

Abstract
Bacterial symbionts are important fitness determinants of insects. Some hosts have independently acquired taxonomically related microbes to meet similar challenges, but whether distantly related hosts that live in tight symbiosis can maintain similar microbial communities has not been investigated. Varying degrees of nest sharing between Megalomyrmex social parasites (Solenopsidini) and their fungus‐growing ant hosts (Attini) from the genera Cyphomyrmex, Trachymyrmex and Sericomyrmex allowed us to address this question, as both ant lineages rely on the same fungal diet, interact in varying intensities and are distantly related. We used tag‐encoded FLX 454 pyrosequencing and diagnostic PCR to map bacterial symbiont diversity across the Megalomyrmex phylogenetic tree, which also contains free‐living generalist predators. We show that social parasites and hosts share a subset of bacterial symbionts, primarily consisting of Entomoplasmatales, Bartonellaceae, Acinetobacter, Wolbachia and Pseudonocardia and that Entomoplasmatales and Bartonellaceae can co‐infect specifically associated combinations of hosts and social parasites with identical 16S rRNA genotypes. We reconstructed in more detail the population‐level infection dynamics for Entomoplasmatales and Bartonellaceae in Megalomyrmex symmetochus guest ants and their Sericomyrmex amabilis hosts. We further assessed the stability of the bacterial communities through a diet manipulation experiment and evaluated possible transmission modes in shared nests such as consumption of the same fungus garden food, eating of host brood by social parasites, trophallaxis and grooming interactions between the ants, or parallel acquisition from the same nest environment. Our results imply that cohabiting ant social parasites and hosts may obtain functional benefits from bacterial symbiont transfer even when they are not closely related.
Keywords: 16S rRNA pyrosequencing, Acinetobacter, Bartonellaceae, Entomoplasmatales, social parasites, symbiosis
Introduction
Since the origin of the eukaryotic cell, bacteria have evolved a variety of strategies to live with host cells (Cavalier‐Smith 2009) or to become associated with tissues after hosts became multicellular. A number of metazoan symbionts have been recognized as mutualists and some have induced bursts of host species diversification (Bordenstein 2003; Moran et al. 2005; Moran 2007). Recent developments in high‐throughput sequencing are facilitating the description of these interrelations and the analysis of their genomic complementarity. The extreme cases have much in common with the tight integration of organelles in the eukaryotic cell (McCutcheon & Moran 2012)—they are obligate primary symbionts that are indispensable for successful host development and reproduction and often live in specialized bacteriomes (Schroder et al. 1996; Takiya et al. 2006; McCutcheon & Moran 2012). However, most are facultative (secondary) symbionts and show wider ranges of adaptation and specificity.
Facultative symbionts are not required for host survival and reproduction and are therefore not necessarily present in every host, but can often modify host phenotypes with fitness effects that span the entire parasitism–mutualism continuum. Examples are Wolbachia, Spiroplasma, Hamiltonella, Regiella and Arsenophonus, which infect a wide variety of insects by manipulating host reproduction or improving host survival (Oliver et al. 2003, 2009; Hansen et al. 2007; Vorburger et al. 2010; Russell et al. 2012). These benefits range from tolerance to environmental stress, protection from natural enemies or facilitation of specific food plant usage (Montllor et al. 2002; Oliver et al. 2003; Tsuchida et al. 2004). Facultative symbionts can rapidly spread in naïve host populations (Jaenike et al. 2010; Himler et al. 2011) and are known to occasionally switch between host lineages on ecological timescales (Haselkorn et al. 2009), sometimes via mites or parasitoid wasps (Heath et al. 1999; Jaenike et al. 2007; Duron et al. 2010).
Not only single bacterial lineages, but also the cumulative gene expression potential of entire bacterial communities can exert major influences on host life histories. Intestinal tracts in particular tend to harbour rich microbial communities. They have been relatively well studied in mammals including humans (Ley et al. 2008; Muegge et al. 2011; Phillips et al. 2012; Delsuc et al. 2014) and are increasingly also inventoried in insects. Here, bacterial symbiont communities often appear to be shaped by specific diets and to vary across host orders (Colman et al. 2012), ant subfamilies (although members of the same subfamily can also be very distinct; Anderson et al. 2012), and between bee genera (Koch & Schmid‐Hempel 2011; Martinson et al. 2011; Engel et al. 2012). Several such insect‐associated bacteria have prevalences that strongly correlate with specific trophic niches, such as the Rhizobiales, which tend to be abundant across unrelated ant species that have direct or indirect (via aphids) herbivorous diets (Russell et al. 2009; Anderson et al. 2012). It has been proposed that these may have roles in fixing or retrieving nitrogen (Van Borm et al. 2002a; Russell et al. 2009), and a recent study proposed that Wolbachia might play similar nutritional roles in Acromyrmex leaf‐cutting ants (Andersen et al. 2012).
The evolution and maintenance of bacterial symbionts in insect hosts has been widely investigated, and cospeciation and horizontal transmission have often been inferred by cophylogenetic methods (Moran et al. 2005; Frost et al. 2010). However, little is known about the proximate mechanisms by which single symbionts switch between host lineages and the extent to which entire bacterial communities converge when hosts live in symbiosis. Recent studies have hypothesized that nest cohabitation by social parasites and their hosts can allow explicit tests of microbiota convergences (Dedeine et al. 2005; Haapaniemi & Pamilo 2015), but only strain‐specific approaches have been used so far (Van Borm et al. 2003; Dedeine et al. 2005; Haapaniemi & Pamilo 2015) and any such results are potentially confounded by hosts and social parasites often being closely related (Emery 1909). In the present study, we set out to disentangle the possible effects of phylogenetic and spatial proximity by investigating whether and to what extent the microbiotas of distantly related ant species may converge when they share the same local habitat and live in the same nest. To address this question, we used the nest‐ and food‐sharing symbioses of socially parasitic Megalomyrmex ants (Formicidae: Solenopsidini) and their fungus‐growing ant hosts (Formicidae: Attini). The genus Megalomyrmex comprises 44 described species (Boudinot et al. 2013), which include a number of free‐living predators and an evolutionarily derived socially parasitic clade that exploits host fungus gardens and brood in various ways across the attine ant phylogeny (Adams 2008; Adams et al. 2012).
Megalomyrmex parasites use alkaloids produced in their venom glands to subdue their hosts and gain access to their resources (Adams et al. 2013, 2015). They are either ‘thief ant’ raiders that nest next to the host colony and periodically steal fungus garden and brood (Adams & Longino 2007; Adams et al. 2015), ‘agro‐predators’ that usurp entire host nests and move to a new host nest once the brood and garden resources are depleted (Adams et al. 2000) or ‘guest ants’ that infiltrate the host nest and exploit it for the entire lifetime of the host colony (Adams et al. 2012, 2013). Megalomyrmex social parasites are very specific in being associated with only one or two host ant species, and in some cases, a single host species can be exploited by two different Megalomyrmex parasites (Boudinot et al. 2013; Adams et al. 2015). This implies that nesting associations between these distantly related ant lineages are sufficiently specific to expect at least some sharing of bacterial symbionts, and that any abundant bacterial lineage that consistently co‐occurs in social parasites and hosts is likely to be of symbiotic significance.
The objectives of our study were to (i) provide comparative phylotype, operational taxonomic unit (OTU) and genotype sequence maps of the bacterial symbionts of the best studied Megalomyrmex species in Central and South America and their attine host ants, (ii) determine whether the microbial community compositions of these ants reflect differences in diet or (non)parasitic lifestyles of the Megalomyrmex species, (iii) assess the identity of shared bacterial lineages and evaluate their prevalence and genotypic diversity, (iv) validate the robustness of shared microbiotas in species pairs sharing the same nest through an artificial diet experiment, (v) unravel the extent to which association specificity between the most prevalent bacterial lineages of Megalomyrmex symmetochus guest ants and their Sericomyrmex amabilis host ants varies across subpopulations and (vi) discuss the possible adaptive significance of shared taxa of prevalent symbionts in the light of what is known about these bacteria from previous work.
Materials and methods
Sampling and DNA extraction
A total of 146 ant colonies belonging to 14 species of the genus Megalomyrmex and seven fungus‐growing ant host species were sampled between 1999 and 2013 in Panama, Ecuador, Costa Rica and Perù (Table S1, Supporting information). Of these colonies, 60 belonged to two species in the genus Sericomyrmex (S. amabilis and S. cf. diego/zacapanus; this genus is currently in revision and formal names have not been confirmed yet; Jesovnik A, personal communication) and 21 of these (all belonging to S. amabilis) were parasitized by a colony of the guest ant M. symmetochus. These Sericomyrmex colony samples were collected between 2010 and 2012 across the Panama Canal Zone at four sampling sites (Plantation Road, Gamboa, Pipeline Road and Barro Colorado Nature Monument). Either ants were collected fresh from the field and stored in 96% EtOH at −20 °C until DNA extraction, or entire colonies were transported to Copenhagen and kept in closed nest boxes with moist plaster bottoms and a dry foraging arena in rearing rooms with a constant temperature of 25 °C and RH 60–70% for 6 months to 2 years, until collection of ants for DNA extraction.
Ants were rinsed in 70% EtOH for 1 min and washed twice in milliQ water. We used this procedure rather than more disruptive protocols, as we aimed to retrieve at least part of the external cuticular microbiota, which can contain important symbiotic bacteria in attine ants (Currie et al. 1999; Sen et al. 2009; Andersen et al. 2013), while trying to remove transient external contaminants. Fungus garden samples were gathered from freshly collected colonies (within the first week from collection, before colonies were fed in the laboratory) and stored at −80 °C without any further treatment until DNA extraction. As we were interested in obtaining colony‐level samples of bacterial lineages, we pooled 2–10 worker ants (Table S1, Supporting information) before extracting total DNA. From one nest (JOA120607‐01), we also obtained DNA from single eggs, larvae, pupae, workers of the guest ant M. symmetochus and its S. amabilis host and from guest ant gynes (virgin queens; not available from the host, as they hardly ever produce gynes when infected by guest ants; Adams RMM & Liberti J, personal observation).
We used three different extraction methods: (i) a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) for the ant samples that were used for 16S rRNA gene amplicon pyrosequencing, diagnostic PCR and Sanger sequencing, (ii) a Chelex 100 (Sigma‐Aldrich) protocol (Ugelvig et al. 2012) for ant samples that were used only in diagnostic PCR screenings followed by sequencing (except Megalomyrmex staudingeri RMMA040609‐05 and Megalomyrmex incisus RMMA060311‐15, which were used for pyrosequencing) and (iii) a CTAB method (Schiøtt et al. 2008) for fungus gardens. Whole ants and fungus gardens were either ground in lysis buffer (according to the extraction method used) with a ceramic bead in a FastPrep homogenizer (Thermo Scientific), or crushed manually with a sterile pestle.
