

ABSTRACT
A key player in the intrinsic resistance against human cytomegalovirus (HCMV) is the interferon-γ-inducible protein 16 (IFI16), which behaves as a viral DNA sensor in the first hours postinfection and as a repressor of viral gene transcription in the later stages. Previous studies on HCMV replication demonstrated that IFI16 binds to the viral protein kinase pUL97, undergoes phosphorylation, and relocalizes to the cytoplasm of infected cells. In this study, we demonstrate that the tegument protein pp65 (pUL83) recruits IFI16 to the promoter of the UL54 gene and downregulates viral replication, as shown by use of the HCMV mutant v65Stop, which lacks pp65 expression. Interestingly, at late time points of HCMV infection, IFI16 is stabilized by its interaction with pp65, which stood in contrast to IFI16 degradation, observed in herpes simplex virus 1 (HSV-1)-infected cells. Moreover, we found that its translocation to the cytoplasm, in addition to pUL97, strictly depends on pp65, as demonstrated with the HCMV mutant RV-VM1, which expresses a form of pp65 unable to translocate into the cytoplasm. Thus, these data reveal a dual role for pp65: during early infection, it modulates IFI16 activity at the promoter of immediate-early and early genes; subsequently, it delocalizes IFI16 from the nucleus into the cytoplasm, thereby stabilizing and protecting it from degradation. Overall, these data identify a novel activity of the pp65/IFI16 interactome involved in the regulation of UL54 gene expression and IFI16 stability during early and late phases of HCMV replication.
IMPORTANCE The DNA sensor IFI16, a member of the PYHIN proteins, restricts HCMV replication by impairing viral DNA synthesis. Using a mutant virus lacking the tegument protein pp65 (v65Stop), we demonstrate that pp65 recruits IFI16 to the early UL54 gene promoter. As a putative counteraction to its restriction activity, pp65 supports the nucleocytoplasmic export of IFI16, which was demonstrated with the viral mutant RV-VM1 expressing a nuclearly retained pp65. These data reveal a dual role of pp65 in IFI16 regulation: in the early phase of HCMV infection, it contributes to viral evasion from IFI16 restriction activity, while at later time points, it promotes the nuclear delocalization of IFI16, thereby stabilizing and protecting it from degradation. In the present work, we further clarify the mechanisms HCMV relies on to overcome intracellular innate immune restriction and provide new insights into the relevance of DNA-sensing restriction factor IFI16 during HCMV infection.
INTRODUCTION
Human cytomegalovirus (HCMV) is a member of the Betaherpesvirinae subfamily of Herpesviridae. It is a widespread pathogen that infects the majority of the world's population by early adulthood (1). The virus establishes a lifelong infection with some cells being latently infected, a state in which the virus resides in a nonproductive form, while other cells show a low-level persistent productive infection in which the virus is not cleared from the organism and is periodically reactivated, leading to the intermittent shedding of infectious virus (2).
Intrinsic immune defenses are mediated by cellular restriction factors (RFs) that are constitutively expressed and active even before a pathogen enters the cell, thus providing the cell's first line of defense. While the interference of retroviral replication by cellular RFs and retroviral evasion strategies have been studied in great detail, our knowledge of the mechanisms through which RFs affect other viral infections remains limited (3–5). In particular, in the case of HCMV at least two cellular components, namely, nuclear domain 10 (ND10) (e.g., promyelocytic leukemia protein [PML], hDaxx, and Sp100) and the interferon-γ-inducible protein 16 (IFI16), have emerged as critical restriction factors involved in mediating intrinsic immunity against this virus (6–9).
The IFI16 protein, a member of the p200 family of proteins, now assigned to the PYHIN family (10–14), contains an N-terminal Pyrin domain and two partially conserved 200-amino-acid domains (HIN domains). In addition to sensing and binding foreign DNA (15–17), IFI16 displays multifaceted activity due to its ability to bind to various target proteins (including transcription factors, signaling proteins, and tumor suppressor proteins) and to modulate a multitude of various cell and viral functions (12, 13, 18–23). IFI16 has been shown to interact with the tegument protein pp65 of HCMV at the major immediate-early promoter/enhancer (MIEP) early during infection, resulting in the upregulation of IE protein expression (20). One of the upregulated IE proteins is IE2, which has been found to block cytokine expression (24). pp65 has since been shown to inhibit this response by interacting with the IFI16 Pyrin domain, blocking its oligomerization upon DNA sensing and the subsequent release of antiviral cytokines via the STING/TBK1/IRF3 signaling pathway (17, 25–28).
We have recently demonstrated that IFI16 restricts HCMV replication by downregulating the transcription of early and late viral mRNAs and, in turn, their protein expression (8). However, late during infection, the viral protein kinase pUL97 coregulates the nuclear export of IFI16 into the cytoplasmic viral assembly complex (vAC), resulting in IFI16 becoming entrapped in mature virions. Our previous studies demonstrated that pUL97 binds to and phosphorylates IFI16, a regulatory step that at least in part contributes to the regulated nucleocytoplasmic export of IFI16, which is postulated to be a viral countermeasure to the IFI16 intranuclear DNA-sensing restriction function. However, it remains unclear whether pUL97 is sufficient for this regulated transport of IFI16 or whether other viral proteins are also required. We have also observed that following the translocation of IFI16 to the assembly complex, it colocalizes with the tegument protein pp65, indicating that pp65 also plays a relevant role in the export of IFI16 from the nucleus and becomes part of the virus tegument during the maturation step (16). Moreover, in contrast to what has been observed for herpes simplex virus 1 (HSV-1), a member of the Alphaherpesvirinae (27, 29, 30), IFI16 does not undergo proteolytic degradation during HCMV infection, suggesting that viral or cellular proteins could stabilize and protect IFI16 during virus infection (16, 31).
To gain more insight into the functional interaction between IFI16 and pp65 and establish whether this interaction is limited to MIEP activity modulation or could be extended to other viral gene promoters, we used a mutant of HCMV entirely lacking pp65 expression (v65Stop) (32) and a mutant unable to export pp65 from the nucleus (RV-VM1) (33). The results of our investigations demonstrate that pp65 is involved in the stabilization of IFI16 and in its nucleocytoplasmic dislocalization. Thus, a refined scenario of our previous work suggests that pp65 activity interferes with the innate restriction capacity of IFI16 and may attenuate the IFI16-mediated suppression of viral gene transcription.
MATERIALS AND METHODS
Cells and viruses.
Primary human foreskin fibroblasts (HFFs; ATCC SCRC-1041), human embryo kidney 293 cells (HEK 293; Microbix Biosystems Inc.), and African green monkey kidney cells (Vero; ATCC CCL-81) were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich) as previously described (34). The HCMVs used in this study all were bacterial artificial chromosome (BAC) clones. The clones of the endotheliotropic HCMV strain TB40/E (wild type), the v65Stop virus (unable to express UL83-encoded pp65), and the corresponding revertant virus (v65Rev) were generated previously (32). The RV-HB5 virus was originally cloned by inserting a BAC vector into the US2-US6 gene region of the AD169 strain (35, 36). The HCMV mutant RV-VM1, expressing nuclear pp65, is a descendant of RV-HB5 and was produced as previously reported (33). Briefly, pp65 in RV-VM1 carries a 30-amino-acid insertion at Arg387 that encompasses an immunodominant HLA-A2-presented peptide from the nonstructural IE1 protein (comprising amino acids 288 to 309) and a myc tag. The sequence responsible for the nuclear egress of pp65 is intact in RV-VM1, yet the protein remains primarily nuclear following RV-VM1 infection. HCMVs were propagated and titrated on HFFs (8). Clinical isolate of HSV-1 was propagated and titrated on Vero cells by standard plaque assay.
Antibodies and reagents.
