

Abstract
Background and purpose
Beta‐interferons (IFNβ) are the most widely prescribed drugs for patients with multiple sclerosis (MS). However, whether or not treatment with IFNβ can delay secondary progressive MS (SPMS) onset remains unknown. Our aim was to examine the association between IFNβ exposure and SPMS onset in patients with relapsing−remitting MS (RRMS).
Methods
A retrospective cohort study using British Columbia (Canada) population‐based clinical and health administrative data (1985–2008) was conducted. RRMS patients treated with IFNβ (n = 794) were compared with untreated contemporary (n = 933) and historical (n = 837) controls. Cohort entry was the first clinic visit during which patients became eligible for IFNβ treatment (baseline). The outcome was time from baseline to SPMS onset. Cox regression models with IFNβ as a time‐dependent exposure were adjusted for sex, and baseline age, disease duration, disability, *socioeconomic status and *comorbidities (*available for the contemporary cohorts only). Additional analyses included propensity score adjustment.
Results
The median follow‐up for the IFNβ‐treated, untreated contemporary and historical controls were 5.7, 3.7 and 7.3 years, and the proportions of patients reaching SPMS were 9.2%, 11.8% and 32.9%, respectively. After adjustment for confounders, IFNβ exposure was not associated with the risk of reaching SPMS when either the contemporary or the historical untreated cohorts were considered (hazard ratio 1.07; 95% confidence interval 0.93–1.48, and hazard ratio 1.04; 95% confidence interval 0.74–1.46, respectively). Further adjustments and the propensity score yielded results consistent with the main analysis.
Conclusions
Amongst patients with RRMS, use of IFNβ was not associated with a delayed onset of SPMS.
Keywords: beta‐interferon, cohort study, multiple sclerosis, progression
Introduction
Multiple sclerosis (MS) is a chronic degenerative disease of the central nervous system and the most common cause of neurological disability in young adults in the western world 1. Despite recent advances in the treatment for MS, the beta‐interferons (IFNβ) remain the most widely prescribed immunomodulatory drugs (IMDs) for patients with relapsing−remitting MS (RRMS) 2, the most common form of MS 3. However, over time, patients with RRMS may transition to secondary progressive MS (SPMS) 4, 5, synonymous with poor health outcomes and a diminished response to the currently licensed IMDs 6.
The IFNβs were licensed for RRMS based on short‐term randomized clinical trials which typically showed a one‐third reduction in relapse rates and benefits on magnetic resonance imaging associated with IFNβ treatment 7, 8. However, the longer‐term impact of the IFNβs has not been well established, especially with respect to delaying or preventing the onset of SPMS 9. To date, three longitudinal cohort studies have examined the role of IFNβ in relation to SPMS in the post‐marketing setting 10, 11, 12. These studies are challenging to conduct and, whilst important, they may have suffered from fundamental biases including immortal time bias (different start times for the treated and untreated groups) 13, 14 and selection bias 14.
Whether current drug treatments for RRMS can delay SPMS onset remains unknown. Indeed, the lack of treatment options to prevent or modify SPMS has been highlighted as a major unmet need by a recent international collaborative group 15. To fill this critical knowledge gap, the impact of IFNβ exposure on SPMS onset in an MS cohort in British Columbia (BC), Canada, was examined.
Methods
Data sources
Study patients were selected from the British Columbia Multiple Sclerosis (BCMS) clinic database (established 1980) which collates information on both incident and prevalent cases, comprising approximately 80% of MS patients 16 in BC (until the end of 2004, the last year all MS clinics in BC participated). This database contains clinical information, including disability [the Expanded Disability Status Scale (EDSS) score], date of symptom onset, disease course, relapses and use of IMDs, and has been extensively used 3, 4, 17, 18.
Additional information was obtained through province‐wide registry linkages: IFNβ use was captured in both the BCMS database and BC's PharmaNet database 19, which contains information on outpatient prescriptions dispensed in BC since 1 January 1996. Comorbidity information was obtained via the Medical Service Plan (MSP) 20 and Discharge Abstract Database (DAD) 21. The MSP files contain fee‐for‐service physician billing records, whilst the DAD contains hospital admission and discharge records; both use the International Classification of Disease system. Socioeconomic status (SES) was obtained from Census Geodata which enables neighbourhood income to be converted to SES estimates using Statistics Canada's algorithm 22, 23. Data linkage was performed at the individual level using each patient's unique personal health number, facilitated by Population Data BC.
Design and setting
This was a retrospective cohort study, using a similar approach to that outlined previously 18. Briefly, the study cohort included adults with RRMS (according to the Poser or McDonald criteria) 24, 25, registered with a BCMS clinic and eligible for IFNβ treatment between April 1985 and December 2004. Eligibility was adapted from the BC government's reimbursement policy (≥18 years, with RRMS and an EDSS ≤6.5). The cohort entry (baseline) was the first clinic visit at which eligibility was reached. Follow‐up was to the study outcome (SPMS, described below) or the last recorded MS clinic visit prior to study end (31 December 2008), whichever came first. Figure 1 outlines the selection of individuals from the BCMS database.
Figure 1.
