

ABSTRACT
Porcine epidemic diarrhea virus (PEDV) is a worldwide-distributed alphacoronavirus, but the pathogenesis of PEDV infection is not fully characterized. During virus infection, type I interferon (IFN) is a key mediator of innate antiviral responses. Most coronaviruses develop some strategy for at least partially circumventing the IFN response by limiting the production of IFN and by delaying the activation of the IFN response. However, the molecular mechanisms by which PEDV antagonizes the antiviral effects of interferon have not been fully characterized. Especially, how PEDV impacts IFN signaling components has yet to be elucidated. In this study, we observed that PEDV was relatively resistant to treatment with type I IFN. Western blot analysis showed that STAT1 expression was markedly reduced in PEDV-infected cells and that this reduction was not due to inhibition of STAT1 transcription. STAT1 downregulation was blocked by a proteasome inhibitor but not by an autophagy inhibitor, strongly implicating the ubiquitin-proteasome targeting degradation system. Since PEDV infection-induced STAT1 degradation was evident in cells pretreated with the general tyrosine kinase inhibitor, we conclude that STAT1 degradation is independent of the IFN signaling pathway. Furthermore, we report that PEDV-induced STAT1 degradation inhibits IFN-α signal transduction pathways. Pharmacological inhibition of STAT1 degradation rescued the ability of the host to suppress virus replication. Collectively, these data show that PEDV is capable of subverting the type I interferon response by inducing STAT1 degradation.
IMPORTANCE In this study, we show that PEDV is resistant to the antiviral effect of IFN. The molecular mechanism is the degradation of STAT1 by PEDV infection in a proteasome-dependent manner. This PEDV infection-induced STAT1 degradation contributes to PEDV replication. Our findings reveal a new mechanism evolved by PEDV to circumvent the host antiviral response.
INTRODUCTION
Porcine epidemic diarrhea virus (PEDV) is an enveloped, positive-stranded RNA virus in the genus Alphacoronavirus, family Coronaviridae, order Nidovirales (1, 2). PEDV is the causative agent of porcine epidemic diarrhea (PED), an acute, highly contagious, and devastating viral enteric disease with a high mortality rate in suckling piglets. Since PED was first reported in England in 1971 (3), the disease has broken out frequently in many pig-producing countries (4–9). Despite the availability of vaccines, outbreaks continue to increase and pose problems for the swine industry, as well as public health concerns (10–12).
During viral infection, the innate immune response is often activated, leading to the induction of type I interferon (IFN-I), or alpha/beta interferon (IFN-α/β). IFN-α/β is a potent cytokine of critical importance in controlling viral infections and priming adaptive immune responses (13). The biological activities of IFN-I are initiated by the binding of IFN-α/β to its cognate receptors on the cell surface (14, 15). The binding of IFN-I to its receptors activates JAK1 and Tyk2, which phosphorylate and activate the signal transducer and activator of transcription (STAT) proteins, STAT1 and STAT2. Upon phosphorylation, STAT1 and STAT2 form heterodimers and then associate with IRF-9 to form a transcription factor complex, termed IFN-stimulated gene factor 3 (ISGF-3). The heterotrimer complexes translocate into the nucleus and bind to the IFN-stimulated response elements to induce the expression of IFN-stimulated genes, which establish an antiviral state (16–20).
To counter innate immune signaling, many viruses, including coronaviruses, have evolved different strategies to prevent the activation of antiviral effectors in host cells, particularly by minimizing IFN production and inhibiting IFN signaling (21, 22). Several viral proteins acting as IFN-I antagonists have been identified in members of the family Coronaviridae, including severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus, and mouse hepatitis virus (MHV) (23–28). Additionally, coronaviruses, such as MHV, feline coronavirus, and infectious bronchitis virus (IBV), have been shown to be relatively resistant to IFN treatment (29–31). As a member of the Coronaviridae, PEDV also encodes some proteins that serve as IFN-I antagonists (32, 33). However, how PEDV responds to interferon is unknown. Our present work describes a mechanism of virus-mediated IFN-I signaling inhibition. We show that PEDV suppresses the IFN-I signaling pathway by inducing STAT1 degradation in a proteasome-dependent manner. STAT1 plays an indispensable role in innate antiviral immunity to PEDV infection. In turn, PEDV subverts the JAK-STAT kinase by inducing STAT1 degradation.
MATERIALS AND METHODS
Cells and viruses.
Vero E6 cells (an African green monkey kidney cell line; ATCC) and IPEC-J2 cells (a porcine small intestinal epithelial cell line, donated by Yanming Zhang of Northwest A&F University, China) (34) were cultured in Dulbecco's minimum essential medium (DMEM; Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher) at 37°C under 5% CO2. The PEDV strain CV777 (GenBank accession number KT323979) was preserved in Harbin Veterinary Research Institute, Harbin, China. This virus was maintained and titrated in Vero E6 cells as described previously (1) and was stored at −80°C. Newcastle disease virus expressing green fluorescent protein (NDV-GFP) (35) was grown in 9-day-old specific-pathogen-free embryonated chicken eggs and was stored at −80°C (36).
Virus infection, drug treatments, and transfection.
Monolayers of Vero E6 and IPEC-J2 cells were infected with PEDV strain CV777 at a multiplicity of infection (MOI) of 0.1 for 1 h at 37°C. Unbound virus was removed, and cells were maintained in complete medium for various times until samples had been harvested. Some cell samples were treated with IFN-α (PBL Assay Science) or IFN-γ (R&D Systems) at the working concentration as indicated. In additional experiments, Vero E6 and IPEC-J2 cells were treated with the proteasome inhibitor MG132 (5 μM; Sigma), bortezomib (80 nM), or lactacystin (20 μM; Sigma), the autophagy inhibitor 3-methyladenine (3-MA) (5 mM; Sigma), the tyrosine kinase inhibitor genistein (100 μM; Sigma), or the carrier control dimethyl sulfoxide (DMSO) for 1 h before they were inoculated with a mock infection control or PEDV. Cells were further cultured in the presence of MG132, bortezomib, lactacystin, genistein, 3-MA, or DMSO for the indicated times. For the overexpression of STAT1, the pcDNA3.1/STAT1 expression vector (2 μg/well) was transfected into Vero E6 cells. Plasmid DNA was transfected with Lipofectamine 2000 (Invitrogen, USA) as recommended by the manufacturer. At 24 h posttransfection, cells were infected with PEDV at an MOI of 0.1 for the indicated times.
