

ABSTRACT
Gibbon ape leukemia virus (GALV) and koala retrovirus (KoRV) most likely originated from a cross-species transmission of an ancestral retrovirus into koalas and gibbons via one or more intermediate as-yet-unknown hosts. A virus highly similar to GALV has been identified in an Australian native rodent (Melomys burtoni) after extensive screening of Australian wildlife. GALV-like viruses have also been discovered in several Southeast Asian species, although screening has not been extensive and viruses discovered to date are only distantly related to GALV. We therefore screened 26 Southeast Asian rodent species for KoRV- and GALV-like sequences, using hybridization capture and high-throughput sequencing, in the attempt to identify potential GALV and KoRV hosts. Only the individuals belonging to a newly discovered subspecies of Melomys burtoni from Indonesia were positive, yielding an endogenous provirus very closely related to a strain of GALV. The sequence of the critical receptor domain for GALV infection in the Indonesian M. burtoni subsp. was consistent with the susceptibility of the species to GALV infection. The second record of a GALV in M. burtoni provides further evidence that M. burtoni, and potentially other lineages within the widespread subfamily Murinae, may play a role in the spread of GALV-like viruses. The discovery of a GALV in the most western part of the Australo-Papuan distribution of M. burtoni, specifically in a transitional zone between Asia and Australia (Wallacea), may be relevant to the cross-species transmission to gibbons in Southeast Asia and broadens the known distribution of GALVs in wild rodents.
IMPORTANCE Gibbon ape leukemia virus (GALV) and the koala retrovirus (KoRV) are very closely related, yet their hosts neither are closely related nor overlap geographically. Direct cross-species infection between koalas and gibbons is unlikely. Therefore, GALV and KoRV may have arisen via a cross-species transfer from an intermediate host whose range overlaps those of both gibbons and koalas. Using hybridization capture and high-throughput sequencing, we have screened a wide range of rodent candidate hosts from Southeast Asia for KoRV- and GALV-like sequences. Only a Melomys burtoni subspecies from Wallacea (Indonesia) was positive for GALV. We report the genome sequence of this newly identified GALV, the critical domain for infection of its potential cellular receptor, and its phylogenetic relationships with the other previously characterized GALVs. We hypothesize that Melomys burtoni, and potentially related lineages with an Australo-Papuan distribution, may have played a key role in cross-species transmission to other taxa.
INTRODUCTION
The evolutionary mechanisms involved in cross-species transmissions (CSTs) of viruses are complex and generally poorly understood. Viral evolution, host contact rates, biological similarity in host defense systems (receptors, viral restriction factors), and host evolutionary relationships have been proposed as key factors in CST rates and outcomes (1). However, there are cases in which the CSTs occur between hosts that are biogeographically separated, distantly related, or both. For example, the koala retrovirus (KoRV) and the gibbon ape leukemia virus (GALV) are very closely related viruses (2) that infect hosts that are neither sympatric nor closely related. GALV is an exogenous gammaretrovirus that has been isolated from captive white-handed gibbons (Hylobates lar) held in or originally from Southeast Asia (3–6). Of the five GALV strains identified so far, four have been isolated in gibbons (3–6) and one—the woolly monkey virus (WMV), formerly referred to as SSAV (7, 8)—in a woolly monkey (Lagothrix lagotricha), probably as the result of a horizontal transmission of GALV from a gibbon. KoRV is a potentially infectious endogenous retrovirus (ERV) of wild koalas (Phascolarctos cinereus) in Australia and captive koalas worldwide (9–11). Both viruses are associated with lymphoid neoplasms in their hosts (12, 13). KoRV and GALV share high nucleotide sequence similarity (80%) and form a monophyletic clade within gammaretroviruses (2). In contrast, the species range of koalas is restricted to Australia and does not overlap that of gibbons, which are endemic to Southeast Asia. The lack of host sympatry suggests that an intermediate host with a less restricted range is responsible for GALV and KoRV CST (9, 14–16).
Mobile species such as bats, birds, or commensal rats have been proposed as potential intermediate hosts of GALV and KoRV (9, 14). Bats can fly and disperse rapidly, they have been linked to the spread of several zoonotic diseases (17), and some Southeast Asian bat species harbor retroviruses related to GALV and KoRV (18). Rodents, however, are plausible intermediate hosts as they have migrated from Southeast Asia to Australia multiple times with several Southeast Asian species having established themselves in Australia (19). Furthermore, endogenous retroviruses related to GALV have been reported to be present in the genome of several Southeast Asian rodents such as Mus caroli, Mus cervicolor, and Vandeleuria oleracea (20–22). However, these reports were based on DNA hybridization techniques, and sequences were not reported. In 2008, the full genome sequence of an endogenous retrovirus found in the genome of Mus caroli (McERV) was reported (23). Despite the relatively high similarity of its genomic sequence to those of GALV and KoRV, McERV has a different host range and uses a different receptor, and therefore it is unlikely to be a progenitor of GALV and KoRV (23). McERV is most closely related to the Mus dunni endogenous virus (MDEV) (24) and the Mus musculus endogenous retrovirus (MmERV) (25), which together form a sister clade to the KoRV/GALV clade (2). Recently, Simmons et al. (16) discovered fragments belonging to a retrovirus closely related to GALV and KoRV in the Australian native rodent Melomys burtoni (MbRV). The MbRV sequence shares 93 and 83% nucleotide identity with those of GALV and KoRV, respectively, and the geographic distribution of Melomys burtoni overlaps that of koalas. However, it is hard to explain how this Australian murid species could have come in contact with gibbons in Southeast Asia. Consequently, it is unlikely that MbRV represents the direct or immediate ancestor virus of KoRV and GALV (16).
The aim of this work was to screen a wide range of rodent species from Southeast Asia for the presence of KoRV- and GALV-like sequences and characterize polymorphisms in their viral receptor proteins in the attempt to identify the intermediate host(s) of KoRV and GALV using a non-PCR-based approach called hybridization capture (26, 27). We focused on Southeast Asian rodent species because 42 Australian vertebrate species were previously screened, with MbRV the only virus identified (16), and because most of the rodent species with GALV-like sequences identified are from Southeast Asia, suggesting that GALVs and KoRVs may be circulating naturally in rodent populations residing there. Twenty-six rodent species were screened, of which only a newly identified Australasian subspecies of Melomys burtoni, in the process of being taxonomically described and geographically reported (P.-H. Fabre, Y. S. Fitriana, M. Pagès, K. Aplin, G. Semiadi, N. Supriatna, and K. M. Helgen, unpublished data), was positive for a GALV sequence distinct from that of MbRV and none were positive for KoRV-like sequences. Specifically, this new subspecies has been discovered in the biogeographical region comprising a group of mainly Indonesian islands between the Asian and Australian continental shelves and called Wallacea (Fabre et al., unpublished). We report the complete nucleotide sequence of the identified GALV-like virus, which we term Melomys woolly monkey virus (MelWMV), its genomic structure, and its phylogenetic relationships with other related gammaretroviruses. We also examine GALV receptor variation among permissive and restrictive hosts, including species belonging to the genus Melomys.
MATERIALS AND METHODS
Sample collection.
The rodents used for the screening for GALV and KoRV were captured using folding rat traps during fieldwork expeditions in Southeast Asia and Asia in the periods of January and February 2010, June and July 2010, and September 2013. Muscle samples were collected and conserved in ethanol. All 49 samples belonging to the 26 species analyzed in the current study are listed in Table 1. For the sequencing of the receptor of GALV, a blood sample was collected from a male white-handed gibbon (Hylobates lar) from the Nuremberg zoo, Germany, during a routine health check on 24 July 1996.
TABLE 1.
Rodent species screened using hybridization capture for the presence of KoRV-like and GALV-like sequences
| Species no. | Species | Country | Code |
|---|---|---|---|
| 1 | Bandicota bengalensis | Bangladesh | 2 |
| 2 | Bandicota indica | Cambodia | 10 |
| 3 | Bandicota savilei | Myanmar | 13 |
| Bandicota savilei | Myanmar | 14 | |
| 4 | Berylmys berdmorei | Laos | 19 |
| Berylmys berdmorei | Laos | 20 | |
| Berylmys berdmorei | Laos | 22 | |
| 5 | Berylmys bowersi | Laos | 27 |
| Berylmys bowersi | Laos | 28 | |
| 6 | Berylmys mackenziei | India | 31 |
| 7 | Chiromyscus chiropus | Laos | 32 |
| Chiromyscus chiropus | Laos | 35 | |
| 8 | Laonastes aenigmamus | Laos | 37 |
| Laonastes aenigmamus | Laos | 41 | |
| 9 | Leopoldamys edwardsi | Laos | 42 |
| 10 | Maxomys moi | Laos | 54 |
| 11 | Maxomys surifer | Laos | 55 |
| 12 | Mus booduga | Bangladesh | 60 |
| Mus booduga | India | 61 | |
| 13 | Mus caroli | Laos | 96 |
| Mus caroli | Cambodia | 99 | |
| 14 | Mus cervicolor | Laos | 103 |
| Mus cervicolor | Laos | 104 | |
| Mus cervicolor | Laos | 106 | |
| Mus cervicolor | Laos | 108 | |
| 15 | Mus cookii | Laos | 115 |
| Mus cookii | Laos | 116 | |
| 16 | Mus fragilicauda | Laos | 118 |
| 17 | Mus lepidoides | Myanmar | 121 |
| Mus lepidoides | Myanmar | 123 | |
| 18 | Mus musculus | Bangladesh | 124 |
| Mus musculus | Bangladesh | 126 | |
| Mus musculus | Bangladesh | 128 | |
| Mus musculus | Bangladesh | 129 | |
| 19 | Mus nitidulus | Myanmar | 133 |
| Mus nitidulus | Myanmar | 134 | |
| 20 | Mus terricolor | Bangladesh | 135 |
| 21 | Niviventer confucianus | Laos | 140 |
| Niviventer confucianus | Laos | 141 | |
| 22 | Niviventer fulvescens | Laos | 143 |
| 23 | Niviventer langbianis | Laos | 150 |
| 24 | Vandeleuria oleracea | Myanmar | 196 |
| 25 | Melomys burtoni subsp. | Indonesia | WD309 |
| Melomys burtoni subsp. | Indonesia | WD282 | |
| Melomys burtoni subsp. | Indonesia | WD283 | |
| Melomys burtoni subsp. | Indonesia | WD310 | |
| Melomys burtoni subsp. | Indonesia | WD144 | |
| Melomys burtoni subsp. | Indonesia | WD279 | |
| 26 | Melomys paveli | Indonesia | YS284 |
Ethics statement.