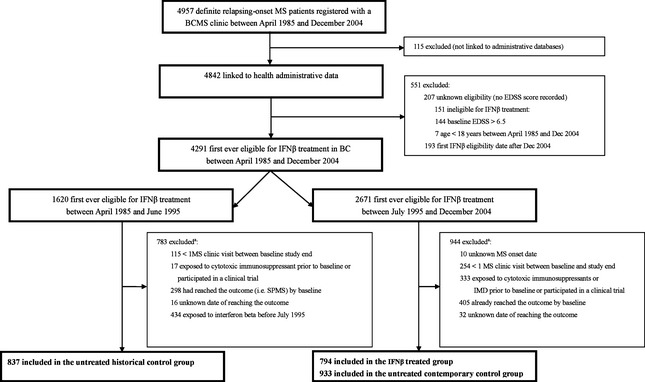
Selection of IFNβ‐treated and untreated cohorts from the BCMS database for the main analysis. BCMS, British Columbia Multiple Sclerosis; IMDs, immunomodulatory drugs; EDSS, Expanded Disability Status Scale; MS, multiple sclerosis; IFNβ, beta‐interferons; SPMS, secondary progressive multiple sclerosis. aThe sum of the individual reasons for exclusion exceeds the total number of patients because some patients met more than 1 condition.
Defining treated and untreated comparison cohorts
The IFNβ‐treated cohort included those first eligible between July 1995 and December 2004 (IFNβ received Canadian approval in 1995). To minimize potential period effects and indication bias, two separate comparator cohorts were used – one contemporary and one historical. The contemporary cohort included patients first eligible but who remained unexposed to IFNβ between July 1995 and December 2004, whilst the historical cohort comprised those first eligible prior to IFNβ approval (April 1985–June 1995) but who remained unexposed to IFNβ during the study period.
Measuring IFNβ exposure
All IFNβs were considered as one therapeutic group; switching was not considered as stopping as long as the prescription gap was ≤3 months (<5% of patients had a gap >3 months).
Measure of study outcome
The study outcome was time from baseline to the onset of SPMS, determined clinically by the treating MS neurologist using the internationally recognized definition of SPMS, i.e. presence of a progressive course with or without superimposed relapses in patients with an initial relapsing−remitting disease course 5, as used in previous studies 4, 10, 11, 26, 27, 28.
Comorbidity
The presence of comorbidity might be associated with both exposure and outcome (e.g. alter the decision surrounding IFNβ utilization or impact MS progression), and so was included as a baseline covariate using the Deyo validated adaptation of the Charlson comorbidity index 29.
Statistical analyses
Basic demographic and clinical characteristics of the three groups were compared using appropriate tests.
Cox proportional hazards regression models, with IFNβ exposure as a time‐dependent variable, were used to assess the hazard of disease progression (time to SPMS onset). This approach allowed the time from baseline to the first IFNβ prescription as well as the time from stopping IFNβ to the end of follow‐up to contribute to the untreated follow‐up time. The models were adjusted for potential confounding factors including age, sex, disease duration, comorbidity index, SES and disability (indicated by the EDSS) at baseline. SES and the comorbidity index were only used to adjust the comparison between the treated and contemporary untreated cohorts as they were not available for the historical cohort. The proportional hazards assumption for baseline covariates was examined using log−log plots; no violations were found.
To investigate whether MS patients with certain baseline characteristics might benefit differently from IFNβ treatment, the association between IFNβ exposure and SPMS onset according to baseline characteristics was further examined. For this analysis, patient subgroups were defined using four clinically relevant baseline characteristics: sex (male, female); age (≤30 years, >30–45 years, >45 years); disease duration (≤2 years, >2–5 years, >5–10 years, >10 years); EDSS (≤1.5, 2–2.5, ≥3). Because relapses may impact MS progression within specific subgroups 17, 30, the association according to annualized relapse rate [ARR (≤1, >1)] in the 2 years pre‐baseline (the onset attack was not counted as a relapse) was also explored. Cox proportional hazards model results were compared with and without the interaction term between IFNβ exposure and the baseline characteristic of interest (all adjusted for the remaining four baseline characteristics).
Supplementary analyses
In order to test key assumptions as well as to fully explore the association between IFNβ exposure and onset of SPMS, several supplementary analyses were conducted (Data S1). Briefly, the association between IFNβ exposure and SPMS onset was also examined: (i) with adjustment for comorbidities using an alternative measure – the Expanded Diagnosis Cluster codes 31; (ii) in relation to relapse history, with the ARR 18 in the 2 years pre‐baseline included as an additional covariate in the Cox proportional hazards models developed in the primary analysis; (iii) using propensity score adjustments. In a final supplementary analysis, (iv) whether there were any underlying temporal trends in the risk of reaching SPMS which might confound the association between IFNβ exposure and the onset of SPMS was explored.
Statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS) (IBM SPSS Statistics for Windows, Version 20.0. Released 2011, IBM Corp., Armonk, NY, USA) and R 32 (version 3.0.2).
The University of British Columbia's Clinical Research Ethics Board approved the study, which includes informed patient consent.
Results
From a cohort of 4957, a total of 2564 patients were eligible for IFNβ treatment between April 1985 and December 2004. In total, 794 formed the IFNβ‐treated cohort, 933 the contemporary control cohort and 837 the historical untreated cohort (Fig. 1). Of those included as contemporary controls, 110 (4%) filled a prescription for glatiramer acetate between cohort entry and end. Demographics were comparable between the excluded and included patients: 73.1% of excluded patients were female with a mean age at MS onset of 31.8 (SD 10.1) years versus 76.0% of included patients whose mean age at MS onset was 32.0 (SD 9.3) years.
Baseline differences between the treated and control groups were largely not clinically significant (Table 1). However, final results produced by multivariable models were adjusted for these baseline characteristics. Compared to the treated cohort, the contemporary untreated group had similar sex and EDSS score distributions, comorbidity status and SES, but were older, had a lower relapse rate in the 2 years pre‐baseline and longer disease duration. The historical untreated group were slightly older, with a lower EDSS score and longer disease duration at baseline (Table 1).
Table 1.