IFA.
An immunofluorescence assay (IFA) was performed as described previously (37). Briefly, at 24 h or 48 h postinoculation, Vero E6 cells were fixed and were stained with a mouse monoclonal antibody (MAb) against the PEDV spike protein (3F12; Median Diagnostics, South Korea) for 1 h. After three washes with phosphate-buffered saline (PBS), the cells were stained with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG for another 1 h. After washing, fluorescence was visualized with an Olympus inverted fluorescence microscope equipped with a camera.
Western blot analysis.
Western blot analysis was performed as described previously with a slight modification (38). Typically, samples were separated by SDS-PAGE under reducing conditions and were transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking, the membranes were incubated first with a primary antibody and then with an appropriate IRDye-conjugated secondary antibody (Li-Cor Biosciences, Lincoln, NE). The membranes were scanned using an Odyssey instrument (Li-Cor Biosciences) according to the manufacturer's instructions. Mouse MAb 2G3 against the PEDV nucleocapsid (N) protein was stocked in our laboratory. The STAT1 rabbit polyclonal antibody, the STAT2 rabbit polyclonal antibody, and a phospho-STAT1 (Tyr701) (D4A7) rabbit MAb were purchased from Cell Signaling Technology, USA. A mouse MAb against β-actin and a rabbit polyclonal antibody against ubiquitin were purchased from Santa Cruz Biotechnology.
Quantitative RT-PCR.
Quantitative reverse transcription-PCR (RT-PCR) analyses were carried out as described previously with a slight modification (39). At 24 h or 48 h post-PEDV infection, total RNA was extracted from cells and was subjected to quantitative RT-PCR using the specific primers listed in Table 1. Relative quantification was performed by the ΔΔCT method (40). Briefly, cycle threshold (CT) values were normalized to that for β-actin mRNA (the internal standard). The normalized values were designated ΔCT, which was calculated, e.g., as CT(STAT1) − CT(β-actin). Fold changes were determined by 2−ΔΔCT, where ΔΔCT is ΔCT(PEDV) − ΔCT(mock infection control).
TABLE 1.
Primers for PCR used in this study
| Primer name | Cells | Primer sequence (5′ → 3′) |
|---|---|---|
| STAT1-F | Vero E6 | TCCGTTTTCATGACCTCCTGT |
| STAT1-R | Vero E6 | CTGAATATTCCCCGACTGAGC |
| STAT1-F | IPEC-J2 | TCCGTTTTCATGACCTTCTGT |
| STAT1-R | IPEC-J2 | CTGAATATTCCCTGACTGAGT |
| β-actin-F | Vero E6 | AGGCTCTCTTCCAACCTTCCTT |
| β-actin-R | Vero E6 | CGTACAGGTCTTTACGGATGTCCA |
| β-actin-F | IPEC-J2 | CAAGCAACCACAGCCACAA |
| β-actin-R | IPEC-J2 | AGGATGGAGCCGCCGATC |
TCID50 assay.
The virus samples collected were clarified by centrifugation at 8,000 × g for 10 min prior to titration. Fifty percent tissue culture infective dose (TCID50) assays were performed on Vero E6 cells according to the method of Reed and Muench, as described previously (41). Briefly, cell monolayers (104 cells per well) in 96-well tissue culture plates (Corning, USA) were inoculated with 100 μl 10-fold serial dilutions of each virus stock and were incubated for 4 days prior to observation of the presence of cytopathic effect.
Immunoprecipitation assay.
Vero E6 cells were infected with PEDV as described above. The infected cells were harvested at 24 h postinfection, washed three times with cold PBS (pH 7.4), and lysed with Pierce IP lysis buffer (Thermo Scientific, Rockford, IL). Clarified extracts were first precleared with protein A/G beads (SC-2003; Santa Cruz) and then incubated with protein A/G beads plus a rabbit polyclonal antibody against STAT1 (Cell Signaling Technology, USA) for 4 h. The beads were first washed with lysis buffer and then boiled in sample buffer. The immunoprecipitated proteins were subjected to reducing SDS-PAGE and were blotted with a rabbit polyclonal antibody against ubiquitin (Santa Cruz, CA).
Statistical analysis.
Variables are expressed as means ± standard deviations (SD). Statistical analyses were performed using Student's t test. A P value of <0.05 was considered significant.
RESULTS
PEDV infection is relatively resistant to IFN-α.
Our previous work demonstrated that PEDV infection fails to activate IFN-I induction in Vero E6 cells (33). In the present study, we tested directly whether IFN-I could inhibit established PEDV infection. Vero E6 cells were infected either with PEDV for 24 h, to establish replication, or with an IFN-sensitive virus, NDV-GFP, as a control (42). The cells were then cultured further in the presence of IFN-α at 1,000 IU/ml or 10,000 IU/ml for 24 h, and the effect of IFN-α on the virus infection was analyzed by an IFA. We observed that a high concentration of IFN-α slightly decreased the level of PEDV infection, whereas NDV-GFP exhibited more sensitivity to IFN-α treatment (Fig. 1A). To investigate the degree of PEDV resistance to IFN-α, we treated Vero E6 cells with increasing concentrations of IFN-α and determined the effect on PEDV infectivity by virus titration. As shown in Fig. 1B, the titer of PEDV decreased only at a high concentration of IFN-α (>1,000 U/ml) (Fig. 1B), a pattern similar to that for other coronaviruses (27, 31, 43, 44). These data suggest that established PEDV infection is resistant to IFN-α treatment.
FIG 1.
