

ABSTRACT
Adaptation of the viral polymerase complex comprising PB1, PB2, and PA is necessary for efficient influenza A virus replication in new host species. We found that PA mutation K356R (PA-K356R) has become predominant since 2014 in avian H9N2 viruses in China as with seasonal human H1N1 viruses. The same mutation is also found in most human isolates of emergent avian H7N9 and H10N8 viruses whose six internal gene segments are derived from the H9N2 virus. We further demonstrated the mammalian adaptive functionality of the PA-K356R mutation. Avian H9N2 virus with the PA-K356R mutation in human A549 cells showed increased nuclear accumulation of PA and increased viral polymerase activity that resulted in elevated levels of viral transcription and virus output. The same mutant virus in mice also enhanced virus replication and caused lethal infection. In addition, combined mutation of PA-K356R and PB2-E627K, a well-known mammalian adaptive marker, in the H9N2 virus showed further cooperative increases in virus production and severity of infection in vitro and in vivo. In summary, PA-K356R behaves as a novel mammalian tropism mutation, which, along with other mutations such as PB2-E627K, might render avian H9N2 viruses adapted for human infection.
IMPORTANCE Mutations of the polymerase complex (PB1, PB2, and PA) of influenza A virus are necessary for viral adaptation to new hosts. This study reports a novel and predominant mammalian adaptive mutation, PA-K356R, in avian H9N2 viruses and human isolates of emergent H7N9 and H10N8 viruses. We found that PA-356R in H9N2 viruses causes significant increases in virus replication and severity of infection in human cells and mice and that PA-K356R cooperates with the PB2-E627K mutation, a well-characterized human adaptive marker, to exacerbate mammalian infection in vitro and in vivo. Therefore, the PA-K356R mutation is a significant adaptation in H9N2 viruses and related H7N9 and H10N8 reassortants toward human infectivity.
INTRODUCTION
The emergence of new pandemic influenza A viruses requires overcoming barriers to cross-species transmission from animal reservoirs to human populations. Influenza viruses of the H9N2 subtype and their reassortants circulating in poultry are a potential source of pandemic viruses and hence represent a significant public health threat (1). Since spring 2013, a novel avian H7N9 reassortant with six internal genes of the H9N2 virus had caused three major outbreaks in humans throughout China (2, 3). During the same period, a newly emergent avian H10N8 virus, also with six H9N2-like internal genes, was reported to cause fatal human infections in China (4). In the meantime, H9N2 viruses continued to cause human infections in China and other countries based on etiological and serological evidence (5–12). We recently found that avian H9N2 viruses have undergone significant genetic evolution, especially in their internal genes, to form a predominant genotype (G57), which in turn provided the internal genes to multiple new subtypes, including H7N9 and H10N8 viruses (13). Thus, the evolving internal genes of H9N2 viruses may be a primary genetic source for emergent H9N2 viruses and their reassortants that can confer human infection. Understanding the mechanisms by which influenza viruses acquire the ability to infect multiple host species is therefore imperative to managing future outbreaks.
Adaptation of viral polymerase is necessary for efficient virus replication in a new host species (14). The viral polymerase complex of the PB1, PB2, and PA proteins assembles with viral RNA and nucleoprotein (NP) to undertake transcription and replication of the viral genome (15). Polymerases of avian origin generally have impaired activity in human and other mammalian cells (16–21). To overcome this natural restriction, avian polymerases need to acquire mutations that lead to improved activity in mammalian hosts. For instance, the well-characterized PB2 mutation E627K (PB2-E627K) mediates a clear preferential switch in enhanced polymerase activity, virus replication, transmission, and, in certain cases, pathogenicity in mammals (22). Furthermore, several other residues, such as PB2-590S/591R, -701N, -591K, and -271A, have been described to increase viral polymerase activity and contribute to increased virus replication and pathogenicity in mammals (16, 23, 24).
PA amino acid residues 224S, 70V, 85I, 86S, 336M, 353R, and 552S are found to contribute to the viral polymerase activity of H1N1/2009, avian H3N2, or H5N1 viruses in mammalian cells that results in enhanced virus replication and pathogenicity in mammals (25). However, little is known about host adaptive mutations in the PA protein of avian H9N2 influenza viruses. Recently, comprehensive phylogenetic and ancestral inference analyses revealed a number of amino acid changes of avian H9N2 viruses, including K356R of PA, which may contribute to the evolutionary path of H7N9 viruses in humans (26). By analyzing the sequence and crystal structure of the PA subunit, Xu et al. also hypothesized that PA-356R is a possible signature of avian H7N9 viruses with bird-to-human transmissibility (27). Yamayoshi et al. tested the adaptive functionality of PA-K356R by reversing the mutation (from a human-like residue to an avian-like residue) to PA-R356K in the context of a human-derived H7N9 virus, which appeared to have little effect on viral polymerase activity and virulence in vitro and in vivo (28). However, the role of the PA-K356R mutation in H9N2 viruses in species tropism and pathogenicity remains far from clear. In this study, we investigated the emergence of the PA-K356R mutation in avian H9N2, H7N9, and H10N8 viruses as well as human H1N1 influenza viruses. We found that PA-K356R has recently emerged in PA of H9N2 avian influenza viruses (AIVs) to become predominant and has been passed on by reassortment to H7N9 and H10N8 viruses. PA-356R and PB2-627K extensively coexist in H7N9 and H10N8 isolates from human rather than avian hosts, which suggests human tropism. Furthermore, PA-K356R in H9N2 viruses showed independent and cooperative effects with PB2-E627K of enhanced replication and severity of infection in mammalian cells and mice.
MATERIALS AND METHODS
Ethical compliance.
All animal work was approved by the Beijing Association for Science and Technology (approval SYXK [Beijing] 2007-0023) and conducted in accordance with Beijing Laboratory Animal Welfare and Ethics guidelines, as issued by the Beijing Administration Committee of Laboratory Animals, and in accordance with China Agricultural University (CAU) Institutional Animal Care and Use Committee guidelines (SKLAB-B-2010-003).
Viruses, plasmids, and cells.
The use of the wild-type (WT) H9N2 virus A/chicken/Hebei/LC/2008 (HB08) was described previously (29). pcDNA-PB1, pcDNA-PB2, pcDNA-PA, and pcDNA-NP protein expression plasmids for the three polymerase subunits and NP of influenza HB08 virus were generated by subcloning of the corresponding coding segments into the pcDNA3.1 vector. Human embryonic kidney (293T) cells, human pulmonary adenocarcinoma (A549) cells, Madin-Darby canine kidney (MDCK) cells, and chicken fibroblast (DF-1) cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37°C in a 5% CO2 atmosphere.
Generation of mutant viruses by reverse genetics.