Baseline demographic and clinical characteristics of the study cohortsa
| Characteristics | IFNβ‐treated patients (n = 794) | Contemporary untreated patients (n = 933)b | P value | Historical untreated patients (n = 837)c | P value |
|---|---|---|---|---|---|
| Sex, n (%) | |||||
| Male | 187 (23.6) | 218 (23.4) | 0.96d | 211 (25.2) | 0.45d |
| Female | 607 (76.4) | 715 (76.6) | 626 (74.8) | ||
| Age at MS onset, years, mean (SD) | 31.8 (9.1) | 33.1 (9.5) | 0.004e | 31.0 (9.2) | 0.07e |
| n (%) | |||||
| <20 | 57 (7.2) | 67 (7.2) | 85 (10.2) | ||
| 20–<30 | 324 (40.8) | 313 (33.5) | 329 (39.3) | ||
| 30–<40 | 266 (33.5) | 337 (36.1) | 0.02d | 287 (34.3) | 0.22d |
| 40–<50 | 121 (15.2) | 169 (18.1) | 110 (13.1) | ||
| ≥50 | 26 (3.3) | 47 (5.0) | 26 (3.1) | ||
| Disease duration, years | |||||
| Mean (SD) | 6.0 (6.9) | 8.5 (8.6) | <0.001e | 7.9 (8.2) | <0.001e |
| Median (IQR) | 3.1 (1.1–8.7) | 5.8 (1.9–12.5) | 5.1 (1.5–12.1) | ||
| Age at baseline, years, mean (SD) | 37.3 (9.1) | 41.1 (10.1) | <0.001e | 38.3 (9.7) | 0.03e |
| n (%) | |||||
| <30 | 169 (21.3) | 120 (12.8) | 151 (18.0) | ||
| 30–<40 | 301 (37.9) | 308 (33.0) | <0.001d | 324 (38.7) | 0.39d |
| 40–<50 | 243 (30.6) | 310 (33.2) | 259 (30.9) | ||
| ≥50 | 81 (10.2) | 195 (20.9) | 103 (12.3) | ||
| EDSS score | |||||
| Mean (SD) | 2.2 (1.3) | 2.1 (1.4) | 0.07e | 2.0 (1.4) | 0.03e |
| Median (range) | 2.0 (0–6.5) | 2.0 (0–6.5) | 0.03f | 1.5 (0–6.5) | 0.001f |
| N (%) | |||||
| 0 | 80 (10.1) | 121 (13.0) | 93 (11.1) | ||
| 1–1.5 | 227 (28.6) | 274 (29.4) | 0.29d | 327 (39.1) | <0.001d |
| 2–2.5 | 270 (34.0) | 319 (34.2) | 197 (23.5) | ||
| 3–3.5 | 140 (17.6) | 136 (14.6) | 139 (16.6) | ||
| 4–4.5 | 39 (4.9) | 35 (3.8) | 36 (4.3) | ||
| 5–5.5 | 13 (1.6) | 14 (1.5) | 18 (2.2) | ||
| 6–6.5 | 25 (3.1) | 34 (3.6) | 27 (3.2) | ||
| Annualized relapse rate in the 2 years before baselineg | |||||
| Mean (SD) | 0.6 (0.6) | 0.4 (0.5) | <0.001e | 0.6 (0.6) | 0.24e |
| Median (IQR) | 0.5 (0–1.0) | 0.5 (0–0.5) | 0.5 (0–1.0) | ||
| Follow‐up time (eligibility date to the end of follow‐upj) | |||||
| Mean (SD) | 5.9 (2.9) | 4.3 (3.1) | <0.001e | 8.4 (6.0) | <0.001e |
| Median (IQR) | 5.7 (3.8–8.0) | 3.7 (1.6–6.6) | 7.3 (2.8–13.2) | ||
| Charlson comorbidity indexh | |||||
| Median (range) | 0 (0–3) | 0 (0–3) | NA | ||
| Score, n (%) | |||||
| 0 (no comorbidity) | 783 (98.6) | 909 (97.4) | 0.09d | NA | |
| ≥1 (at least one comorbid condition) | 11 (1.4) | 24 (2.6) | |||
| Neighbourhood income quintile,i n (%) | |||||
| 1 (lowest income) | 144 (18.8) | 166 (18.7) | |||
| 2 | 130 (17.0) | 180 (20.3) | |||
| 3 | 168 (21.6) | 178 (20.1) | 0.49f | NA | |
| 4 | 166 (21.7) | 181 (20.4) | |||
| 5 (highest income) | 157 (20.5) | 182 (20.5) | |||
IFNβ, beta interferons; EDSS, Expanded Disability Status Scale; IQR, interquartile range; NA, data not available/incomplete. aBaseline was considered as the first date a patient became eligible for IFNβ treatment; buntreated patients who first became eligible for treatment in the ‘IFNβ era’ (between July 1995 and December 2004); cuntreated patients who first became eligible for treatment in the ‘pre‐IFNβ era’ (between April 1985 and June 1995); dPearson's chi‐squared test; eStudent's t test; fMann–Whitney–Wilcoxon test; gif this period included multiple sclerosis symptom onset, this first attack was not included as a relapse; hDeyo adaptation of the Charlson comorbidity index 24, based on hospital admissions or physician visits in the 2 years prior to baseline and derived from the International Classification of Diseases, 9th Revision, Clinical Modification (ICD‐9‐CM) codes, excluding hemiplegia, paraplegia and dementia to avoid misclassifying complications of MS as comorbidity. All relevant comorbidities are aggregated into a single variable theoretically ranging from 0 to 33; higher scores indicate greater burden of comorbidity; iused as a proxy for socioeconomic status. Data were missing for 20 patients in the IFNβ‐treated cohort and 30 patients in the contemporary control cohort; jpatients were followed until they reached the outcome of secondary progressive multiple sclerosis, or until their last MS clinic visit or start of participation in an MS ‘disease‐modifying drug’ related clinical trial.