Primary antibodies were obtained from various sources, as shown in Table 1. Conjugated secondary antibodies included Alexa Fluor 488 anti-mouse or Alexa Fluor 555 anti-rabbit antibodies (Life Technologies) and horseradish peroxidase-labeled anti-mouse and anti-rabbit antibodies (GE Healthcare). Protein synthesis was inhibited by using cycloheximide (CHX; Sigma-Aldrich) at a concentration of 120 μg/ml (37).
TABLE 1.
Antibodies used in this study
| Antigen | Species and isotype | Source | Dilutiona |
|
|---|---|---|---|---|
| WB | IFL and PLA | |||
| IFI16 | Rabbit polyclonal anti-N-terminal | Santo Landolfo | 1:1,000 | 1:100 |
| Rabbit polyclonal anti-C-terminal | Santo Landolfo | 1:1,000 | 1:100 | |
| Mouse IgG1 | Santa Cruz Biotechnology | 1:1,000 | 1:100 | |
| HSV-1 ICP0 | Mouse IgG2b | Santa Cruz Biotechnology | 1:1,000 | |
| HCMV IEA | Mouse IgG1k | Argene | 1:1,000 | |
| HCMV pp65 | Mouse IgG2a | Virusys | 1:1,000 | 1:200 |
| HCMV pp28 | Mouse IgG2a | Virusys | 1:1,000 | |
| HCMV MCP | Mouse | Jens Von Einem | 1:2.5 | |
| HCMV gB | Mouse IgG1k | Virusys | 1:200 | |
| Sp3 | Rabbit polyclonal | Santa Cruz Biotechnology | 1:200 | |
| TBP | Mouse IgG1 | Abcam | 1:1,000 | |
| α-Tubulin | Mouse IgG2b | Active-Motive | 1:4,000 | |
| β-Actin | Mouse IgG1 | Sigma | 1:5,000 | |
WB, Western blot; IFL, immunofluorescence; PLA, proximity ligation assay.
Virion gradient purification.
Virion gradient purification was performed as previously described (38). Briefly, medium was collected from fully infected flasks (v65Rev and v65Stop), and cellular debris was removed by low-speed centrifugation (6,000 rpm, 4°C, 15 min). Virus particles were pelleted by ultracentrifugation for 1 h at 4°C at 23,000 rpm in an SW-28 rotor (Beckman). Pelleted virus particles were resuspended in 0.04 M sodium-phosphate buffer (pH 7.4) and layered onto a glycerol-tartrate gradient formed in 0.04 M sodium phosphate. Virus particles were then separated by centrifugation for 90 min at 6°C at 23,000 rpm in an SW-41 rotor. Virions extracted from the gradient were diluted in sodium-phosphate buffer and centrifuged once again at 23,000 rpm for 1 h. The extracted virions were then processed for Western blot analysis by adding radioimmunoprecipitation assay (RIPA) buffer and 4× loading buffer to the pellet.
Luciferase assay.
HFFs were electroporated with luciferase reporter plasmid driven by MIEP or the UL54 gene promoter and pRL-SV40 (Promega) plasmid as previously described (8). HFFs were infected with AdVIFI16 or the control indicator vector, AdVLacZ (multiplicity of infection [MOI] of 50), and then 24 h later they were infected with v65Rev (MOI of 1). Following a further 24 h postinfection (hpi), firefly and Renilla luciferase activities were measured (as previously described by Baggetta et al. [34]) using the Dual-Luciferase reporter assay system kit (Promega) and a Lumino luminometer (Stratec Biomedical Systems, Birkenfeld, Germany). Firefly luciferase activity from the luciferase reporter vector was normalized to the Renilla luciferase activity from the pRL-SV40 vector. Data report the ratio of relative light units (RLU) measured for firefly luciferase activity to the RLU measured for Renilla luciferase activity.
Immunoprecipitation assay.
Uninfected or HCMV-infected cells (MOI of 1) were washed with 1× phosphate-buffered saline (PBS) and lysed in RIPA buffer (50 mM Tris, pH 7.4; 150 mM NaCl; 1 mM EDTA; 1% Nonidet P-40; 0.1% SDS; 0.5% deoxycholate; protease inhibitors). Two hundred micrograms of proteins was incubated with 2 μg of specific or control antibody for 1 h at room temperature with rotation, followed by an overnight incubation at 4°C with protein G-Sepharose (Sigma-Aldrich). Immune complexes were collected by centrifugation and washed with RIPA buffer. The Sepharose beads were pelleted and washed three times with RIPA buffer, resuspended in reducing sample buffer (50 mM Tris, pH 6.8; 10% glycerol; 2% SDS; 1% 2-mercaptoethanol), boiled for 5 min, and resolved on an SDS-PAGE gel to assess protein binding by Western blotting.
Western blot analysis.
Nuclear and cytoplasmic extracts, collected using a nuclear extract kit (Active Motif), and total cell protein extracts were subjected to immunoblot analysis as previously described (16, 37). Briefly, equal amounts of cell extracts were fractionated by electrophoresis on sodium dodecyl sulfate polyacrylamide gels and transferred to Immobilon P membranes (Bio-Rad). After blocking with 5% nonfat dry milk in TBS–0.05% Tween, membranes were incubated overnight at 4°C with the appropriate primary antibodies (Table 1). Membranes were then washed and incubated for 1 h at room temperature with secondary antibodies. Proteins were detected using an enhanced chemiluminescence detection kit (SuperSignal West Pico chemiluminescent substrate; Thermo Scientific). Scanning densitometry of the bands was performed using ImageJ software (Image Processing and Analysis in Java). Background values were subtracted from each calculated value.
Immunofluorescence microscopy.
Immunofluorescence analysis was performed as previously described (24), using the appropriate dilution of primary antibodies (Table 1) for 1 h at room temperature, followed by 1 h with conjugated secondary antibodies in the dark at room temperature. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). Finally, images were taken using a fluorescence microscope (Leica Microsystems).
PLA.
Proximity ligation assay (PLA; DuoLink; Sigma-Aldrich) was performed using the DuoLink PLA kit to detect protein-protein interactions using fluorescence microscopy per the manufacturer's protocol. Briefly, HFF cells were cultured and infected with v65Rev or v65Stop and with RV-HB5 or RV-VM1 at an MOI of 1 for 48 h and 144 h, fixed for 15 min at room temperature, permeabilized with 0.2% Triton X-100, and blocked with 10% HCMV-negative human serum for 30 min at room temperature. Cells were then incubated with primary antibodies diluted in TBS–0.05% Tween for 1 h, washed, and then incubated for another hour at 37°C with species-specific PLA probes under hybridization conditions and in the presence of 2 additional oligonucleotides to facilitate the hybridization only in close proximity (∼40 nm). A ligase was then added to join the two hybridized oligonucleotides, forming a closed circle. Using the ligated circle as the template, rolling-circle amplification was initiated by adding an amplification solution, generating a concatemeric product extending from the oligonucleotide arm of the PLA probe. Lastly, a detection solution consisting of fluorophore-labeled oligonucleotides was added, and the labeled oligonucleotides were hybridized to the concatemeric products. The signal was detected as distinct fluorescent dots in the Texas red channel and analyzed by fluorescence microscopy (Leica Microsystems). Negative controls consisted of mock-infected cells that were otherwise treated in the same way as that described for the infected cells.
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay was performed as previously described (39). Briefly, HFFs were infected with v65Rev or v65Stop at an MOI of 1 for 6 h and 36 h. ChIP assays were performed using the shearing optimization kit and the OneDay ChIP kit (Diagenode Europe). Extracts were sonicated using a BioruptorH Twin (Diagenode) for 10 cycles (30 s on, 30 s off) on the high power setting. Immunoprecipitation was performed using 5 μg of antibody. One microliter of DNA solution per reaction mixture was used for qPCR using HCMV- or human-specific primers (the primer sequences are available on request).
Adenoviral vectors.