All animal experiments were performed according to the directive 2010/63/EEC on the Protection of Animals Used for Experimental and Other Scientific Purposes. The animal work also complied with the French law (nu 2012–10 dated 5 January 2012 and 2013-118 dated 2 January 2013). The rodents were captured using Sherman traps; the study of the species used in this project did not require the approval of an ethics committee (European directives 86-609 CEE and 2010/63/EEC). The species used are not protected, and no experiment was performed on living animals. No permit approval was needed, as the species were trapped outside any preserved areas (national parks or natural reserves). The rodents were euthanized by vertebrate dislocation immediately after capture in agreement with the legislation and the ethical recommendations (2010/63/EEC annexe IV) (see also the protocol available on http://www.ceropath.org/references/rodent_protocols_book). All experimental protocols involving animals were carried out by qualified personnel (accreditation number of the Center of Biology and Management of the Populations [CBGP] for wild and inbred animal manipulations, A34-1691). For the samples from Laos and Thailand, approval notices for the trapping and investigation of rodents were provided by the Ministry of Health Council of Medical Sciences, National Ethics Committee for Health Research (NHCHR) Lao PDR, number 51/NECHR, and by the Ethical Committee of Mahidol University, Bangkok, Thailand, number 0517.1116/661. Oral agreements for trappings were obtained from local community leaders and land owners. Aplin's rodent sampling in Southeast Asia was carried out under CSIRO Sustainable Ecosystems Animal Ethics Committee Approval Numbers 00/01-27, 00/01-28, and 02/03-18. For the samples from Indonesia, rodent capture and handling in the field followed animal care and use guidelines recommended by the American Society of Mammalogists (28). Permits to collect scientific specimens were requested and provided by the State Ministry of Research and Technology (RISTEK) and the Ministry of Forestry, Republic of Indonesia. Specimens were prepared in the field by Museum Zoologicum Bogoriense personnel.
Cell lines, viruses, and DNA extraction.
GALV DNA for hybridization capture bait generation (26, 27) was obtained from the following productively infected cell lines: SEATO-88, GALV-SEATO-infected Tb 1 Lu bat lung fibroblasts (ATCC CCL-88); GALV-4-88, GALV-Brain infected Tb 1 Lu bat lung fibroblasts (ATCC CCL-88); 71-AP-1, WMV-infected marmoset fibroblasts; 6G1-PB, GALV-Hall's Island-infected lymphocytes; HOS (ATCC CRL-1543) GALV-SF-infected human osteosarcoma cells. Genomic DNA extraction from the cell lines was performed using the Wizard Genomic DNA purification kit (Promega) according to the manufacturer's protocol. Rodent tissue samples were first homogenized using a Precellys 24 (Bertin Technologies), with genomic DNA then extracted using the QIAamp DNA minikit (Qiagen) according to the manufacturer's instructions. The genomic DNA of the white-handed gibbon was extracted following the method described by Sambrook and Russell (29). For all DNA extracts, DNA concentration was determined using the double-stranded DNA (dsDNA) High Sensitivity Assay kit on a Qubit 2.0 fluorometer (Invitrogen).
Rodent species identification.
The rodent species were taxonomically identified using genetic barcoding methodologies based on both mitochondrial markers—the cytochrome c oxydase I (COI) and cytochrome b (cytb) genes—and a nuclear marker—the interphotoreceptor retinoid binding protein gene (IRBP). The primer sets and cycling conditions used to amplify these markers are described by Pages et al. (30). The obtained sequences were compared with the database of reference sequences of Southeast Asian rodents of CIRAD, Montpellier, France. Furthermore, the rodent species were also identified using different morphological characteristics by Ken Aplin, an expert rodent taxonomist. The individuals of Melomys burtoni and Melomys paveli were identified using morphological characteristics (skin and skull measurements).
Illumina library preparation.
All rodent sample DNA extracts were sheared using a Covaris S220 (Covaris) to an average size of 300 bp prior to building Illumina sequencing libraries. Libraries were generated as described by Meyer and Kircher (31) with the modifications described by Alfano et al. (32), except for using a variable starting amount of DNA extract according to each sample availability and using 1 μl Illumina adapter mix (20 μM) in the adapter ligation step. Each library contained a unique combination of index adapters, one at each end of the library molecule (double indexing) (33), to allow for subsequent discrimination among samples after the sequencing of pooled libraries. Negative-control extraction libraries were also prepared and indexed separately to monitor for experimental cross contamination. Each library was amplified in three replicate reactions to minimize amplification bias in individual PCRs. The amplifications of the libraries were performed using Herculase II Fusion DNA polymerase (Agilent Technologies) in 50-μl reaction volumes, with the cycling conditions of 95°C for 5 min, followed by 7 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 40 s and finally 72°C for 7 min. After pooling the three replicate PCR products for each sample, amplified libraries were purified using the QIAquick PCR Purification kit (Qiagen) and quantified using a 2200 TapeStation (Agilent Technologies) on D1K ScreenTapes. Additional amplification cycles were performed for some of the libraries, when needed to balance library concentrations, using Herculase II Fusion DNA polymerase with P5 and P7 Illumina library outer primers under the same cycling conditions.
Hybridization capture baits.
Two different approaches were used to amplify the genomes of GALV and KoRV for hybridization capture bait production (26, 27). The KoRV genome was amplified in 38 500-bp overlapping products as described by Tsangaras et al. (27) using the DNA of a northern Australian koala (PCI-SN248) from the San Diego Zoo. The 38 amplicons were then pooled in equimolar ratios. In contrast, the genomes of the five isolated GALV strains (SEATO, SF, Brain, Hall's Island, WMV) were amplified in two ca. 4.3-kb-long overlapping PCR products using primers designed on an alignment of the recently published genomes of the GALV strains (accession numbers KT724047 to KT724051) (2). The amplicons were produced from five different GALV-infected cell lines. Primers U5 (5′-CAGGATATCTGTGGTCAT-3′) and PolR1 (5′-GTCGAGTTCCAGTTTCTT-3′) amplify the first 4.3 kb of the GALV genome (5′ long terminal repeat [LTR], gag, and part of the pol gene), and primers PolF1 (5′-CTCATTACCAGAGCCTGCTG-3′) and U3 (5′-GGATGCAAATAGCAAGAGGT-3′) amplify the second 4.3-kb (part of pol gene, gag gene, and 3′ LTR). Primer U3_SEATO (5′-GGATGCAATCAGCAAGAGGT-3′) was used instead of primer U3 for the SEATO strain to account for a 2-nucleotide (nt) difference existing in that region for GALV-SEATO. The GALV PCRs were performed in a volume of 23 μl using approximately 200 ng of DNA extract, 0.65 μM final concentration of each primer, 12.5 μl of 2× MyFi Mix (Bioline), and sterile distilled water. The thermal cycling conditions were 95°C for 4 min, 35 cycles at 95°C for 30 s, at 54 to 62°C (based on the best PCR product yield per strain determined empirically) for 30 s, and at 72°C for 6 min, followed by 72°C for 10 min. An aliquot of each PCR product was visualized on 1.5% (wt/vol) agarose gels stained with Midori Green Direct (Nippon Genetics Europe). PCR products were purified using the MSB Spin PCRapace kit (Stratec Molecular GmbH), quantified using a Qubit 2.0 fluorometer (Invitrogen), and Sanger sequenced at LGC Genomics (Berlin, Germany) to verify that the correct target had been amplified. The PCR products from each GALV strain were then pooled in equimolar concentrations and sheared to obtain a fragment size of approximately 350 bp using a Covaris S220. The mixed sheared GALV amplicons were then pooled with the mixed KoRV amplicons at a 1:6 KoRV-to-GALV ratio to balance the one KoRV amplicon set with the 5 GALV strains in the final bait pool. The GALV-KoRV mixed amplicons were then blunt ended using the Quick Blunting kit (New England BioLabs), ligated to a biotin adaptor using the Quick Ligation kit (New England BioLabs), and immobilized in separated individual tubes on streptavidin-coated magnetic beads as described previously (26).
Hybridization capture.