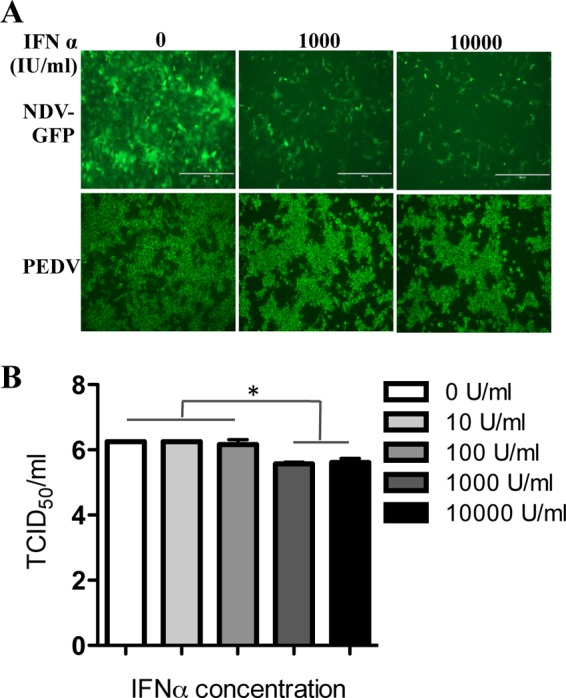
PEDV infection is resistant to IFN-α. (A) Vero E6 cells were infected either with NDV-GFP at an MOI of 0.01, as a control, or with PEDV at an MOI of 0.1 for 24 h. Subsequently, 1,000 IU/ml or 10,000 IU/ml of IFN-α was added to the medium. At 24 h post-IFN-α treatment, PEDV-infected cells were stained with a MAb to PEDV spike protein as described in Materials and Methods. NDV-GFP-infected cells were observed directly under a fluorescence microscope. (B) PEDV-infected Vero E6 cells were grown for 24 h in the presence of various concentrations of IFN-α as indicated. Supernatant samples were collected, and titers were determined. Data represent the means of duplicate measurements (±SD) of virus titers from three independent experiments. *, P < 0.05. The P value was calculated using Student's t test.
PEDV replication induces STAT1 degradation.
The basic signaling pathway activated in response to IFN-I has been reviewed comprehensively (45). The key component in this signaling pathway is JAK-STAT (46). To examine the effect of PEDV infection on the JAK-STAT system, Vero E6 and IPEC-J2 cells were infected with PEDV. At 24 h and 48 h postinfection, endogenous STAT1 and STAT2 proteins were detected by Western blotting. As shown in Fig. 2A, STAT1 levels in PEDV-infected Vero E6 cells were significantly lower than those in uninfected cells, a reduction that seems to depend on viral replication, while STAT2 levels remained constant throughout the experiment. Similar results were obtained using IPEC-J2 cells (Fig. 2B). These data indicate that PEDV infection induced STAT1 downregulation but not STAT2 downregulation.
FIG 2.

STAT1, but not STAT2, is downregulated in PEDV-infected cells. (A and B) PEDV infection reduces the expression levels of STAT1, but not STAT2, in Vero E6 and IPEC-J2 cells. Cells were infected with PEDV at an MOI of 0.1 for 24 h or 48 h. Detergent lysates collected from Vero E6 (A) or IPEC-J2 (B) cells were directly subjected to reducing SDS-PAGE and immunoblotting with antibodies to STAT1, STAT2, or β-actin (loading control). Densitometric data for STAT1/actin, STAT2/actin, and STAT1/N ratios from three independent experiments are expressed as means ± SD. (C) An IFA and a Western blot assay verified that UV-inactivated PEDV was replication defective. PEDV S protein was stained with MAb 3F12, followed by FITC-conjugated goat anti-mouse IgG. Vero E6 cells were inoculated with a mock infection control, PEDV, or UV-inactivated PEDV at an MOI of 1 for 24 h. Cells were lysed and were analyzed by Western blotting with a MAb against PEDV N protein. (D) The levels of STAT1 protein were not reduced by UV-inactivated PEDV in cells. Vero E6 and IPEC-J2 cells were inoculated with UV-inactivated PEDV at an MOI of 0.1 for 24 h or 48 h. Western blotting was used to detect the levels of STAT1 protein. Densitometric data for STAT1/actin ratios from three independent experiments are expressed as means ± SD. *, P < 0.05. The P value was calculated using Student's t test.
The downregulation of STAT1 by PEDV infection could be caused by incoming virions or by viral replication products. To test whether viral replication is needed for the interference effect, we inactivated PEDV virions by UV illumination and verified the inactivation by IFA and Western blotting after inoculation of Vero E6 cells (Fig. 2C). When the UV-inactivated virus was used to inoculate Vero E6 cells, STAT1 expression levels were similar to those in mock-inoculated cells (Fig. 2D). Similar results were observed in IPEC-J2 cells. These results indicate that active PEDV replication was needed for the downregulation of STAT1 in Vero E6 and IPEC-J2 cells.
To further examine the effect of PEDV on STAT1 and to address the possibility that PEDV infection downregulates STAT1 by inhibiting STAT1 transcription, relative levels of STAT1 mRNA were assessed by quantitative RT-PCR. IFN-α, which is known to stimulate STAT1 transcription (47, 48), was used as a positive control. We observed that IFN-α appropriately induced increases in STAT1 mRNA levels in Vero E6 and IPEC-J2 cells (Fig. 3A and B). The levels of STAT1 mRNA were also increased in PEDV-inoculated Vero E6 cells at 24 h and 48 h postinoculation (Fig. 3A). Similar results were obtained with IPEC-J2 cells (Fig. 3B). These data indicate that PEDV replication leads to the degradation of STAT1. However, we cannot formally rule out the possibility that posttranscriptional regulation of STAT1 gene expression may be involved in STAT1 downregulation, and this possibility should be further investigated.
FIG 3.