The adenovirus transfer vector pAC-CMV IFI16 was constructed as previously described (37). For cell transduction, HFF cells were washed once with 1× PBS and incubated with AdVIFI16 or AdVLacZ at an MOI of 50 in Dulbecco's modified Eagle's medium. After 2 h at 37°C, the virus was washed off and fresh medium applied.
Inhibition of IFI16 expression.
HFF cells were transiently transfected using a MicroPorator (Digital Bio) according to the manufacturer's instructions (1,200 V, 30-ms pulse width, one impulse), with a pool of IFI16 small interfering RNAs (siIFI16) or control siRNA (siCTRL) as a negative control (final concentration, 300 nM; FlexiTube siRNAs SI04373726, SI04341092, SI04156005, SI00445697, SI05101278, SI05101285, and SI03650318; Qiagen). IFI16 siRNA-induced blockade of IFI16 expression was checked by Western blotting and by reverse transcription-quantitative PCR (RT-qPCR).
Quantitative nucleic acid analysis and RT-qPCR.
Total RNA was extracted with the NucleoSpin RNA kit (Macherey-Nagel), and 1 μg was retrotranscribed using the RevertAid H Minus first-strand cDNA synthesis kit (Fermentas) according to the manufacturer's protocol. Comparison of mRNA expression between samples (i.e., infected versus mock infected) was performed using SYBR green (Fermentas) 2−ΔΔCT-based semiquantitative RT-qPCR with the Mx3000P apparatus (Stratagene). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as the reference housekeeping gene. To determine the number of viral DNA genomes per nanogram of cellular reference DNA (GAPDH gene), viral DNA levels were measured by quantitative PCR as described in Luganini et al. (40). HCMV DNA copy numbers were normalized by dividing by the amount of human GAPDH gene amplified per reaction mixture. A standard curve of serially diluted genomic DNA mixed with an IE1-encoding plasmid (from 107 to 1 copy) was created in parallel with each analysis. The primers used in this study were the following: IFI16, 5′-ACTGAGTACAACAAAGCCATTTGA-3′ and 5′-TTGTGACATTGTCCTGTCCCCAC-3′; UL54, 5′-CGGCTACAGTATCTGCGTCA-3′ and 5′-AGCCACCAGGTCAGAGACAT-3′; IE1, 5′-TCAGTGCTCCCCTGATGAGA-3′ and 5′-GATCAATGTGCGTGAGCACC-3′; GAPDH, 5′-AGTGGGTGTCGCTGTTGAAGT-3′ and 5′-AACGTGTCAGTGGTGGACCTG-3′.
Statistical analysis.
All statistical tests were performed using GraphPad Prism version 5.00 for Windows (www.graphpad.com; GraphPad Software, San Diego, CA). The data are presented as the means ± standard deviations (SD). Means between two groups were compared using unpaired t test. Differences were considered statistically significant for P < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
RESULTS
IFI16 differentially regulates viral gene promoters.
We have previously demonstrated that IFI16 overexpression downregulates HCMV UL54 expression at the transcriptional level (8). However, in contrast with this result, Cristea et al. (20) demonstrated that IFI16 stimulates the activity of the HCMV major immediate-early promoter/enhancer (MIEP), indicating that IFI16 is able to differentially regulate viral promoter activity. To test this possibility, we first compared the capability of IFI16 to modulate the activity of the MIEP and the UL54 gene promoter under identical conditions during HCMV infection. For this purpose, HFFs were transfected with plasmids containing the luciferase gene driven by either MIEP or the UL54 gene promoter. Twenty-four hours later, the cells, transduced with recombinant adenovirus vector AdVIFI16 or control vector AdVLacZ at an MOI of 50 for 24 h or left uninfected, were infected with HCMV v65Rev at an MOI of 1. Luciferase activity was then assessed following an additional 24 h of incubation. As shown in Fig. 1A, overexpression of IFI16 significantly increased luciferase activity driven by the MIEP upon HCMV infection, while the activity of the UL54 gene promoter was significantly decreased compared with that observed in cells infected with v65Rev alone or v65Rev preceded by AdVLacZ infection. As shown by Western blotting (Fig. 1B), AdVLacZ, at an MOI of 50, did not modulate IFI16 expression levels, excluding the possibility that AdVLacZ affects IFI16 activity. These results demonstrate that IFI16 differentially regulates viral promoters (MIEP versus UL54 gene promoter) during HCMV infection, in agreement with earlier findings reported by our group (8).
FIG 1.
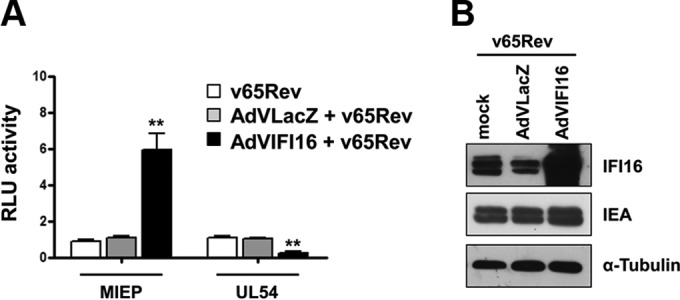
Effects of IFI16 overexpression on the activity of HCMV MIEP and UL54 gene promoter. (A) HFF cells were transiently electroporated with luciferase plasmids encoding MIEP or the UL54 gene promoter. Twenty-four hours later, the cells were transduced with AdVIFI16 (black bar) or AdVLacZ (gray bar) at an MOI of 50 or left uninfected (white bar). Afterwards, cells were infected with v65Rev (MOI of 1). Following a further 24 h, firefly and Renilla luciferase activities were measured. Luciferase activity in whole-cell lysates was normalized to Renilla luciferase activity. The mean value of v65Rev-infected cells (white bar) was arbitrarily defined as 1. Data represent the mean relative activities from three independent experiments, each performed in duplicate. A statistically significant difference compared to v65Rev-infected cells is indicated by ±SD (**, P < 0.01 by unpaired t test). RLU, relative light units. (B) HFFs were transduced with AdVIFI16 or AdVLacZ (MOI of 50) or were mock infected. After 24 h, cells were infected with v65Rev at an MOI of 1. Total cell extracts were prepared and subjected to Western blot analysis using anti-IFI16 polyclonal Ab. HCMV-IEA was employed as a positive control for viral infection, and α-tubulin served as the internal control.
To evaluate whether the different modulation of promoter activity involves the interaction of IFI16 in complex with HCMV pp65, ChIP analysis was performed on HFFs infected with either v65Rev virus, expressing wild-type pp65, or v65Stop virus, unable to express pp65, at an MOI of 1. HFFs infected with HCMV v65Rev for 6 h or 36 h were cross-linked with formaldehyde and sonicated to obtain chromatin fragments for immunoprecipitation using anti-IFI16 polyclonal or anti-pp65 monoclonal antibodies (MAb). Immunoprecipitated proteins were examined by Western blotting using antibodies recognizing IFI16 or pp65 (Fig. 2A). IFI16 was similarly detected, with bands of identical molecular masses, in the extracts of cells infected with v65Rev as well as with v65Stop (Fig. 2A, left). As expected, a pp65-specific band could be observed solely in the extracts of cells infected with v65Rev but not in the extracts of cells infected with v65Stop (Fig. 2A, right). Other affinity-purified polyclonal antibodies (CTRL), used as negative controls, failed to immunoprecipitate IFI16 and pp65. The DNA released from the immunocomplexes was then analyzed by quantitative PCR (qPCR) for the presence of the following DNA sequences: MIEP, UL54, UL69, and GAPDH (Fig. 2B to E). Consistent with the results reported by Cristea et al. (20) and Gariano et al. (8), DNA sequences from the MIEP and UL54 gene promoter were found in pp65-specific as well as in IFI16-specific immune complexes obtained from v65Rev-infected cell extracts, both at 6 and 36 hpi. In contrast, a strong decrease of MIEP and UL54 gene promoter DNA sequences was observed in immune complexes from cell extracts derived from v65Stop virus at 36 hpi, indicating that the binding of IFI16 to these sequences requires pp65. The proteins were not detected at the UL69 viral gene promoter at 6 hpi, although they were present to a limited extent at the later time point (36 hpi) (Fig. 2D). Finally, to demonstrate further the specificity of the interaction of IFI16 and pp65 with MIEP and the UL54 gene promoter, immune complexes were analyzed for the presence of cellular GAPDH DNA sequences. As shown in Fig. 2E, there were no differences in the levels of GADPH DNA in different virus strains and at different times of infection.