The treated cohort contributed 2774 patient‐years of IFNβ exposed time and 1882 patient‐years of unexposed time. The contemporary and historical untreated cohorts contributed 4017 and 7033 patient‐years of unexposed time, respectively. The follow‐up time (eligibility date to the end of follow‐up) differed between groups; it was considerably longer for the historical untreated cohort [median 7.3 years, interquartile range (IQR) 2.8–13.2 years]. The median follow‐up time was 5.7 years (IQR 3.8–8.0 years) for the IFNβ‐treated cohort and 3.7 years (IQR 1.6–6.6 years) for the contemporary untreated cohort.
In total, 9.2% of treated patients reached SPMS, whilst 11.8% of contemporary and 32.9% of historical untreated patients reached SPMS during the follow‐up period. The median time to SPMS onset was 3.7 years for the treated cohort, 2.0 years for the contemporary untreated cohort and 4.4 years for the historical untreated cohort.
Exposure to IFNβ was not associated with the hazard of reaching SPMS when the IFNβ‐treated and contemporary control groups were compared [adjusted hazard ratio 1.07; 95% confidence interval (CI) 0.93–1.48) (Fig. 2]. Similar findings were observed when comparing the treated and the historical untreated group (adjusted hazard ratio 1.04; 95% CI 0.74–1.46) (Fig. 2). Findings remained virtually unchanged when SES and the comorbidity index were also included as covariates in the contemporary analysis (Fig. 3).
Figure 2.
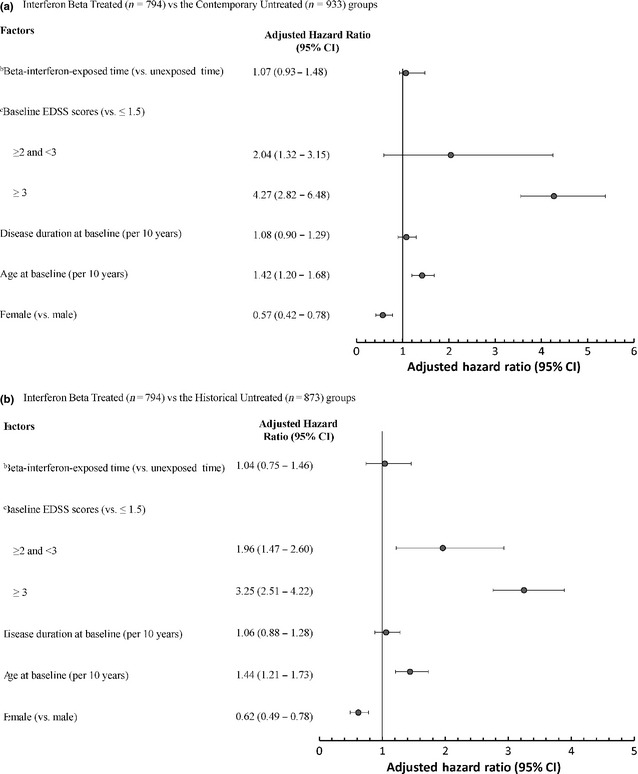
Multivariable time‐dependent Cox regression analysis of potential factors associated with time to SPMS a onset for IFNβ‐treated versus untreated cohorts. (a) IFNβ‐treated (n = 794) versus the contemporary untreated (n = 933) group. (b) IFNβ‐treated (n = 794) versus the historical untreated (n = 873) group. a SPMS, secondary progressive multiple sclerosis. b IFNβ, beta‐interferons; IFNβ exposure was modelled as a time‐dependent variable. c EDSS, Expanded Disability Status Scale score which ranged from 0 to 6.5 at baseline.
Figure 3.
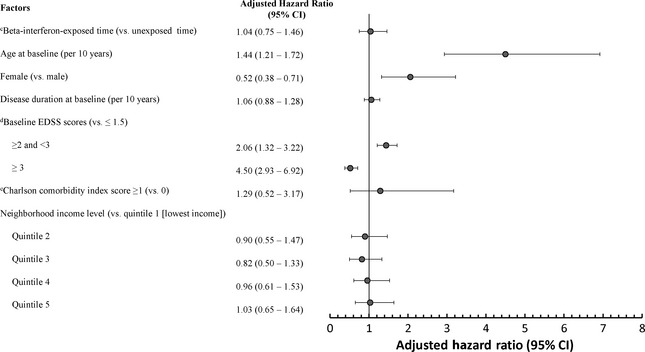
Time‐dependent Cox regression analysis of potential factors associated with time to SPMS a onset for IFNβ‐treated versus the contemporary untreated cohortb (with additional adjustments for socioeconomic status and the Charlson comorbidity index). a SPMS, secondary progressive multiple sclerosis. b IFNβ, beta‐interferons; IFNβ‐treated patients, n = 794; contemporary untreated patients, n = 933. cIFNβ exposure was modelled as a time‐dependent variable. d EDSS, Expanded Disability Status Scale score which ranged from 0 to 6.5 at baseline. eDeyo adaptation of the Charlson comorbidity index 24, based on hospital admissions or physician visits in the 2 years prior to baseline and derived from the International Classification of Diseases, 9th Revision, Clinical Modification (ICD‐9‐CM) codes, excluding hemiplegia, paraplegia and dementia to avoid misclassifying complications of MS as comorbidity. All relevant comorbidities are aggregated into a single variable theoretically ranging from 0 to 33; higher scores indicate greater burden of comorbidity.