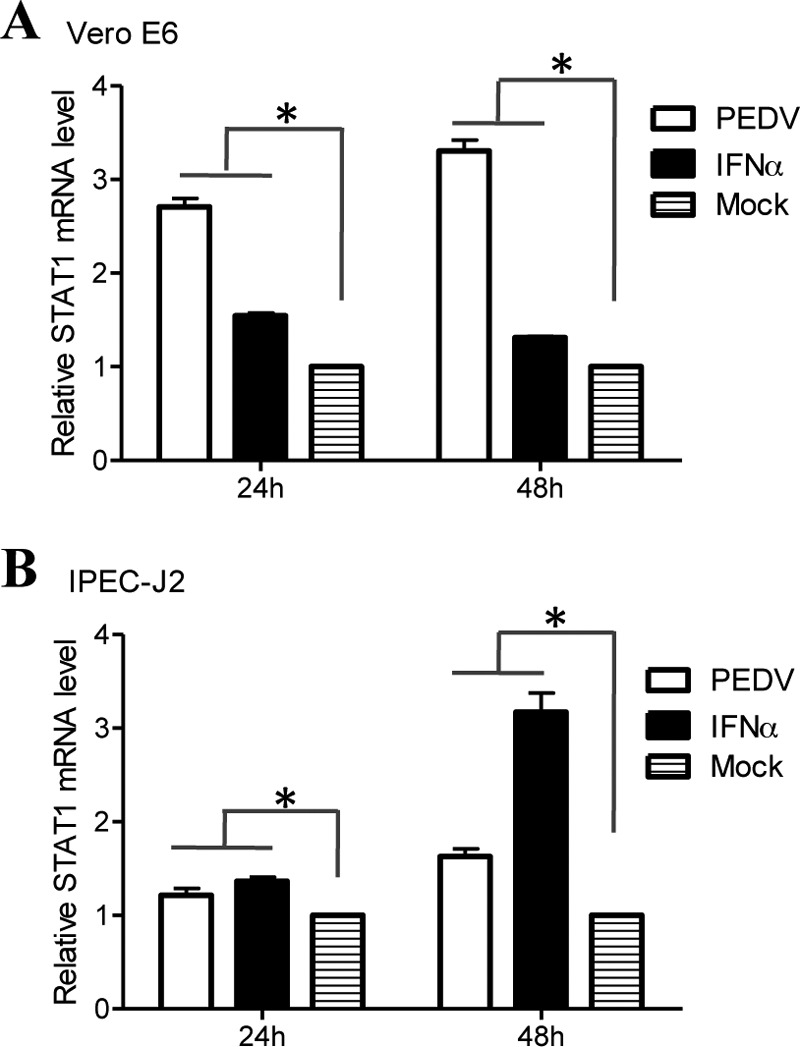
PEDV infection activates the transcription of STAT1. Vero E6 and IPEC-J2 cells were infected with PEDV at an MOI of 0.1 for 24 h or 48 h. Cells were treated with IFN-α at 10 IU/ml as a positive control. Total RNA was extracted from cells, and STAT1 mRNA levels were assessed by quantitative RT-PCR using the primers listed in Table 1. Three independent experiments were performed in triplicate, and values are means ± SD for all three experiments. *, P < 0.05. The P value was calculated using Student's t test.
PEDV promotes STAT1 degradation via the ubiquitin-proteasome system.
In eukaryotic cells, there are two major intracellular protein degradation pathways: the ubiquitin-proteasome system and autophagy (49). The proteasomal degradation pathway has high selectivity, and the proteasome generally recognizes ubiquitinated substrates (50). In contrast, autophagy is a highly conserved process for degrading redundant cellular components by encircling them with a membrane, followed by fusion of the vesicle with lysosomes (51). Therefore, to determine the mechanism by which PEDV induces STAT1 degradation, the expression levels of STAT1 protein were examined in cells treated with a protease inhibitor, MG132 (52–54). As shown in Fig. 4A, treatment with MG132 blocked STAT1 degradation in PEDV-infected Vero E6 and IPEC-J2 cells, suggesting proteasome-mediated degradation of STAT1 by PEDV. Since ubiquitinated proteins are targeted for proteasomal degradation (55), we examined protein ubiquitination in PEDV-infected cells. Lysates from Vero E6 cells infected with PEDV were immunoprecipitated for STAT1 and immunoblotted for ubiquitin. Actin, an endogenous cell protein, served as an input control. We observed that PEDV infection enhanced the levels of ubiquitinated STAT1 (Fig. 4B). Additionally, we analyzed whether autophagy was involved in STAT1 degradation. To accomplish this, we exposed the cells to 3-MA, which is commonly used to inhibit autophagy (56, 57). We observed STAT1 degradation in virus-infected Vero E6 and IPEC-J2 cells, and 3-MA treatment did not inhibit STAT1 degradation (Fig. 4C). These data indicate that PEDV induces STAT1 degradation through the ubiquitin-proteasome system.
FIG 4.

PEDV-induced STAT1 downregulation occurs through proteasome-mediated degradation but not through an autophagy mechanism. (A) Cells were first treated with the proteasome inhibitor MG132 or the carrier control DMSO for 1 h and were then either infected with PEDV at an MOI of 0.1 or left uninfected. Cells were further cultured in the absence or presence of MG132 for various times as indicated. Detergent lysates were collected from cells and were subjected to reducing SDS-PAGE and immunoblotting with a STAT1 antibody. Densitometric data for STAT1/actin ratios from three independent experiments are expressed as means ± SD. (B) PEDV infection induced the ubiquitination (Ub) of STAT1. Vero E6 cells were infected with PEDV at an MOI of 0.1 for 24 h. Detergent lysates were first immunoprecipitated with a STAT1 antibody as described in Materials and Methods and then subjected to reducing SDS-PAGE and immunoblotting with a rabbit polyclonal anti-Ub antibody. Detergent lysates blotted with the actin antibody were used as an input protein control. Densitometric data for Ub STAT1/actin ratios from three independent experiments are expressed as means ± SD. (C) Vero E6 and IPEC-J2 cells were treated either with 3-MA (5 mM) or with the carrier control DMSO for 4 h prior to PEDV infection. At 24 h or 48 h postinfection, cell lysates were subjected to blotting with a STAT1 antibody. Densitometric data for STAT1/actin ratios from three independent experiments are expressed as means ± SD. *, P < 0.05. The P value was calculated using Student's t test.
Established PEDV infection interrupts STAT1 phosphorylation.
Phosphorylation of STAT1 on tyrosine 701 is required for full activation of STAT1 (58, 59). To determine the effect of PEDV infection on STAT1 activation, phosphorylated STAT1 (p-STAT1) was initially examined in PEDV-infected cells at different time points. Western blot analysis showed that phosphorylated STAT1 levels were increased at early time points (1 to 24 h postinfection) and decreased after 24 h postinfection (Fig. 5A). These results suggested that the IFN response is activated at an early stage of virus infection and is then decreased at late stages of infection. We thus examined whether established PEDV infection can inhibit STAT1 phosphorylation to block the IFN signaling pathway. Exogenous administration of recombinant IFN-α was used as the positive control, since the ability of IFN-α to induce STAT1 phosphorylation has been well documented previously (60). Vero E6 and IPEC-J2 cells were infected with PEDV for 36 h and were then treated with IFN-α for 30 min. As shown in Fig. 5B, IFN-α-driven p-STAT1 was barely detectable in PEDV-infected cells, in contrast to a strong p-STAT1 band in uninfected IFN-α-treated cells, indicating that PEDV-infected cells failed to activate STAT1 in IFN-α-treated cells. The results indicate that established PEDV infection interrupts the IFN-I-mediated JAK/STAT1 signaling pathway.