FIG 2.
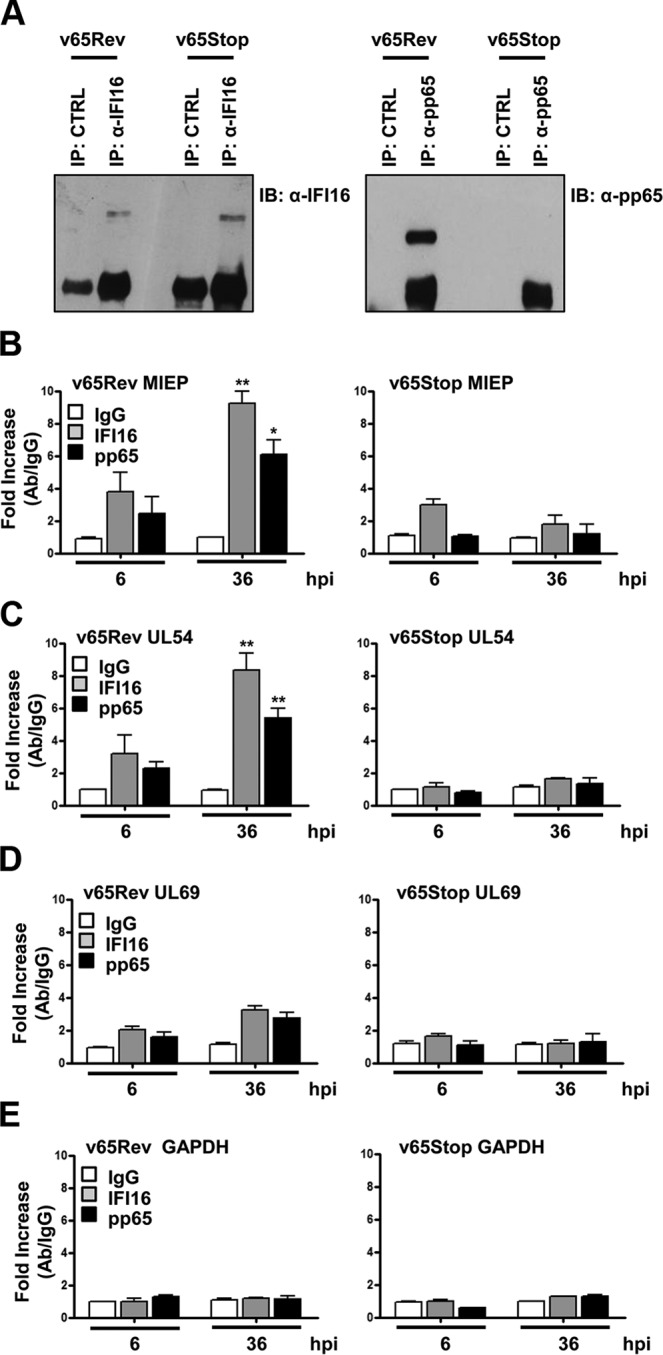
pp65 and IFI16 are present at the MIEP and UL54 gene promoter. HFF cells were infected at an MOI of 1 with v65Rev or v65Stop virus and processed for ChIP assay 36 h later. Cells were cross-linked with formaldehyde, and then the IFI16, pp65, or appropriate control antibody (CTRL) immune complex was isolated by immunoprecipitation. (A) Immunoprecipitated proteins were examined by Western blotting with antibodies for IFI16 or pp65. (B to E) Purified ChIP DNA was analyzed by qPCR for the presence of the following specific DNA sequences: MIEP (B), UL54 (C), UL69 (D), and GAPDH (E). The mean value of control antibody (IgG) was arbitrarily defined as 1. A statistically significant difference compared to IgG is indicated by one (P < 0.05) or two (P < 0.01) asterisks (unpaired t test).
To definitively answer the key question as to whether IFI16 recruits pp65 to the UL54 gene promoter or whether pp65 recruits IFI16, HFFs were transfected with control siRNA (siCTRL) or a pool of siRNAs targeting IFI16 (siIFI16) in order to knock down its expression. As shown in Fig. 3A, IFI16 levels were efficiently decreased in cells treated with specific siIFI16 compared with siCTRL-treated cells. Twenty-four hours after siRNA transfection, cells were infected with v65Rev or v65Stop. After a further 36 h, ChIP analysis was performed to evaluate the binding of pp65 and IFI16 on MIEP or the UL54 gene promoter. As shown in Fig. 3B and C, DNA sequences encompassing both MIEP and the UL54 gene promoter were found in pp65-specific immune complexes obtained from extracts of IFI16 knockdown cells infected with v65Rev. As expected, no DNA sequences were found in pp65-specfic immune complexes from IFI16 knockdown cells infected with v65Stop. In addition, no DNA sequences were found in IFI16 immune complexes from extracts of cells transfected with either siCTRL or siIFI16 and then infected with v65Stop or from extracts of IFI16 knockdown cells infected with v65Rev. Altogether, these results demonstrate that pp65 can bind MIEP and the UL54 gene promoter in the absence of IFI16, whereas IFI16 is unable to bind to these promoters in the absence of pp65.
FIG 3.

IFI16 requires the presence of pp65 for targeting MIEP and UL54 gene promoter. HFFs were electroporated with either pools of siRNAs targeting IFI16 (siIFI16) or scrambled control siRNA (siCTRL), and 24 h later they were infected with v65Rev or v65Stop at an MOI of 1 and examined at 36 hpi. (A) The efficiency of IFI16 depletion was assayed by Western blotting. (B and C) Cells were cross-linked with formaldehyde, and then IFI16 and pp65 immune complexes were isolated by immunoprecipitation. Purified ChIP DNA was analyzed by qPCR for the presence of the following specific DNA sequences: MIEP (B) and UL54 gene promoter (C). The mean value of control antibody (IgG) was arbitrarily defined as 1. A statistically significant difference compared to IgG is indicated by one (P < 0.05) or two (P < 0.01) asterisks (unpaired t test).
To test the functional consequences of the IFI16 interaction with pp65 at the UL54 gene promoter during the early stage of HCMV infection, we compared the growth properties of v65Rev with those of v65Stop virus in cells depleted of IFI16. Primary human fibroblasts were electroporated with either a pool of siRNAs targeting IFI16 (siIFI16) or scrambled control siRNA (siCTRL). Twenty-four hours posttransfection, cells were infected with v65Rev or v65Stop virus and examined at 48 hpi. As shown in Fig. 4A, IFI16 mRNA levels were efficiently decreased in cells treated with specific siIFI16 compared with siCTRL-treated cells. Figure 4B shows a significant increase in the level of UL54 mRNA expression in v65Stop virus-infected cells depleted for IFI16 compared with siCTRL-treated cells, suggesting that repression of the UL54 gene promoter depends on the presence of both proteins. Interestingly, under conditions in which either IFI16 or pp65 was missing, regulation of the UL54 gene promoter was observed to a lesser extent. In addition, the number of viral genome copies in total cellular DNA was quantified using qPCR. Consistent with elevated levels of UL54 mRNA, viral DNA copies were strongly elevated in cells depleted of IFI16 and infected with v65Stop compared with siCTRL-treated cells (Fig. 4C). Moreover, these same results (as reported in Fig. 4C) also were obtained using a second pool of independent siRNA (data not shown). A significant increase of viral DNA copies was observed in cells depleted of IFI16 and infected with v65Stop compared with siCTRL-treated cells.