The 50 rodent indexed libraries were pooled in groups of 5 in order to reach a library input of 2 μg for each capture reaction mixture. The negative controls for library preparation were also included in the capture reaction mixtures. Each indexed library pool was mixed with blocking oligonucleotides (200 μM) to prevent cross-linking of Illumina library adapters, Agilent 2× hybridization buffer, Agilent 10× blocking agent, and heated at 95°C for 3 min to separate the DNA strands (26). Each hybridization mixture was then combined with the biotinylated bait bound streptavidin beads. Samples were incubated in a mini rotating incubator (Labnet) for 48 h at 65°C. After 48 h, the beads were washed to remove off-target DNA as described previously (26), and the hybridized libraries were eluted by incubation at 95°C for 3 min. The DNA concentration for each captured sample was measured using the 2200 TapeStation on D1K ScreenTapes and further amplified accordingly using P5 and P7 Illumina outer primers (31). The enriched amplified libraries were then pooled in equimolar amounts to a final library concentration of 4.5 nM for paired-end sequencing (2 × 250) on an Illumina MiSeq platform with the v2 reagents kit at the Berlin Centre for Genomics in Biodiversity Research (BeGenDiv).
Genome sequence assembly.
A total of 12,502,407 paired-end sequence reads 250 bp long were generated (average, 250,046.8 paired-end reads per sample; standard deviation [SD], 113,859.9) and sorted by their double index sequences. Cutadapt v1.2.1 (34) and Trimmomatic v0.27 (35) were used to remove adaptor sequences and low-quality reads using a quality cutoff of 20 and a minimal read length of 30 nt. After trimming, 97.6% of the sequences were retained. Thereafter, reads were aligned to the NCBI nucleotide database using BLASTn (36) and the taxonomic profile of BLAST results were visualized using Krona (37) in order to assess the taxonomic content of the captured libraries. Reads were then mapped (i.e., aligned to a reference based on sequence similarity) to the genome sequences of GALV strains (KT724047 to KT724051), KoRV (AF151794), and closely related gammaretroviruses (McERV, KC460271; MDEV, AF053745; MmERV, AC005743) using BWA v0.7.10 with default parameters (BWA-MEM algorithm) (38). The alignments were further processed using Samtools v1.2 (39) and Picard (http://broadinstitute.github.io/picard) for sorting and removal of potential duplicates, respectively. Mapping was used as a preliminary screen to identify samples potentially positive or negative for viral sequences. Only samples that produced reads mapping across the genome of a viral reference were considered positive and subjected to further analyses. Samples that exhibited reads mapping only to limited portions of the reference, likely due to random homology of part of the bait to host genomic regions, were not further considered. Reads from positive samples were mapped to the reference of interest, and the resulting alignments were visualized and manually curated using Geneious v7.1.7 (Biomatters, Inc.).
PCR amplifications.
Two primer pairs based on the GALV consensus sequences generated from the hybridization capture data were designed to fill in gaps found in the bioinformatics assembly. Primers GagF1 (5′-TGAGTAGCGAGCAGACGTGTT-3′) and GagR1 (5′-GGCAAAATCACAGTGGAGTCA-3′) were used to amplify a region encompassing part of the gag gene and the interspace fragment between 5′ LTR and gag, while primers EnvF1 (5′-CAGTTGACCATTCGCTTGGA-3′) and EnvR1 (5′-CCGAGGGTGAGCAACAGAA-3′) were used to amplify part of the env gene. The PCR mix comprised 12.5 μl of 2× MyFi Mix (Bioline), 0.6 μM (final concentration) of forward primer, 0.6 μM (final concentration) of reverse primer, approximately 100 ng of DNA template, and sterile distilled water to a final volume of 22 μl. Thermal cycling conditions were 95°C for 3 min, 40 cycles of 95°C for 15 s, 59°C for 20 s, and 72°C for 30 s, and finally 72°C for 30 s. For EnvF1-EnvR1, the annealing temperature was set to 61°C instead of 59°C and the extension time to 40 s instead of 30 s.
Five primer sets were designed based on the alignment of the phosphate transporter 1 (PiT1 or SLC20A1) and the phosphate transporter 2 (PiT2 or SLC20A2) sequences available in GenBank for Mus musculus, Rattus norvegicus, Cricetulus griseus, Homo sapiens, Macaca mulatta, and Nomascus leucogenys to sequence region A of PiT1 and PiT2 from Hylobates lar, Melomys sp., Melomys paveli, and Mus caroli. Primers PiT1-F1long (5′-AGATCCTTACAGCCTGCTTTGG-3′) and PiT1-R1 (5′-TCCTTCCCCATRGTCTGGAT-3′) were designed to amplify a region approximately 600 bp long and encompassing the exons 7 and 8 of PiT1, which contains region A, compared to the M. musculus sequence (800 bp long and targeting exons 8 and 9 compared to the H. sapiens sequence). Primers PiT1-F1short (5′-CCTCTGGTTGCTTTGTATCTTGTT-3′) for the rodent templates and PiT1-F1short_apes for the gibbon template (5′-GGCCTCTGGTTGCTTTATATTTG-3′), both in combination with the above-mentioned PiT1-R1, were designed to amplify a 150-bp-long fragment including region A. Two primer pairs, PiT2-F1 (5′-TGCTATTGGTCCCCTTGTGG-3′) and PiT2-R1 (5′-CCCCAAACCCAGAGACCTGT-3′) for the rodents and PiT2-F1_apes (5′-CCTGGTAGCCTTGTGGCTGA-3′) and PiT2-R1_apes (5′-TGATGGGAGTGAGGTCCTTC-3′) for the gibbon, were designed to amplify a fragment approximately 150 bp long including PiT2 region A. The PCRs were performed using approximately 100 ng of DNA extract, a 0.6 μM final concentration of each primer, 12.5 μl of 2× MyFi Mix (Bioline), and sterile distilled water to a final volume of 22 μl. Cycling conditions were 95°C for 3 min, 35 cycles at 95°C for 15 s, 57°C for 20 s, and 72°C for 10 s, and finally 72°C for 10 s. For PiT1-F1long and PiT1-R1, the extension at 72°C was prolonged to 30 s.
An aliquot of each PCR product was visualized on 1.5% (wt/vol) agarose gels stained with Midori Green Direct (Nippon Genetics Europe). PCR products were purified using the MSB Spin PCRapace kit (STRATEC Molecular GmbH), quantified using a Qubit 2.0 fluorometer (Invitrogen), and Sanger sequenced at LGC Genomics (Berlin, Germany). Sequences were then screened against the NCBI nucleotide database using the BLAST online search tool (https://blast.ncbi.nlm.nih.gov/).
Evolutionary analyses.
To characterize the phylogenetic relationships among the identified viral consensus sequences, the known GALV strains, MbRV, and other related gammaretroviruses, phylogenetic trees were inferred based on the viral nucleotide sequences. The following reference sequences were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/GenBank): GALV-SEATO (KT724048), GALV-SF (KT724047), GALV-Brain (KT724049), GALV-Hall's Island (KT724050), woolly monkey virus (WMV; KT724051), and Melomys burtoni retrovirus (MbRV; KF572483 to KF572486). KoRV (AF151794) was used as an outgroup. Genomic sequences and individual gene (env, gag, and pol) sequences were aligned using MAFFT (40). Phylogenetic analysis was performed using the maximum-likelihood (ML) method available in RAxML v8 (41), including 500 bootstrap replicates to determine the node support. The general time-reversible substitution model (42) with among-site rate heterogeneity modeled by the Γ distribution and four rate categories (43) were used. Nucleotide sequences of env, gag, and pol were concatenated and analyzed in a partitioned framework, whereby each partition was allowed to evolve under its own substitution model. In order to infer the phylogenetic trees, the nucleotide sequences of env, gag, and pol were both analyzed separately and concatenated, including noncoding LTRs and spacers, and analyzed in a partitioned framework.
Accession numbers.
The complete sequence and annotations of the MelWMV genome was deposited in GenBank under accession number KX059700. Illumina reads mapping to WMV for the six Melomys burtoni subsp. samples were deposited in the NCBI Sequence Read Archive as BioProject PRJNA318360.
RESULTS
Screening for GALV and KoRV in rodents using hybridization capture.
Twenty-six rodent species (1 to 6 individuals per species) were screened for the presence of KoRV- and GALV-like sequences (Table 1). None of the 26 species yielded sequences mapping to KoRV. Only the six samples belonging to a Wallacean Melomys burtoni subspecies that has not yet been reported in the literature produced reads mapping uniformly across the genome of the woolly monkey virus (WMV), which is considered a strain of GALV. All of the tested species of Mus produced sequence reads mapping to one of the GALV-related murine retroviruses (MmERV, McERV, MDEV). These sequences were likely captured by GALV/KoRV baits based on the homology of these ERVs with GALV and KoRV. Specifically, we recovered portions of the genome of MmERV from the samples belonging to Mus musculus. Mus nitidulus and Mus booduga samples demonstrated the presence of a virus similar to MmERV. Mus nitidulus and Mus terricolor yielded sequences similar to those of MDEV. Sequences similar to those of McERV were also found in Mus caroli, M. cervicolor, M. cookii, M. fragilicauda, and M. lepidoides.
Melomys woolly monkey virus.