16S rRNA tag‐encoded FLX 454 pyrosequencing and data analyses
A total of 33 samples (31 ant and two fungus garden samples) were chosen for pyrosequencing. We sequenced fragments of the bacterial 16S rRNA gene flanking the V3 and V4 regions with universal bacterial primers 341F and 806R (Hansen et al. 2012) in four one‐region 454 runs following the protocol used in Andersen et al. (2013). Raw sequence data have been deposited at the Sequence Read Archive under Accession no. SRP052752. The data generated from the sequencing runs were initially analysed using mothur v.1.33.2 (Schloss et al. 2009), applying a standardized operating procedure (SOP) developed by Schloss et al. (2011). The raw sequences were denoised with pyronoise within mothur, using a default of 450 flows. Sequences were trimmed to a minimum length of 200 bp with default parameters (allowing one mismatch in the barcodes, two mismatches in the primer sequences and a maximum of eight homopolymers), after which 645 696 sequences were retained. We then identified and subsequently aligned 75 433 unique sequences against the silva database. Sequences that did not cover positions 6428–23444 of the full‐length SILVA alignment (V3–V4 hypervariable regions of the 16S rRNA gene) were then removed, which retained 49 240 sequences that were trimmed to match this window. Following the remaining steps of the 454 SOP protocol, we ended up with 6199 unique sequences (after chimera checking), which were classified using the Greengenes reference set (May 2013 release; McDonald et al. 2012) with a threshold of 80% bootstrap confidence. We then removed any Archaea, Eukaryota, chloroplast, mitochondrial and unknown sequence, retaining 5912 unique bacterial sequences.
Operational taxonomic units were identified at 97% sequence similarity using the nearest neighbour option, and singletons (i.e. OTUs with only one read across the entire data set) were removed from the alignments. At this stage, misalignments in the sequence data matrix may induce inflation of OTU numbers (Hu et al. 2014), so we iterated a series of steps by: (i) picking representative sequences for all the OTUs, (ii) realigning them with the MUSCLE algorithm (Edgar 2004) within mega v.5.1 (Tamura et al. 2011) and (iii) reclustering OTUs at 97% identity in mothur, until repeating this procedure did not further reduce the total number of OTUs and no misalignments could be detected through manual inspection. The final data set consisted of 542 866 curated sequences, containing 4882 unique sequences, which clustered into 923 OTUs at 97% identity (Table S2, Supporting information).
We next produced two biom‐format tables, containing information on taxonomical assignment and abundance at the unique sequence and 97% OTU levels across all 31 ant samples, and continued the analysis with qiime v.1.7.0 (Caporaso et al. 2010a). The representative sequences of the 97% OTUs and the unique sequences were realigned with pynast (Caporaso et al. 2010b) to the Greengenes core reference alignment (DeSantis et al. 2006), and two phylogenetic trees were constructed with fasttree (Price et al. 2010) to be used in phylogenetically informed diversity analyses. We then computed rarefaction curves using 97% OTUs in R (package vegan, R Core Team 2013) to assess whether sequencing depth was adequate to produce reasonable estimates of the total bacterial diversity in the samples. megan4 (Huson et al. 2011) was used to lump OTUs with identical classification into phylotypes. Finally, we investigated clustering patterns of ant microbiotas using network‐ and phylogeny‐based approaches (UniFrac; Lozupone & Knight 2005), testing for significant differences between: (i) all Megalomyrmex vs. all attine host ants, (ii) the five sampled pairs of specific attine hosts and their Megalomyrmex parasites (S. amabilis–M. symmetochus; Trachymyrmex zeteki–Megalomyrmex adamsae; Cyphomyrmex longiscapus–Megalomyrmex wettereri; Cyphomyrmex costatus–Megalomyrmex mondaboroides and Megalomyrmex silvestrii; Cyphomyrmex cornutus–Megalomyrmex mondabora) and all the free‐living Megalomyrmex as a sixth category, and (iii) social parasites, attine hosts and free‐living Megalomyrmex as three categories. Detailed methods of clustering analyses are provided in Appendix S1 (Supporting information).
Primer design, diagnostic PCR screenings and sequencing
To investigate the distribution of specific phylotypes across samples, we designed diagnostic primers from 16S rRNA sequences generated through pyrosequencing and previously published work (Table S3, Supporting information). Using the MUSCLE algorithm (Edgar 2004) in geneious v.4.8.5, we aligned sequences belonging to OTUs with identical taxonomical assignments and other closely related OTUs. Primers were then designed to be specific to OTUs with identical classification and to include the two hypervariable regions V3 and V4 in the amplified fragment. The efficiency of the designed primers was then tested with perlprimer v.1.1.21 (Marshall 2004).
For M. symmetochus guest ants and Sericomyrmex host ants, we first generated sequencing data for 384 bp of mitochondrial cytochrome oxidase I (mtCOI) and approximately 630 bp of the nuclear wingless (wgls) gene (Table S3, Supporting information) to confirm the identity of the ant species, as recent studies have revealed a number of cryptic species in closely related fungus‐growing ants (Mehdiabadi et al. 2012; De Fine Licht & Boomsma 2014). We amplified mtCOI fragments using Jerry as the forward primer and either Ben (Simon et al. 1994) or George (after reverse translation; Sperling et al. 1995) as reverse primers.
PCRs were performed with both a negative and a positive control (for PCR conditions, see Table S3, Supporting information), and PCR products were visualized by 2% agarose gel electrophoresis. PCRs targeting the nuclear wgls gene (Tables S1 and S3, Supporting information) were used as controls of DNA quality and to ensure that fungus garden extractions were not contaminated by ant tissues. Samples from a subset of ant nests that tested positively for specific bacterial lineages were then selected for new PCRs using the designed forward primers and the universal 1513R primer (Russell et al. 2009) or primers for the Wolbachia wsp gene (Braig et al. 1998). PCR products were purified with EXO‐SAP IT (USB Corporation, Cleveland, OH, USA) and sequenced at BGI (Copenhagen, Denmark) or Eurofins MWG Operon (Ebersberg, Germany).
Assessing within‐OTU variation of abundant and shared bacterial lineages
The five most abundant bacterial lineages were further analysed for lower‐level genotypic variation, to assess whether associated hosts and social parasites shared identical 16S rRNA genotypes. We extracted the raw 16S rRNA gene sequences generated through pyrosequencing and then performed an analysis with oligotyping (Eren et al. 2013), which concatenates only the high‐information nucleotide positions in the 16S rRNA gene fragments using an entropy criterion (Shannon 1948), after which sequencing data can be partitioned into meaningful genotypes while discarding redundant information and sequencing noise.
We further built phylogenetic trees from the c. 970 bp of the 16S rRNA gene amplified with the designed forward primers for Entomoplasmatales and Bartonellaceae bacteria and the universal 1513R primer, and from the c. 520 bp of the Wolbachia wsp gene, aligned with additional published sequences from GenBank using MUSCLE (Edgar 2004) within geneious v.4.8.5. Trees were built in mega v.5.1 (Tamura et al. 2011), selecting models of nucleotide substitution (K2 + G for Entomoplasmatales, K2 + G + I for Bartonellaceae, T92 + G for Wolbachia wsp and Sericomyrmex mtCOI) under the Bayesian information criterion (BIC) and then performing maximum‐likelihood analyses with 1000 bootstrap replicates.
Assessing the stability of bacterial communities
Because ant social parasites and hosts physically interact and feed on the same fungal resources, part of the discovered bacterial communities may represent transient acquisitions from the nest environment or from the food source rather than stable associations. We therefore performed a diet manipulation experiment to assess the stability of the microbiotas of ant species sharing the same nest over time with and without their normal food.
Four nests of S. amabilis hosts and M. symmetochus guest ants, one colony of M. wettereri agro‐predators and one colony of M. mondaboroides thief ants were chosen for the diet manipulations. We sampled ants and fungus gardens at the beginning of the experiment, then placed 20 workers from each colony in petri dishes of 35 mm diameter lined with moistened filter paper and provided the ants with either a piece of their original garden or 10% sucrose–2% yeast water ad libitum in sterile caps of 0.2‐ml PCR tubes.
When mortality rates approached an average of 70% in sucrose treatments and 50% in garden treatments after 2 weeks, pools of whole live ants (3–5 per colony; Table S1, Supporting information) were collected, rinsed with 70% EtOH and MilliQ water as previously explained, and total DNA was extracted with the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), while DNA from fungus garden fragments was extracted using the CTAB method (Schiøtt et al. 2008). Bacterial DNA was amplified with primers 341F/806R and sequenced on a MiSeq Illumina platform after library preparation (for detailed methods, see Appendix S1, Supporting information).
Raw sequencing data (deposited at the Sequence Read Archive under Accession no. SRP052752) were analysed following mothur MiSeq SOP (Kozich et al. 2013). Paired‐end reads were assembled using make.contigs in mothur, sequences longer than 450 bp were removed with screen.seqs, and the remaining sequences were trimmed to remove primers and barcodes, after which a similar procedure was followed as for the 454 pyrosequencing data set, until producing a 97% OTU table. A heatmap and rarefaction plot of the 97% OTUs were subsequently obtained with r (packages gplots and vegan, R Core Team 2013). We also calculated the inverse Simpson diversity indexes for each ant sample based on a rarefied OTU table selecting 1839 sequences (the number of sequences in the sample with the lowest sequencing depth) and tested for differences in these alpha diversity estimates between the treatment groups by repeated‐measures anova in Paleontological Statistics v.3.04 (past; Hammer et al. 2004). Finally, we performed ordination analyses (nonmetric multidimensional scaling (NMDS) of Bray–Curtis dissimilarities) and matching statistical tests (permutational manova) on the clustering patterns of ant samples in the different treatment groups with past.