FIG 4.

Interplay between IFI16 and pp65 regulates HCMV replication. (A) HFFs were electroporated with either pools of siRNAs targeting IFI16 (siIFI16) or scrambled control siRNA (siCTRL), and 24 h later they were infected with v65Rev or v65Stop at an MOI of 1 and examined at 48 hpi. The efficiency of IFI16 RNA depletion (A) and UL54 mRNA viral transcripts (B) was assayed by RT-qPCR. The relative levels of IFI16 and UL54 mRNA are normalized to the levels of cellular GAPDH. The data shown are averages from three experiments. Statistical significance is indicated by one (P < 0.05), two (P < 0.01), or three (P < 0.001) asterisks (unpaired t test). (C) Cells were infected as described for panel A. Viral DNA was isolated at 48 hpi and analyzed by qPCR. The primers amplified a segment of the IE1 gene to determine the number of viral DNA genomes per nanogram of cellular reference DNA (GAPDH gene). The data shown are averages (±SD) from three experiments, each performed in duplicate (*, P < 0.05; ***, P < 0.001; unpaired t test). (D) HFFs were electroporated with a pool of IFI16 small interfering RNAs (siIFI16) or scrambled control siRNA (siCTRL) and then infected with v65Rev or v65Stop (MOI of 0.1). The extent of virus replication was measured at the indicated times postinfection by titrating the infectivity of supernatants and cell suspension on HFFs by standard plaque assay. Results are expressed as means ± SD.
Finally, we examined the replication kinetics of HCMV in cells lacking both pp65 and IFI16. In accordance with previously published data (8), multistep growth analysis of v65Rev replication revealed that knockdown of IFI16 expression caused an increase in the accumulation of infectious progeny at 72 and 144 hpi (Fig. 4D) and that it was more evident when IFI16-depleted cells were infected with v65Stop. Taken together, these results suggest that the recruitment of IFI16 to the UL54 gene promoter impairs viral replication efficiency and is regulated by pp65.
The IFI16 protein is stably expressed during HCMV infection.
Previous studies have shown that IFI16 levels decrease with progression of HSV-1 infection (27, 41, 42). To investigate whether IFI16 is similarly regulated during HCMV infection, HFFs were mock infected or infected with HSV-1 (MOI of 1) or HCMV (MOI of 1). At different time points postinfection, total cell extracts were analyzed for IFI16 and viral protein levels (Fig. 5A and C). HSV-1-infected cells displayed a substantial increase in IFI16 within 2 hpi that gradually started to decrease at 4 hpi, becoming barely detectable by 8 hpi (Fig. 5A). In contrast, in HCMV-infected cells, IFI16 expression significantly increased at 24 hpi, peaked at 72 hpi, and slightly decreased at 144 hpi (Fig. 5C). Consistent with the Western blot results, RT-qPCR analysis confirmed that IFI16 expression upon HSV-1 or HCMV infection is the result of the modulation in mRNA expression (Fig. 5B and D). Taken together, these results support the hypothesis that IFI16 is stabilized during HCMV infection.
FIG 5.

IFI16 protein is stably expressed during HCMV infection. (A) HFF cells were mock infected or infected with HSV-1 at an MOI of 1. (Left) Protein levels of IFI16 and ICP0 were assessed at different times postinfection (hpi) by Western blotting. (Right) IFI16 protein was subjected to densitometry and normalized to α-tubulin, serving as a loading control. The data show the mean values ± SD from three experiments (*, P < 0.05 by unpaired t test). (B) Kinetics analysis of IFI16 mRNA expression in HFFs upon HSV-1 infection at an MOI of 1 by RT-qPCR. IFI16 mRNA expression was normalized to that of GAPDH and is shown as the mean ± SD fold changes following HSV-1 versus mock infection (*, P < 0.05 by unpaired t test). (C) HFFs were mock infected or infected with HCMV v65Rev (MOI of 1). (Left) Lysates were prepared at the indicated time points and subjected to Western blot analysis for IFI16, IEA, and pp28. IFI16 protein was subjected to densitometry and normalized to α-tubulin, which served as a loading control. The data show the mean values ± SD from three experiments (***, P < 0.001 by unpaired t test). (D) Kinetics analysis of IFI16 mRNA expression in HFFs upon HCMV infection at an MOI of 1 by RT-qPCR. IFI16 mRNA expression was normalized to that of GAPDH and is shown as the mean ± SD fold change following HCMV versus mock infection (**, P < 0.05 by unpaired t test).
Viral pp65 interacts with IFI16 during late stages of HCMV infection.
Previous studies have demonstrated that the tegument protein pp65 interacts with IFI16 through the Pyrin domain very early during infection (17). To extend and confirm this observation, we first attempted to monitor the pp65-IFI16 interaction during HCMV infection in situ using a proximity ligation assay (PLA). PLA allows the detection of adjacent proteins through the use of antibodies able to recognize two proteins located within a maximum of 40 nm of each other, as demonstrated in Fig. 6C (IFI16-pp65), or two different epitopes of the same protein, as shown in Fig. 6B (IFI16-IFI16) (42). Mock-, HCMV-v65Rev-, or v65Stop-infected HFFs (MOI of 1) were fixed, stained with anti-pp65 and anti-IFI16 antibodies, and analyzed by indirect immunofluorescence (IFL) or PLA at 48 and 144 hpi (Fig. 6A and C, respectively). Consistent with previous reports, IFI16 and pp65 colocalized in the nucleus and nucleoli of infected cells at 48 hpi. In contrast, at 144 hpi, IFL and PLA signals for the two proteins were mostly found in the cytoplasm. As expected, in cells infected with v65Stop, no signal was generated by PLA. We can conclude that IFI16-pp65 colocalization is specific because when v65Rev HFFs were stained at 144 hpi with HCMV gB, IFI16 PLA did not generate any positive signal despite the presence of both proteins, as shown by indirect IFL (Fig. 6D, upper and lower, respectively). This conclusion is supported further by the PLA results obtained using another nuclear protein, namely, Sp3. Once again, no positive signal was obtained despite the confirmed presence of both IFI16 and Sp3, as shown by indirect IFL analysis (Fig. 6E, upper and lower, respectively). Finally, interaction was also confirmed by immunoprecipitating pp65 from lysates of infected cells at 48 hpi and 144 hpi using either an antibody directed against IFI16 or an antibody against pp65 (Fig. 6F). The specificity of this interaction was verified by the fact that neither protein could be detected in immunoprecipitates using control antibodies.
FIG 6.

Confirmation of the pp65-IFI16 interaction. (A) Kinetics of IFI16-pp65 subcellular localization upon HCMV infection. HFF cells were mock infected or infected with HCMV v65Rev at an MOI of 1 for 48 or 144 h and then subjected to immunofluorescence analysis. pp65 (green) and IFI16 (red) were visualized using primary antibodies followed by secondary antibody staining in the presence of 10% HCMV-negative human serum. Nuclei were counterstained with DAPI (blue). (B to E) PLA was performed to detect protein-protein interactions using fluorescence microscopy. The signal was detected as distinct fluorescent dots in the Texas red channel when cells reacted with the indicated pairs of primary antibodies followed by PLA to assess the interactions between IFI16-IFI16 (B), IFI16-pp65 (C), IFI16-gB (D, upper), and IFI16-Sp3 (E, upper). (D and E, lower) HFF cells were infected with HCMV v65Rev virus at an MOI of 1 for 144 h and then subjected to immunofluorescence analysis. (F) Coimmunoprecipitation from virus-infected or mock-infected cell lysates. HFFs were infected with v65Rev virus (MOI of 1) and harvested at 48 or 144 hpi. Immunoprecipitations were performed using antibodies against IFI16 or pp65 and the appropriate control antibody (CTRL). Immunoprecipitated proteins were detected by Western blot analyses using antibodies against IFI16 and pp65. Nonimmunoprecipitated whole-cell extracts (Input) were immunoblotted using anti-IFI16 or anti-pp65 antibodies.