Seven Melomys species samples were screened, of which six were from a new subspecies of Melomys burtoni from Wallacea, which is in the process of being described (Fabre et al., unpublished) (here referred to as Melomys burtoni subsp.). In addition, a sample of Melomys paveli from Seram Island (Moluccas, Indonesia) was included. Only Melomys burtoni subsp. yielded GALV-like sequences, with reads mapping to the woolly monkey virus (WMV) detected in all six Melomys burtoni subsp. samples. For most of the samples, only few reads were found, from a minimum of 24 to a maximum of 1,008 mapping reads, but in each case they were distributed evenly across the WMV genome. However, in sample WD279, almost-full coverage of the viral genome was obtained with an average per-base 18× coverage. The enrichment (proportion of on-target reads mapping to WMV) was low (below 1%) in all samples, similarly to what was seen in our previous experiments (2). The negative control generated few sequence reads, none mapping to GALV.
Based on the hybridization capture Illumina reads, we determined that the viral sequences were identical in the 6 Melomys burtoni subsp. samples. The identified virus was characterized by the common genetic structure of simple mammalian gammaretroviruses with a 5′ LTR-gag-pol-env-3′ LTR organization (Fig. 1). The 5′ and 3′ LTRs were identical. Nevertheless, the virus lacked approximately 60% of pol, with the whole reverse transcriptase domain missing, and almost one-half of the surface unit gp70 (SU) and most of the transmembrane subunit p15E (TM) of env (Fig. 1). The remaining protein domains of Pol, i.e., the protease (PR) and integrase (IN), and all Gag protein domains, i.e., the matrix p15 (MA), p12, capsid p30 (CA), and nucleocapsid p10 (NC), were intact. However, the open reading frame (ORF) of gag was truncated by a premature stop codon. Therefore, the Gag protein was 324 amino acids long, instead of the 521 residues expected for WMV. The same regulatory motifs found in WMV and in the other GALVs (2) were identified: a tRNAPro primer binding site, a CAAT box, a TATA box, a Cys-His box, a polypurine tract, and a polyadenylation signal (Fig. 1). Furthermore, no differences between MelWMV and WMV were observed in the domains known to affect GALV and KoRV differential infectivity: the CETTG motif (44) of the Env protein (residues 167 to 171) and the PRPPIY and PPPY motifs (44, 45) of the Gag protein (residues 123 to 128 and 140 to 143). In addition, compared to WMV, MelWMV was conserved in the variable regions A and B (VRA and VRB) (Fig. 1) of the Env protein (residues 86 to 153 and 192 to 203, respectively), which are known to influence receptor specificity (46): only 6 of 80 residues differed between the two viruses.
FIG 1.

MelWMV genomic assembly and structure. Alignment of WMV, MbRV, and MelWMV consensus sequences generated from hybridization capture data combined with the PCR products that were produced to fill in the gaps in the bioinformatics assembly, shown as continuous black bars. The Illumina reads obtained by hybridization capture and mapping to WMV are shown below the PCR products. The gaps representing deletions in the MelWMV genome are indicated by brown bars labeled “deletion.” Nucleotide positions identical among the strains are indicated in light gray, while mismatches are shown in black. Gaps in the sequence alignment, including those separating the four fragments constituting the MbRV genome, are shown as dashes. The green and red bar above the alignment indicates the percent identity among the sequences (green, highest identity; red, lowest identity). The positions of proviral genes (gag, pol, and env) and protein domains of WMV are indicated in yellow and sky blue, respectively, and are used as reference also for MelWMV. The truncated ORF of MelWMV gag is indicated as an orange thin bar. The following structural regions are shown: the 5′ and 3′ long terminal repeats (LTRs) with the typical U3-R-U5 structure (in light blue), the CAAT box and TATA box (in red), the polyadenylation [poly(A)] signal (in violet), the primer binding site (PBS) (in green), the Cys-His box (in gray), and the polypurine tract (PPT) (in pink). The variable regions A and B (VRA and VRB) of the envelope of WMV are indicated in dark blue. Protein domain abbreviations: MA, matrix; CA, capsid; NC, nucleocapsid; Pro, protease; RT, reverse transcriptase; IN, integrase; SU, surface unit; TM, transmembrane subunit.
Two primer sets (GagF1-GagR1 and EnvF1-EnvR1) based on the mapped reads were designed to fill gaps in the assembly to WMV. The generated PCR products were used both to complete the viral genomic sequence and to confirm the bioinformatics assembly of the sequences obtained by hybridization capture. Primers EnvF1-EnvR1 were specifically designed to cover a gap in the assembly in the env gene of the virus, but the resulting Sanger sequences confirmed that this portion of env, corresponding to positions 6,777 to 7,758 in the WMV sequence, is not present in the viral genome. The primers were applied to the Melomys paveli sample as well and confirmed the absence of GALV-like sequences suggested by the hybridization capture experiment. Identical amplification products from each primer set were produced for all 6 Melomys burtoni subsp. samples. A schematic representation of the genome assembly based on captured sequences and of the PCR products is shown in Fig. 1.
The integration sites, which were captured for 4 of 6 Melomys burtoni subsp. samples, were identical in each sample. Only a single 5′ and 3′ integration site was found. The genomic sequences of Melomys burtoni subsp. flanking MelWMV 5′ and 3′ integration sites were queried by BLAST against the NCBI nucleotide database and returned a hit to bacterial artificial chromosome (BAC) clone RP23-133I8 from chromosome 1 of Mus musculus (accession number AC124760), the closest relative of Melomys burtoni with a genome sequence available in GenBank. The 5′ and 3′ flanking sequences were found to match contiguous regions of the genome of Mus musculus, suggesting that the two flanks correspond to the genomic sequence of Melomys burtoni subsp. on either side of the integration site of MelWMV. Comparing the 5′ and 3′ host genomic flanks also allowed the identification on both sides of the provirus of the target site duplication, a segment of host DNA that is replicated during retroviral integration and that appears as an identical sequence immediately upstream and downstream of the integrated provirus. The duplicated sequence for MelWMV was GTCAC flanking both the 5′ and 3′ ends of the virus. This is consistent with the length of target site duplications of retroviruses, which is generally 4 to 6 bp (47). For example, for KoRV the target site duplication is 4 bp (48).
The detection in all the Melomys burtoni subsp. individuals tested and the identification of identical integration sites in each sample suggest that the virus is endogenous. To estimate a maximum age of endogenization, we used a molecular clock relying on the divergence between the 5′ and 3′ LTR sequences within the same provirus, as described by Ishida et al. (48). No differences were observed in the 1,012 bp of 5′ and 3′ LTRs (each 506 bp long). Using a mouse mutation rate of approximately 4.5 × 10−9 mutations per site per year (49–51) to estimate the nuclear mutation rate of Melomys burtoni, we calculated that the first mutation anywhere within the LTRs would be expected to occur within 219,600 years of integration. Since no mutations were detected in LTRs, this would represent a maximum age estimate for the integration of the virus.
The newly identified virus shared 98% nucleotide identity with WMV and 96.7% with the Melomys burtoni retrovirus (MbRV). A phylogenetic analysis was performed, including sequences from the genomes of the GALV strains and MbRV, using KoRV as an outgroup. The evolutionary relationships among these viruses were robust regardless of the type of data analyzed, full-genome (Fig. 2) or individual-gene (gag, pol, and env) nucleotide sequences (data not shown). The new virus formed a sister taxon to WMV, which together formed a monophyletic group with MbRV (Fig. 2). These three viruses in turn constituted a sister clade to the other GALV strains. The evolutionary relationship between the new virus and WMV was well supported (bootstrap, 88 to 91%) using both concatenated partitioned nucleotide sequences (Fig. 2) and gag and env nucleotide sequences (data not shown). Therefore, the new virus can be considered a strain of GALV and is here designated Melomys woolly monkey virus (MelWMV). Lower support was found using pol nucleotide sequences (bootstrap, 51%), likely due to the large deletion of the gene in MelWMV, which reduced the number of phylogenetically informative sites (data not shown). The support for the relationship among the WMV-MelWMV clade and MbRV was not very robust (bootstrap, 61 to 75%), since only partial sequences of pol and env were recovered for MbRV (Fig. 2; data not shown for pol and env trees).
FIG 2.