Results
Correlated bacterial communities of Megalomyrmex social parasites and attine host ants
Rarefaction analyses (Fig. S1A, Supporting information) showed that sequencing depth was adequate to approximate true bacterial diversity, with the possible exception of a single sample, S. amabilis JOA120528‐05. We performed three types of clustering analyses: (i) network‐based analysis, where samples are clustered based on the number and relative weight of shared OTUs or unique sequences; (ii) unweighted UniFrac, where comparisons are based on estimates of phylogenetic distances between the bacterial communities (i.e. in pairwise comparisons across all ant samples, the fraction of branch lengths that each pair of ant microbiotas shared within the phylogenetic tree built from all 97% OTUs or unique sequences identified); (iii) weighted UniFrac, where the latter distances are weighted according to the number of reads within OTUs or unique sequences. We then tested for significant differences with the methods used in Costello et al. (2009) and implemented in qiime (make_distance_boxplots.py script with the nonparametric options) for the comparisons based on the UniFrac metrics and an equivalent statistical approach for the network analyses (statistical analyses are further explained in Appendix S1, Supporting information) after grouping the ant samples in three different combinations (Megalomyrmex vs. attine ants; five host–parasite associations and the free‐living Megalomyrmex as sixth category; hosts vs. parasites vs. free‐living predators) and repeated each test at the 97% OTU level and at the unique sequence level. Hence, we performed a total of 18 tests, the results of which are presented in Table S4 (Supporting information) and summarized in the paragraphs below.
The network analysis showed a significant clustering of ant microbiotas across the five naturally associated hosts and parasites (after pooling all free‐living Megalomyrmex into a sixth association), both at the 97% OTU level (one‐tailed t‐test, P = 0.017) and at the unique sequence level (one‐tailed t‐test, P = 0.007; Fig. 1), indicating that more OTUs and 16S rRNA genotypes were shared within each pair of attine hosts and their specific Megalomyrmex social parasites and within the free‐living Megalomyrmex predators than between these categories.
Figure 1.
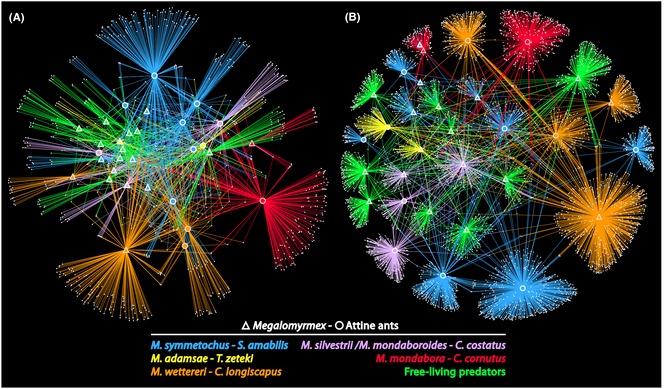
Network analyses of ant microbiotas based on 97% operational taxonomic units (OTUs) (A) or unique sequences (B), with Megalomyrmex samples shown as triangles and attine host samples as circles. The six colours separate the five categories of specifically associated host and parasite species and the sixth category of free‐living Megalomyrmex predators (plotted in green). Lines connect each sample to the particular OTUs (A) or unique sequences (B) that ants harboured (represented as small white dots) so that samples that share more OTUs or unique sequences cluster closer together after adjusting for the number of reads found within each OTU or unique sequence.
In spite of sharing specific bacterial lineages in associated hosts and parasites, the two ant lineages (all Megalomyrmex vs. all attine hosts) harboured bacterial communities with distinct phylogenetic signatures based on the unweighted UniFrac metric (one‐tailed t‐tests, P = 0.025 at the 97% OTUs level and P = 0.001 at the unique sequences level; Fig. 2). Alternative statistical analyses gave comparable results (Adonis test performed with compare_categories.py within qiime, 10 000 permutations; R 2 = 0.053, P = 0.012 at the 97% OTU level and R 2 = 0.054, P = 0.008 at the unique sequence level). These differences did not appear to be mainly driven by the free‐living Megalomyrmex species, as the same analysis was not significant when comparing attine hosts, Megalomyrmex social parasites and Megalomyrmex free‐living predators (one‐tailed t‐tests, P = 0.109 at the 97% OTU level and P = 0.068 at the unique sequence level; Fig. S2A, B, Supporting information).
Figure 2.
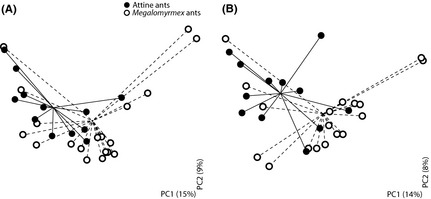
Principal coordinate analyses (PCoA) of unweighted UniFrac metrics based on 97% operational taxonomic units (OTUs) (A) or unique sequences (B) showing the separation in the phylogenetic signatures of the microbiotas harboured by attine ants (filled circles) and Megalomyrmex ants (open circles). Solid lines (for attine ants) and dashed lines (for Megalomyrmex ants) connect individual samples to group centroids. Colour‐coded versions of these plots are given as Fig. S2 (Supporting information), allowing direct comparisons between hosts, parasites and free‐living predators as three categories, and between associated hosts and parasites and free‐living Megalomyrmex as six categories.
We further compared the five distinct pairs of attine hosts and their specific Megalomyrmex parasites, and the sixth category of free‐living Megalomyrmex, in an additional unweighted UniFrac analysis. While different nest‐sharing species pairs harboured phylogenetically similar microbial communities when considering 97% OTUs (one‐tailed t‐test, P = 0.182), the genotypes within these OTUs varied more systematically between the different host–parasite pairs (one‐tailed t‐test, P = 0.030; Fig. S2C, D, Supporting information), suggesting some association specificity in microbiotas at this level. However, none of these overall comparisons was significant when using weighted UniFrac, as clustering patterns became strongly affected by a few dominant bacterial lineages that were abundantly infecting samples across the different groups, mainly Entomoplasmatales, Bartonellaceae, Acinetobacter, Pseudonocardia and Wolbachia (Fig. S2E, F, Supporting information).
Microbiota composition of Megalomyrmex and their fungus‐growing ant hosts
The bacterial communities of Megalomyrmex and attine ants appeared similar at the bacterial order level (71% overlap; 49 of the 69 identified orders were shared across the two ant lineages), with members of the Rhizobiales, Entomoplasmatales, Actinomycetales, Pseudomonadales, Xanthomonadales, Legionellales, Enterobacteriales, Rickettsiales, Bacillales and Lactobacillales being particularly abundant (Fig. S3, Supporting information). However, when comparing bacterial phylotypes, OTUs and unique sequences, the differences between Megalomyrmex and attine ants became clearer (58% shared phylotypes, 20% shared OTUs, 3% shared unique sequences), even though they were scattered across the bacterial orders (Fig. S4, Supporting information).
Symbiont sharing between social parasites and hosts collected from joint nests was strikingly evident for two OTUs classified as Entomoplasmatales and one Bartonellaceae OTU, which were frequently present and sometimes dominant in M. symmetochus guest ants and their S. amabilis hosts (Fig. 3), but sharing was also observed for Acinetobacter, Wolbachia, Pseudonocardia, Bradyrhizobiaceae, Phyllobacteriaceae, Bacteroidetes, Mycobacterium and other lower abundance phylotypes (Fig. S5, Supporting information). A PCR screening with specific primers for Entomoplasmatales, Bartonellaceae and Wolbachia revealed that Entomoplasmatales were widespread across almost all the parasitic Megalomyrmex species and their fungus‐growing ant hosts, with the exception of the Costa Rican thief ant M. mondabora and its C. cornutus host (Fig. 3).
Figure 3.
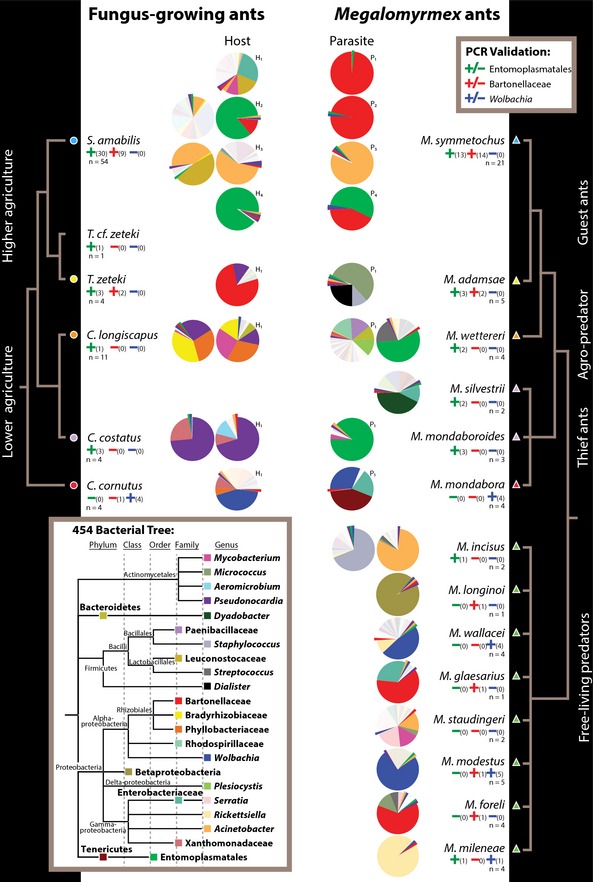
Phylotype‐level microbiotas of Megalomyrmex predators and social parasites (right) and fungus‐growing ant hosts (left) as revealed by tag‐encoded FLX 454 pyrosequencing, with PCR validations for Entomoplasmatales, Bartonellaceae and Wolbachia given as +/− signs in, respectively, green, red and blue, with the number of confirmed infected nests in parentheses relative to the total number of nests screened (n). Simplified phylogenetic trees obtained from Schultz & Brady (2008) for the attine host ants and from morphological and molecular studies (Brandão 1990; Adams 2008; Longino 2010) for Megalomyrmex are given towards the left and right side. The terminal branches of the phylogenies are coded with triangles and circles and coloured corresponding to Fig. 1. Pie charts represent ant microbiotas, plotting only the 25 bacterial phylotypes that were either most abundant across all samples or particularly abundant in single samples (for complete operational taxonomic unit (OTU) assignments, see Table S2, Supporting information), with phylogenetic relationships given as inset towards the bottom left. The top five bacterial phylotypes were enlarged and pulled out of the pies when only present at low relative abundances. Shades of white or light grey are rare bacterial taxa that are not included in the tree of the 25 most abundant phylotypes. Hosts and parasites that were inhabiting the same nest upon sample collection are marked with either H (hosts) or P (parasites) and numbered with subscripts to identify specific joint nests. Pies without numbered P and H annotations represent ant colonies that were not associated upon collection, hosts that were not parasitized, or free‐living Megalomyrmex predators.