FIG 5.

Established PEDV infection inhibits STAT1 phosphorylation. (A) Vero E6 and IPEC-J2 cells were infected with PEDV at an MOI of 0.1 for 12, 24, 30, 36, 48, or 55 h. Cell lysates were collected and were subjected to blotting with a phospho-STAT1 (Tyr701) antibody. Densitometric data for p-STAT1/actin ratios from three independent experiments are expressed as means ± SD. (B) Vero E6 and IPEC-J2 cells were first infected with PEDV at an MOI of 0.1 for 36 h and then treated with IFN-α (10 U/ml). At 30 min post-IFN-α treatment, cells were lysed and were subjected to Western blotting with the antibody against phospho-STAT1 (Tyr701). Densitometric data for p-STAT1/actin ratios from three independent experiments are expressed as means ± SD. *, P < 0.05. The P value was calculated using Student's t test.
Both phosphorylated and nonphosphorylated forms of STAT1 are degraded by PEDV.
Previous research demonstrated that the level of phosphorylated STAT1 is usually turned down by ubiquitin-proteasome system-mediated degradation (61). Given that phosphorylated STAT1 expression is reduced by PEDV infection at late stages, we determined whether only phosphorylated forms of STAT1 were targeted for degradation. IFN-α was used as a positive control (Fig. 6A and B). For this purpose, cells were pretreated with genistein, a Src family-selective tyrosine kinase inhibitor (62), to inhibit STAT1 phosphorylation in response to IFN-α. It was clear from the results that both phosphorylated and nonphosphorylated forms of STAT1 were degraded in PEDV-infected Vero E6 (Fig. 6A) and IPEC-J2 (Fig. 6B) cells.
FIG 6.

Both phosphorylated and nonphosphorylated forms of STAT1 are degraded by PEDV. Vero E6 and IPEC-J2 cells were pretreated with the kinase inhibitor genistein (100 μM) or the carrier control DMSO for 1 h and were then incubated either with PEDV at an MOI of 0.1 or with a mock infection control. Cells were cultured in the presence or absence of genistein for a further 36 h. Detergent lysates collected from Vero E6 (A) and IPEC-J2 (B) cells were directly subjected to reducing SDS-PAGE and immunoblotting with a STAT1 antibody or a phospho-STAT1 (Tyr701) antibody. Densitometric data for p-STAT1/actin or STAT1/actin ratios from three independent experiments are expressed as means ± SD. *, P < 0.05. The P value was calculated using Student's t test.
PEDV inhibits IFN-α signaling by reducing the levels of phosphorylated STAT1.
To determine whether IFN signaling transduction was inhibited in PEDV-infected cells, we performed a Western blot analysis of STAT1 phosphorylation in Vero E6 cells (Fig. 7A). As expected, treatment with IFN-α induced STAT1 phosphorylation in Vero E6 cells. p-STAT1 levels in response to IFN-α were significantly lower in PEDV-infected cells than in uninfected cells. In contrast, p-STAT1 levels in response to IFN-γ were slightly reduced in Vero E6 cells at early time points; these signals might be transmitted through a distinct but overlapping pathway (63). In order to ensure that these observations were not limited to the specific cell type used, we repeated these experiments with IPEC-J2 cells. We found that p-STAT1 levels were reduced in PEDV-infected cells in response to IFN-α as well as in response to IFN-γ (Fig. 7B). These data indicate that the STAT1-related components of the IFN signal transduction pathway are targets for PEDV inhibition.
FIG 7.

PEDV inhibits the type I interferon pathway. Vero E6 and IPEC-J2 cells were either mock infected or infected with PEDV at an MOI of 0.1 for 36 h; they were then treated with IFN-α (10 U/ml) or IFN-γ (10 U/ml) for different times as indicated. Vero E6 (A) and IPEC-J2 (B) cells were collected and lysed and were then subjected to Western blot analysis with an anti-STAT1 antibody or an anti-phospho-STAT1 (Tyr701) antibody. Densitometric data for p-STAT1/actin ratios from three independent experiments are expressed as means ± SD. *, P < 0.05. The P value was calculated using Student's t test.
STAT1 plays a critical role in the control of PEDV replication.
Given that STAT1 degradation is induced by PEDV infection, and since the degradation of STAT1 has been shown to contribute to the blockage of IFN signaling (54, 60), we investigated whether STAT1 has an antiviral effect on PEDV replication. To accomplish this, we treated Vero E6 cells with the protease inhibitor MG132, which can significantly inhibit STAT1 degradation in PEDV-infected cells (Fig. 4A). Using an IFA, we observed that the percentage of PEDV-infected Vero E6 cells was lower for MG132-treated cells than for cells treated with the carrier DMSO (Fig. 8A). The reduction in the titers of progeny virus in MG132-treated cells was confirmed by measuring the TCID50 (Fig. 8B). Additionally, Vero E6 cells were treated with two other proteasome inhibitors, bortezomib and lactacystin (64), followed by infection with PEDV. As shown in Fig. 8C and D, the titers of progeny virus in PEDV-infected Vero E6 cells were significantly lower in cells treated with either inhibitor than in control-treated cells. To further confirm the role of STAT1 in PEDV replication, we overexpressed STAT1 in Vero E6 cells and then infected cells with PEDV. We observed that STAT1 overexpression significantly inhibited PEDV replication (Fig. 8E). These observations suggest that ubiquitin-proteasome-mediated STAT1 degradation promotes PEDV replication.
FIG 8.