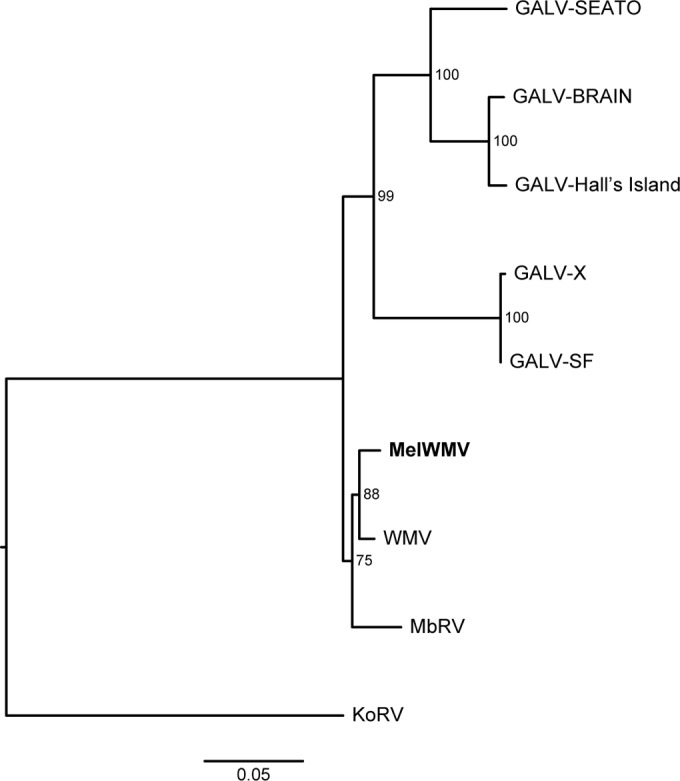
Maximum likelihood phylogenetic tree of GALVs inferred using concatenated partitioned full-genome nucleotide sequences. Coding sequences, noncoding LTRs, and intergene spacers were included in the analysis. The sequences obtained from GenBank with the corresponding accession codes are GALV-SEATO (KT724048), GALV-SF (KT724047), GALV-Brain (KT724049), GALV-Hall's Island (KT724050), woolly monkey virus (WMV; KT724051), and Melomys burtoni retrovirus (MbRV; KF572483 to KF572486). The MelWMV sequence generated in study is shown in bold. KoRV (AF151794) was used as the outgroup. Node support was assessed with 500 rapid bootstrap pseudoreplicates and is indicated at each node. The scale bar indicates 0.05 nucleotide substitutions per site. The tree is midpoint rooted for purposes of clarity.
Sequencing of region A of PiT1 and PiT2.
Residues present in the C-terminal region of the fourth extracellular domain of PiT1, the receptor used by GALV to infect host cells (52), have been identified as critical for receptor function and therefore GALV infection (53–56). This nine-residue region, designated region A, has been extensively analyzed by mutational analysis and by comparative alignment of PiT1 orthologs that function as GALV receptors to PiT1 orthologs that fail to support GALV entry. Substitution of region A residues of PiT1 for the corresponding residues of two proteins that do not support GALV entry, Pit2 (a PiT1 paralog) (55) and the distantly related phosphate transporter Pho-4 from the filamentous fungus Neurospora crassa (56), renders these proteins functional as GALV receptors. Five primer sets were designed to sequence region A of PiT1 and PiT2 from Hylobates lar, Melomys burtoni subsp., Melomys paveli, and Mus caroli. The region A of PiT2 was also sequenced since it is used by GALV to infect Chinese hamster and Japanese feral mouse cells (54, 57). An amplification product was obtained from each of the five primer sets. Sanger sequencing of the amplicons and the subsequent BLAST search confirmed the amplification of the region A of PiT1 and PiT2. The sequences were then aligned with the reference sequences of Mus musculus, Rattus norvegicus, Cricetulus griseus, Homo sapiens, Macaca mulatta, and Nomascus leucogenys available in GenBank and translated into amino acid sequences. The amino acid sequences were then aligned and compared with the amino acid sequences of region A of PiT1 and PiT2 (residues at positions 550 to 558 and 522 to 530, respectively) of all the species known to be permissive (Homo sapiens, Rattus norvegicus, Mus musculus molossinus, Felis catus, Canis lupus, Bos taurus, Macaca mulatta, Cricetulus griseus) or resistant (Mus musculus musculus and Mus dunni) to GALV infection according to the literature (Table 2) (52, 54, 57–59).
TABLE 2.
Residues of PiT1 and PiT2 region A of species permissive and resistant to GALV infectiona
| Receptor and species | Residue in region A position:b |
GALV recognition | Method | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 550/522 | 551/523 | 552/524 | 553/525 | 554/526 | 555/527 | 556/528 | 557/529 | 558/530 | |||
| PiT1 | |||||||||||
| Felis catus | D− | T | G | D− | V | S | S | K | V | + | In vitro |
| Canis lupus | D− | T | G | D− | V | S | S | K | V | + | In vitro |
| Bos taurus | D− | T | G | D− | V | S | S | K | V | + | In vitro |
| Macaca mulatta | D− | T | G | D− | V | S | S | K | V | + | In vitro |
| Homo sapiens | D− | T | G | D− | V | S | S | K | V | + | In vitro |
| Hylobates lar | D− | T | G | D− | V | S | S | K | V | + | Infection |
| Nomascus leucogenys | D− | T | G | D− | V | S | S | K | V | ? (+) | Sequence |
| Rattus norvegicus | E− | T | R | D− | V | T | T | K | E | + | In vitro |
| Mus musculus molossinus | I | T | G | D− | V | S | S | K | M | + | In vitro |
| Melomys burtoni subsp. | E− | T | G | D− | V | S | T | K | A | + | Infection |
| Melomys paveli | E− | T | — | D− | V | S | T | K | A | ? (+) | Sequence |
| Mus musculus musculus | K | Q | — | E− | A | S | T | K | A | − | In vitro |
| Mus dunni | K | Q | — | D− | A | S | T | K | A | − | In vitro |
| Mus caroli | K | Q | — | D− | A | S | T | K | A | ? (−) | Sequence |
| PiT2 | |||||||||||
| Cricetulus griseus | E− | Q | G | G | V | M | Q | E− | A | + | In vitro |
| Felis catus | E− | Q | G | A | V | L | Q | E− | A | ? (+) | Sequence |
| Canis lupus | E− | Q | G | A | V | L | Q | E− | A | ? (+) | Sequence |
| Bos taurus | E− | Q | G | A | V | L | Q | E− | A | ? (+) | Sequence |
| Melomys burtoni subsp. | E− | Q | G | G | V | T | Q | E− | A | + | Infection |
| Melomys paveli | E− | Q | G | G | V | T | Q | E− | A | ? (+) | Sequence |
| Mus musculus molossinus | Q | Q | G | G | V | T | Q | E− | A | + | In vitro |
| Mus caroli | Q | Q | G | G | V | T | Q | E− | A | ? (+) | Sequence |
| Homo sapiens | K | Q | G | G | V | T | Q | E− | A | − | In vitro |
| Rattus norvegicus | K | Q | G | G | V | T | Q | E− | A | − | In vitro |
| Hylobates lar | K | Q | G | G | V | M | Q | E− | A | ? (−) | Sequence |
| Macaca mulatta | K | Q | G | G | V | M | Q | E− | A | ? (−) | Sequence |
Lys (K) is bold when present at the first position of PiT1 or PiT2 A regions, which prevent GALV infection. Asp (D) and Glu (E), which are acidic and negatively charged residues, are italicized and followed by a superscript minus sign (−). The last column specifies for each species the method used to determine GALV recognition: in vitro, experimental validation; infection, natural infection with GALV; sequence, inference based on region A sequence similarity with experimentally tested or infected species. A question mark (?) is used for those species that were never found to be infected with GALV or never experimentally tested for susceptibility to GALV infection. A plus sign (+) or minus sign (−) after the question mark indicates if the species is potentially susceptible or resistant, respectively, to GALV infection based on the region A sequence.
The region A positions for PiT1 are in roman lightface font; the positions for PiT2 are in bold italics. The dashes at position 552 of PiT1 region A indicate a gap in the sequence alignment.
The sequence of PiT1 region A of Melomys burtoni subsp. was very similar to the sequence carried by the known susceptible species. Melomys burtoni subsp. had a Glu (E) at position 550 and an Asp (D) at position 553, identical to what is seen in rat. These two positions, and the corresponding sites in PiT2 (522 and 529), are thought to be crucial for receptor function (54–56), with functional GALV receptors having an acidic residue, either Asp (D) or Glu (E), at one or both of these positions. The Thr (T), Val (V), and Lys (K) at positions 551, 554, and 557, respectively, were invariant among Melomys burtoni subsp. and the other permissive species, with the Lys (K)-557 shared with both resistant and permissive species. The residues at positions 555, 556, and 558 of PiT1 varied randomly among resistant and susceptible species, while the residue at position 552 was missing in the resistant ones. The PiT2 sequence of Melomys burtoni subsp. had a Glu (E) at position 522 and differed in only one residue, Met (M) to Thr (T) at position 527, compared to C. griseus (60), which is also susceptible to GALV infection. Except for the first residue, the sequence was identical to that of Mus musculus molossinus PiT2, which is also considered a functional GALV receptor (59). The PiT1 and PiT2 region A sequences of Melomys paveli were almost identical to those of Melomys burtoni subsp., but the PiT1 sequence of Melomys paveli lacked the residue, a Gly (G) in Melomys burtoni subsp., at position 552, like in the resistant species. The PiT1 sequence of M. caroli was identical to that of M. dunni, the cells of which are resistant to GALV infection (59). They both have a Lys (K) at position 550, which is known to abrogate receptor function. The sequence of PiT2 of M. caroli was identical to that of Mus musculus molossinus, which serves as a functional GALV receptor (59): they both have a Gln (Q) at position 522 and a Glu (E) at position 529. The sequence of H. lar PiT1 region A had an Asp (D) at both positions 550 and 553 and was identical to the human sequence (52), whereas PiT2 displayed one amino acid difference, Thr (T) to Met (M) at position 527, compared to the human PiT2 ortholog (58). Both human cells and gibbons are permissive to GALV infection, but human PiT2, which has a Lys (K) at positions 522, like gibbon PiT2, does not function as a GALV receptor. Indeed, similarly to PiT1, a Lys (K) at position 522 in PiT2 is believed to abrogate receptor function (54, 61).