Together, these results suggest an enduring interaction between pp65 and IFI16, which begins in the nucleus (48 hpi) and then continues in the cytoplasm during late stages of HCMV infection.
Stabilization of IFI16 in the presence of pp65.
The interaction of IFI16 with pp65 has been reported to modulate its functional activity, but other potential consequences, such as the stability of IFI16, have not been investigated. To determine whether pp65 has an effect on IFI16 protein levels, lysates of HFFs infected with the v65Stop HCMV were compared with those of the revertant virus, v65Rev, or the wild-type parental strain TB40/E at different times postinfection by Western blotting. As shown in Fig. 7A (upper), IFI16 levels started to increase at 12 hpi and remained high until 48 hpi in both wild-type and v65Rev-infected cells. Although the expression level of IFI16 in v65Stop-infected cells was similar to that observed in wild-type- and v65Rev-infected cells until 24 hpi, by 48 hpi it was barely detectable. IEA expression, used as the positive control, was maintained at similar levels throughout the course of infection. Quantitative analysis performed by densitometry confirmed this conclusion (Fig. 7A, lower).
FIG 7.

Stabilization of IFI16 in the presence of pp65. (A) HFFs were infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1. (Upper) Lysates were prepared at the indicated time points and subjected to Western blot analysis for IFI16, IEA, pp65, and α-tubulin. (Lower) IFI16 was subjected to densitometry normalized to α-tubulin, which served as a loading control. Data present the means ± SD from three experiments (*, P < 0.05 by unpaired t test). (B) HFFs were infected with wild-type, v65Rev, and v65Stop viruses at an MOI of 1. Nuclear and cytoplasmic fractions were prepared at the indicated time points and subjected to Western blot analysis for IFI16 and pp65. α-Tubulin and TBP were used as purity and loading controls for the cytoplasmic and nuclear fractions, respectively. HCMV-IEA was employed as a positive control for viral infection. (C) Protein extracts were obtained from purified virions (v65Rev and v65Stop) and subjected to Western blot analysis using antibodies against IFI16 and MCP. MCP was employed to confirm infection and the isolation of virions. (D) HFFs were infected with either the v65Rev or v65Stop HCMV strain, and at 36 hpi they were incubated with cycloheximide (CHX) for 2 h. Lysates were subjected to Western blot analysis for IFI16, pp65, and α-tubulin, which served as a loading control.
To investigate whether nuclear egress of IFI16 is dependent on the presence of pp65, nuclear and cytoplasmic fractions prepared at 24 hpi, 96 hpi, and 144 hpi from HFFs infected with wild-type, v65Rev, or v65Stop virus at an MOI of 1 were analyzed by Western blotting with immunostaining for pp65, IFI16, nuclear TATA-binding protein (TBP), and cytoplasmic α-tubulin expression. Anti-IEA antibodies were used as a positive control of infection. As shown in Fig. 7B, the IFI16 protein was expressed at much lower levels in the nuclei of v65Stop-infected cells, starting at 96 hpi, compared with the expression levels observed in the nuclei of wild-type- or v65Rev-infected cells. Interestingly, IFI16 was detectable in the nuclear fraction but not in the cytoplasm of v65Stop-infected cells, unlike fractions of wild-type- or v65Rev-infected cell lysates. Consistent with these results, we failed to detect any IFI16 protein entrapped in the virions purified from v65Stop-infected cells, while it was evident in virions from v65Rev-infected cells. Detection of the major capsid protein (MCP) was verified by Western blotting to confirm virion purification (Fig. 7C).
To test whether pp65 regulates IFI16 protein levels by modulating its stability, we determined the half-life of IFI16 in the presence versus the absence of pp65 expression. HFFs were infected with either v65Rev or v65Stop viruses and incubated with cycloheximide (CHX) for 2 h at 36 hpi. IFI16 showed a half-life of less than 1 h in the absence of pp65, whereas IFI16 protein levels were stable for up to 2 h in the presence of pp65, indicating that IFI16 is quickly degraded in the absence of pp65 (Fig. 7D).
Taken together, these results show that HCMV pp65 stabilizes IFI16 by increasing its half-life and allowing it to translocate first into the cytoplasm and, subsequently, into virions.
Translocation of IFI16 from the nucleus into the cytoplasm at late stages of infection requires pp65.
To corroborate the finding that pp65 stabilizes and delocalizes IFI16 from the nucleus, experiments were performed with the RV-VM1 HCMV strain encoding a pp65 protein with a 30-amino-acid insertion at Arg 387 (33) that retains pp65 in the nucleus. In line with previous studies, phase-contrast images of cells infected with RV-VM1, but not with the wild-type parental strain RV-HB5, showed that NIBs (nuclear inclusion bodies) were undetectable in the majority of the infected nuclei and replaced by large globular structures (LGS) (Fig. 8A), both of which are considered sites of virus replication, transcription, and capsid assembly (33). Indirect immunofluorescence staining using a pp65 MAb demonstrated the nuclear retention of mutant pp65, especially at 144 hpi in cells infected with RV-VM1 (Fig. 8B). Mutant pp65 within the nucleus was detected at the edges of LGS but not within the body of these structures. A similar pattern of distribution was observed when RV-VM1-infected cells were stained with anti-IFI16 antibodies. As expected, in HFFs infected with RV-HB5 at 144 hpi, both pp65 and IFI16 presented cytoplasmic localization. Consistent with these observations, nuclear colocalization of IFI16 with pp65-, RV-VM1-, or RV-HB5-infected cells was supported by PLA analysis. As shown in Fig. 8C, PLA signals in RV-VM1-infected cells were exclusively observed in the nucleus for both proteins at 48 and 144 hpi, consistent with the finding that nuclear IFI16 delocalization is driven by pp65. In contrast, RV-HB5-infected HFFs displayed cytoplasmic colocalization of the two proteins at 144 hpi.
FIG 8.

IFI16 is retained in the nucleus during late stages of RV-VM1 infection. (A) Performance of the pp65 mutant RV-VM1 HCMV in fibroblasts compared with wild-type virus (RV-HB5). The arrows indicate large globular structures (LGS) in the nuclei typically associated with RV-VM1 replication. (B) HFF cells were mock infected or infected with RV-HB5 or RV-VM1 viruses at an MOI of 1 for 48 or 144 h and then subjected to immunofluorescence analysis. pp65 (green) and IFI16 (red) were visualized using primary antibodies followed by secondary antibody staining in the presence of 10% HCMV-negative human serum. Nuclei were counterstained with DAPI (blue). (C) PLA were performed to detect protein-protein interactions using fluorescence microscopy in HFF cells mock infected or infected with RV-HB5 or RV-VM1 virus at an MOI of 1 for 48 or 144 h. A positive signal was detected as distinct fluorescent dots in the Texas red channel when IFI16 and pp65 were in close proximity (∼40 nm). (D) HFFs were infected with RV-HB5 or RV-VM1 at an MOI of 1. Nuclear and cytoplasmic fractions were prepared at the indicated time points and subjected to Western blot analysis for IFI16, pp65, α-tubulin, and TBP. HCMV-IEA was employed as a positive control for viral infection.
To provide definitive proof of the nuclear retention of pp65 together with IFI16, Western blot analysis of nuclear or cytoplasmic extracts derived from RV-HB5- or RV-VM1-infected cells was performed at different time points postinfection, ranging from 24 h to 144 h (Fig. 8D). Consistent with the above-described results, in extracts from RV-VM1-infected HFFs, pp65 and IFI16 could be detected only in the nuclear extracts at all time points indicated. Interestingly, in extracts from RV-HB5-infected cells, pp65 and IFI16 were observed in nuclear extracts from 24 hpi through 144 hpi, and from 96 hpi to 144 hpi they were also detectable in cytoplasmic extracts.