Focusing on the widely sampled population of M. symmetochus guest ants and their S. amabilis hosts, we were able to further investigate the consistency of the pattern of OTU sharing for Entomoplasmatales and Bartonellaceae. Entomoplasmatales had a 56% infection frequency in the host ants (n = 54) and 62% in the guest ants (n = 21) and always co‐occurred in host and guest ants when both were present in a nest (r = 1, P < 0.0001, n = 21), as well as in fungus garden samples as far as we screened them (6/6 Entomoplasmatales‐infected nests), except for nest JOA120525‐03 where the garden was infected and the ants were not. For the Bartonellaceae, co‐infection patterns of guest and host ants were less consistent (r = 0.224, P = 0.329, n = 21). Prevalences in the guest ants were high (67%, n = 21), but much lower in the hosts (17%, n = 54). Hosts were only infected in five of 14 cases in which the guest ants harboured Bartonellaceae, and we found Bartonellaceae DNA to be amplifiable in three cases where we could also screen the fungus gardens and both ant species were infected (Fig. 4; see also Fig. S6, Supporting information, for additional comparisons of S. amabilis hosts, M. symmetochus guest ants and their shared gardens based on the 454 16S rRNA pyrosequencing samples).
Figure 4.

Distribution of Entomoplasmatales and Bartonellaceae bacteria within and across the sampled subpopulations of Sericomyrmex amabilis hosts and their Megalomyrmex symmetochus guest ants. (A) The four sampling sites in the Panama Canal Zone: Pipeline Road (blue filled square), Barro Colorado Nature Monument (blue open square), Gamboa (blue filled circle), Plantation Road (blue open circle). (B) Maximum‐likelihood phylogeny based on 384 bp of Sericomyrmex mtCOI sequence with Sericomyrmex cf. diego/zacapanus (the closest sympatric relative) as outgroup. Values above the nodes represent support after 1000 bootstrap replicates. Blue symbols behind sample numbers refer to the different sampling locations (panel A; open space means that exact location was unknown). +/− indicates presence/absence of Entomoplasmatales (green) and Bartonellaceae (red) in the ants and fungus gardens.
We further screened individual eggs, larvae, pupae and workers of hosts and guest ants, and guest ant gynes, from a single nest (JOA120607‐01) for Entomoplasmatales prevalence. There was no significant difference in overall infection prevalence between hosts and guest ants, but there were prevalence differences between developmental stages (Binomial GLM, L‐R χ2 = 39.96, P < 0.0001), as workers were always infected with Entomoplasmatales while pupae and larvae had intermediate infection prevalences (larvae: 60% hosts and 70% guest ants; pupae: 100% hosts and 40% guest ants; n = 10 each) that differed between hosts and guest ants (Binomial GLM, L‐R χ2 = 10.69, P = 0.0135). Some host eggs were infected (four of 10), whereas none of the guest ant eggs were (n = 10). However, every guest ant gyne that we analysed (n = 10) was infected, suggesting that Entomoplasmatales could be transmitted across generations of guest ants with mechanisms other than transovarial transmission.
Assessing genotypic variation of the five major bacterial lineages
Our network analysis suggested that associated hosts and social parasites shared not only some of the same 97% OTUs, but also a subset of 16S rRNA genotypes within these OTUs. To better estimate the genotypic diversity and the distribution of specific genotypes of the main bacterial lineages across our samples, we employed a more stringent approach that resolves true genotypic diversity by making use of only a few variable positions in the 16S rRNA gene fragments, so that the introduction of false positives due to sequencing errors is largely avoided. This analysis was performed with oligotyping (Eren et al. 2013) on all reads belonging to the five most abundant bacterial lineages in our 454 16S rRNA gene pyrosequencing data set: Entomoplasmatales, Bartonellaceae, Wolbachia, Pseudonocardia and Acinetobacter. This revealed the presence of four dominant Entomoplasmatales genotypes (four main colours in the stacked bars of Fig. 5A), and another 22 low abundance genotypes (Table S5, Supporting information). In two cases where we had sampled M. symmetochus guest ants and their S. amabilis hosts from the same nest, the two ant species shared the same genotype, and in one of these cases where we also had data for the fungus garden, the same dominant genotype was found in the garden alongside other genotypes in lower abundance (Fig. 5A).
Figure 5.
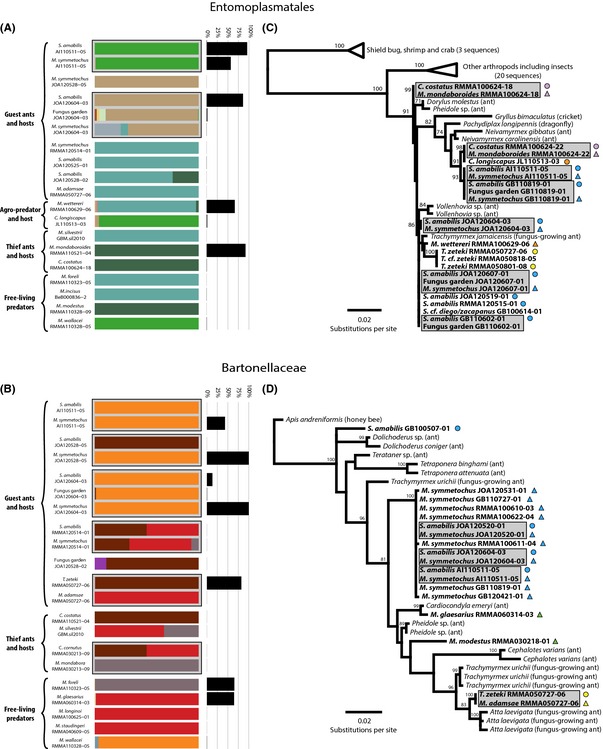
Distribution of 16S rRNA genotypes as identified by oligotyping from the reads extracted from the 454 16S rRNA pyrosequencing data set (A and B), and phylogenetic relationships (C and D) of Entomoplasmatales and Bartonellaceae. Colours in the stacked bars represent the relative proportions of the different genotypes found in each sample for Entomoplasmatales (A) and Bartonellaceae (B), while black bar charts show the overall abundance of Entomoplasmatales (A) and Bartonellaceae (B) relative to the total microbiota in each sample. Maximum‐likelihood phylogenies of Entomoplasmatales (C) and Bartonellaceae (D) are based on c. 970 bp of 16S rRNA amplified from samples collected for the present study (bold faced text) or closely related strains from GenBank (normal text), and coded with triangles and circles coloured by specific host–parasite associations with free‐living Megalomyrmex in green as in Fig. 1. Values above the nodes represent support ≥50 after 1000 bootstrap replicates. In all panels, grey‐shaded cells highlight hosts, parasites and fungus gardens that were collected from the same nest.
Similarly, four dominant Bartonellaceae genotypes were found among a total of 19 genotypes identified (Fig. 5B and Table S5, Supporting information). In three cases, hosts and parasites collected from the same nest showed almost identical genotypes, which were also found in the fungus garden in the single instance where the garden was sampled and the Bartonellaceae bacteria were present in the nest. However, in three other cases (one M. symmetochus–S. amabilis nest, one M. adamsae–T. zeteki nest and one M. mondabora–C. cornutus nest), the associated ant species did not share the same genotypes, although in each of these cases at least one of the ant species was infected only in trace amounts (as shown by the black bars of Fig. 5B).
We assessed the sharing of Entomoplasmatales and Bartonellaceae genotypes and their relationships with other known strains of these bacteria by building phylogenies based on c. 970 bp of 16S rRNA from a subsample of colonies that tested positively in our diagnostic PCR screening. This confirmed that identical genotypes of these bacteria were shared between M. symmetochus and S. amabilis collected from several shared nests (Fig. 5C, D) and that M. mondaboroides thief ants and their C. costatus hosts were also infected with identical Entomoplasmatales genotypes, in spite of having less strict nest sharing. These genotypes were different from the ones associated with M. symmetochus guest ants and their S. amabilis hosts (Fig. 5C) and, similarly, M. adamsae guest ants shared the same Bartonellaceae genotype with their T. zeteki host ants, which fell into a well‐supported clade that included strains from other Trachymyrmex and Atta fungus‐growing ants (Fig. 5D).
Comparable analyses of genotypic distributions of Pseudonocardia, Acinetobacter and Wolbachia did not show clear co‐infection patterns. For example, although S. amabilis hosts and M. symmetochus guest ants from nest RMMA120514‐01 were abundantly co‐infected with Acinetobacter, only some minor genotypes were shared (Fig. S7 and Table S5, Supporting information). Similarly, the only associated hosts and parasites that were infected with Wolbachia in abundance, C. cornutus and M. mondabora, did not share the same 16S rRNA genotypes (Fig. S7, Supporting information), and this was further confirmed by sequencing 520 bp of the wsp gene from the same and additional samples (hosts and parasites from four nests; Fig. S8, Supporting information). Finally, Pseudonocardia, a cuticular antibiotic‐producing mutualist of fungus‐growing ants, used to keep the fungus garden free of infections (Cafaro et al. 2011), was found in some abundance in the agro‐predator M. wettereri, and with genotypes that partly overlapped with its C. longiscapus host (Fig. S7 and Table S5, Supporting information).
Assessing the stability of bacterial communities
We conducted a diet manipulation experiment on a subset of Megalomyrmex socially parasitic species and their hosts to further assess the extent to which the shared bacterial lineages represented stable associations in both hosts and social parasites. We obtained 1 360 636 curated 16S rRNA sequences from 36 samples, including six fungus garden samples collected at the start of the experiment, clustering into 852 (97% cut‐off) OTUs (Table S6, Supporting information).
Rarefaction analyses showed that, with few exceptions (S. amabilis JOA120516‐01 from the sucrose treatment, and fungus gardens JOA120604‐01 and RMMA120514‐01), samples had a proper sequencing depth (Fig. S1B, Supporting information). There was no significant difference between inverse Simpson diversity indexes for the three treatment groups (repeated‐measures anova, F 2,27 = 0.617, P = 0.551), suggesting that feeding the ants a sucrose diet did not lead to a general loss of bacterial diversity. This does not necessarily mean that all bacteria identified in our study have functional symbiotic roles, but it reinforced our confidence that the sharing patterns are not driven by external contaminants introduced through our sampling and DNA extraction procedures, as such OTUs would likely have been lost after we isolated the ants and changed their diet.
After 2 weeks of diet manipulation, none of the abundant bacterial lineages was clearly lost in the sucrose treatment (Fig. 6), although there was some variation in presence/absence of OTUs with low relative abundance when the same ant colonies were resampled for the different treatment groups. These inconsistencies were likely due to subsampling of only few workers (3–5) per treatment and possible prevalence variation between nestmate workers, alongside biases that might have been introduced by PCR and sequencing. In spite of this, several of the most abundant OTUs remained consistently shared in all treatment groups between M. symmetochus guest ants and their S. amabilis hosts in at least one nest. These included not only Entomoplasmatales and Bartonellaceae, but also OTUs classified as Acinetobacter, Stenotrophomonas, Enterobacteriaceae, Serratia, Pseudomonas, Pseudomonadaceae, Mycobacterium, Erwinia and Burkholderia among other lower abundance OTUs (Fig. 6 and Table S7, Supporting information).