DISCUSSION
KoRV and GALV are closely related retroviruses (2). However, their respective hosts, koalas and gibbons, have neither a recent common ancestor nor overlapping geographic distributions. Thus, KoRV and GALV may have arisen from a cross-species transmission that involved an intermediate host (9, 14–16). In order to identify such a vector, Simmons et al. (16) screened 42 Australian vertebrate species (birds and mammals, including rodents and bats) for KoRV- and GALV-like sequences. An ERV closely related to GALV (MbRV) was found in the Australian subspecies of the murid species Melomys burtoni, but even if related to GALVs, particularly WMV, it does not represent an ancestor of GALV because the distributions of Melomys burtoni and gibbons do not overlap (16). Because GALV-like viruses have been identified in Southeast Asian rodents (20–22), we screened rodent species from this geographic area in an attempt to identify potential intermediate hosts and retrieve ancestral viral strains of KoRV and GALV. Twenty-six rodent species were screened (Table 1). Some of the species tested (Bandicota savilei, Bandicota indica, Bandicota bengalensis, Berylmys berdmorei, Mus musculus) had been reported as negative for GALV and KoRV by Simmons et al. (16), consistent with the absence of GALV and KoRV from the Southeast Asian samples from the same species in this study. None of the species tested in the current study or in that of Simmons et al. (16) was positive for KoRV-like sequences, while only two different subspecies of Melomys burtoni, one from Australia from Simmons et al. (16) and one from Wallacea from the current study, were found positive for GALV-like sequences. Based on the homology to WMV (98%) and phylogenetic affinity, the Melomys woolly monkey virus (MelWMV) that we discovered in the Wallacean Melomys burtoni subsp. is a subtype of WMV, whereas MbRV from the Australian subspecies is a sister taxon (Fig. 2).
We cannot rule out low-titer infections by exogenous retroviruses in the rodent samples that we analyzed, especially considering that muscle, i.e., the sample type obtained for this study, is not the usual target of exogenous retroviral infections. However, we are confident that we have not missed GALV- or KoRV-like endogenous retroviruses in the species tested, since hybridization capture has been shown to be equal to or superior to PCR-based methods in retrieving nuclear DNA sequences, including endogenous retroviruses. Several studies (2, 27, 62) have shown that samples that failed by PCR for retroviral detection worked by hybridization capture because hybridization capture is less sensitive to sequence mismatch or DNA degradation. Melomys paveli, which did not yield GALV-like sequences by hybridization capture, was also negative by PCR, indicating that the results do not represent a false negative.
Only one integration site was found for MelWMV. Therefore, there may be only a single copy of MelWMV in the genome of Melomys burtoni subsp., and this would explain the low hybridization capture coverage. Furthermore, MelWMV was detected in all 6 individuals of Melomys burtoni subsp. tested and the integration site was identical in all 4 individuals for which they were identified by hybridization capture. This result, the premature stop codon in gag, and the deletions in pol and env (Fig. 1) strongly indicate that MelWMV is an endogenous retrovirus. Furthermore, we estimate that MelWMV has recently (within the last 200,000 years) integrated into the genome of the Wallacean Melomys burtoni subsp., based on the identical 5′ and 3′ LTR sequences and the mutation rate of a murid host (48). MelWMV is not present in M. paveli, tested in this study, or in the Australian M. burtoni subspecies and the endemic Australian Melomys cervinipes, tested by Simmons et al. (16). The different species of Melomys diverged from a common ancestor between 1 and 2 million years ago (63), consistent with the date of integration of MelWMV into the Melomys burtoni genome based on the LTR sequences.
MelWMV, WMV, and MbRV represent the most basal clade of the GALV phylogeny described to date (Fig. 2), so it can be argued that the WMV-like viruses are the most ancestral GALV strains currently known to be circulating and most likely the closest viruses to the progenitor of GALV and KoRV. Such close GALV relatives were found in only two different populations of the murine species Melomys burtoni among 68 total species tested in Australia (16) and Southeast Asia. Furthermore, the more distantly related GALV-like ERVs are found in rodents belonging to the genus Mus (20, 22). Taken together, this suggests an overall rodent origin of the clade, more specifically an Australo-Papuan murine origin. However, since MelWMV is an ERV in Melomys burtoni subsp. but M. paveli did not yield any GALV-like sequences, it is not clear whether Melomys is a reservoir or a susceptible host for GALVs. Thus, it is formally possible that GALV did not originate in Melomys and the two Melomys burtoni subspecies were independently infected with GALV in Wallacea and Australia from an unknown reservoir species. As the vast majority of samples in the current study were from Southeast Asia and those of Simmons et al. (16) were exclusively from Australia, species in Wallacea and Papua New Guinea remain largely unexplored. In addition, only 3 species of Melomys have been tested of a total of 22 Melomys species, 19 of which are found in the Moluccas, Melanesia, and Papua New Guinea (The IUCN red list of threatened species, version 2015-4, IUCN 2015 [http://www.iucnredlist.org]), suggesting that many more GALVs, including potentially exogenous GALVs, and possibly KoRV-like sequences may be present. Of particular relevance to the current host range of GALV, Melomys species are found in both Australia and Wallacea. Since Wallacea is a transitional zone between Asia and Australia, the discovery of MelWMV in a Wallacean subspecies of M. burtoni represents the most proximate record of GALV to the Asian continent and to the distribution of gibbons. However, even if the genus Melomys is one of the most widespread murine genera in the Australo-Papuan region, and specifically one of those that have dispersed furthest to the West (to the Moluccas), it has never been reported, not even from the fossil record, in Sulawesi or the Sunda Shelf (mainland Southeast Asia) (64), and thus it has probably never been in direct contact with gibbons. White-handed gibbons are distributed in the wild from northern Sumatra (Indonesia) to mainland Southeast Asia, including Malaysia, Thailand, Myanmar, and Laos (65) (The IUCN red list of threatened species, version 2015-4, IUCN 2015 [http://www.iucnredlist.org]). GALV has thus far been isolated only from captive gibbons, from the colony housed at the SEATO Laboratory in Bangkok, Thailand (12), or from colonies in the U.S. primate laboratories (San Francisco Medical Center, Hall's Island in Bermuda, Gulf South Primate Center at the National Institutes of Health, Louisiana), which were derived from gibbons shipped from Southeast Asia, mainly the SEATO colony. Therefore, it is still not clear how the virus moved from Australia and Wallacea to mainland Southeast Asia, crossing the Wallace Line, a line running between the islands of Bali and Lombok and dividing the Australian and the Asian biogeographic zones. An intermediate and mobile host that is distributed across the Wallace Line must have played a critical role in the viral transmission. However, our study suggests that any intermediate host that eventually infected captive gibbons in Southeast Asia came in contact with M. burtoni in Wallacea. It is possible that several intermediate hosts, including Melomys burtoni, have been involved in the cross-species transmission among koalas and gibbons, in a stepwise process that finally led to the outbreaks of GALV in gibbons and to the emergence of KoRV in koalas. Rattus species would be good candidates as GALV and KoRV hosts, given their widespread distribution in this region (Australia, Papua New Guinea, and both insular and mainland Southeast Asia). However, nine Rattus species, including both species that are endemic to Australia and species with an Australo-Papuan distribution, were tested and reported negative for GALV and KoRV by Simmons et al. (16). Similarly, in a preliminary screening of Rattus exulans and Rattus rattus from Southeast Asia using a single GALV strain and KoRV as hybridization capture baits, we did not identify any GALV-like sequences (data not shown). Other candidate hosts are lineages belonging to the same molecular tribe of Melomys, such as Hydromys and Uromys genera, which display a similarly wide Australo-Papuan distribution but have not been included in this study and have not been extensively sampled by Simmons et al. (16). Gibbons in particular are surprising hosts: the fact that GALVs have been isolated only from captive and not wild gibbons suggests that they may be accidental hosts and that they have had infrequent but regular contact with a GALV reservoir or host species but only in captive facilities. This is particularly relevant for the gibbon colony housed at the SEATO Laboratory in Bangkok, Thailand (12), from which the other non-Asian gibbon colonies originated.
GALV infects cells using a ubiquitous transmembrane protein that functions as sodium-dependent phosphate transporter called PiT1 or SLC20A1 (52). GALV can alternatively infect cells using a related phosphate transporter, PiT2 or SLC20A2, originally recognized as the amphotropic murine leukemia virus (A-MuLV) and 10A1 MuLV receptor, to infect Chinese hamster and Japanese feral mouse cells (54, 57, 58). This similarity of receptor usage is consistent with the phylogenetic relationship of GALVs and MuLVs, which belong to the same overall retroviral group (2).
Mutagenesis studies have shown that region A of PiT1, a stretch of nine residues corresponding to residues 550 to 558 of human PiT1, which is highly polymorphic among species, is crucial for GALV entry into cells (53, 54). Because of its highly polymorphic nature, it is not clear which of the residues of region A are essential for GALV infection. We have sequenced PiT1 and PiT2 A regions from species that were positive (Melomys burtoni subsp.) and negative (Melomys paveli and Mus caroli) to our GALV screening, and also from Hylobates lar, another host of GALV. When they were compared with the previously reported GALV receptors sequences obtained from both permissive cells, i.e., from humans (Homo sapiens), rat (Rattus norvegicus), Japanese feral mouse (Mus musculus molossinus), Chinese hamster (Cricetulus griseus), dog (Canis lupus), cat (Felis catus), cow (Bos taurus), rhesus macaque (Macaca mulatta), and from nonpermissive cells (Mus musculus, Mus dunni) for GALV infection (Table 2), the sequences generated here were consistent with the findings of previous functional studies (53, 54, 61). Functional GALV receptors have an acidic residue at either position 550 or 553 of PiT1 (522 or 529 of PiT2) or both, but lysine at position 550 (522 in PiT2) abrogates GALV receptor function, even when an acidic residue is present at position 553 (529 in PiT2).