All eight gene segments amplified by reverse transcription-PCR (RT-PCR) from the HB08 virus were individually cloned into a dual-promoter plasmid, pHW2000 (29). Mutations K356R and E627K were introduced into the PA and PB2 genes, respectively, by using a site-directed QuikChange mutagenesis kit (Agilent, Santa Clara, CA) according to the manufacturer's instructions. Primer sequences are available upon request. The rescued viruses possessing the single mutations PA-K356R and PB2-E627K and the combined mutations (double mutant) PA-K356R and PB2-E627K were designated PA-K356R, PB2-E627K, and PA-K356R/PB2-E627K, respectively. The WT HB08 virus, the PA-K356R and PB2-E627K single mutant viruses, and the PA-K356R/PB2-E627K double mutant virus were generated by reverse genetics as previously described (29). Viral RNA was extracted and amplified by RT-PCR, and each viral segment was sequenced to confirm the mutational identity of each virus.
Virus detection by immunofluorescence.
A549 cells were grown on glass-bottom dishes and infected with the indicated viruses at a multiplicity of infection (MOI) of 2.0. At the specified time points postinfection, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min and permeabilized with 0.5% Triton X-100 in PBS for 30 min. After blocking with 5% bovine serum albumin (BSA) in PBS, the cells were incubated with rabbit antisera against PA (GeneTex, USA) for 12 h at 4°C. The cells were then washed three times with PBS and incubated with fluorescein isothiocyanate (FITC)-coupled goat anti-rabbit (Abcam, United Kingdom) secondary antibodies for 1 h at 37°C. The cells were subsequently washed three times with PBS and incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Cells were imaged with a laser scanning confocal microscope (Leica). The frequency of nuclear localization of the PA protein was determined by cell counting (n = 100).
Viral ribonucleoprotein polymerase assay.
Ribonucleoprotein (RNP) polymerase (minigenome luciferase) assays were based on the cotransfection of four pcDNA3.1 expression plasmids harboring PB2, PB1, PA, and NP (125 ng each) from individual H9N2 recombinant viruses (WT, PA-K356R, PB2-E627K, and PA-K356R/PB2-E627K) into human 293T or chicken DF-1 cells, together with a pYH-NS1-Luci plasmid expressing a reporter firefly luciferase gene under the control of the human or chicken RNA polymerase I promoter (10 ng) and an internal control plasmid expressing renilla luciferase (2.5 ng). Cultures were incubated at 33°C, 37°C, or 39°C. After 24 h of transfection, cell lysates were prepared with the Dual Luciferase Reporter Assay system (Promega), and luciferase activity was determined with a GloMax 96 microplate luminometer (Promega).
Western blotting.
Total cell protein lysates were extracted from transfected 293T or DF-1 cells with radioimmunoprecipitation assay (RIPA) lysis buffer, and the total protein concentration was determined by using a bicinchoninic acid (BCA) protein assay kit (Beyotime, China). In nuclear import experiments with plasmids, cytoplasmic and nuclear fractions of cell lysates were separated by using a nuclear and cytoplasmic extraction kit (Beyotime, China) according to the manufacturer's instructions. Protein samples derived from cell lysates were heated at 100°C for 10 min and then separated on a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel, transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad, USA), and subsequently incubated with a primary antibody. Primary antibodies were specific for influenza A virus PA (diluted 1:3,000; GeneTex, USA) and PB2 (diluted 1:3,000; Thermo Fisher, USA), β-actin (diluted 1:5,000; Beyotime, China), or PCNA (diluted 1:2,000; Abcam, United Kingdom). The secondary antibody used was horseradish peroxidase (HRP)-conjugated anti-rabbit or -mouse antibody (diluted 1:10,000; Beyotime, China). HRP was detected by using a Western Lightning chemiluminescence kit (Amersham Pharmacia, Freiburg, Germany) according to the manufacturer's protocols.
Quantitative real-time PCR.
Levels of mRNA and viral RNA (vRNA) were determined in A549 cells infected with different H9N2 viruses at an MOI of 1.0 or 0.01. Total RNA was extracted from infected A549 cells by using TRIzol reagent according to the manufacturer's instructions (Invitrogen). For the detection of mRNA and vRNA, the oligo(dT) primer and primer uni-12 (5′-AGCAAACGACC-3′), respectively, were used to generate cDNAs with 1 μg of total RNA per sample by using Superscript III first-strand synthesis supermix (Invitrogen). The quantitative real-time PCR (qRT-PCR) mixture for each reaction sample consisted of 10 μl of 2× SYBR green PCR master mix (Applied Biosystems), 7 μl of nuclease-free water, 0.5 μl of each primer, and 2 μl of the cDNA template. mRNA and vRNA expression levels for the β-actin, matrix protein (M1), and nucleoprotein (NP) genes were quantified by using the 7500 real-time PCR system (Applied Biosystems) with the following program: 1 cycle at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Expression values for each gene, relative to β-actin, were calculated by using the 2−ΔΔCT method. Each experiment contained three technical replicates for each sample, and two experimental replicates were performed. Primers for the amplification of the β-actin, M1, and NP genes are as follows: forward primer 5′-AGAGCTACGAGCTGCCTGAC-3′ and reverse primer 5′-CGTGGATGCCACAGGACT-3′ for β-actin, forward primer 5′-CGCACAGAGACTTGAGGATG-3′ and reverse primer 5′-TGGGTCTCCATTCCCATTTA-3′ for M1, and forward primer 5′-CAGTGAAGGGGATAGGGACA-3′ and reverse primer 5′-CCAGGATTTCTGCTCTCTCG-3′ for NP.
Virus titration and replication kinetics.
The 50% tissue culture infectious dose (TCID50) was determined in MDCK cells with 10-fold serially diluted viruses inoculated at 37°C for 72 h. The TCID50 value was calculated by the Reed-Muench method (30). Multistep replication kinetics was determined by inoculating MDCK or A549 cells with viruses at an MOI of 0.001 or 0.01, respectively. After 1 h of incubation at 37°C, the cells were washed twice and further incubated in serum-free DMEM containing 1.0 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin. Supernatants were sampled at 6, 12, 24, 36, 48, 60, and 72 h postinoculation (hpi). Analysis of single-replication-cycle kinetics was similarly conducted except with a starting inoculation at an MOI of 1.0. Supernatants were sampled at 2, 4, 6, 8, 10, and 12 hpi. All collected supernatants were titrated on MDCK cells in 96-well plates; three independent experiments were performed.
Mouse challenge study.