Figure 6.
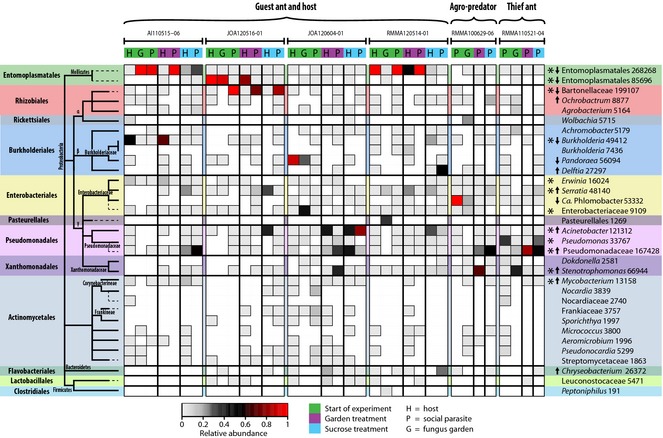
Heatmap of all 97% operational taxonomic units (OTUs) that had at least 5% relative abundance in one sample of the diet experiment data set generated on the Illumina MiSeq platform. Colours in each cell reveal relative abundances of the identified OTUs within each sample (columns), and cells are not outlined if the OTUs were absent. Sample columns are ordered by species pair, nest ID and treatment group, while OTUs are listed in rows and ordered based on the bacterial phylogenetic tree given towards the left. OTU classifications and the total number of assigned reads per OTU across the 36 samples are given towards the right and are colour‐coded by bacterial order, as shown in the bacterial phylogeny. Asterisks show the OTUs that were consistently shared by hosts and parasites across all treatment groups in a least one nest. Arrow direction highlights the OTUs that substantially increased or decreased between the start of the experiment and the end of the sucrose treatment as shown by the similarity percentages test (SIMPER; for complete results see Table S9, Supporting information).
We further explored whether the dietary manipulation and isolation of the ants produced significant shifts in the ant microbiotas by NMDS of Bray–Curtis dissimilarities between the bacterial communities (Fig. S9, Supporting information). We found an overall significant difference between the treatment groups in a permutational manova (permanova) with 10 000 permutations (F = 1.627, P = 0.044), driven by the difference between the start of the experiment and the end of the sucrose treatment (Bonferroni adjusted P = 0.0009; Fig. S9A and Table S8, Supporting information), as the Bray–Curtis dissimilarities between samples were reduced in the sucrose diet treatment group in spite of consistent nest‐specific signatures in the bacterial communities of hosts and parasites (Fig. S9B, Supporting information). This difference did not mainly depend on significant changes in community memberships, as a permanova test on Bray–Curtis dissimilarities from the OTU table transformed into a presence/absence matrix was not significant (F = 1.244, P = 0.069; Fig. S9C, D, Supporting information). Rather, it depended primarily on consistent changes in relative abundance of particular OTUs as shown by a similarity percentages test (SIMPER; Table S9, Supporting information). In particular, there was a decrease across samples in the relative abundance of the two main Entomoplasmatales OTUs, the Bartonellaceae OTU, one Burkholderia and one Pandorea OTU (both Burkholderiales), and a Candidatus Phlomobacter OTU (Enterobacteriales) in the sucrose treatment. By contrast, Acinetobacter and Pseudomonadaceae OTUs (both Pseudomonadales), Ochrobactrum (Rhizobiales), Serratia (Enterobacteriales), Delftia (Burkholderiales), Stenotrophomonas (Xanthomonadales), Mycobacterium (Actinomycetales) and Chryseobacterium (Flavobacteriales) OTUs substantially increased in the sucrose treatment compared to the experiment start (Fig. 6 and Table S9, Supporting information).
Discussion
Our results show a significant degree of convergence of microbiotas between Megalomyrmex social parasite species and their fungus‐growing host ants. These similarities were most pronounced for species pairs that permanently share the same nest, feeding on the same fungal food and being exposed to the same microhabitat. These results raise the possibility that symbiont sharing may have facilitated evolutionary transitions to social parasitism in Megalomyrmex ants, as would be expected if these bacteria assist in the digestion of fungal material, in overcoming dietary deficiencies, or in resisting pathogens in the shared nest environment. Despite these microbiota convergences, we found an overall signal of phylogenetic community divergence between the two ant lineages. This is likely due to attine and Megalomyrmex ants having had distinct evolutionary histories. Furthermore, not only the free‐living Megalomyrmex predators, but also the socially parasitic species maintain clear ecological differences compared to the attine ants, such as a partially carnivorous diet and not foraging outside the nest. The microbiota convergences that we found raise further questions about mutual transmission and acquisition modes and the possible benefits of the bacteria involved. We address both issues in the sections below, reviewing what is known about these bacteria and their associations with other insect hosts and outlining approaches by which hypotheses about acquisition and function can be tested experimentally.
Transmission dynamics of bacterial symbionts
While producing substantial insights on the identity, structure and taxonomic overlap of the ant microbiotas, our comparative approach cannot provide conclusive evidence for the underlying transmission mechanisms of every bacterial symbiont, because 16S rRNA can be conserved in some bacterial lineages and therefore does not allow unambiguous characterization of strain‐level diversity for each OTU. A multilocus approach targeting the individual bacterial lineages that we have now identified, coupled with transmission experiments, would be required to further clarify the mechanisms and directionality of symbiont acquisition in hosts and social parasites inhabiting the same nest. Nevertheless, considering the biology of these ants and the insights provided by our data, we can offer some hypotheses on the proximate mechanisms of symbiont sharing.
Co‐infections could derive from shared ancestry, but this is unlikely as these ant lineages have been separated for c. 80 million years, with the attine ants depending exclusively on a fungal diet for the last c. 50 million years (Schultz & Brady 2008; Ward et al. 2015). Another possibility is that phylogenetically distinct ant species acquire the same bacteria from the joint nest environment. This may be a viable explanation for Acinetobacter bacteria, which were in some cases similar to environmental strains found in soil (Table S5, Supporting information), and some Pseudonocardia, the nondominant strains of which have been suggested to be environmentally acquired (Sen et al. 2009; Andersen et al. 2013). However, environmental acquisition seems unlikely for the other three dominant bacterial lineages. The Bartonellaceae that we identified fall into an ant‐specific clade (Russell et al. 2009). The Entomoplasmatales have been shown to be generally prevalent across the ants and to form several ant‐associated clades (Funaro et al. 2011; Kautz et al. 2013). Similarly, Wolbachia are arthropod‐ and nematode‐specific symbionts that have never been found in the environment. Therefore, the co‐infection patterns for these three major symbionts are likely due to horizontal transmission, either directly or via unknown mites or parasitoid wasps (Heath et al. 1999; Jaenike et al. 2007; Duron et al. 2010).
Previous studies have found mixed evidence for social parasitism being a viable transmission route for specific bacterial symbionts. Dedeine et al. (2005) showed that Solenopsis hosts and their inquiline social parasite Solenopsis daguerrei shared similar or identical Wolbachia strains, suggesting possible horizontal transmission. A similar pattern has been found for Acromyrmex insinuator and its A. echinatior host, although this parasite also harbours additional Wolbachia strains (Van Borm et al. 2003; Dedeine et al. 2005). However, a recent study has found no such evidence for Spiroplasma, Entomoplasma and Wolbachia in the slave‐making ant Formica sanguinea and its Serviformica hosts (Haapaniemi & Pamilo 2015). Our data suggest that horizontal transmission is likely for Entomoplasmatales and Bartonellaceae, for which identical 16S rRNA genotypes were shared in joint nests, and that for Entomoplasmatales it may occur during the larval stage. In contrast, the abundant sharing of nonidentical Wolbachia in M. mondabora thief ants and their C. cornutus hosts may be better explained by the parasite being independently infected via parasitoids as suggested by our phylogenetic reconstruction (Fig. S8, Supporting information).
Several mechanisms could mediate direct transmission between associated hosts and social parasites. First, socially parasitic Megalomyrmex lineages consume the fungus gardens that the attine ants maintain and their gut bacteria may therefore reach the garden when host ants mix fresh leaf material with faecal fluid (Andersen et al. 2012). However, our finding that identical genotypes of Entomoplasmatales and Bartonellaceae are present in both ant species and fungus gardens merely shows that DNA from these bacteria is present in the garden and not that these bacteria are alive there. Further studies involving microscopy will thus be needed to clarify whether fungus gardens offer a viable transmission route. Second, Megalomyrmex parasites may acquire host ant bacteria through brood predation, a transmission mode that has previously been suggested for Wolbachia in a predatory mite and its prey (Hoy & Jeyaprakash 2004). Third, co‐infections might be mediated by oral–anal trophallaxis, which has been proposed as a transmission mechanism for bacterial symbionts that are gut residents (Wheeler 1984; Russell et al. 2009; see also Stuart 1981). Attine host workers have been observed grooming Megalomyrmex workers focusing on the abdominal area (Adams et al. 2012), but transmission of endosymbionts through this behaviour remains to be tested.
Putative functional roles of the dominant bacterial lineages
The overall phylogenetic variation of attine ant microbiotas is just beginning to be unravelled, so the functional significance of these bacteria remains largely unknown. However, the cultivation of a fungal crop as the main nutritional resource may imply nitrogen retrieval as a possible function, because this diet is likely to be protein limited (Pinto‐Tomás et al. 2009). Similarly, the partial specialization of Megalomyrmex social parasites on a fungivorous diet might have imposed selection for facultative bacteria conferring benefits related to digestion, recycling or acquisition of nutrients from fungal material. The Bartonellaceae that we found may be the most promising targets for testing nitrogen preservation functions as these ant‐specific Rhizobiales have been shown to be exceptionally frequent in herbivorous ant lineages that suffer from nitrogen limitations (Russell et al. 2009).
The most common bacterial lineage that we have identified belongs to the order Entomoplasmatales, more specifically to the genus Spiroplasma (Table S5, Supporting information). The presence of these bacteria had already been reported in the fungus‐growing ant Trachymyrmex septentrionalis (Ishak et al. 2011), while Acromyrmex octospinosus leaf‐cutting ants are known to be infected with different Entomoplasmatales bacteria (Van Borm et al. 2002b). Spiroplasma are widespread insect endosymbionts that have important effects on host survival or reproductive success as they can either be reproductive manipulators through male‐killing or defensive mutualistic symbionts (Jiggins et al. 2000; Xie et al. 2014), but no study has yet measured such relationships in ants. The abundant and widespread prevalence of these bacteria across fungus‐growing ant hosts and their Megalomyrmex social parasites suggests a long history of association with these ants, but genome‐level studies of these symbionts will be needed to test hypotheses of this kind.