No statistically significant differences in the associations between IFNβ exposure and SPMS onset were found across subgroups based on likelihood ratio tests (Fig. 4). However, for patients with an ARR > 1, there was a trend towards a lower hazard of reaching SPMS for IFNβ exposed versus unexposed time (hazard ratio 0.62; 95% CI 0.24–1.55 for the contemporary approach; hazard ratio 0.60; 95% CI 0.26–1.36 for the historical approach). This reduction was not observed in those with an ARR ≤ 1.
Figure 4.
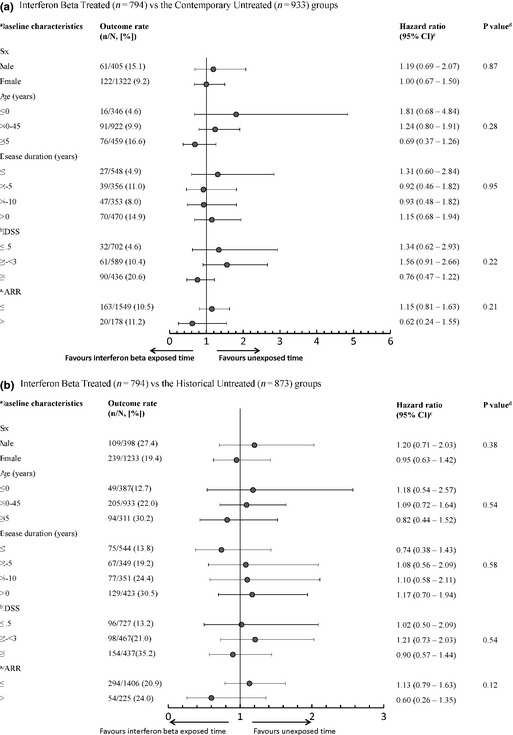
Association between IFNβ exposure and onset of SPMS amongst IFNβ‐treated patients and untreated patients according to each baseline characteristic. (a) IFNβ‐treated (n = 794) versus the contemporary untreated (n = 933) group. (b) IFNβ‐treated (n = 794) versus the historical untreated (n = 873) group. aBaseline was considered as the first clinic visit when a patient became eligible for beta interferon (IFNβ) treatment. b EDSS, Expanded Disability Status Scale; ARR, annualized relapse rate. cThe hazard ratio of IFNβ exposure for the corresponding baseline characteristics level estimated using Cox regression models adjusting for the main effects of all the baseline characteristics, the IFNβ exposure as well as the interaction term between IFNβ exposure and the baseline characteristic of interest. d P values for testing the interaction between IFNβ exposure and the baseline characteristic of interest based on the likelihood ratio test which compares the models with and without the interaction term. eIf this period included the onset attack, this was not counted as a relapse. fOutcome rates indicate how many patients were observed to reach the outcome (onset of SPMS) for each level of the baseline characteristic.
Results of supplementary analyses are detailed online (Table S1; Figs S1–S3). Briefly, no statistically significant association between IFNβ exposure and SPMS onset was found when either individual comorbidities or the ARR were included as additional covariates, or in the propensity score adjusted model. Finally, time from MS onset to SPMS was not significantly different across the different time periods explored (Table S1; Figures S2, S3).
Discussion
It was found that exposure to IFNβ was not associated with the clinical onset of SPMS in the 5–6 years following treatment initiation in a cohort of patients with RRMS from British Columbia, Canada. This finding was consistent regardless of whether a contemporary or a historical (pre‐ versus post‐IFNβ era) untreated comparison cohort was considered or if propensity score adjustment was performed. Further, none of the baseline characteristics considered could be identified as indicating a response to drug treatment with respect to reaching SPMS, including sex, age, disease duration, disability (EDSS) or ARR. Findings could also be considered as reassuring given that concerns were raised after the pivotal clinical trials of IFNβ were published that they might hasten SPMS onset 33, 34. No evidence was found to support this concern.
A limited number of previous post‐marketing, longitudinal observational studies have also investigated the relationship between IFNβ exposure and time to reach SPMS and have suggested a beneficial association 10, 11, 12. However, given the challenges in conducting rigorous pharmaco‐epidemiological studies, these studies could be prone to particular methodological shortcomings. One study 10 followed 1504 patients with RRMS for up to 7 years, but the design was susceptible to immortal time bias due to differing start times for the treatment and control groups 13, which was not accounted for in the propensity score analysis. An independent re‐analysis that corrected for this reported no significant impact of IFNβ exposure on SPMS onset 13, and others have shown that a propensity score alone cannot address this imbalance in immortal time 13, 35. A more recent study examined the risk of reaching SPMS in 730 contemporary patients treated with IFNβ or glatiramer acetate from the Swedish MS Registry (MS onset 1995–2004) and compared them to 186 historical untreated patients from the Gothenburg Incidence Cohort (MS onset between 1950 and 1964) 11. They reported a lower risk of reaching SPMS in the contemporary treated cohort compared to the historical untreated cohort, although the authors reported that it was ‘difficult to disentangle’ period effects from possible drug effects 11. The differences in time periods between the two groups was compounded further by their selection from disparate populations 11; the earlier incidence cohort was population‐based from one region in Sweden, whereas the later treated cohort represented patients from different regions within a registry which was not population‐based, covering less than 60% of the Swedish MS population 11, 36. Finally, the IFNβ‐treated and the historical untreated groups were imbalanced in terms of follow‐up; the treated group was followed from the initiation of drug whereas the control group was followed from the onset of disease. This imbalance is a major problem in pharmaco‐epidemiological studies and may bias the association between IFNβ exposure and SPMS 13.