STAT1 degradation promotes PEDV replication. (A) Immunofluorescent assay to detect PEDV infection. Vero E6 cells were treated with MG132 at 5 μM for 1 h before inoculation with PEDV CV777. Cells were further cultured in the absence or presence of MG132 as described above. At 24 h or 48 h after inoculation, the cell monolayers were fixed and were examined for PEDV infection by an IFA with a MAb (3F12) against PEDV spike protein. (B) Blocking STAT1 degradation with MG132 inhibits PEDV replication. Vero E6 cells were treated with MG132 as mentioned above. At 24 h or 48 h postinfection, the progeny viruses were recovered from culture fluids. The virus titer was determined by the TCID50 assay. Results represent means ± SD for three independent experiments. (C and D) Blocking STAT1 degradation with bortezomib (C) or lactacystin (D) inhibits PEDV replication. Vero E6 cells were treated with bortezomib at 80 nM or lactacystin at 20 μM during PEDV infection. The virus titer was determined by the TCID50 assay. (E) Overexpression of STAT1 decreases PEDV replication. Vero E6 cells were transfected with the pcDNA3.1/STAT1 or pcDNA3.1 vector prior to PEDV inoculation. At 24 h posttransfection, Vero E6 cells were infected with PEDV for 24 h or 48 h. Results represent means ± SD for three independent experiments. *, P < 0.05. The P value was calculated using Student's t test.
DISCUSSION
The first line of host defense against viruses is the innate immune system, in which the interferons are a group of secreted cytokines that exert antiviral effects. To establish productive infection, viruses must circumvent the powerful immune defense mechanisms, including those induced by interferons. PEDV, like other coronaviruses (24, 28, 33), appears to have evolved several mechanisms to circumvent the host innate immune response. These antagonistic strategies have developed on at least three levels: inhibition of RIG-I-mediated IFN production pathways (65), disruption of IFN induction cascades (12, 66), and inhibition of IRF-3, a protein that itself induces IFN gene transcription (33, 67). The experiments described in this paper demonstrate that PEDV infection inhibits the IFN response signaling pathway by inducing the degradation of STAT1. This is a unique example of a coronavirus that directly degrades an IFN response protein to enable its survival, highlighting the multifaceted control of host innate immunity by coronaviruses.
To circumvent the IFN response, different viruses have evolved a great diversity of molecular mechanisms. We and others have previously reported that PEDV infection does not stimulate the production of IFN-I in Vero E6 cells (33, 65, 67), which may be crucial to the pathogenesis of this virus. Another way in which viruses evade the IFN response is to block the actions of IFNs (47, 63). In this study, we found that PEDV is relatively resistant to treatment with IFN-I, suggesting that this virus has also developed another strategy to prevent the biological activities of IFNs. These activities are initiated by the binding of IFN-I to its cognate receptors, resulting in the activation of the JAK/STAT pathway (15). The data from our virus infection model indicate that STAT1 is an antiviral signal transduction molecule against PEDV infection and that PEDV replication blocks IFN signaling by degrading STAT1. Similarly, several members of the virus family Paramyxoviridae target STAT proteins as a means to evade IFN antiviral signaling (52, 68–70). Therefore, we concluded that PEDV specifically inhibits IFN-I signaling by degrading the expression of STAT1 protein. However, differential STAT targeting has also been observed with some rubulaviruses. For example, human parainfluenza virus 2 most frequently targets STAT2 (71). Moreover, it has been observed that mumps virus has acquired a unique ability to target both STAT1 and STAT3 for destruction (72).
Currently, there are two major distinct intracellular degradation pathways, i.e., autophagy and the ubiquitin-proteasome system (73). Thus, to determine whether both pathways are involved in the degradation of STAT1, we pharmacologically inhibited the formation of autophagy but did not block STAT1 degradation; proteasomal inhibition with MG132 prevented the degradation of STAT1. Previous research demonstrated that hepatitis C virus and simian virus 5 selectively degrade STAT1 in a proteasome-dependent manner (52, 54, 60). Although STAT1 degradation was confirmed in PEDV-infected cells, we do not know whether this coronavirus utilizes its proteins to induce STAT1 degradation directly. Further work is needed to answer this question conclusively. However, whatever the mechanism for STAT1 degradation by PEDV, it is independent of the IFN signaling pathway, since both phosphorylated and nonphosphorylated forms of STAT1 are degraded in PEDV-infected cells. Overall, our findings clearly demonstrate that PEDV infection-induced STAT1 degradation is mediated by the ubiquitin-proteasome system.
Previous reports showed that STAT1 is involved in the upregulation of genes due to signaling by type I or type II interferons (74). Therefore, to determine whether STAT1 degradation affects both types of IFN signaling pathways, we first infected cells with PEDV and then treated them either with IFN-α or with IFN-γ. We found that STAT1 degradation in PEDV-infected cells blocked both IFN-I signaling and IFN-II signaling. The observation that STAT1 degradation occurs in PEDV-infected cells suggests that this will result in the ability of the virus to partially overcome the IFN response. Thus, we inhibited STAT1 degradation with three proteasome inhibitors—MG132, bortezomib, and lactacystin—to confirm our hypothesis. We observed that blocking STAT1 degradation rescues the ability of the host to suppress PEDV replication. Additionally, STAT1 overexpression also rescued the antiviral ability of host cells. Similarly, the replication of several other coronaviruses, including MHV, SARS-CoV, IBV, and feline enteric coronavirus, is affected upon impairment of proteasomal activity (31, 75–78). Other viruses, such as hepatitis C virus and simian virus 5, have evolved similar mechanisms for the evasion of host defenses (52, 79). Our data suggest that different viruses may use similar mechanisms to circumvent IFN action and inhibit the JAK/STAT signaling pathway.
In conclusion, we have shown that STAT1 expression is reduced in PEDV-infected Vero E6 and IPEC-J2 cells. We have also provided evidence of PEDV-mediated ubiquitination and proteasomal degradation of STAT1, outlining a basic viral immune evasion mechanism used by PEDV to block the IFN receptor-mediated JAK/STAT pathway in virus-infected cells. This PEDV-mediated immune evasion strategy may highlight a potential target pathway for therapeutic restoration of the antiviral immune response.
ACKNOWLEDGMENTS
We thank Qian Yang (College of Veterinary Medicine, Nanjing Agricultural University) for technical support.