Therefore, Mus caroli PiT1, which has a Lys (K) at position 550, is unlikely to serve as a GALV receptor. This is consistent with the lack of detection of any GALV-like sequence in this species. McERV sequences were detected, but this virus uses a different receptor from the one used by GALV (23). However, GALV could potentially infect Mus caroli using PiT2, given its identity to Mus musculus molossinus PiT2, which is a functional GALV receptor. The A regions of human and gibbon PiT1 are identical, and both humans and gibbons have a Lys (K) at the first position of PiT2 region A. Human PiT1 functions as a GALV receptor, while PiT2 does not. Given the similarity between human and gibbon PiT receptors, captive gibbons were likely infected via PiT1.
Both PiT1 and PiT2 of Melomys burtoni subsp. are potentially functional GALV receptors, consistent with our discovery of MelWMV in this species. However, MelWMV and WMV are highly similar in the VRA and VRB domains of the envelope, with only 6 polymorphic amino acids of the 80 constituting VRA and VRB (data not shown), and WMV is known to be unable to use the PiT2 receptor to infect hamster cells due to a block mediated by WMV envelope, specifically VRA and VRB (46). Therefore, it is likely that Melomys burtoni subsp. was infected by WMV via the PiT1 receptor. Melomys paveli is also potentially susceptible to GALV infection, since its PiT1 and PiT2 A regions are identical to Melomys burtoni subsp., with the exception that residue 552 is missing in PiT1, as observed in resistant species (Mus musculus musculus, Mus dunni). Since the lack of this residue was never taken into account as a determinant of resistance to GALV in former functional studies, we cannot draw conclusions on the effect of this deletion on receptor functionality. However, we detected GALV in Melomys burtoni subsp. only. As only one Melomys paveli sample was analyzed, we cannot rule out that GALVs may be circulating at low abundance in this species. Furthermore, it is also possible that M. paveli never came into contact with a GALV, since its distribution is restricted to Seram Island. Therefore, the absence of GALV may be biogeographically determined rather than driven by a receptor restriction for this species.
In conclusion, our screen of Southeast Asian rodents identified MelWMV in a Melomys burtoni subspecies from Wallacea. MelWMV represents the most closely related retrovirus to GALV identified from rodents to date and the second GALV relative identified from two different subspecies of Melomys burtoni, suggesting either that Melomys burtoni is a host of GALVs or that more species within the genus Melomys are sympatric with the reservoir. With the current data, we cannot distinguish between the two possibilities that (i) MelWMV derives from MbRV and represents a single infection of M. burtoni with subsequent evolution or (ii) the two viruses represent independent infections. However, WMV itself must represent a distinct infection event because Melomys species do not overlap gibbons geographically. The PiT1 and PiT2 region A sequences of the Melomys species tested in the current study are consistent with the general susceptibility of these species to GALV infection. Further screening of GALV and KoRV in Melomys across the range of this genus, in older Melomys-related lineages of the Murinae subfamily, and in potential host species that have crossed the Wallace Line would be promising for identifying additional GALV sequences.
ACKNOWLEDGMENTS
We thank Karin Hönig (Leibniz Institute for Zoo and Wildlife Research) and Susan Mbedi of the Berlin Centre for Genomics in Biodiversity Research (BeGenDiv) for sequencing support. We thank the State Ministry of Research and Technology (RISTEK, permit number 028/SIP/FRP/SMII/2012) and the Ministry of Forestry of the Republic of Indonesia for providing permits to carry out fieldwork in the Moluccas. Likewise, we thank the Research Center for Biology, Indonesian Institute of Sciences (RCB-LIPI), and the Museum Zoologicum Bogoriense for providing staff and support to carry out fieldwork in the Moluccas. We thank Christian Roos of the Deutsche Primatenzentrum (DPZ) in Göttingen, Germany, for providing the gibbon sample.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the National Institutes of Health.
Funding Statement
This work, including the efforts of Yasuko Ishida, Kristofer Helgen, Alfred Roca, and Alex David Greenwood, was funded by HHS | NIH | National Institute of General Medical Sciences (NIGMS) (R01GM092706). This work, including the efforts of Maribeth Eiden, was funded by HHS | NIH | National Institute of Mental Health (NIMH) (ZIAMH002592). Niccolò Alfano was supported by the International Max Planck Research School for Infectious Diseases and Immunology (IMPRS-IDI) at the Interdisciplinary Center of Infection Biology and Immunity (ZIBI) of Humboldt University Berlin (HU). Pierre-Henri Fabre was funded by a Marie Curie Fellowship (PIOF-GA-2012-330582-CANARIP-RAT). He also acknowledges Carsten Rahbek (Center for Macroecology Evolution and Climate, University of Copenhagen, Copenhagen, Denmark) for generously funding part of his recent field research in the Moluccas. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Streicker DG, Turmelle AS, Vonhof MJ, Kuzmin IV, McCracken GF, Rupprecht CE. 2010. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329:676–679. doi: 10.1126/science.1188836. [DOI] [PubMed] [Google Scholar]
- 2.Alfano N, Kolokotronis SO, Tsangaras K, Roca AL, Xu W, Eiden MV, Greenwood AD. 2016. Episodic diversifying selection shaped the genomes of gibbon ape leukemia virus and related gammaretroviruses. J Virol 90:1757–1772. doi: 10.1128/JVI.02745-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami TG, Huff SD, Buckley PM, Dungworth DL, Synder SP, Gilden RV. 1972. C-type virus associated with gibbon lymphosarcoma. Nat New Biol 235:170–171. [DOI] [PubMed] [Google Scholar]
- 4.DePaoli A, Johnsen DO, Noll MD. 1973. Granulocytic leukemia in white handed gibbons. J Am Vet Med Assoc 163:624–628. [PubMed] [Google Scholar]
- 5.Reitz MS Jr, wong-Staal F, Haseltine WA, Kleid DG, Trainor CD, Gallagher RE, Gallo RC. 1979. Gibbon ape leukemia virus-Hall's Island: new strain of gibbon ape leukemia virus. J Virol 29:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Todaro GJ, Lieber MM, Benveniste RE, Sherr CJ. 1975. Infectious primate type C viruses: three isolates belonging to a new subgroup from the brains of normal gibbons. Virology 67:335–343. doi: 10.1016/0042-6822(75)90435-3. [DOI] [PubMed] [Google Scholar]
- 7.Theilen GH, Gould D, Fowler M, Dungworth DL. 1971. C-type virus in tumor tissue of a woolly monkey (Lagothrix spp.) with fibrosarcoma. J Natl Cancer Inst 47:881–889. [PubMed] [Google Scholar]
- 8.Wolfe LG, Smith RK, Deinhardt F. 1972. Simian sarcoma virus, type 1 (Lagothrix): focus assay and demonstration of nontransforming associated virus. J Natl Cancer Inst 48:1905–1908. [PubMed] [Google Scholar]
- 9.Hanger JJ, Bromham LD, McKee JJ, O'Brien TM, Robinson WF. 2000. The nucleotide sequence of koala (Phascolarctos cinereus) retrovirus: a novel type C endogenous virus related to Gibbon ape leukemia virus. J Virol 74:4264–4272. doi: 10.1128/JVI.74.9.4264-4272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shojima T, Yoshikawa R, Hoshino S, Shimode S, Nakagawa S, Ohata T, Nakaoka R, Miyazawa T. 2013. Identification of a novel subgroup of Koala retrovirus from Koalas in Japanese zoos. J Virol 87:9943–9948. doi: 10.1128/JVI.01385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu W, Stadler CK, Gorman K, Jensen N, Kim D, Zheng H, Tang S, Switzer WM, Pye GW, Eiden MV. 2013. An exogenous retrovirus isolated from koalas with malignant neoplasias in a US zoo. Proc Natl Acad Sci U S A 110:11547–11552. doi: 10.1073/pnas.1304704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami TG, Kollias GV Jr, Holmberg C. 1980. Oncogenicity of gibbon type-C myelogenous leukemia virus. Int J Cancer 25:641–646. doi: 10.1002/ijc.2910250514. [DOI] [PubMed] [Google Scholar]
- 13.Tarlinton R, Meers J, Hanger J, Young P. 2005. Real-time reverse transcriptase PCR for the endogenous koala retrovirus reveals an association between plasma viral load and neoplastic disease in koalas. J Gen Virol 86:783–787. doi: 10.1099/vir.0.80547-0. [DOI] [PubMed] [Google Scholar]