Fourteen mice (6-week-old female BALB/c mice; Vital River Laboratory, Beijing, China) per group were anesthetized with tiletamine-zolazepam (Zoletil; Virbac SA, Carros, France) (20 mg/g) and inoculated intranasally with 50 μl of 106 TCID50 of each test virus diluted in PBS. Five mice from each group were monitored daily for 14 days, and mice that lost >30% of their original body weight were humanely euthanized. Three mice from each group were euthanized at 1, 3, and 5 days postinoculation (dpi) for the determination of virus titers, histology, and cytokine levels. Lungs and nasal turbinates were collected and homogenized in 1 ml of cold PBS. Virus titers were determined by a TCID50 assay. A portion of the lung from each euthanized mouse at 5 dpi was fixed in 10% phosphate-buffered formalin for histopathological examination, which was performed as described previously (31).
Quantification of cytokines in murine lungs.
Levels of cytokine proteins, including tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), were determined by a using cytometric bead array method (BD Cytometric Bead Array mouse inflammation kit; BD Bioscience, San Diego, CA, USA). Briefly, a 50 μl of a mouse inflammation capture bead suspension and 50 μl of detection reagent were added to an equal amount of sample, and the sample was incubated in the dark for 2 h at room temperature. Subsequently, each sample was washed with 1 ml wash buffer and then centrifuged at 200 × g at room temperature for 5 min. Supernatants were discarded, and a further 300 μl of wash buffer was added. Samples were analyzed on a BD fluorescence activated cell sorting (FACS) Array bioanalyzer (BD Bioscience). Data were analyzed by using BD CBA software (BD Bioscience). Levels of each cytokine were computed as picograms per milliliter of homogenate.
Statistical analyses.
All statistical analyses were performed by using GraphPad Prism software version 5.00 (GraphPad Software Inc., San Diego, CA, USA). Statistically significant differences between experimental groups were determined by analysis of variance (ANOVA). Differences were considered statistically significant at a P value of <0.05.
RESULTS
Potential mammalian adaptive mutation K356R is predominant in the PA protein of avian H9N2 virus.
Previous computational studies suggested that K356R in PA is a potential mammalian adaptive mutation (27). The PA-356R residue is conserved among pandemic H1N1/2009 (pH1N1/2009) and seasonal human H1N1/H2N2/H3N2 viruses but is rarely found in avian influenza viruses (27). Our sequence searches revealed an adaptive mutation trace of PA-K356R in seasonal human H1N1 influenza viruses (Fig. 1A). The human-like signature of PA-356R had steadily replaced the avian-like PA-356K signature from the early prevalence of avian-origin H1N1 viruses in humans from 1918 to 1976 to become the predominant mutation in 1977 to 2008. Coincidentally, we found that all avian H9N2 strains isolated in 2014 in China, unlike previous years, also carried the human-like residue PA-356R (Fig. 1A). The percentage of H9N2 viruses with PA-356R had increased sharply from 4.76% in 2011 to 100% in 2014. H9N2 viruses before 2010 were nearly all avian-like PA-356K viruses. As H9N2 viruses provide six internal genes to emergent H7N9 and H10N8 viruses that have been causing severe human infections since 2013, we examined the prevalence of PA-356R in avian- and human-derived H7N9/H10N8 strains. As shown in Fig. 1B, human H7N9 and H10N8 isolates showed higher frequencies of human-like residue 356R than did avian H7N9 and H10N8 isolates. Furthermore, the proportion of human H7N9 isolates with both PA-356R and PB2-627K was much higher than that of the corresponding avian isolates. With further screening of H9N2 viruses, we also found the coexistence of PA-K356R and PB2-E627K mutations in two naturally occurring avian H9N2 isolates (A/chicken/Jinan/3952/2013 and A/chicken/Jinan/4225/2013). These findings suggest that the PA-K356R mutation in avian H9N2 viruses, by itself or in coordination with PB2-E627K, might confer an adaptive change to the polymerase complex of avian influenza virus for cross-species transmission to facilitate virus replication and/or pathogenicity in humans. The biological contribution of the PA-K356R mutation to H9N2 avian influenza virus is not known.
FIG 1.

Prevalence of amino acid residues PA-356K and PA-356R in seasonal human H1N1 and avian H9N2 influenza viruses (A) and prevalence of PA-356R alone or PA-356R/PB2-627K in H7N9 and H10N8 influenza viruses during 2013 to 2014 (B). For each column from left to right, the actual numbers of viruses are 22, 21, 1,320, 138, 590, 84, 101, 241, and 29, respectively (A) and 399, 77, 399, 77, 54, 2, 54, and 2, respectively (B). In panel A, the proportion of viruses with PA-356R (0.33%) or other residues (0.30%) is too low to be seen between 2001 and 2010. In panel B, “Others” refers to viruses without the two residues PA-356R and PB2-627K; the number of influenza viruses with PA-356R/PB2-627K is 2 for avian H7N9 viruses.
PA-K356R increases nuclear accumulation of PA protein.
During influenza virus replication, the PA protein, synthesized in the cytoplasm, is imported into the nucleus to form a functional polymerase complex with PB2, PB1, and NP (15). We determined if the K356R mutation in the PA protein affects the nuclear transport of PA. Human A549 cells were infected with parental WT (HB08) and mutant PA-K356R viruses at an MOI of 2.0, and the cellular localization of PA was determined at different time points postinfection (Fig. 2A and B). For each virus, the accumulation of PA beyond 4 h of infection shifted from the cytoplasm to the nucleus. However, PA from the mutant virus showed a significantly higher rate of nuclear accumulation at 6, 8, and 10 hpi than the corresponding WT virus (P < 0.05). To further determine whether the enhanced nuclear import was the result of an increase in PA-K356R protein nuclear affinity, Western blotting was performed to detect the relative distribution of PA in the cytoplasmic and nuclear compartments of transfected (WT PA and PA-K356R expression plasmids) human 293T cells (Fig. 2C). WT PA and PA-K356R protein levels were similarly detected in each compartment. Thus, the increase in the nuclear entry rate of PA-K356R relative to that of WT PA during infection (Fig. 2A and B) was likely the result of increased viral replication rather than a change in its nuclear affinity (Fig. 2C).
FIG 2.
Nuclear import of PA proteins of H9N2 viruses. (A) Nuclear localization of the PA protein in A549 cells infected with H9N2 viruses. PA proteins in A549 cells infected with the indicated H9N2 viruses at an MOI of 2.0 were localized by immunofluorescence (green) at 4, 6, 8, and 10 hpi. Nuclei were stained with DAPI (blue). (B) Relative nuclear localization of PA protein. A hundred cells (blue nuclei) per microscopic field were randomly selected, from which the percentage of intranuclear PA (green) was determined. Data are presented as means ± standard deviations of results from three independent experiments. * indicates that the value is significantly different from that for the wild-type virus (P < 0.05 as determined by ANOVA). (C) Protein levels of the PA protein in the cytoplasm and nucleus of transfected 293T cells. 293T cells were transfected with a plasmid expressing wild-type PA (PA-WT) or a PA mutant (PA-K356R). Cells were harvested 24 h later, and the expression levels of cytoplasmic and nuclear PA protein were detected by immunoblotting. PA-N and PA-C represent the expression levels of PA in the nucleus and cytoplasm, respectively. The density of each band on the immunoblot was normalized to the density of β-actin in the cytoplasm or PCNA in the nucleus.