Apart from epidemiological research arising from public concern about their human pathogenicity (reviewed in Dijkshoorn et al. 2007), not much is known about the role of Acinetobacter bacteria in insect hosts. One study has suggested that these bacteria may be vectored by ants in tropical regions (Moreira et al. 2005), and another has shown that two of three Acinetobacter isolates harboured by Apis mellifera larvae inhibit a bacterial pathogen of honey bees, Paenibacillus larvae (Evans & Armstrong 2006). The fact that Acinetobacter bacteria were widely distributed in our samples and consistently present across colonies of S. amabilis suggests that they could be important symbionts. However, also for this lineage of putative mutualists, further functional experiments involving culturing and competition trials with known fungal and bacterial pathogens would be needed to substantiate whether they have similar protective functions in the ants as they have in the honey bee.
The cuticular Actinobacteria associated with many genera of attine ants have been the target of several recent studies (e.g. Sen et al. 2009; Andersen et al. 2013) as the ants use the antibiotics produced by these bacteria to keep their gardens free of infection (Cafaro et al. 2011). Interestingly, the agro‐predator M. wettereri harboured some of the same Pseudonocardia genotypes as their hosts. As these ants are able to keep gardens viable for several weeks after having usurped and chased away the host ants, it is possible that they might have co‐opted these bacteria to maintain prophylactic defences of the crop fungus in the absence of the original ant farmers.
Diet‐induced shifts in the relative abundance of specific bacterial lineages have previously been documented in the guts of Cephalotes varians ants (Hu et al. 2014), although only a specific pollen diet among three tested diets exerted these effects. The causes of these changes remained unresolved, but the similar results of our artificial diet experiment suggest that abundance shifts may be related to how bacterial symbionts influence host ant digestive physiology. In our data set, Entomoplasmatales and Bartonellaceae consistently decreased in relative abundance, whereas Acinetobacter and some other members of the Pseudomonadales had increased following 2 weeks of sucrose feeding (Fig. 6).
A striking example was offered by the paired colonies of M. symmetochus and S. amabilis (RMMA120514‐01), which were sampled in the field in May 2012 and resampled in May 2014. In these 2 years, both host and guest ants had naturally shifted from being dominated by Acinetobacter and moderately infected with Entomoplasmatales (H3‐P3 pair in Fig. 3), to Entomoplasmatales dominance and low Acinetobacter infection (start of diet experiment, nest RMMA120514‐01; Fig. 6). After 2 weeks of sucrose diet, the balance was reverted to favour Acinetobacter, albeit more consistently for the host ants than for the guest ants (Fig. 6). Sericomyrmex amabilis workers are frequently found aggregating and feeding on ripe fruits such as fallen mangos (Liberti J & Adams RMM, personal observation) rather than searching for leaf fragments, flowers and seeds (De Fine Licht & Boomsma 2010), which is their normal forage when provisioning gardens. Such dramatic but temporary shifts between ingesting sugars rather than fungal material could well explain the observed abundance variation between bacterial lineages and the rapid pace by which such responses occur. The ants may thus maintain several adaptive symbionts with mutually exclusive abundances that vary in response to diet shifts. The results of our artificial diet experiment were consistent with this reasoning as none of the putative symbionts was lost, but their relative abundances changed significantly (Fig. 6).
Among the symbionts affected by the diet manipulations, one OTU classified as Candidatus Phlomobacter (Greengenes reference set) decreased its relative abundance in the sucrose treatment. This OTU was further found to be related to Arsenophonus symbionts of aphids and other Hemiptera (blastn; 100% identity, 0.0 E value to Arsenophonus endosymbionts of Aphis melosae, Philaenarcys bilineata, among others). Arsenophonus bacteria had never been recorded from attine ants but are known to have a broad range of fitness effects, to be widespread across the insects and to have transmitted horizontally between distantly related host species (Jousselin et al. 2013), so they represent a lineage worthy of further investigation.
Conclusions
Exploiting the potential of 16S rRNA pyrosequencing, we have detailed partial convergences in microbiota compositions between attine fungus‐growing host ants and Megalomyrmex social parasites. Bacterial symbiont sharing in hosts and parasites is likely to be at least partly dependent on the intensity of nest and food sharing between these distantly related ants. The significantly nonrandom patterns of association and their maintenance after a diet manipulation experiment suggest that at least some of these bacteria are likely to have mutualistic or parasitic significance, but the mechanisms mediating such associations deserve further study.
J.L., J.J.B., P.S. and R.M.M.A. designed the study and J.L. and R.M.M.A. did the fieldwork. H.L. and S.J.S. performed sequencing runs on the Roche Life Sciences 454 GS FLX Titanium and MiSeq Illumina platforms. J.L. carried out the laboratory work and data analyses with contributions from P.S. and R.M.M.A. J.L. drafted the manuscript and J.J.B., P.S. and R.M.M.A. contributed to the interpretation of the sequencing results and helped completing the manuscript.
Data accessibility
The NGS data sets were deposited at the NCBI Sequence Read Archive under the Accession no. SRP052752. COI (LC025532–LC025612), wingless (LC027779–LC027860), wsp (LC027861–LC027874) and 16S rRNA gene fragments for Entomoplasmatales (LC027737–LC027760) and Bartonellaceae (LC027761–LC027778) were deposited at the DNA Data Bank of Japan (DDBJ).
Sequence alignments and tree files, mapping file with barcode sequences for the 454 data set, OTU tables, taxonomical assignments of the OTUs and unique sequences, representative sequences for all OTUs and unique sequences and input alignments for the oligotyping analyses are accessible from the Dryad Digital Repository (doi: 10.5061/dryad.ft36k).
Supporting information
Fig. S1 Rarefaction curves of tag‐encoded FLX 454 pyrosequencing samples (A) and Illumina MiSeq diet experiment samples (B).
Fig. S2 Principal Coordinates Analyses (PCoA) of unweighted UniFrac metrics based on 97% OTUs (A and C) or unique sequences (B and D) and PCoA bi‐plots of weighted UniFrac metrics based on 97% OTUs (E) or unique sequences (F).
Fig. S3 Phylogenetic trees of all 69 bacterial orders identified across attine and Megalomyrmex ants from the 454 16S rRNA pyrosequencing dataset.
Fig. S4 Phylogenetic trees of all 255 bacterial phylotypes identified across attine and Megalomyrmex ants from the 454 16S rRNA pyrosequencing dataset.
Fig. S5 Proportions of shared phylotypes in hosts and social parasites from joint nests as estimated from the 454 16S rRNA pyrosequencing dataset.
Fig. S6 Taxonomic assignment of 454 16S rRNA gene amplicons of Sericomyrmex amabilis hosts, Megalomyrmex symmetochus guest ants, and their fungus gardens as produced by megan4 (Huson et al. 2011).
Fig. S7 Distribution of 16S rRNA genotypes as identified by oligotyping from the 454 16S rRNA pyrosequencing dataset based on all reads identified as Pseudonocardia, Wolbachia and Acinetobacter.
Fig. S8 Maximum Likelihood phylogeny based on c. 520 bp of the Wolbachia wsp gene from a subsample of positive PCRs in our diagnostic screening, aligned with closely related sequences obtained from GenBank.
Fig. S9 Non‐metric multidimensional scaling of Bray‐Curtis dissimilarities between the bacterial communities identified in the diet manipulation experiment samples, calculated in two separate analyses from a matrix of relative abundances (A and B) and from a presence‐absence matrix (C and D).
Table S1 DNA extractions with details of the species sampled, numbers of individuals pooled in each extraction, nest IDs, sampling locations, GPS coordinates and diagnostic PCR results for the wingless (wgls) host nuclear gene, Entomoplasmatales, Bartonellaceae and Wolbachia.
Table S2 Taxonomic assignment and number of reads per sample of all the 97% OTUs and unique sequences identified in the 454 rRNA gene pyrosequencing dataset.
Table S3 Primers, lengths of amplified fragments, and PCR conditions used in this study.
Table S4 Summary of statistical results from network and UniFrac clustering analyses at both the 97% OTU and unique sequence levels.
Table S5 Number of reads per sample of all genotypes identified by Oligotyping of the five most abundant bacterial lineages in the 454 rRNA gene pyrosequencing dataset, with the top blastn hit for each genotype reported towards the right of the tables.
Table S6 Taxonomic assignment and number of reads per sample of all the 852 (97% cutoff) OTUs identified in the diet experiment MiSeq Illumina dataset.
Table S7 List of OTUs per nest that were consistently shared in all treatment groups between M. symmetochus guest ants and S. amabilis host ants samples from the diet experiment Miseq Illumina dataset.
Table S8 Results of permanova comparisons between the treatment groups based on Bray‐Curtis dissimilarities between the bacterial communities identified from the ant samples of the diet experiment.
Table S9 SIMPER results showing OTUs with differences in relative abundance between the start of the experiment and the sucrose treatment.
Appendix S1 Additional methodologies and list of GenBank Accessions used to build the bacterial phylogenies.
Acknowledgements
We thank the Smithsonian Tropical Research Institute for providing facilities to work in Panama and the Autoridad Nacional del Ambiente y el Mar for issuing sampling and export permits. Sebastian Gottfried assisted in the field sampling and helped with the laboratory work in Panama. Gaspar Bruner and Hermógenes Fernández‐Marín provided part of the ant and fungal samples. Michael Poulsen, Pepijn W. Kooij, Saria Otani and Morten Schiøtt gave valuable suggestions for data analyses. Waleed A. Al‐Soud and Karin P. Vestberg assisted with the sequencing runs and Ana Jesovnik with the nomenclature of the S. cf. diego/zacapanus samples. David Nash and Gösta Nachman helped with statistical analyses, Bjørn Hermansen with creating maps of the sampling sites, Luigi Pontieri with editing some of the figures, Rozlyn E. Haley with the ant drawings and Andreas Schramm, E. Allen Herre and William T. Wcislo with insightful comments and discussion. This work was funded by a Marie Curie International Incoming Fellowship (237266) to R.M.M.A., a grant from the Danish National Research Foundation (DNRF57) to J.J.B. and an ERC Advanced Grant (323085) to J.J.B.