Altogether, these data reveal a dual role for pp65: during early infection, it modulates IFI16 activity at the promoter of immediate-early and early genes; subsequently, it delocalizes IFI16 from the nucleus into the cytoplasm, thereby stabilizing and protecting it from degradation (Fig. 9).
FIG 9.

Proposed model for the functional role of pp65 in modulating IFI16 activity during HCMV infection. IFI16 and pp65 cooperate at the level of viral genes, and the effect of this interaction (downregulation versus upregulation) depends on the viral promoter. In the early phases of infection, HCMV-pp65 hijacks IFI16 to activate MIEP expression; later on, IFI16 inhibits HCMV replication by blocking the UL54 gene promoter. Moreover, pp65 interacts with, stabilizes, and protects IFI16 from proteolytic degradation during HCMV infection. Finally, pp65 moves the protein into the cytoplasm, and then IFI16 becomes entrapped within newly assembled virions.
DISCUSSION
Viral replication and maturation rely on a complex interplay between viral and cellular proteins. During the replication cycles of herpesviruses, a number of host immune defense mechanisms have to be overcome and, in particular, innate immune barriers that are poorly defined so far have to be circumvented in order to achieve high efficiencies of replication and dissemination in host tissues. Previous work by Cristea et al. (20) has shown that very early during infection, pp65 and IFI16 interact at the HCMV MIEP, thereby triggering an increase in IE protein expression, which is accompanied by a concomitant decrease in antiviral cytokine production. Consistent with this observation, an increase in IFI16 expression during the first steps of HCMV infection has been reported previously by our group (16), confirming that HCMV triggers IFI16 expression with the scope of increasing IE gene expression early during infection.
Based on these observations, the aim of the present study was to investigate whether the interaction between the two proteins continued at the late stages of infection and whether it played a role in the regulation of other viral promoters. Here, we provide evidence that IFI16 is recruited by pp65, as shown by ChIP assay, and downregulates UL54 gene promoter activity during the early steps of HCMV infection. We also demonstrated robust virus DNA synthesis accompanied by an increase in virus yield when using the v65Stop virus lacking pp65 expression to infect cells knocked down in IFI16. Of note, the interplay between pp65 and IFI16 was not limited to the very early stage of infection but remained functionally relevant throughout the later stages of viral replication, including the central process of viral DNA synthesis. A second important observation was that the outcome of the pp65-IFI16 interaction on promoter activity (i.e., downregulation versus upregulation) depends on the type of promoter the two proteins interact with, as observed here for the MIEP versus UL54 gene promoter. Finally, the observation that robust DNA synthesis takes place in IFI16 knockdown HFFs infected with the v65Stop virus suggests that their interaction is required for the fine regulation of the UL54 gene promoter. Additional support for the biological relevance of this interaction came from the outcome of virus yield experiments, which show that in the absence of both proteins the yield of the viral progeny was greater.
Of note, the interaction between pp65 and IFI16 was not limited to the regulation of UL54 gene promoter activity. We have previously shown that during the early phase of HCMV infection, IFI16 levels decrease inside the nucleus, and this is accompanied by a parallel increase in its presence in the cytoplasmic viral assembly complex (vAC). The nucleocytoplasmic egress of IFI16 in HCMV-infected cells is driven, at least in part, by the viral protein kinase pUL97, which binds and phosphorylates nuclear IFI16. Later on, IFI16 moves from the vAC and becomes entrapped within newly assembled virions (8). Interestingly, Kamil and Coen (43) have demonstrated, by means of coimmunoprecipitation experiments in lysates of infected cells, an interaction between pUL97 and pp65 that leads to incorporation of these proteins into HCMV particles. In this scenario, it emerges that both proteins are required for IFI16 to undergo complete nuclear delocalization and entrapment in the mature virus particles. To corroborate the requirement of pp65 for the translocation of IFI16 into the cytoplasm, similar experiments were performed using RV-VM1 virus, which expresses a mutant pp65 previously characterized for its nuclear retention. In RV-VM1-infected HFFs, IFI16 was confirmed to accumulate in the nucleus, bound to pp65, a finding that suggested pUL97 is not the only viral protein involved in the regulation of IFI16 nucleocytoplasmic dislocalization but that pp65 contributes to this activity.
The functional relevance of this interaction can be inferred by the finding that proteolytic IFI16 degradation at late time points does not occur, in contrast to what has been observed during HSV-1 infection (27, 29, 41, 42). Thus, IFI16 nuclear delocalization may contribute to preventing the detection of HCMV DNA and the suppression of viral gene transcription, in turn enhancing viral replication. The following observations allow us to draw our working model: pp65 initially exploits IFI16 to increase IE gene expression (20) and then, to prevent its restriction activity on early genes, e.g., UL54, stabilizes and exports IFI16 into the cytoplasm. In addition, since inhibition of viral gene expression following v65Stop treatment with UV irradiation prevented IFI16 degradation (data not shown), we can argue that one or more genes must be responsible for IFI16 degradation occurring in the absence of pp65. Thus, IFI16 stabilization and its entrapment in the virions is an active process carried out by pp65 and bona fide pUL97.
These findings are at variance with the observations made in HFFs infected with HSV-1. In fact, HSV-1 blocks the IFI16-mediated immune response by catalyzing its degradation, in part via the contribution of ICP0. IFI16 degradation leads to an attenuation of antiviral cytokine expression (17, 28) that contributes to virus evasion of innate immune defenses. Interestingly, Kerur et al. (18) demonstrated that during Kaposi sarcoma-associated herpesvirus (KSHV) infection of endothelial cells, IFI16 interacts with the adaptor molecule ASC (apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain [CARD]) and procaspase-1 to form a functional inflammasome. IFI16 was still detectable at 48 hpi, suggesting that KSHV, similar to what we observed here with HCMV, does not trigger IFI16 proteasomal degradation but rather exploits IFI16 to trigger an inflammatory response.
A central question posed by these studies is why HCMV would stabilize and export IFI16 in the cytoplasm and then include it in mature virions. Although a definitive hypothesis cannot be formulated yet, Cristea et al. (20) demonstrated that, in the first 6 h of infection, pp65 and IFI16 cooperate to increase the MIEP transcriptional activity, accompanied by a concomitant decrease in antiviral cytokine production. Consistent with these results, we had previously shown that replication of murine cytomegalovirus (MCMV) is indeed decreased in cells containing a dominant-negative form of the murine p200 family member p204, the homologue of the human IFI16 (44). The replication defect was found to include delayed IE gene expression, suggesting that regulation of viral IE genes by p200 family members is a conserved feature of CMV infection. Thus, the Trojan horse mechanism of IFI16 entrapment may render it readily available to the infected cells and accelerate virus replication.
Considering all these findings together, we suggest that individual viruses have developed distinct strategies resulting in the maintenance or degradation of IFI16, depending on the role that IFI16 plays in the physiology of the virus's life in terms of replication, immune evasion, suppression of antiviral cytokine production, and inflammasome activation.