PA-K356R enhances polymerase activity alone and with PB2-E627K in human cells.
RNP polymerase activity has been shown to catalyze viral transcription and genomic replication, which correlate with viral replication and pathogenicity in the host (15). We examined the effect of PA-356R on RNP polymerase activity in human 293T cells (Fig. 3A). PA-K356R showed a significant increase in polymerase activity in 293T cells at 33°C and 37°C (P < 0.05) compared to the wild-type polymerase complex. As mutations PA-K356R and PB2-E627K coexist in avian H9N2 and human H7N9/H10N8 viruses, we also examined the polymerase activity of the combined PA-K356R/PB2-E627K mutations. Mutation PB2-E627K alone or double mutation PA-K356R/PB2-E627K induced higher polymerase activity than did PA-K356R; the PA-K356R/PB2-E627K double mutation elicited the highest polymerase activity (P < 0.05) (Fig. 3A). The observed increase in the polymerase activity of the mutated complexes might be associated with altered protein production. Western blotting with protein lysates derived from 293T cells transfected with RNP plasmids indicated that PA and PB2 mutations did not quantitatively affect protein expression (Fig. 3A). Thus, the enhanced polymerase activities detected with PA and PB2 mutations were likely due to increased enzymatic activities. In contrast to the results with 293T cells, RNP polymerase activity remained unchanged with all three mutations (PA-K356R, PB2-E627K, and PA-K356R/PB2-E627K) in DF-1 cells at 37°C and 39°C (Fig. 3B). These results demonstrate that PA-K356R is a species-specific regulator that enhances the polymerase activity of avian H9N2 viruses in human 293T but not chicken DF-1 cells and exhibits cooperative polymerase enhancement with mutation PB2-E627K.
FIG 3.

RNP polymerase activities of wild-type and mutant H9N2 influenza viruses. Polymerase activities in 293T cells (A) or DF-1 cells (B) were determined by minigenome replication assays and are expressed as mean percent activities ± standard deviations, with the activity of the corresponding wild-type PA protein being set at 100% from three independent experiments. Western blotting was performed on protein lysates from 293T or DF-1 cells transfected with wild-type and mutant polymerase plasmids (PA and PB2). The density of each band on the immunoblot was normalized to the density of β-actin. * indicates that the value is significantly different from that for the wild-type virus (P < 0.05 as determined by ANOVA), and # indicates that the value is significantly different from that for the virus with single mutation PB2-E627K (P < 0.05 as determined by ANOVA).
PA-K356R augments viral transcription and genomic replication.
The performance of viral RNP polymerase in viral transcription (mRNA) and genomic replication (vRNA) was evaluated in A549 cells with the WT, PA-K356R, PB2-E627K, and double mutant PA-K356R/PB2-E627K H9N2 viruses at 2, 6, 8, 24, and 48 hpi (Fig. 4). qRT-PCR analyses with mRNA- or vRNA-specific primers indicated that all three mutant viruses produced significantly higher levels of both viral M1 and NP mRNA and vRNA at 2 hpi than did the WT H9N2 virus (P < 0.05). The promotional effect of PA-K356R was similar to that of PB2-E627K. Combined PA-K356R and PB2-E627K mutations showed cooperativity at 6 and 8 hpi to significantly increase viral transcription and replication (P < 0.05). These data demonstrate that PA-356R (like PB2-627K) in avian H9N2 viruses increases viral transcription and replication in human A549 cells.
FIG 4.
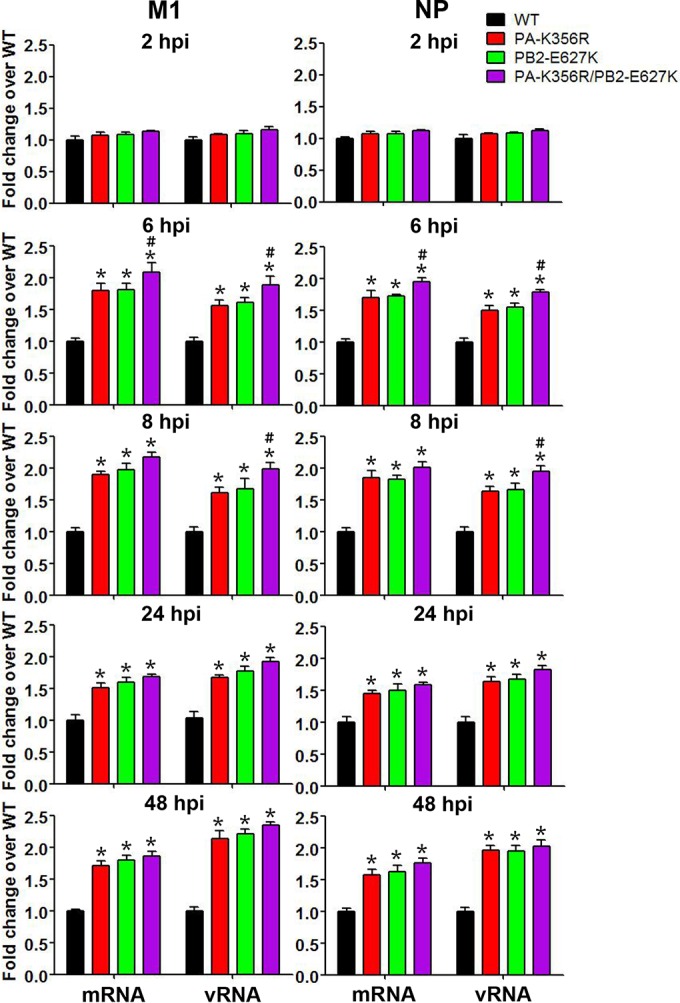
Quantitative estimation of mRNA and vRNA levels of M1 and NP in H9N2 virus-infected A549 cells. A549 cells were infected with the indicated H9N2 mutant viruses at an MOI of 1.0 for 2, 6, and 8 h or at an MOI of 0.01 for 24 and 48 h. The mRNA and vRNA expression levels of M1 and NP from mutant viruses are expressed as fold changes in relation to the wild-type group. Data are presented as means ± standard deviations of results from three independent experiments. * indicates that the value is significantly different from that for the wild-type virus (P < 0.05 as determined by ANOVA), and # indicates that the value is significantly different from that for the virus with single mutation PB2-E627K (P < 0.05 as determined by ANOVA).