Our study was designed to address these limitations. First, the cohort entry (start of follow‐up) was comparable between groups and IFNβ exposure was treated as a time‐dependent variable, taking into account the drug unexposed time period during follow‐up and ensuring the same start times across groups 13. Secondly, it was possible to select both the treated and untreated cohorts from the same population and explore whether a period effect could have influenced our findings. Thirdly, the inclusion of two separate untreated cohorts – a historical group and contemporary group (i.e. pre‐ and post‐ IFNβ era) − was another study strength; the historical cohort minimized the potential for indication bias, whilst the contemporary cohort provided another means of addressing potential period effects that can be difficult to measure. Fourthly, the longitudinal data with observations spanning up to 20 years and a population‐based cohort of MS patients allowed sufficient follow‐up 37. Finally, it was possible to link our MS clinic cohort with provincial health administrative databases, providing a rich, reliable and objective data source that allowed for adjustment of important potential confounders such as comorbidity and SES.
The reasons why patients who were eligible for IFNβ but chose not to initiate therapy are probably complex and multifactorial, but may include perceived stable disease, unwillingness to receive a non‐curative treatment, or needle phobia. Eligible patients who did not start IFNβ were found to be at a somewhat lower risk of reaching SPMS in both the main and supplementary analyses (findings did not reach significance), despite model adjustment for patients' characteristics, such as disease duration and disability (EDSS). This suggests residual confounding due to ‘indication bias’ 18, i.e., more rapidly progressing patients were more likely to be exposed to drug.
A trend towards a lower hazard of reaching SPMS for IFNβ exposed versus unexposed time was observed in patients with an ARR > 1, which suggests that IFNβ treatment may benefit patients whose disease is in an active inflammatory phase (i.e. a higher relapse rate). However, this finding did not reach statistical significance and further confirmatory studies are needed.
Our overall findings do concur with our previous study in which no association was observed between IFNβ exposure and disability progression indicated by time to confirmed and sustained EDSS scores of 6.0 and 4.0 30. In addition, findings are consistent with results from the 16‐year follow‐up of MS patients who were randomized to receive placebo or IFNβ treatment in a 2‐year clinical trial which did not demonstrate a significant impact of IFNβ exposure on SPMS 26.
Our study has some limitations. Patients eligible for IFNβ treatment during the post‐ IFNβ era but who did not receive treatment may have a better clinical status compared to those who received treatment 18. This confounding by indication was demonstrated in a re‐analysis of the Italian study 10, 13, which used a contemporary comparison cohort. However, by including a historical untreated group it was possible to minimize this potential issue. It was not possible to consider neutralizing antibodies, which when titres are high may be associated with reduced IFNβ effectiveness 38. Although a variety of confounding factors were considered, the possibility cannot be ruled out that unmeasured confounders still existed. Only patients attending a BCMS clinic could be considered in our study population and it is possible that the presence of very mild or very severe disease would differentially prevent attendance at clinic for untreated patients. However, a systematic occurrence of one of these scenarios seems unlikely. Our findings were based on reaching SPMS. Although this is considered a relevant outcome and important landmark in the disease course of MS 26, 27, 39, it is primarily determined clinically and after a retrospective evaluation of disease activity. Although no formal validation of this outcome could be found, it is reassuring that the median time to SPMS from disease onset, as estimated by survival analyses, is comparable between studies from different regions and countries (averaging around 18–20 years across cohorts) 3, 4. Finally, the duration of follow‐up was modest for the contemporary cohorts and the disease duration at entry was relatively short which may contribute to the relatively low occurrence of SPMS compared to that reported in previous population‐based natural history studies (in which 40%–60% of patients developed SPMS over 10–20 years) 4.
In conclusion, no association was found between IFNβ exposure and SPMS onset in patients with RRMS in the 5–6 years following start of treatment. Our findings address concerns that were raised in the mid‐1990s that these medications might hasten SPMS onset 33, 34. Further ‘real‐world’ longitudinal studies are encouraged to confirm our findings, ideally with additional metadata such as genomic or biomarker information that might prove fruitful in identifying whether specific patient subgroups benefit from regular IFNβ use. Our findings also encourage the continued investigation of novel therapeutics to prevent or delay progressive MS.
Disclosure of conflicts of interest
Messrs Duggan and Zhu were compensated through study research grants. Dr Shirani is funded through a postdoctoral fellowship from the Multiple Sclerosis Society of Canada and grants from the CIHR (MOP‐93646) and the NMSS (RG 4202‐A‐2). Dr Zhao receives research funding from the CIHR, the Multiple Sclerosis Society of Canada and the NMSS. Dr Evans is funded through grants from the CIHR (MOP‐93646), the NMSS (RG 4202‐A‐2), and the Michael Smith Foundation for Health Research. Dr Kingwell is supported by postdoctoral fellowships from the Multiple Sclerosis Society of Canada and the Michael Smith Foundation for Health Research. Dr Oger receives support from the Christopher Foundation and the University of British Columbia (UBC). He receives fees for service from the Medical Services Commission of British Columbia. Dr Gustafson is supported by the Natural Sciences and Engineering Research Council of Canada. Dr Petkau holds research grants from the CIHR, the Multiple Sclerosis Society of Canada, the NMSS, and the Natural Sciences and Engineering Research Council of Canada. Dr Tremlett is funded by the Multiple Sclerosis Society of Canada (Don Paty Career Development Award), is a Michael Smith Foundation for Health Research Scholar, and is the Canada Research Chair for Neuroepidemiology and Multiple Sclerosis. She has also received research support from the NMSS, CIHR and UK Multiple Sclerosis Trust. The BCMS database has been funded from various sources (including the above) and also by an unrestricted grant from Donald Paty, MD, FRCPC, University of British Columbia, and the MS/MRI Research Group. Dr Zhang reports no disclosure.