REFERENCES
- 1.Chen J, Wang C, Shi H, Qiu H, Liu S, Chen X, Zhang Z, Feng L. 2010. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch Virol 155:1471–1476. doi: 10.1007/s00705-010-0720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Chen J, Shi D, Shi H, Zhang X, Yuan J, Jiang S, Feng L. 2016. Immunogenicity and antigenic relationships among spike proteins of porcine epidemic diarrhea virus subtypes G1 and G2. Arch Virol 161:537–547. doi: 10.1007/s00705-015-2694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood EN. 1977. An apparently new syndrome of porcine epidemic diarrhoea. Vet Rec 100:243–244. doi: 10.1136/vr.100.12.243. [DOI] [PubMed] [Google Scholar]
- 4.Song D, Park B. 2012. Porcine epidemic diarrhoea virus: a comprehensive review of molecular epidemiology, diagnosis, and vaccines. Virus Genes 44:167–175. doi: 10.1007/s11262-012-0713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Temeeyasen G, Srijangwad A, Tripipat T, Tipsombatboon P, Piriyapongsa J, Phoolcharoen W, Chuanasa T, Tantituvanont A, Nilubol D. 2014. Genetic diversity of ORF3 and spike genes of porcine epidemic diarrhea virus in Thailand. Infect Genet Evol 21:205–213. doi: 10.1016/j.meegid.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Masuda T, Murakami S, Takahashi O, Miyazaki A, Ohashi S, Yamasato H, Suzuki T. 2015. New porcine epidemic diarrhoea virus variant with a large deletion in the spike gene identified in domestic pigs. Arch Virol 160:2565–2568. doi: 10.1007/s00705-015-2522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sung MH, Deng MC, Chung YH, Huang YL, Chang CY, Lan YC, Chou HL, Chao DY. 2015. Evolutionary characterization of the emerging porcine epidemic diarrhea virus worldwide and 2014 epidemic in Taiwan. Infect Genet Evol 36:108–115. doi: 10.1016/j.meegid.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang YW, Dickerman AW, Pineyro P, Li L, Fang L, Kiehne R, Opriessnig T, Meng XJ. 2013. Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States. mBio 4(5):e00737-13. doi: 10.1128/mBio.00737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mole B. 2013. Deadly pig virus slips through US borders. Nature 499:388. doi: 10.1038/499388a. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q. 2012. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353. doi: 10.3201/eid1808.120002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun RQ, Cai RJ, Chen YQ, Liang PS, Chen DK, Song CX. 2012. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis 18:161–163. doi: 10.3201/eid1801.111259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding Z, Fang L, Jing H, Zeng S, Wang D, Liu L, Zhang H, Luo R, Chen H, Xiao S. 2014. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol 88:8936–8945. doi: 10.1128/JVI.00700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Bon A, Tough DF. 2002. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol 14:432–436. doi: 10.1016/S0952-7915(02)00354-0. [DOI] [PubMed] [Google Scholar]
- 14.Darnell JE Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 15.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu Rev Biochem 67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 16.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horvath CM, Stark GR, Kerr IM, Darnell JE Jr. 1996. Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol Cell Biol 16:6957–6964. doi: 10.1128/MCB.16.12.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 21.Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450. doi: 10.1038/nrmicro2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fehr AR, Perlman S. 2015. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Totura AL, Baric RS. 2012. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Ye F, Zhu N, Wang W, Deng Y, Zhao Z, Tan W. 2015. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci Rep 5:17554. doi: 10.1038/srep17554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Zhang L, Geng H, Deng Y, Huang B, Guo Y, Zhao Z, Tan W. 2013. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niemeyer D, Zillinger T, Muth D, Zielecki F, Horvath G, Suliman T, Barchet W, Weber F, Drosten C, Muller MA. 2013. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J Virol 87:12489–12495. doi: 10.1128/JVI.01845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koetzner CA, Kuo L, Goebel SJ, Dean AB, Parker MM, Masters PS. 2010. Accessory protein 5a is a major antagonist of the antiviral action of interferon against murine coronavirus. J Virol 84:8262–8274. doi: 10.1128/JVI.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Y, Hauns K, Langland JO, Jacobs BL, Hogue BG. 2007. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J Virol 81:2554–2563. doi: 10.1128/JVI.01634-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou T, Wang H, Luo D, Rowe T, Wang Z, Hogan RJ, Qiu S, Bunzel RJ, Huang G, Mishra V, Voss TG, Kimberly R, Luo M. 2004. An exposed domain in the severe acute respiratory syndrome coronavirus spike protein induces neutralizing antibodies. J Virol 78:7217–7226. doi: 10.1128/JVI.78.13.7217-7226.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth-Cross JK, Martinez-Sobrido L, Scott EP, Garcia-Sastre A, Weiss SR. 2007. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J Virol 81:7189–7199. doi: 10.1128/JVI.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kint J, Dickhout A, Kutter J, Maier HJ, Britton P, Koumans J, Pijlman GP, Fros JJ, Wiegertjes GF, Forlenza M. 2015. Infectious bronchitis coronavirus inhibits STAT1 signaling and requires accessory proteins for resistance to type I interferon activity. J Virol 89:12047–12057. doi: 10.1128/JVI.01057-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK, Li K, Mesecar AD, Baker SC. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 84:4619–4629. doi: 10.1128/JVI.02406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xing Y, Chen J, Tu J, Zhang B, Chen X, Shi H, Baker SC, Feng L, Chen Z. 2013. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J Gen Virol 94:1554–1567. doi: 10.1099/vir.0.051169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu F, Li G, Wen K, Bui T, Cao D, Zhang Y, Yuan L. 2010. Porcine small intestinal epithelial cell line (IPEC-J2) of rotavirus infection as a new model for the study of innate immune responses to rotaviruses and probiotics. Viral Immunol 23:135–149. doi: 10.1089/vim.2009.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Ge J, Xie X, Ding Y, Bu Z. 2008. Impact of modification of cleavage site of fusion protein and foreign gene insertion on the virulence of Newcastle disease virus LaSota vaccine strain. Wei Sheng Wu Xue Bao 48:362–368. (In Chinese.) [PubMed] [Google Scholar]
- 36.Ge J, Deng G, Wen Z, Tian G, Wang Y, Shi J, Wang X, Li Y, Hu S, Jiang Y, Yang C, Yu K, Bu Z, Chen H. 2007. Newcastle disease virus-based live attenuated vaccine completely protects chickens and mice from lethal challenge of homologous and heterologous H5N1 avian influenza viruses. J Virol 81:150–158. doi: 10.1128/JVI.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo L, Niu J, Yu H, Gu W, Li R, Luo X, Huang M, Tian Z, Feng L, Wang Y. 2014. Modulation of CD163 expression by metalloprotease ADAM17 regulates porcine reproductive and respiratory syndrome virus entry. J Virol 88:10448–10458. doi: 10.1128/JVI.01117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu W, Guo L, Yu H, Niu J, Huang M, Luo X, Li R, Tian Z, Feng L, Wang Y. 2015. Involvement of CD16 in antibody-dependent enhancement of porcine reproductive and respiratory syndrome virus infection. J Gen Virol 96:1712–1722. doi: 10.1099/vir.0.000118. [DOI] [PubMed] [Google Scholar]