PA-K356R enhances production of H9N2 virus in mammalian cells.
To compare the replication of H9N2 viruses with a PA-K356R, PB2-E627K, and combined PA-K356R/PB2-E627K mutations, we determined their multistep replication kinetics at 6, 12, 24, 36, 48, 60, and 72 hpi in MDCK and A549 cells infected at MOIs of 0.001 and 0.01, respectively. All three mutant viruses showed similar virus outputs that were significantly higher (up to 10-fold higher) than those from the wild-type virus over 12 to 36 hpi (P < 0.05) (Fig. 5A and B). To determine if increased virus replication took place early during infection, we conducted assays of single-replication-cycle kinetics at an MOI of 1.0 over 12 hpi (Fig. 5C and D). All three mutant viruses generated more progeny virus (up to nearly 100-fold higher) than did the wild-type virus in MDCK and A549 cells. The H9N2 virus with double mutation PA-K356R/PB2-E627K showed a sustained elevation of virus output that was higher than the output from the PB2-E627K mutant in A549 cells at 6, 8, and 12 hpi (Fig. 5D). Taken together, these data show that the PA-K356R mutation alone or in combination with PB2-E627K enhances the progeny output of H9N2 virus in MDCK and A549 cells, particularly during the initial 12 h of infection.
FIG 5.

Virus output of mutant and wild-type H9N2 viruses from infected MDCK and A549 cells. (A and B) Multistep growth curves of H9N2 viruses were determined in MDCK (A) and A549 (B) cells. MDCK and A549 cells were inoculated with viruses at MOIs of 0.001 and 0.01, respectively. (C and D) Single replication cycles of H9N2 viruses were also examined in MDCK (C) and A549 cells (D) at an MOI of 1.0. The supernatants of infected cells were collected at the indicated time points. The virus titers are means ± standard deviations (n = 3). * indicates that the value is significantly different from that for the wild-type virus (P < 0.05 as determined by ANOVA), and # indicates that the value is significantly different from that for the virus with single mutation PB2-E627K (P < 0.05 by ANOVA).
PA-K356R increases pathogenicity and replication of H9N2 virus in mice.
We further determined the functionality of PA-K356R alone or in conjunction with PB2-E627K in pathogenicity and replication in mice. All mice infected with mutant viruses (harboring PA-K356R, PB2-E627K, or both) at 106 TCID50 died by 8 dpi (Fig. 6A). With the PA-K356R/PB2-E627K double mutant virus, complete lethality occurred sooner, at 5 dpi. In contrast, all mice infected with the wild-type virus survived infection. Expectedly, the weight loss of infected mice corresponded to relative virus lethality (Fig. 6B). Viruses with the PB2-E627K or PA-K356R/PB2-E627K mutation produced the greatest weight loss. Lung tissues were collected at 5 dpi for histopathology. Severe interstitial pneumonia and bronchopneumonia, characterized by alveolar interstitial consolidation and extensive inflammatory cell infiltration, were evident in PA-K356R, PB2-E627K, and PA-K356R/PB2-E627K virus-infected lung sections (Fig. 6C). The most severe pathology was observed for the PA-K356R/PB2-E627K virus-infected group. In summary, PA-356R in the H9N2 virus caused severe pathology similar to that of PB2-627K, with a further increase in severity being evident when both mutations were present in the virus.
FIG 6.

Pathogenicity of H9N2 mutant viruses in mice. Five BALB/c mice per group were inoculated with 106 TCID50 of the indicated viruses or mock infected with PBS. (A) Survival rate of mice. Mice that lost >30% of their baseline weight were euthanized. (B) Body weight changes of mice. The values represent the average body weights compared to the baseline weight ± standard deviations from five mice. (C) Hematoxylin- and eosin-stained lung sections from mice infected with the indicated viruses at 5 dpi. Bar, 200 μm.
Virus titers in nasal turbinates and lungs from infected mice (three in each group) were evaluated at 1, 3, and 5 dpi (Table 1). Consistent with the pathogenicity results, the virus carrying combined mutation PA-K356R/PB2-E627K produced the highest titers in the nasal turbinates and lungs. The PA-K356R and PB2-E627K single mutants generated significantly higher titers than did the wild-type virus at 3 and/or 5 dpi (P < 0.05). Collectively, PA-356R alone or cooperatively with PB2-627K in the H9N2 virus conferred elevated pathogenicity and virus replication in mice.
TABLE 1.
Average virus titers in organs of infected mice
| Virus | Avg virus titer (mean log10 TCID50/ml ± SD)a |
|||||
|---|---|---|---|---|---|---|
| 1 dpi |
3 dpi |
5 dpi |
||||
| Nasal turbinate | Lung | Nasal turbinate | Lung | Nasal turbinate | Lung | |
| Wild type | 2.08 ± 0.29 | 3.08 ± 0.58 | 4.12 ± 0.38 | 5.83 ± 0.38 | 3.42 ± 0.14 | 5.58 ± 0.14 |
| PA-K356R | 2.33 ± 0.14 | 3.33 ± 0.14 | 4.67 ± 0.14 | 6.25 ± 0.43 | 4.25 ± 0.66* | 6.42 ± 0.58* |
| PB2-E627K | 2.42 ± 0.14 | 3.50 ± 0.25 | 4.83 ± 0.38 | 6.75 ± 0* | 4.75 ± 0.66* | 6.42 ± 0.58* |
| PA-K356R/PB2-E627K | 2.67 ± 0.14 | 3.83 ± 0.38 | 5.83 ± 0.14*# | 6.83 ± 0.38* | 5.08 ± 0.29* | 6.50 ± 0.25* |
Lower limits of detection were 101.25 TCID50/ml for nasal turbinates or lungs. * indicates that the virus titer of the corresponding mutant strain was significantly higher than that of the wild-type virus, and # indicates that the virus titer of the corresponding mutant strain was significantly higher than that of the PB2-E627K virus, with a P value of <0.05, as determined by one-way ANOVA.
H9N2 virus with the PA-K356R mutation induces an elevated proinflammatory response in mice.
Severe influenza virus infection in humans and animal models is associated with abnormally elevated pulmonary expression levels of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β (32–34). To compare the proinflammatory impacts of PA-K356R, PB2-E627K, and PA-K356R/PB2-E62K, we determined the protein expression levels of the three cytokines in the lungs of infected mice at 1, 3, and 5 dpi (Fig. 7). All three mutant viruses induced higher levels of production of TNF-α, IL-6, and IL-1β over all three time points than did the wild-type virus (P < 0.05). Furthermore, the PA-356R/PB2-627K mutant elicited higher levels of production of TNF-α and IL-6 at 1 dpi and of IL-1β at 5 dpi than did the PB2-E627K virus (P < 0.05) (Fig. 7). These results show that individual and combined substitutions of PA-K356R and PB2-E627K in H9N2 viruses increase proinflammation in mice, consistent with increased pathogenicity and virus replication.