- 14.Tarlinton R, Meers J, Young P. 2008. Biology and evolution of the endogenous koala retrovirus. Cell Mol Life Sci 65:3413–3421. doi: 10.1007/s00018-008-8499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiebig U, Hartmann MG, Bannert N, Kurth R, Denner J. 2006. Transspecies transmission of the endogenous koala retrovirus. J Virol 80:5651–5654. doi: 10.1128/JVI.02597-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simmons G, Clarke D, McKee J, Young P, Meers J. 2014. Discovery of a novel retrovirus sequence in an Australian native rodent (Melomys burtoni): a putative link between gibbon ape leukemia virus and koala retrovirus. PLoS One 9:e106954. doi: 10.1371/journal.pone.0106954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong S, Lau S, Woo P, Yuen KY. 2007. Bats as a continuing source of emerging infections in humans. Rev Med Virol 17:67–91. doi: 10.1002/rmv.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cui J, Tachedjian G, Tachedjian M, Holmes EC, Zhang S, Wang LF. 2012. Identification of diverse groups of endogenous gammaretroviruses in mega- and microbats. J Gen Virol 93:2037–2045. doi: 10.1099/vir.0.043760-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin J, Herniou E, Cook J, O'Neill RW, Tristem M. 1999. Interclass transmission and phyletic host tracking in murine leukemia virus-related retroviruses. J Virol 73:2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lieber MM, Sherr CJ, Todaro GJ, Benveniste RE, Callahan R, Coon HG. 1975. Isolation from the Asian mouse Mus caroli of an endogenous type C virus related to infectious primate type C viruses. Proc Natl Acad Sci U S A 72:2315–2319. doi: 10.1073/pnas.72.6.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callahan R, Meade C, Todaro GJ. 1979. Isolation of an endogenous type C virus related to the infectious primate type C viruses from the Asian rodent Vandeleuria oleracea. J Virol 30:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benveniste RE, Callahan R, Sherr CJ, Chapman V, Todaro GJ. 1977. Two distinct endogenous type C viruses isolated from the Asian rodent Mus cervicolor: conservation of virogene sequences in related rodent species. J Virol 21:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller AD, Bergholz U, Ziegler M, Stocking C. 2008. Identification of the myelin protein plasmolipin as the cell entry receptor for Mus caroli endogenous retrovirus. J Virol 82:6862–6868. doi: 10.1128/JVI.00397-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolgamot G, Bonham L, Miller AD. 1998. Sequence analysis of Mus dunni endogenous virus reveals a hybrid VL30/gibbon ape leukemia virus-like structure and a distinct envelope. J Virol 72:7459–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bromham L, Clark F, McKee JJ. 2001. Discovery of a novel murine type C retrovirus by data mining. J Virol 75:3053–3057. doi: 10.1128/JVI.75.6.3053-3057.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maricic T, Whitten M, Paabo S. 2010. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One 5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsangaras K, Siracusa MC, Nikolaidis N, Ishida Y, Cui P, Vielgrader H, Helgen KM, Roca AL, Greenwood AD. 2014. Hybridization capture reveals evolution and conservation across the entire Koala retrovirus genome. PLoS One 9:e95633. doi: 10.1371/journal.pone.0095633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikes RS, Gannon WL, Animal Care and Use Committee of the American Society of Mammalogists . 2011. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J Mammal 92:235–253. doi: 10.1644/10-MAMM-F-355.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. 2006. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc 2006(1):pii:pdb.prot4455. doi: 10.1101/pdb.prot4455. [DOI] [PubMed] [Google Scholar]
- 30.Pages M, Chaval Y, Herbreteau V, Waengsothorn S, Cosson JF, Hugot JP, Morand S, Michaux J. 2010. Revisiting the taxonomy of the Rattini tribe: a phylogeny-based delimitation of species boundaries. BMC Evol Biol 10:184. doi: 10.1186/1471-2148-10-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc 2010(6):pdb.prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 32.Alfano N, Courtiol A, Vielgrader H, Timms P, Roca AL, Greenwood AD. 2015. Variation in koala microbiomes within and between individuals: effect of body region and captivity status. Sci Rep 5:10189. doi: 10.1038/srep10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kircher M, Sawyer S, Meyer M. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res 40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin M. 2012. Cutadapt removes adapter sequences from high-throughput sequencing reads. Bioinformatics Action 17:10–12. [Google Scholar]
- 35.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ondov BD, Bergman NH, Phillippy AM. 2011. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997. [Google Scholar]
- 39.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanave C, Preparata G, Saccone C, Serio G. 1984. A new method for calculating evolutionary substitution rates. J Mol Evol 20:86–93. doi: 10.1007/BF02101990. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39:306–314. doi: 10.1007/BF00160154. [DOI] [PubMed] [Google Scholar]
- 44.Oliveira NM, Satija H, Kouwenhoven IA, Eiden MV. 2007. Changes in viral protein function that accompany retroviral endogenization. Proc Natl Acad Sci U S A 104:17506–17511. doi: 10.1073/pnas.0704313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shojima T, Hoshino S, Abe M, Yasuda J, Shogen H, Kobayashi T, Miyazawa T. 2013. Construction and characterization of an infectious molecular clone of Koala retrovirus. J Virol 87:5081–5088. doi: 10.1128/JVI.01584-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ting YT, Wilson CA, Farrell KB, Chaudry GJ, Eiden MV. 1998. Simian sarcoma-associated virus fails to infect Chinese hamster cells despite the presence of functional gibbon ape leukemia virus receptors. J Virol 72:9453–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serrao E, Ballandras-Colas A, Cherepanov P, Maertens GN, Engelman AN. 2015. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology 12:39. doi: 10.1186/s12977-015-0167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishida Y, Zhao K, Greenwood AD, Roca AL. 2015. Proliferation of endogenous retroviruses in the early stages of a host germ line invasion. Mol Biol Evol 32:109–120. doi: 10.1093/molbev/msu275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar S, Subramanian S. 2002. Mutation rates in mammalian genomes. Proc Natl Acad Sci U S A 99:803–808. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 51.Xue Y, Wang Q, Long Q, Ng BL, Swerdlow H, Burton J, Skuce C, Taylor R, Abdellah Z, Zhao Y, MacArthur DG, Quail MA, Carter NP, Yang H, Tyler-Smith C. 2009. Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol 19:1453–1457. doi: 10.1016/j.cub.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Hara B, Johann SV, Klinger HP, Blair DG, Rubinson H, Dunn KJ, Sass P, Vitek SM, Robins T. 1990. Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ 1:119–127. [PubMed] [Google Scholar]
- 53.Johann SV, van Zeijl M, Cekleniak J, O'Hara B. 1993. Definition of a domain of GLVR1 which is necessary for infection by gibbon ape leukemia virus and which is highly polymorphic between species. J Virol 67:6733–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneiderman RD, Farrell KB, Wilson CA, Eiden MV. 1996. The Japanese feral mouse Pit1 and Pit2 homologs lack an acidic residue at position 550 but still function as gibbon ape leukemia virus receptors: implications for virus binding motif. J Virol 70:6982–6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pedersen L, Johann SV, van Zeijl M, Pedersen FS, O'Hara B. 1995. Chimeras of receptors for gibbon ape leukemia virus/feline leukemia virus B and amphotropic murine leukemia virus reveal different modes of receptor recognition by retrovirus. J Virol 69:2401–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pedersen L, van Zeijl M, Johann SV, O'Hara B. 1997. Fungal phosphate transporter serves as a receptor backbone for gibbon ape leukemia virus. J Virol 71:7619–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson CA, Farrell KB, Eiden MV. 1994. Properties of a unique form of the murine amphotropic leukemia virus receptor expressed on hamster cells. J Virol 68:7697–7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Zeijl M, Johann SV, Closs E, Cunningham J, Eddy R, Shows TB, O'Hara B. 1994. A human amphotropic retrovirus receptor is a second member of the gibbon ape leukemia virus receptor family. Proc Natl Acad Sci U S A 91:1168–1172. doi: 10.1073/pnas.91.3.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson CA, Farrell KB, Eiden MV. 1994. Comparison of cDNAs encoding the gibbon ape leukaemia virus receptor from susceptible and non-susceptible murine cells. J Gen Virol 75(Part 8):1901–1908. [DOI] [PubMed] [Google Scholar]
- 60.Eiden MV, Farrell KB, Wilson CA. 1996. Substitution of a single amino acid residue is sufficient to allow the human amphotropic murine leukemia virus receptor to also function as a gibbon ape leukemia virus receptor. J Virol 70:1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaudry GJ, Eiden MV. 1997. Mutational analysis of the proposed gibbon ape leukemia virus binding site in Pit1 suggests that other regions are important for infection. J Virol 71:8078–8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cui P, Lober U, Alquezar-Planas DE, Ishida Y, Courtiol A, Timms P, Johnson RN, Lenz D, Helgen KM, Roca AL, Hartman S, Greenwood AD. 2016. Comprehensive profiling of retroviral integration sites using target enrichment methods from historical koala samples without an assembled reference genome. PeerJ 4:e1847. doi: 10.7717/peerj.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bryant LM, Donnellan SC, Hurwood DA, Fuller SJ. 2011. Phylogenetic relationships and divergence date estimates among Australo-Papuan mosaic-tailed rats from the Uromys division (Rodentia: Muridae). Zoologica Scripta 40:433–447. doi: 10.1111/j.1463-6409.2011.00482.x. [DOI] [Google Scholar]
- 64.Breed WG, Aplin KP. 2008. The ‘Uromys Group’: Papuan old endemics. In Van Dyck SM, Strahan R (ed), The mammals of Australia, 3rd ed Reed New Holland, Sydney, Australia. [Google Scholar]
- 65.Macdonald D. 2001. Gibbons. The encyclopaedia of mammals, p 398–405. Oxford University Press, Oxford, United Kingdom. [Google Scholar]