References
- Adams RMM, Mueller UG, Schultz TR, Norden B (2000) Agro‐predation: usurpation of attine fungus gardens by Megalomyrmex ants. Naturwissenschaften, 87, 549–554. [DOI] [PubMed] [Google Scholar]
- Adams RMM, Longino JT (2007) Nesting biology of the arboreal fungus‐growing ant Cyphomyrmex cornutus and behavioral interactions with the social‐parasitic ant Megalomyrmex mondabora . Insectes Sociaux, 54, 136–143. [Google Scholar]
- Adams RMM (2008) Unraveling the origins of social parasitism in Megalomyrmex ants. PhD Dissertation, The University of Texas, Austin. [Google Scholar]
- Adams RMM, Shah K, Antonov LD, Mueller UG (2012) Fitness consequences of nest infiltration by the mutualist‐exploiter Megalomyrmex adamsae . Ecological Entomology, 37, 453–462. [Google Scholar]
- Adams RMM, Liberti J, Illum AA, Jones TH, Nash DR, Boomsma JJ (2013) Chemically armed mercenary ants protect fungus‐farming societies. Proceedings of the National Academy of Sciences of the United States of America, 110, 15752–15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RMM, Jones TH, Longino JT, Weatherford RG, Mueller UG (2015) Alkaloid venom weaponry of three Megalomyrmex thief ants and the behavioral response of Cyphomyrmex costatus host ants. Journal of Chemical Ecology, doi:10.1007/s10886‐015‐0565‐y. [DOI] [PubMed] [Google Scholar]
- Andersen SB, Boye M, Nash DR, Boomsma JJ (2012) Dynamic Wolbachia prevalence in Acromyrmex leaf‐cutting ants: potential for a nutritional symbiosis. Journal of Evolutionary Biology, 25, 1340–1350. [DOI] [PubMed] [Google Scholar]
- Andersen SB, Hansen LH, Sørensen SJ, Boomsma JJ (2013) Specificity and stability of the Acromyrmex‐Pseudonocardia symbiosis in changing environments. Molecular Ecology, 22, 4307–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KE, Russell JA, Moreau CS et al (2012) Highly similar microbial communities are shared among related and trophically similar ant species. Molecular Ecology, 21, 2282–2296. [DOI] [PubMed] [Google Scholar]
- Bordenstein SR (2003) Symbiosis and the origin of species In: Insect Symbiosis (eds Bourtzis K, Miller TA.), vol. 1, pp. 283–304. CRC Press, Boca Raton, Florida. [Google Scholar]
- Boudinot BE, Sumnicht TP, Adams RMM (2013) Central American ants of the genus Megalomyrmex Forel (Hymenoptera: Formicidae): six new species and keys to workers and males. Zootaxa, 3732, 001–082. [DOI] [PubMed] [Google Scholar]
- Braig HR, Zhou W, Dobson SL, O'Neill S (1998) Cloning and characterization of a gene encoding the major surface protein of the bacterial endosymbiont Wolbachia pipientis . Journal of Bacteriology, 180, 2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandão CRF (1990) Systematic revision of the Neotropical ant genus Megalomyrmex Forel (Hymenoptera: Formicidae: Myrmicinae), with the description of thirteen new species. Arquivos de Zoologia, 31, 411–481. [Google Scholar]
- Cafaro MJ, Poulsen M, Little AE et al (2011) Specificity in the symbiotic association between fungus‐growing ants and protective Pseudonocardia bacteria. Proceedings of the Royal Society of London, Series B: Biological Sciences, 278, 1814–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J, Kuczynski J, Stombaugh J et al (2010a) QIIME allows analysis of high throughput community sequencing data. Nature Methods, 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD et al (2010b) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics, 26, 266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier‐Smith T (2009) Predation and eukaryote cell origins: a coevolutionary perspective. The International Journal of Biochemistry & Cell Biology, 41, 307–322. [DOI] [PubMed] [Google Scholar]
- Colman DR, Toolson EC, Takacs‐Vesbach CD (2012) Do diet and taxonomy influence insect gut bacterial communities? Molecular Ecology, 21, 5124–5137. [DOI] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R (2009) Bacterial community variation in human body habitats across space and time. Science, 326, 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie CR, Scott JA, Summerbell RC, Malloch D (1999) Fungus‐growing ants use antibiotic producing bacteria to control garden parasites. Nature, 398, 701–704. [Google Scholar]
- Dedeine F, Ahrens M, Calcaterra L, Shoemaker DD (2005) Social parasitism in fire ants (Solenopsis spp.): a potential mechanism for interspecies transfer of Wolbachia . Molecular Ecology, 14, 1543–1548. [DOI] [PubMed] [Google Scholar]
- De Fine Licht HH, Boomsma JJ (2010) Forage collection, substrate preparation, and diet composition in fungus‐growing ants. Ecological Entomology, 35, 259–269. [Google Scholar]
- De Fine Licht HH, Boomsma JJ (2014) Variable interaction specificity and symbiont performance in Panamanian Trachymyrmex and Sericomyrmex fungus‐growing ants. BMC Evolutionary Biology, 14, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc F, Metcalf JL, Parfrey LW, Song SJ, González A, Knight R (2014) Convergence of gut microbiomes in myrmecophagous mammals. Molecular Ecology, 23, 1301–1317. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N et al (2006) Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology, 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkshoorn L, Nemec A, Seifert H (2007) An increasing threat in hospitals: multidrug‐resistant Acinetobacter baumannii . Nature Reviews Microbiology, 5, 939–951. [DOI] [PubMed] [Google Scholar]
- Duron O, Wilkes TE, Hurst GDD (2010) Interspecific transmission of a male‐killing bacterium on an ecological timescale. Ecology Letters, 13, 1139–1148. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery C (1909) Über den ursprung der dulotischen, parasitischen und myrmekophilen ameisen. Biologisches Centralblatt, 29, 352–362. [Google Scholar]
- Engel P, Martinson V, Moran N (2012) Functional diversity within the simple gut microbiota of the honey bee. Proceedings of the National Academy of Sciences of the United States of America, 109, 11002–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Maignien L, Sul WJ et al (2013) Oligotyping: differentiating between closely related microbial taxa using 16S rRNA gene data. Methods in Ecology and Evolution, 4, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Armstrong TN (2006) Antagonistic interactions between honey bee bacterial symbionts and implications for disease. BMC Ecology, 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost CL, Fernández‐Marín H, Smith JE, Hughes WOH (2010) Multiple gains and losses of Wolbachia symbionts across a tribe of fungus‐growing ants. Molecular Ecology, 19, 4077–4085. [DOI] [PubMed] [Google Scholar]
- Funaro CF, Kronauer DJ, Moreau CS et al (2011) Army ants harbor a host‐specific clade of Entomoplasmatales bacteria. Applied and Environmental Microbiology, 77, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapaniemi K, Pamilo P (2015) Social parasitism and transfer of symbiotic bacteria in ants (Hymenoptera: Formicidae). Myrmecological News, 21, 49–57. [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD (2004) Past: paleontological statistics software package for education and data analysis. Palaeontologia Electronica, 4, art. 4. [Google Scholar]
- Hansen AK, Jeong G, Paine TD, Stouthamer R (2007) Frequency of secondary symbiont infection in an invasive psyllid relates to parasitism pressure on a geographic scale in California. Applied and Environmental Microbiology, 73, 7531–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CHF, Krych L, Nielsen DS et al (2012) Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia, 55, 2285–2294. [DOI] [PubMed] [Google Scholar]
- Haselkorn TS, Markow TA, Moran NA (2009) Multiple introductions of the Spiroplasma bacterial endosymbiont into Drosophila . Molecular Ecology, 18, 1294–1305.19226322 [Google Scholar]
- Heath BD, Butcher RDJ, Whitfield WGF, Hubbard SF (1999) Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Current Biology, 9, 313–316. [DOI] [PubMed] [Google Scholar]
- Himler AG, Adachi‐Hagimori T, Bergen JE et al (2011) Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science, 332, 254–256. [DOI] [PubMed] [Google Scholar]
- Hoy MA, Jeyaprakash A (2004) Microbial diversity in the predatory mite Metaseiulus occidentalis (Acari: Phytoseiidae) and its prey, Tetranychus urticae (Acari: Tetranychidae). Biological Control, 32, 427–441. [Google Scholar]
- Hu Y, Łukasik P, Moreau CS, Russell JA (2014) Correlates of gut community composition across an ant species (Cephalotes varians) elucidate causes and consequences of symbiotic variability. Molecular Ecology, 23, 1284–1300. [DOI] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Research, 21, 1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak HD, Miller JL, Sen R, Dowd SE, Meyer E, Mueller UG (2011) Microbiomes of ant castes implicate new microbial roles in the fungus‐growing ant Trachymyrmex septentrionalis . Scientific Reports, 1, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J, Polak M, Fiskin A, Helou M, Minhas M (2007) Interspecific transmission of endosymbiotic Spiroplasma by mites. Biology Letters, 3, 23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike J, Unckless R, Cockburn SN, Boelio LM, Perlman SJ (2010) Adaptation via symbiosis: recent spread of a Drosophila defensive symbiont. Science, 329, 212–215. [DOI] [PubMed] [Google Scholar]
- Jiggins FM, Hurst GDD, Jiggins CD, Majerus M (2000) The butterfly Danaus chrysippus is infected by a male‐killing Spiroplasma bacterium. Parasitology, 120, 439–446. [DOI] [PubMed] [Google Scholar]
- Jousselin E, Cœur d'Acier A, Vanlerberghe‐Massuti F, Duron O (2013) Evolution and diversity of Arsenophonus endosymbionts in aphids. Molecular Ecology, 22, 260–270. [DOI] [PubMed] [Google Scholar]
- Kautz S, Rubin BER, Moreau CS (2013) Bacterial infections across the ants: frequency and prevalence of Wolbachia, Spiroplasma, and Asaia . Psyche, Article ID 936341. doi: 10.1155/2013/936341. [Google Scholar]
- Koch H, Schmid‐Hempel P (2011) Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proceedings of the National Academy of Sciences of the United States of America, 108, 19288–19292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual‐index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and Environmental Microbiology, 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Hamady M, Lozupone C et al (2008) Evolution of mammals and their gut microbes. Science, 320, 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longino JT (2010) A taxonomic review of the ant genus Megalomyrmex Forel (Hymenoptera: Formicidae) in Central America. Zootaxa, 2720, 35–58. [Google Scholar]
- Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology, 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall OJ (2004) PerlPrimer: cross‐platform, graphical primer design for standard, bisulphite and real‐time PCR. Bioinformatics, 20, 2471–2472. [DOI] [PubMed] [Google Scholar]