Supporting information
Data S1. Supplementary analyses.
Figure S1. Time‐dependent Cox regression analysis of potential factors associated with time to SPMS a onset for IFNβ‐treated versus the contemporary untreated cohortb (with additional adjustment of SES and high impact/prevalence comorbidities).
Figure S2. Selection of the study population from the British Columbia Multiple Sclerosis clinic database: patients with relapsing‐onset MS with symptom onset between 1975 and 1995.
Figure S3. Survival curves of time from MS symptom onset to SPMS occurrence within 10 years' disease duration, by 5‐year onset intervals.
Table S1. Multivariable Cox regression analysis of potential factors affecting time to reach SPMS within 10 years from disease onset.
Acknowledgements
We gratefully acknowledge the BCMS clinic neurologists who contributed to the study through patient examination and data collection (current members have been listed in alphabetical order). UBC MS Clinic: A. Traboulsee, MD, FRCPC (UBC Hospital MS Clinic Director and Head of the UBC MS Programs); A.‐L. Sayao, MD, FRCPC; V. Devonshire, MD, FRCPC; S. Hashimoto, MD, FRCPC (UBC and Victoria MS Clinics); J. Hooge, MD, FRCPC (UBC and Prince George MS Clinic); L. Kastrukoff, MD, FRCPC (UBC and Prince George MS Clinic); J. Oger, MD, FRCPC. Kelowna MS Clinic: D. Adams, MD, FRCPC; D. Craig, MD, FRCPC; S. Meckling, MD, FRCPC. Prince George MS Clinic: L. Daly, MD, FRCPC. Victoria MS Clinic: O. Hrebicek, MD, FRCPC; D. Parton, MD, FRCPC; K Atwell‐Pope, MD, FRCPC. We also thank P. Rieckmann, MD (Sozialstiftung Bamberg Hospital, Bamberg, Germany) for helpful revisions of the original Canadian Institutes of Health Research grant. None has received compensation for their role in the study. We also thank the UBC MS clinic nurses and staff and the UBC's Clinical Trials Group. We are grateful to Tom Duggan, BA, University of British Columbia, for significant help with data manipulation and conversion and the Pharmacoepidemiology in MS Research Group for research support. We are thankful to Population Data BC and the British Columbia Ministry of Health for support with linkage to British Columbia administrative health care and health services data (hospital separations and medical service plan payment information), as well as PharmaNet for drug information. Finally, we are indebted to all MS patients who participated in this study.
References
- 1. Leary SM, Porter B, Thompson AJ. Multiple sclerosis: diagnosis and the management of acute relapses. Postgrad Med J 2005; 81: 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The Multiple Sclerosis International Federation . An Atlas of MS. http://www.atlasofms.org/ (accessed 19/09/2013).
- 3. Tremlett H, Zhao Y, Rieckmann P, Hutchinson M. New perspectives in the natural history of multiple sclerosis. Neurology 2010; 74: 2004–2015. [DOI] [PubMed] [Google Scholar]
- 4. Tremlett H, Yinshan Z, Devonshire V. Natural history of secondary‐progressive multiple sclerosis. Mult Scler 2008; 14: 314–324. [DOI] [PubMed] [Google Scholar]
- 5. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996; 46: 907–911. [DOI] [PubMed] [Google Scholar]
- 6. Ann Marrie R, Rudick RA. Drug insight: interferon treatment in multiple sclerosis. Nat Clin Pract Neurol 2006; 2: 34–44. [DOI] [PubMed] [Google Scholar]
- 7. McFarland HF, Barkhof F, Antel J, Miller DH. The role of MRI as a surrogate outcome measure in multiple sclerosis. Mult Scler 2002; 8: 40–51. [DOI] [PubMed] [Google Scholar]
- 8. Filippini G, Munari L, Incorvaia B, et al Interferons in relapsing remitting multiple sclerosis: a systematic review. Lancet 2003; 361: 545–552. [DOI] [PubMed] [Google Scholar]
- 9. La Mantia L, Vacchi L, Di Pietrantonj C, et al Interferon beta for secondary progressive multiple sclerosis. Cochrane Database Syst Rev 2012; 1: CD005181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Trojano M, Pellegrini F, Fuiani A, et al New natural history of interferon‐beta‐treated relapsing multiple sclerosis. Ann Neurol 2007; 61: 300–306. [DOI] [PubMed] [Google Scholar]
- 11. Tedeholm H, Lycke J, Skoog B, et al Time to secondary progression in patients with multiple sclerosis who were treated with first generation immunomodulating drugs. Mult Scler 2012; 19: 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergamaschi R, Quaglini S, Tavazzi E, et al Immunomodulatory therapies delay disease progression in multiple sclerosis. Mult Scler 2012; PMID: 22653657 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 13. Renoux C, Suissa S. Immortal time bias in the study of effectiveness of interferon‐beta in multiple sclerosis . Ann Neurol 2008; 64: 109–110. [DOI] [PubMed] [Google Scholar]
- 14. Koch M, Mostert J, De Keyser J, Tremlett H, Filippini G. Interferon‐beta treatment and the natural history of relapsing−remitting multiple sclerosis. Ann Neurol 2008; 63: 125–126. [DOI] [PubMed] [Google Scholar]
- 15. Fox RJ, Thompson A, Baker D, et al Setting a research agenda for progressive multiple sclerosis: the International Collaborative on Progressive MS. Mult Scler 2012; 18: 1534–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sadovnick AD, Ebers GC, Wilson RW, Paty DW. Life expectancy in patients attending multiple sclerosis clinics. Neurology 1992; 42: 991–994. [DOI] [PubMed] [Google Scholar]
- 17. Tremlett H, Yousefi M, Devonshire V, Rieckmann P, Zhao Y. Impact of multiple sclerosis relapses on progression diminishes with time. Neurology 2009; 73: 1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shirani A, Zhao Y, Karim ME, et al Association between use of interferon beta and progression of disability in patients with relapsing−remitting multiple sclerosis. JAMA 2012; 308: 247–256. [DOI] [PubMed] [Google Scholar]
- 19. BC Ministry of Health [creator] (2010): PharmaNet. BC Ministry of Health [publisher]. Data Extract. Data Stewardship Committee (2010). http://www.popdata.bc.ca/data (accessed 19/03/2015).