ACKNOWLEDGMENTS
We acknowledge Salvatore Oliviero and Stefania Rapelli, Human Genetics Foundation (HuGeF)–Turin, for their technical assistance.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2702–2772. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA, USA. [Google Scholar]
- 2.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 3.Dempsey A, Bowie AG. 2015. Innate immune recognition of DNA: a recent history. Virology 479-480:146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson WE. 2013. Rapid adversarial co-evolution of viruses and cellular restriction factors. Curr Top Microbiol Immunol 371:123–151. [DOI] [PubMed] [Google Scholar]
- 5.Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat Immunol 5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- 6.Adler M, Tavalai N, Müller R, Stamminger T. 2011. Human cytomegalovirus immediate-early gene expression is restricted by the nuclear domain 10 component Sp100. J Gen Virol 92:1532–1538. doi: 10.1099/vir.0.030981-0. [DOI] [PubMed] [Google Scholar]
- 7.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahn JH, Hayward GS. 2000. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 274:39–55. doi: 10.1006/viro.2000.0448. [DOI] [PubMed] [Google Scholar]
- 10.Connolly DJ, Bowie AG. 2014. The emerging role of human PYHIN proteins in innate immunity: implications for health and disease. Biochem Pharmacol 92:405–414. doi: 10.1016/j.bcp.2014.08.031. [DOI] [PubMed] [Google Scholar]
- 11.Gariglio M, Mondini M, De Andrea M, Landolfo S. 2011. The multifaceted interferon-inducible p200 family proteins: from cell biology to human pathology. J Interferon Cytokine Res 31:159–172. doi: 10.1089/jir.2010.0106. [DOI] [PubMed] [Google Scholar]
- 12.Diner BA, Lum KK, Cristea IM. 2015. The emerging role of nuclear viral DNA sensors. J Biol Chem 290:26412–26421. doi: 10.1074/jbc.R115.652289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakobsen MR, Paludan SR. 2014. IFI16: at the interphase between innate DNA sensing and genome regulation. Cytokine Growth Factor Rev 25:649–655. doi: 10.1016/j.cytogfr.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Schattgen SA, Fitzgerald KA. 2011. The PYHIN protein family as mediators of host defenses. Immunol Rev 243:109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- 15.Stratmann S, Morrone S, van Oijen AM, Sohn J. 2015. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. eLife 4:e11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dell'Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Lo Cigno I, Biolatti M, Vallino M, Marschall M, Gariglio M, Landolfo S. 2014. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol 88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Chen J, Cristea IM. 2013. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B. 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9:363–375. doi: 10.1016/j.chom.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bawadekar M, De Andrea M, Gariglio M, Landolfo S. 2015. Mislocalization of the interferon inducible protein IFI16 by environmental insults: implications in autoimmunity. Cytokine Growth Factor Rev 26:213–219. doi: 10.1016/j.cytogfr.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O'Keefe ES, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol 84:7803–7814. doi: 10.1128/JVI.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ansari MA, Dutta S, Veettil MV, Dutta D, Iqbal J, Kumar B, Roy A, Chikoti L, Singh VV, Chandran B. 2015. Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-β responses. PLoS Pathog 11:e1005019. doi: 10.1371/journal.ppat.1005019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, Chikoti L, Kumar B, Johnson KE, Chandran B. 2015. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLoS Pathog 11:e1005030. doi: 10.1371/journal.ppat.1005030. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Dell'Oste V, Gatti D, Giorgio AG, Gariglio M, Landolfo S, De Andrea M. 2015. The interferon-inducible DNA-sensor protein IFI16: a key player in the antiviral response. New Microbiol 38:5–20. [PubMed] [Google Scholar]
- 24.Taylor RT, Bresnahan WA. 2006. Human cytomegalovirus immediate-early 2 protein IE86 blocks virus-induced chemokine expression. J Virol 80:920–928. doi: 10.1128/JVI.80.2.920-928.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM, Fitzgerald KA, Kurt-Jones EA. 2014. Interferon γ-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 289:23568–23581. doi: 10.1074/jbc.M114.554147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diner BA, Lum KK, Javitt A, Cristea IM. 2015. Interactions of the antiviral factor IFI16 mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol Cell Proteomics 14:2341–2356. doi: 10.1074/mcp.M114.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanfranca MP, Mostafa HH, Davido DJ. 2014. HSV-1 ICP0: an E3 ubiquitin ligase that counteracts host intrinsic and innate immunity. Cells 3:438–454. doi: 10.3390/cells3020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biolatti M, Pautasso S, Dell'Oste V, De Andrea M, von Einem J, Plachter B, Marschall M, Gariglio M, Landolfo S. 2015. ID: 37. The IFI16 restriction factor cooperates with HCMV pUL83 to down-regulate UL54 gene expression and viral DNA synthesis. Cytokine 76:70–71. [Google Scholar]
- 32.Chevillotte M, Landwehr S, Linta L, Frascaroli G, Lüske A, Buser C, Mertens T, von Einem J. 2009. Major tegument protein pp65 of human cytomegalovirus is required for the incorporation of pUL69 and pUL97 into the virus particle and for viral growth in macrophages. J Virol 83:2480–2490. doi: 10.1128/JVI.01818-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becke S, Fabre-Mersseman V, Aue S, Auerochs S, Sedmak T, Wolfrum U, Strand D, Marschall M, Plachter B, Reyda S. 2010. Modification of the major tegument protein pp65 of human cytomegalovirus inhibits virus growth and leads to the enhancement of a protein complex with pUL69 and pUL97 in infected cells. J Gen Virol 91:2531–2541. doi: 10.1099/vir.0.022293-0. [DOI] [PubMed] [Google Scholar]
- 34.Baggetta R, De Andrea M, Gariano GR, Mondini M, Rittà M, Caposio P, Cappello P, Giovarelli M, Gariglio M, Landolfo S. 2010. The interferon-inducible gene IFI16 secretome of endothelial cells drives the early steps of the inflammatory response. Eur J Immunol 40:2182–2189. doi: 10.1002/eji.200939995. [DOI] [PubMed] [Google Scholar]
- 35.Reyda S, Tenzer S, Navarro P, Gebauer W, Saur M, Krauter S, Büscher N, Plachter B. 2014. The tegument protein pp65 of human cytomegalovirus acts as an optional scaffold protein that optimizes protein uploading into viral particles. J Virol 88:9633–9646. doi: 10.1128/JVI.01415-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borst E-M, Hahn G, Koszinowski UH, Messerle M. 1999. Cloning of the human cytomegalovirus (HCMV) genome as an infectious bacterial artificial chromosome in Escherichia coli: a new approach for construction of HCMV mutants. J Virol 73:8320–8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gugliesi F, Mondini M, Ravera R, Robotti A, de Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2005. Up-regulation of the interferon-inducible IFI16 gene by oxidative stress triggers p53 transcriptional activity in endothelial cells. J Leukoc Biol 77:820–829. doi: 10.1189/jlb.0904507. [DOI] [PubMed] [Google Scholar]
- 38.Cappadona I, Villinger C, Schutzius G, Mertens T, von Einem J. 2015. Human cytomegalovirus pUL47 modulates tegumentation and capsid accumulation at the viral assembly complex. J Virol 89:7314–7328. doi: 10.1128/JVI.00603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lo Cigno I, De Andrea M, Borgogna C, Albertini S, Landini MM, Peretti A, Johnson KE, Chandran B, Landolfo S, Gariglio M. 2015. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J Virol 89:7506–7520. doi: 10.1128/JVI.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luganini A, Caposio P, Landolfo S, Gribaudo G. 2008. Phosphorothioate-modified oligodeoxynucleotides inhibit human cytomegalovirus replication by blocking virus entry. Antimicrob Agents Chemother 52:1111–1120. doi: 10.1128/AAC.00987-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cuchet-Lourenço D, Anderson G, Sloan E, Orr A, Everett RD. 2013. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol 87:13422–13432. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Everett RD. 2015. The dynamic response of IFI16 and PML nuclear body components to HSV-1 infection. J Virol 14:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamil JP, Coen DM. 2007. Human cytomegalovirus protein kinase UL97 forms a complex with the tegument phosphoprotein pp65. J Virol 81:10659–10668. doi: 10.1128/JVI.00497-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hertel L, De Andrea M, Azzimonti B, Rolle A, Gariglio M, Landolfo S. 1999. The interferon-inducible 204 gene, a member of the Ifi 200 family, is not involved in the antiviral state induction by IFN-alpha, but is required by the mouse cytomegalovirus for its replication. Virology 262:1–8. doi: 10.1006/viro.1999.9885. [DOI] [PubMed] [Google Scholar]