- Martinson VG, Danforth BN, Minckley RL et al (2011) A simple and distinctive microbiota associated with honey bees and bumble bees. Molecular Ecology, 20, 619–628. [DOI] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA (2012) Extreme genome reduction in symbiotic bacteria. Nature Reviews Microbiology, 10, 13–26. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J et al (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME Journal, 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdiabadi NJ, Mueller UG, Brady SG, Himler AG, Schultz TR (2012) Symbiont fidelity and the origin of species in fungus‐growing ants. Nature Communications, 3, 840. [DOI] [PubMed] [Google Scholar]
- Montllor C, Maxmen A, Purcell A (2002) Facultative bacterial endosymbionts benefit pea aphids Acyrtosiphon pisum under heat stress. Ecological Entomology, 27, 189–195. [Google Scholar]
- Moran NA, Tran P, Gerardo NM (2005) Symbiosis and insect diversification: an ancient symbiont of sap‐feeding insects from the bacterial phylum Bacteroidetes. Applied and Environmental Microbiology, 71, 8802–8810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA (2007) Symbiosis as an adaptive process and source of phenotypic complexity. Proceedings of the National Academy of Sciences of the United States of America, 104, 8627–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira DDO, Morais V, Vieira‐da‐Motta O, Campos‐Farinha AEC, Tonhasca A (2005) Ants as carriers of antibiotic‐resistant bacteria in hospitals. Neotropical Entomology, 34, 999–1006. [Google Scholar]
- Muegge BD, Kuczynski J, Knights D et al (2011) Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science, 332, 970–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KM, Russell JA, Moran NA, Hunter MS (2003) Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proceedings of the National Academy of Sciences of the United States of America, 100, 1803–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KM, Degnan PH, Hunter MS, Moran NA (2009) Bacteriophages encode factors required for protection in a symbiotic mutualism. Science, 325, 992–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CD, Phelan G, Dowd SE et al (2012) Microbiome analysis among bats describes influences of host phylogeny, life history, physiology and geography. Molecular Ecology, 21, 2617–2627. [DOI] [PubMed] [Google Scholar]
- Pinto‐Tomás AA, Anderson MA, Suen G et al (2009) Symbiotic nitrogen fixation in the fungus gardens of leaf‐cutter ants. Science, 326, 1120–1123. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP (2010) FastTree 2 – approximately maximum‐likelihood trees for large alignments. PLoS One, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3‐900051‐07‐0, URL http://www.R-project.org/. [Google Scholar]
- Russell JA, Moreau CS, Goldman‐Huertas B et al (2009) Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proceedings of the National Academy of Sciences of the United States of America, 106, 21236–21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA, Funaro CF, Giraldo YM et al (2012) A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLoS One, 7, e51027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiøtt M, De Fine Licht HH, Lange L, Boomsma JJ (2008) Towards a molecular understanding of symbiont function: identification of a fungal gene for the degradation of xylan in the fungus gardens of leaf‐cutting ants. BMC Microbiology, 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T et al (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA‐based studies. PLoS One, 6, e27310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder D, Deppisch H, Obermayer M et al (1996) Intracellular endosymbiotic bacteria of Camponotus species (carpenter ants): systematics, evolution and ultrastructural characterization. Molecular Microbiology, 21, 479–489. [DOI] [PubMed] [Google Scholar]
- Schultz TR, Brady SG (2008) Major evolutionary transitions in ant agriculture. Proceedings of the National Academy of Sciences of the United States of America, 105, 5435–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R, Ishak HD, Estrada D, Dowd SE, Hong E, Mueller UG (2009) Generalized antifungal activity and 454‐screening of Pseudonocardia and Amycolatopsis bacteria in nests of fungus growing ants. Proceedings of the National Academy of Sciences of the United States of America, 106, 17805–17810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948) A mathematical theory of communication. Bell System Technical Journal, 27, 379–423. [Google Scholar]
- Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P (1994) Evolution, weighting, and phylogenetic utility of mitochondrial gene‐sequences and a compilation of conserved polymerase chain‐reaction primers. Annals of the Entomological Society of America, 87, 651–701. [Google Scholar]
- Sperling FAH, Landry JF, Hickey DA (1995) DNA‐based identification of introduced ermine moth species in North America (Lepidoptera: Yponomeutidae). Annals of the Entomological Society of America, 88, 155–162. [Google Scholar]
- Stuart RJ (1981) Abdominal trophallaxis in the slave‐making ant, Harpagoxenus americanus (Hymenoptera: Formicidae). Psyche, 88, 331–334. [Google Scholar]
- Takiya DM, Tran PL, Dietrich CH, Moran NA (2006) Co‐cladogenesis spanning three phyla: leafhoppers (Insecta: Hemiptera: Cicadellidae) and their dual bacterial symbionts. Molecular Ecology, 15, 4175–4191. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, Koga R, Fukatsu T (2004) Host plant specialization governed by facultative symbiont. Science, 303, 1989. [DOI] [PubMed] [Google Scholar]
- Ugelvig LV, Andersen A, Boomsma JJ, Nash DR (2012) Dispersal and gene flow in the rare, parasitic Large Blue butterfly Maculinea arion . Molecular Ecology, 21, 3224–3236. [DOI] [PubMed] [Google Scholar]
- Van Borm S, Buschinger A, Boomsma JJ, Billen J (2002a) Tetraponera ants have gut symbionts related to nitrogen‐fixing root‐nodule bacteria. Proceedings of the Royal Society of London, Series B: Biological Sciences, 269, 2023–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Borm S, Billen J, Boomsma JJ (2002b) The diversity of microorganisms associated with Acromyrmex leaf‐cutting ants. BMC Evolutionary Biology, 2, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Borm S, Wenseleers T, Billen J, Boomsma JJ (2003) Cloning and sequencing of wsp encoding gene fragments reveals a diversity of co‐infecting Wolbachia strains in Acromyrmex leafcutter ants. Molecular Phylogenetics and Evolution, 26, 102–109. [DOI] [PubMed] [Google Scholar]
- Vorburger C, Gehrer L, Rodriguez P (2010) A strain of the bacterial symbiont Regiella insecticola protects aphids against parasitoids. Biology Letters, 6, 109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Brady SG, Fisher BL, Schultz TR (2015) The evolution of myrmicine ants: phylogeny and biogeography of a hyperdiverse ant clade (Hymenoptera: Formicidae). Systematic Entomology, 40, 61–81. [Google Scholar]
- Wheeler DE (1984) Behavior of the ant, Procryptocerus scabriusculus (Hymenoptera: Formicidae), with comparisons to other Cephalotines. Psyche, 91, 171–192. [Google Scholar]
- Xie J, Butler S, Sanchez G, Mateos M (2014) Male killing Spiroplasma protects Drosophila melanogaster against two parasitoid wasps. Heredity, 112, 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Rarefaction curves of tag‐encoded FLX 454 pyrosequencing samples (A) and Illumina MiSeq diet experiment samples (B).
Fig. S2 Principal Coordinates Analyses (PCoA) of unweighted UniFrac metrics based on 97% OTUs (A and C) or unique sequences (B and D) and PCoA bi‐plots of weighted UniFrac metrics based on 97% OTUs (E) or unique sequences (F).
Fig. S3 Phylogenetic trees of all 69 bacterial orders identified across attine and Megalomyrmex ants from the 454 16S rRNA pyrosequencing dataset.
Fig. S4 Phylogenetic trees of all 255 bacterial phylotypes identified across attine and Megalomyrmex ants from the 454 16S rRNA pyrosequencing dataset.
Fig. S5 Proportions of shared phylotypes in hosts and social parasites from joint nests as estimated from the 454 16S rRNA pyrosequencing dataset.
Fig. S6 Taxonomic assignment of 454 16S rRNA gene amplicons of Sericomyrmex amabilis hosts, Megalomyrmex symmetochus guest ants, and their fungus gardens as produced by megan4 (Huson et al. 2011).
Fig. S7 Distribution of 16S rRNA genotypes as identified by oligotyping from the 454 16S rRNA pyrosequencing dataset based on all reads identified as Pseudonocardia, Wolbachia and Acinetobacter.
Fig. S8 Maximum Likelihood phylogeny based on c. 520 bp of the Wolbachia wsp gene from a subsample of positive PCRs in our diagnostic screening, aligned with closely related sequences obtained from GenBank.
Fig. S9 Non‐metric multidimensional scaling of Bray‐Curtis dissimilarities between the bacterial communities identified in the diet manipulation experiment samples, calculated in two separate analyses from a matrix of relative abundances (A and B) and from a presence‐absence matrix (C and D).
Table S1 DNA extractions with details of the species sampled, numbers of individuals pooled in each extraction, nest IDs, sampling locations, GPS coordinates and diagnostic PCR results for the wingless (wgls) host nuclear gene, Entomoplasmatales, Bartonellaceae and Wolbachia.
Table S2 Taxonomic assignment and number of reads per sample of all the 97% OTUs and unique sequences identified in the 454 rRNA gene pyrosequencing dataset.
Table S3 Primers, lengths of amplified fragments, and PCR conditions used in this study.
Table S4 Summary of statistical results from network and UniFrac clustering analyses at both the 97% OTU and unique sequence levels.
Table S5 Number of reads per sample of all genotypes identified by Oligotyping of the five most abundant bacterial lineages in the 454 rRNA gene pyrosequencing dataset, with the top blastn hit for each genotype reported towards the right of the tables.
Table S6 Taxonomic assignment and number of reads per sample of all the 852 (97% cutoff) OTUs identified in the diet experiment MiSeq Illumina dataset.
Table S7 List of OTUs per nest that were consistently shared in all treatment groups between M. symmetochus guest ants and S. amabilis host ants samples from the diet experiment Miseq Illumina dataset.
Table S8 Results of permanova comparisons between the treatment groups based on Bray‐Curtis dissimilarities between the bacterial communities identified from the ant samples of the diet experiment.
Table S9 SIMPER results showing OTUs with differences in relative abundance between the start of the experiment and the sucrose treatment.
Appendix S1 Additional methodologies and list of GenBank Accessions used to build the bacterial phylogenies.
Data Availability Statement
The NGS data sets were deposited at the NCBI Sequence Read Archive under the Accession no. SRP052752. COI (LC025532–LC025612), wingless (LC027779–LC027860), wsp (LC027861–LC027874) and 16S rRNA gene fragments for Entomoplasmatales (LC027737–LC027760) and Bartonellaceae (LC027761–LC027778) were deposited at the DNA Data Bank of Japan (DDBJ).
Sequence alignments and tree files, mapping file with barcode sequences for the 454 data set, OTU tables, taxonomical assignments of the OTUs and unique sequences, representative sequences for all OTUs and unique sequences and input alignments for the oligotyping analyses are accessible from the Dryad Digital Repository (doi: 10.5061/dryad.ft36k).