FIG 7.

Detection of cytokine proteins in lungs of mice infected with H9N2 mutant viruses. Nine mice per group were either inoculated with 106 TCID50 of the indicated mutant viruses or mock infected with PBS, and protein levels of cytokines in the lungs of three infected mice were determined at 1, 3, and 5 dpi. Data are presented as means ± standard deviations of results from three independent experiments. * indicates that the value is significantly different from that for the wild-type virus (P < 0.05 as determined by ANOVA), and # indicates that the value is significantly different from that for the virus with single mutation PB2-E627K (P < 0.05 as determined by ANOVA).
DISCUSSION
Adaptive mutations in PA proteins of AIVs are recognized as being crucial for cross-species transmission to mammals (35). In the present study, we characterized and compared one such adaptive mutation, PA-K356R, in an avian H9N2 virus, which promoted PA nuclear accumulation and viral polymerase activity in mammalian cells. Importantly, mice infected with this mutant PA-K356R virus exhibited increased virus output, severe lung pathology, and an elevated proinflammatory cytokine response that culminated in complete lethality by 8 dpi.
The PA protein, which is 716 amino acids long, is the third subunit of the viral polymerase complex. The functions of the PA protein are associated with its interactions with the PB1 and PB2 proteins and with its endonuclease and protease activities (35). It can be cleaved by limited tryptic digestion into N-terminal (PA-N) and C-terminal (PA-C) domains (36). Known mammalian adaptive mutations are found mainly in the PA-N region (35), with some being found in the PA-C domain, such as PA-T552S in human isolates (25, 37). PA-356 is located in the PA-C domain and is close to the loop spanning amino acid positions 350 to 355 (i.e., the 350–355 loop) (38). The loops from two PA monomers are antiparallel to each other and have been proposed to form multiple interactions (38). Besides PA-356, residues located in the 350–355 loop are found to confer mammalian tropism and pathogenicity. For example, the I353R mutation is a primary determinant of enhanced polymerase activity, virus replication, and increased virulence of an avian H5N1 virus strain in a mouse model (39). Liedmann et al. found that a single mutation (K351E) in the PA protein contributes to the inhibition of beta interferon (IFN-β) induction and virulence of the A/Puerto Rico/8/1934 (A/PR/8/34) virus in mice (40). Therefore, the 350–355 loop and its proximal residues may perform a critical role in determining cross-species transmission and mammalian adaptation.
Neumann et al. recently identified a number of critical amino acid changes in the PA gene of ancestral H9N2 lineages, including the PA-K356R mutation, that contributed to the emergent avian H7N9 viruses that readily cause severe human infections (26). However, in the A/Anhui/1/2013 (H7N9) virus background, the reverse (avian-like) mutant PA-R356K appears to have little effect on viral polymerase activity and virulence in vitro and in vivo (28). The H7N9 human isolate used in that study would have contained multiple mammalian adaptive markers, including the well-characterized PB2-627K marker. In the present study, we used an earlier wild-type H9N2 isolate to provide a viral background with no known mammalian adaptive markers. Indeed, the PA-K356R mutation in an H9N2 virus background exhibited significant increases in virus replication and pathogenicity in mice, underlining the functional significance of this mutation.
To better understand the natural selection of mutation PA-K356R, we investigated its emergence and prevalence in avian and human influenza viruses. We found that PA-356K and PA-356R are presently conserved in avian (except for H9N2) and human viruses, respectively, suggesting that this residue has a critical role in host species tropism. Since the first avian-origin H1N1 virus entered humans in 1918 to become established as a human H1N1 seasonal virus, its avian-like signature PA-356K has been gradually replaced with the human-like PA-356R mutation. Strikingly, in the last few years, the same human-like residue PA-356R has emerged and become predominant in avian H9N2 viruses in chickens. The PA-356R signature, along with the well-known mammalian adaptive marker PB2-E627K, has further spread to avian H7N9 and H10N8 reassortants, which harbor the PA gene of H9N2 origin. The functionality of the PA-K356R mutation appears cooperative with PB2-E627K. These mutations individually and collectively increased viral transcription and replication, progeny virus production, and severity of infection in mammalian cells and mice. Conceivably, viruses that harbor both mutations could pose a severe risk to human health. Indeed, hundreds of severe human cases of H7N9 and several H10N8 virus infections were recently documented (2–4). The combined mutations PA-356R and PB2-627K in H7N9 and H10N8 viruses appear to act in concert to enhance infection and pathogenicity in humans.
In this study, we found that the PA-K356R mutation could contribute to the possible transfer from avian-like tropism to human-like tropism. We found that this mutation increased viral replication and pathogenicity in mice. However, the PA-K356R mutation did not alter viral polymerase activity in avian cells, suggesting that it could be a mammalian species-specific regulator. There is epidemiological evidence of avian viruses with mutations, akin to PA-K356R, that confer mammalian tropism and that have emerged or become predominant in avian strains before transmission to mammals. Similarly to PA-K356R, hemagglutinin (HA) mutation Q226L, which is a human-like mutation and confers binding to the human-like sialic acid (SA) α-2,6-Gal receptor, has presently become predominant in H9N2 avian viruses since appearing in the 1990s (1). Wan and Perez demonstrated that an HA-Q226L mutant promotes both cell tropism and replication of H9N2 viruses in human airway epithelial cells (41). However, Vines et al. showed that this single residue has little or no effect on viral replication in ducks (42). Likewise, the PA-I353R mutation in avian H5N1 viruses contributes to viral infection only in mice but not in avian hosts (39, 43). Thus, these mutations also seem to be mammal-specific regulators. Some mutations, however, are found to confer improved infection of both birds and mammals. For example, our previous study revealed that H9N2 viruses with an HA-A316S substitution and/or a shorter neuraminidase (NA) stalk are predominantly prevalent in avian strains and can increase virulence in chickens and mice (44). Thus, emerging or even established mutations in avian strains can exhibit increased viral infectivity in mammalian host species, which in turn could be different in avian species. The prevalence of these mammalian tropism mutations in large avian populations poses great concerns to public health. In summary, PA-K356R behaves as a mammalian tropism mutation, which, along with other mutations such as PB2-E627K, could render avian H9N2 viruses or their reassortants more adapted to human infection.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31270046 and 31430086) and the Program for New Century Excellent Talents in University (NCET-13-0559).