- 20. British Columbia Ministry of Health [creator] (2010): Medical Services Plan (MSP) Payment Information File. Population Data BC [publisher]. Data Extract. MOH (2010). http://www.popdata.bc.ca/data (accessed 19/03/2015).
- 21. British Columbia Ministry of Health [creator] (2010): Discharge Abstract Database (Hospital Separations). Population Data BC [publisher]. Data Extract. MOH (2010). http://www.popdata.bc.ca/data (accessed 19/03/2015).
- 22. British Columbia Ministry of Health [creator] (2009): Consolidation File (MSP Registration and Premium Billing). Population Data BC [publisher]. Data Extract. MOH (2009). http://www.popdata.bc.ca/data (accessed 19/03/2015).
- 23. Wilkins R, Berthelot JM, Ng E. Trends in mortality by neighbourhood income in urban Canada from 1971 to 1996. Health Rep 2002; 13(Suppl): 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poser CM, Paty DW, Scheinberg L, et al New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 1983; 13: 227–231. [DOI] [PubMed] [Google Scholar]
- 25. McDonald WI, Compston A, Edan G, et al Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol 2001; 50: 121–127. [DOI] [PubMed] [Google Scholar]
- 26. Ebers GC, Traboulsee A, Li D, et al Analysis of clinical outcomes according to original treatment groups 16 years after the pivotal IFNB‐1b trial. J Neurol Neurosurg Psychiatry 2010; 81: 907–912. [DOI] [PubMed] [Google Scholar]
- 27. European Study Group on interferon beta‐1b in secondary progressive MS . Placebo‐controlled multicentre randomised trial of interferon beta‐1b in treatment of secondary progressive multiple sclerosis. Lancet 1998; 352: 1491–1497. [PubMed] [Google Scholar]
- 28. Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon‐Beta‐1a in MS (SPECTRIMS) Study Group . Randomized controlled trial of interferon‐ beta‐1a in secondary progressive MS: clinical results. Neurology 2001; 56: 1496–1504. [DOI] [PubMed] [Google Scholar]
- 29. Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD‐9‐CM administrative databases. J Clin Epidemiol 1992; 45: 613–619. [DOI] [PubMed] [Google Scholar]
- 30. Shirani A, Zhao Y, Karim ME, et al Investigation of heterogeneity in the association between interferon beta and disability progression in multiple sclerosis: an observational study. Eur J Neurol 2013; 21: 835–844. [DOI] [PubMed] [Google Scholar]
- 31. British Columbia Ministry of Health [creator] (2009): Adjusted Clinical Groups. Population Data BC [publisher]. Data Extract. Version 5. MOH (2011). http://www.popdata.bc.ca/data (accessed 19/03/2015).
- 32. R Core Team . R: A Language and Environment for Statistical Computing, Version 3.0.2. Vienna, Austria: R Foundation for Statistical Computing, 2013. http://www.R-project.org/. [computer program]. [Google Scholar]
- 33. Goodkin DE. Interferon beta treatment for multiple sclerosis: persisting questions. Mult Scler 1996; 1: 321–324. [DOI] [PubMed] [Google Scholar]
- 34. Rolak LA. IFNB in MS. Neurology 1996; 47: 1110. [DOI] [PubMed] [Google Scholar]
- 35. Dafni U. Landmark analysis at the 25‐year landmark point. Circ Cardiovasc Qual Outcomes 2011; 4: 363–371. [DOI] [PubMed] [Google Scholar]
- 36. Marrie RA. Observational studies of treatment effectiveness: useful, useless or somewhere in between? Mult Scler 2013; 19: 707–708. [DOI] [PubMed] [Google Scholar]
- 37. Haut ER, Pronovost PJ. Surveillance bias in outcomes reporting. JAMA 2011; 305: 2462–2463. [DOI] [PubMed] [Google Scholar]
- 38. Sorensen PS. Neutralizing antibodies against interferon‐beta. Ther Adv Neurol Disord 2008; 1: 125–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol 2006; 5: 343–354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary analyses.
Figure S1. Time‐dependent Cox regression analysis of potential factors associated with time to SPMS a onset for IFNβ‐treated versus the contemporary untreated cohortb (with additional adjustment of SES and high impact/prevalence comorbidities).
Figure S2. Selection of the study population from the British Columbia Multiple Sclerosis clinic database: patients with relapsing‐onset MS with symptom onset between 1975 and 1995.
Figure S3. Survival curves of time from MS symptom onset to SPMS occurrence within 10 years' disease duration, by 5‐year onset intervals.
Table S1. Multivariable Cox regression analysis of potential factors affecting time to reach SPMS within 10 years from disease onset.