- 39.Li R, Guo L, Gu W, Luo X, Zhang J, Xu Y, Tian Z, Feng L, Wang Y. 2016. Production of porcine TNFα by ADAM17-mediated cleavage negatively regulates porcine reproductive and respiratory syndrome virus infection. Immunol Res 64:711–720. doi: 10.1007/s12026-015-8772-8. [DOI] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Chua BH, Phuektes P, Sanders SA, Nicholls PK, McMinn PC. 2008. The molecular basis of mouse adaptation by human enterovirus 71. J Gen Virol 89:1622–1632. doi: 10.1099/vir.0.83676-0. [DOI] [PubMed] [Google Scholar]
- 42.Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol 77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuri T, Eriksson KK, Putics A, Zust R, Snijder EJ, Davidson AD, Siddell SG, Thiel V, Ziebuhr J, Weber F. 2011. The ADP-ribose-1″-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J Gen Virol 92:1899–1905. doi: 10.1099/vir.0.031856-0. [DOI] [PubMed] [Google Scholar]
- 44.Wathelet MG, Orr M, Frieman MB, Baric RS. 2007. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol 81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 46.Murray PJ. 2007. The JAK-STAT signaling pathway: input and output integration. J Immunol 178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 47.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- 48.Najjar I, Fagard R. 2010. STAT1 and pathogens, not a friendly relationship. Biochimie 92:425–444. doi: 10.1016/j.biochi.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizushima N, Komatsu M. 2011. Autophagy: renovation of cells and tissues. Cell 147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 50.Hershko A, Ciechanover A. 1986. The ubiquitin pathway for the degradation of intracellular proteins. Prog Nucleic Acid Res Mol Biol 33:19–56. doi: 10.1016/S0079-6603(08)60019-7. [DOI] [PubMed] [Google Scholar]
- 51.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Didcock L, Young DF, Goodbourn S, Randall RE. 1999. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J Virol 73:9928–9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Didcock L, Young DF, Goodbourn S, Randall RE. 1999. Sendai virus and simian virus 5 block activation of interferon-responsive genes: importance for virus pathogenesis. J Virol 73:3125–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, Chung RT. 2005. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology 128:1034–1041. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. 1994. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 78:773–785. doi: 10.1016/S0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 56.Seglen PO, Gordon PB. 1982. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A 79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. 2010. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shuai K, Stark GR, Kerr IM, Darnell JE Jr. 1993. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science 261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 59.Wen Z, Zhong Z, Darnell JE Jr. 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 60.Andrejeva J, Young DF, Goodbourn S, Randall RE. 2002. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J Virol 76:2159–2167. doi: 10.1128/jvi.76.5.2159-2167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim TK, Maniatis T. 1996. Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science 273:1717–1719. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- 62.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem 271:695–701. [DOI] [PubMed] [Google Scholar]
- 63.Goodbourn S, Didcock L, Randall RE. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol 81:2341–2364. doi: 10.1099/0022-1317-81-10-2341. [DOI] [PubMed] [Google Scholar]
- 64.Schneider M, Ackermann K, Stuart M, Wex C, Protzer U, Schatzl HM, Gilch S. 2012. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J Virol 86:10112–10122. doi: 10.1128/JVI.01001-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cao L, Ge X, Gao Y, Herrler G, Ren Y, Ren X, Li G. 2015. Porcine epidemic diarrhea virus inhibits dsRNA-induced interferon-β production in porcine intestinal epithelial cells by blockade of the RIG-I-mediated pathway. Virol J 12:127. doi: 10.1186/s12985-015-0345-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, Chen H, Li K, Xiao S. 2016. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol 90:2090–2101. doi: 10.1128/JVI.02514-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Q, Shi K, Yoo D. 2016. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology 489:252–268. doi: 10.1016/j.virol.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. 2003. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parisien JP, Lau JF, Horvath CM. 2002. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol 76:6435–6441. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. 2002. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol 76:4190–4198. doi: 10.1128/JVI.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parisien JP, Lau JF, Rodriguez JJ, Sullivan BM, Moscona A, Parks GD, Lamb RA, Horvath CM. 2001. The V protein of human parainfluenza virus 2 antagonizes type I interferon responses by destabilizing signal transducer and activator of transcription 2. Virology 283:230–239. doi: 10.1006/viro.2001.0856. [DOI] [PubMed] [Google Scholar]
- 72.Ulane CM, Rodriguez JJ, Parisien JP, Horvath CM. 2003. STAT3 ubiquitylation and degradation by mumps virus suppress cytokine and oncogene signaling. J Virol 77:6385–6393. doi: 10.1128/JVI.77.11.6385-6393.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kraft C, Peter M, Hofmann K. 2010. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 74.Katze MG, He Y, Gale M Jr. 2002. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 75.Cheng W, Chen S, Li R, Chen Y, Wang M, Guo D. 2015. Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin-dependent proteosomal degradation of N-Myc (and STAT) interactor. Virol Sin 30:153–161. doi: 10.1007/s12250-015-3581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim Y, Liu H, Galasiti Kankanamalage AC, Weerasekara S, Hua DH, Groutas WC, Chang KO, Pedersen NC. 2016. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog 12:e1005531. doi: 10.1371/journal.ppat.1005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raaben M, Posthuma CC, Verheije MH, te Lintelo EG, Kikkert M, Drijfhout JW, Snijder EJ, Rottier PJ, de Haan CA. 2010. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J Virol 84:7869–7879. doi: 10.1128/JVI.00485-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu GY, Lai MM. 2005. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J Virol 79:644–648. doi: 10.1128/JVI.79.1.644-648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foy E, Li K, Wang C, Sumpter R Jr, Ikeda M, Lemon SM, Gale M Jr. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]