1460-9568.2006.05029.x) [DOI] [PubMed] [Google Scholar]
- 69.Hall AC, Lucas FR, Salinas PC. 2000. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell 100, 525–535. (doi:10.1016/S0092-8674(00)80689-3) [DOI] [PubMed] [Google Scholar]
- 70.Krylova O, Herreros J, Cleverley KE, Ehler E, Henriquez JP, Hughes SM, Salinas PC. 2002. WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron 35, 1043–1056. (doi:10.1016/S0896-6273(02)00860-7) [DOI] [PubMed] [Google Scholar]
- 71.Li L, Hutchins BI, Kalil K. 2010. Wnt5a induces simultaneous cortical axon outgrowth and repulsive turning through distinct signaling mechanisms. Sci. Signal. 3, pt2. (doi:10.1126/scisignal.3147pt2) [DOI] [PubMed] [Google Scholar]
- 72.Lu W, Yamamoto V, Ortega B, Baltimore D. 2004. Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119, 97–108. (doi:10.1016/j.cell.2004.09.019) [DOI] [PubMed] [Google Scholar]
- 73.Morris EJ, Geller HM. 1996. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J. Cell Biol. 134, 757–770. (doi:10.1083/jcb.134.3.757) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schreiber RC, Krivacic K, Kirby B, Vaccariello SA, Wei T, Ransohoff RM., Zigmond RE. 2001. Monocyte chemoattractant protein (MCP)-1 is rapidly expressed by sympathetic ganglion neurons following axonal injury. Neuroreport 12, 601–606. (doi:10.1097/00001756-200103050-00034) [DOI] [PubMed] [Google Scholar]
- 75.Tanaka T, Minami M, Nakagawa T, Satoh M. 2004. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci. Res. 48, 463–469. (doi:10.1016/j.neures.2004.01.004) [DOI] [PubMed] [Google Scholar]
- 76.Flugel A, Hager G, Horvat A, Spitzer C, Singer GM, Graeber MB, Kreutzberg GW, Schwaiger F-W. 2001. Neuronal MCP-1 expression in response to remote nerve injury. J. Cereb. Blood Flow Metab. 21, 69–76. (doi:10.1097/00004647-200101000-00009) [DOI] [PubMed] [Google Scholar]
- 77.Kalehua AN, Nagel JE, Whelchel LM, Gides JJ, Pyle RS, Smith RJ, Kusiak JW, Taub DD. 2004. Monocyte chemoattractant protein-1 and macrophage inflammatory protein-2 are involved in both excitotoxin-induced neurodegeneration and regeneration. Exp. Cell Res. 297, 197–211. (doi:10.1016/j.yexcr.2004.02.031) [DOI] [PubMed] [Google Scholar]
- 78.Gilchrist M, Henderson WR Jr, Clark AE, Simmons RM, Ye X, Smith KD, Aderem A. 2008. Activating transcription factor 3 is a negative regulator of allergic pulmonary inflammation. J. Exp. Med. 205, 2349–2357. (doi:10.1084/jem.20072254) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolfgang CD, Liang G, Okamoto Y, Allen AE, Hai T. 2000. Transcriptional autorepression of the stress-inducible gene ATF3. J. Biol. Chem. 275, 16 865–16 870. (doi:10.1074/jbc.M909637199) [DOI] [PubMed] [Google Scholar]
- 80.Hollis ER 2nd, Zou Y. 2012. Expression of the Wnt signaling system in central nervous system axon guidance and regeneration. Front. Mol. Neurosci. 5, 5 (doi:10.3389/fnmol.2012.00005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jing X, Wang T, Huang S, Glorioso JC, Albers KM. 2012. The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp. Neurol. 233, 221–232. (doi:10.1016/j.expneurol.2011.10.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Palkovits M. 1995. Neuropeptide messenger plasticity in the CNS neurons following axotomy. Mol. Neurobiol. 10, 91–103. (doi:10.1007/BF02740669) [DOI] [PubMed] [Google Scholar]
- 83.Zigmond RE, Sun Y. 1997. Regulation of neuropeptide expression in sympathetic neurons. Paracrine and retrograde influences. Ann. N. Y. Acad. Sci. 814, 181–197. (doi:10.1111/j.1749-6632.1997.tb46157.x) [DOI] [PubMed] [Google Scholar]
- 84.Park M, Baek IJ, Kim H, Woo DK, Park YJ, Shim S. 2015. CCN3 overexpression inhibits growth of callosal projections via upregulation of RAB25. Biochem. Biophys. Res. Commun. 461, 456–462. (doi:10.1016/j.bbrc.2015.04.016) [DOI] [PubMed] [Google Scholar]
- 85.Francis JS, Dragunow M, During MJ. 2004. Over expression of ATF-3 protects rat hippocampal neurons from in vivo injection of kainic acid. Brain Res. Mol. Brain Res. 124, 199–203. (doi:10.1016/j.molbrainres.2003.10.027) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.