REFERENCES
- 1.Li X, Shi J, Guo J, Deng G, Zhang Q, Wang J, He X, Wang K, Chen J, Li Y, Fan J, Kong H, Gu C, Guan Y, Suzuki Y, Kawaoka Y, Liu L, Jiang Y, Tian G, Li Y, Bu Z, Chen H. 2014. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 avian influenza viruses. PLoS Pathog 10:e1004508. doi: 10.1371/journal.ppat.1004508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, Chen J, Jie Z, Qiu H, Xu K, Xu X, Lu H, Zhu W, Gao Z, Xiang N, Shen Y, He Z, Gu Y, Zhang Z, Yang Y, Zhao X, Zhou L, Li X, Zou S, Zhang Y, Li X, Yang L, Guo J, Dong J, Li Q, Dong L, Zhu Y, Bai T, Wang S, Hao P, Yang W, Zhang Y, Han J, Yu H, Li D, Gao GF, Wu G, Wang Y, Yuan Z, Shu Y. 2013. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 3.Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, Ma C, Lycett SJ, Leung CY, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LL, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JS, Rambaut A, Zhu H, Guan Y. 2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502:241–244. doi: 10.1038/nature12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Yuan H, Gao R, Zhang J, Wang D, Xiong Y, Fan G, Yang F, Li X, Zhou J, Zou S, Yang L, Chen T, Dong L, Bo H, Zhao X, Zhang Y, Lan Y, Bai T, Dong J, Li Q, Wang S, Zhang Y, Li H, Gong T, Shi Y, Ni X, Li J, Zhou J, Fan J, Wu J, Zhou X, Hu M, Wan J, Yang W, Li D, Wu G, Feng Z, Gao GF, Wang Y, Jin Q, Liu M, Shu Y. 2014. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet 383:714–721. doi: 10.1016/S0140-6736(14)60111-2. [DOI] [PubMed] [Google Scholar]
- 5.Sun Y, Liu J. 2015. H9N2 influenza virus in China: a cause of concern. Protein Cell 6:18–25. doi: 10.1007/s13238-014-0111-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blair PJ, Putnam SD, Krueger WS, Chum C, Wierzba TF, Heil GL, Yasuda CY, Williams M, Kasper MR, Friary JA, Capuano AW, Saphonn V, Peiris M, Shao H, Perez DR, Gray GC. 2013. Evidence for avian H9N2 influenza virus infections among rural villagers in Cambodia. J Infect Public Health 6:69–79. doi: 10.1016/j.jiph.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coman A, Maftei DN, Krueger WS, Heil GL, Friary JA, Chereches RM, Sirlincan E, Bria P, Dragnea C, Kasler I, Gray GC. 2013. Serological evidence for avian H9N2 influenza virus infections among Romanian agriculture workers. J Infect Public Health 6:438–447. doi: 10.1016/j.jiph.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Gray GC, Ferguson DD, Lowther PE, Heil GL, Friary JA. 2011. A national study of US bird banders for evidence of avian influenza virus infections. J Clin Virol 51:132–135. doi: 10.1016/j.jcv.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okoye J, Eze D, Krueger WS, Heil GL, Friary JA, Gray GC. 2013. Serologic evidence of avian influenza virus infections among Nigerian agricultural workers. J Med Virol 85:670–676. doi: 10.1002/jmv.23520. [DOI] [PubMed] [Google Scholar]
- 10.Uyeki TM, Nguyen DC, Rowe T, Lu X, Hu-Primmer J, Huynh LP, Hang NL, Katz JM. 2012. Seroprevalence of antibodies to avian influenza A (H5) and A (H9) viruses among market poultry workers, Hanoi, Vietnam, 2001. PLoS One 7:e43948. doi: 10.1371/journal.pone.0043948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Ju L, Liu P, Zhou J, Lv X, Li L, Shen H, Su H, Jiang L, Jiang Q. 2015. Serological and virological surveillance of avian influenza A virus H9N2 subtype in humans and poultry in Shanghai, China, between 2008 and 2010. Zoonoses Public Health 62:131–140. doi: 10.1111/zph.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawar SD, Tandale BV, Raut CG, Parkhi SS, Barde TD, Gurav YK, Kode SS, Mishra AC. 2012. Avian influenza H9N2 seroprevalence among poultry workers in Pune, India, 2010. PLoS One 7:e36374. doi: 10.1371/journal.pone.0036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pu J, Wang S, Yin Y, Zhang G, Carter RA, Wang J, Xu G, Sun H, Wang M, Wen C, Wei Y, Wang D, Zhu B, Lemmon G, Jiao Y, Duan S, Wang Q, Du Q, Sun M, Bao J, Sun Y, Zhao J, Zhang H, Wu G, Liu J, Webster RG. 2015. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc Natl Acad Sci U S A 112:548–553. doi: 10.1073/pnas.1422456112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naffakh N, Tomoiu A, Rameix-Welti MA, van der Werf S. 2008. Host restriction of avian influenza viruses at the level of the ribonucleoproteins. Annu Rev Microbiol 62:403–424. doi: 10.1146/annurev.micro.62.081307.162746. [DOI] [PubMed] [Google Scholar]
- 15.Eisfeld AJ, Neumann G, Kawaoka Y. 2015. At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 13:28–41. doi: 10.1038/nrmicro3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labadie K, Dos Santos Afonso E, Rameix-Welti MA, van der Werf S, Naffakh N. 2007. Host-range determinants on the PB2 protein of influenza A viruses control the interaction between the viral polymerase and nucleoprotein in human cells. Virology 362:271–282. doi: 10.1016/j.virol.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 17.Mehle A, Doudna JA. 2009. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc Natl Acad Sci U S A 106:21312–21316. doi: 10.1073/pnas.0911915106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rameix-Welti MA, Tomoiu A, Dos Santos Afonso E, van der Werf S, Naffakh N. 2009. Avian Influenza A virus polymerase association with nucleoprotein, but not polymerase assembly, is impaired in human cells during the course of infection. J Virol 83:1320–1331. doi: 10.1128/JVI.00977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salomon R, Franks J, Govorkova EA, Ilyushina NA, Yen HL, Hulse-Post DJ, Humberd J, Trichet M, Rehg JE, Webby RJ, Webster RG, Hoffmann E. 2006. The polymerase complex genes contribute to the high virulence of the human H5N1 influenza virus isolate A/Vietnam/1203/04. J Exp Med 203:689–697. doi: 10.1084/jem.20051938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steel J, Lowen AC, Mubareka S, Palese P. 2009. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLoS Pathog 5:e1000252. doi: 10.1371/journal.ppat.1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Hoeven N, Pappas C, Belser JA, Maines TR, Zeng H, Garcia-Sastre A, Sasisekharan R, Katz JM, Tumpey TM. 2009. Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc Natl Acad Sci U S A 106:3366–3371. doi: 10.1073/pnas.0813172106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou B, Ma J, Liu Q, Bawa B, Wang W, Shabman RS, Duff M, Lee J, Lang Y, Cao N, Nagy A, Lin X, Stockwell TB, Richt JA, Wentworth DE, Ma W. 2014. Characterization of uncultivable bat influenza virus using a replicative synthetic virus. PLoS Pathog 10:e1004420. doi: 10.1371/journal.ppat.1004420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehle A, Doudna JA. 2008. An inhibitory activity in human cells restricts the function of an avian-like influenza virus polymerase. Cell Host Microbe 4:111–122. doi: 10.1016/j.chom.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatta M, Hatta Y, Kim JH, Watanabe S, Shinya K, Nguyen T, Lien PS, Le QM, Kawaoka Y. 2007. Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog 3:1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bussey KA, Desmet EA, Mattiacio JL, Hamilton A, Bradel-Tretheway B, Bussey HE, Kim B, Dewhurst S, Takimoto T. 2011. PA residues in the 2009 H1N1 pandemic influenza virus enhance avian influenza virus polymerase activity in mammalian cells. J Virol 85:7020–7028. doi: 10.1128/JVI.00522-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neumann G, Macken CA, Kawaoka Y. 2014. Identification of amino acid changes that may have been critical for the genesis of A(H7N9) influenza viruses. J Virol 88:4877–4896. doi: 10.1128/JVI.00107-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu W, Sun Z, Liu Q, Xu J, Jiang S, Lu L. 2013. PA-356R is a unique signature of the avian influenza A (H7N9) viruses with bird-to-human transmissibility: potential implication for animal surveillances. J Infect 67:490–494. doi: 10.1016/j.jinf.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Yamayoshi S, Yamada S, Fukuyama S, Murakami S, Zhao D, Uraki R, Watanabe T, Tomita Y, Macken C, Neumann G, Kawaoka Y. 2014. Virulence-affecting amino acid changes in the PA protein of H7N9 influenza A viruses. J Virol 88:3127–3134. doi: 10.1128/JVI.03155-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Y, Qin K, Wang J, Pu J, Tang Q, Hu Y, Bi Y, Zhao X, Yang H, Shu Y, Liu J. 2011. High genetic compatibility and increased pathogenicity of reassortants derived from avian H9N2 and pandemic H1N1/2009 influenza viruses. Proc Natl Acad Sci U S A 108:4164–4169. doi: 10.1073/pnas.1019109108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reed LJ, Muench HA. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493–497. [Google Scholar]
- 31.Zhang Y, Sun Y, Sun H, Pu J, Bi Y, Shi Y, Lu X, Li J, Zhu Q, Gao GF, Yang H, Liu J. 2012. A single amino acid at the hemagglutinin cleavage site contributes to the pathogenicity and neurovirulence of H5N1 influenza virus in mice. J Virol 86:6924–6931. doi: 10.1128/JVI.07142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog 4:e1000115. doi: 10.1371/journal.ppat.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng H, Belser JA, Goldsmith CS, Gustin KM, Veguilla V, Katz JM, Tumpey TM. 2015. A(H7N9) virus results in early induction of proinflammatory cytokine responses in both human lung epithelial and endothelial cells and shows increased human adaptation compared with avian H5N1 virus. J Virol 89:4655–4667. doi: 10.1128/JVI.03095-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao H, Sun Y, Hu J, Qi L, Wang J, Xiong X, Wang Y, He Q, Lin Y, Kong W, Seng LG, Sun H, Pu J, Chang KC, Liu X, Liu J. 2015. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci Rep 5:8262. doi: 10.1038/srep08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu J, Liu X. 2015. Crucial role of PA in virus life cycle and host adaptation of influenza A virus. Med Microbiol Immunol 204:137–149. doi: 10.1007/s00430-014-0349-y. [DOI] [PubMed] [Google Scholar]
- 36.Hara K, Schmidt FI, Crow M, Brownlee GG. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J Virol 80:7789–7798. doi: 10.1128/JVI.00600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehle A, Dugan VG, Taubenberger JK, Doudna JA. 2012. Reassortment and mutation of the avian influenza virus polymerase PA subunit overcome species barriers. J Virol 86:1750–1757. doi: 10.1128/JVI.06203-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang S, Sun D, Liang H, Wang J, Li J, Guo L, Wang X, Guan C, Boruah BM, Yuan L, Feng F, Yang M, Wang L, Wang Y, Wojdyla J, Li L, Wang J, Wang M, Cheng G, Wang HW, Liu Y. 2015. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 A resolution. Mol Cell 57:925–935. doi: 10.1016/j.molcel.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 39.Hu J, Hu Z, Song Q, Gu M, Liu X, Wang X, Hu S, Chen C, Liu H, Liu W, Chen S, Peng D, Liu X. 2013. The PA-gene-mediated lethal dissemination and excessive innate immune response contribute to the high virulence of H5N1 avian influenza virus in mice. J Virol 87:2660–2672. doi: 10.1128/JVI.02891-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, McCullers JA, Ludwig S, Ehrhardt C. 2014. Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun 5:5645. doi: 10.1038/ncomms6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wan H, Perez DR. 2007. Amino acid 226 in the hemagglutinin of H9N2 influenza viruses determines cell tropism and replication in human airway epithelial cells. J Virol 81:5181–5191. doi: 10.1128/JVI.02827-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vines A, Wells K, Matrosovich M, Castrucci MR, Ito T, Kawaoka Y. 1998. The role of influenza A virus hemagglutinin residues 226 and 228 in receptor specificity and host range restriction. J Virol 72:7626–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu J, Hu Z, Mo Y, Wu Q, Cui Z, Duan Z, Huang J, Chen H, Chen Y, Gu M, Wang X, Hu S, Liu H, Liu W, Liu X, Liu X. 2013. The PA and HA gene-mediated high viral load and intense innate immune response in the brain contribute to the high pathogenicity of H5N1 avian influenza virus in mallard ducks. J Virol 87:11063–11075. doi: 10.1128/JVI.00760-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun Y, Tan Y, Wei K, Sun H, Shi Y, Pu J, Yang H, Gao GF, Yin Y, Feng W, Perez DR, Liu J. 2013. Amino acid 316 of hemagglutinin and the neuraminidase stalk length influence virulence of H9N2 influenza virus in chickens and mice. J Virol 87:2963–2968. doi: 10.1128/JVI.02688-12. [DOI] [PMC free article] [PubMed] [Google Scholar]