

Abstract
Recent studies have identified oncogenic lesions in Ph+ ALL and ABL1 kinase mutations that confer resistance to tyrosine kinase inhibitors. We sought to determine the prevalence and clinical impact of these lesions in patients on CALGB 10001, a previously reported phase II study of imatinib, chemotherapy, and hematopoietic cell transplant in adult Ph+ ALL. Of the 58 enrolled, 22 relapsed. By direct sequencing, an ABL1 kinase mutation known to induce imatinib resistance was present at relapse in 13 of 20. Using quantitative PCR assays, the mutations were detectable at diagnosis or early during treatment in most (62%) relapsed patients. Aberrations in IKZF1, CDKN2A/B, and PAX5 were assessed in 28 samples using SNP arrays and genomic DNA sequencing. Of these, 22 (79%) had IKZF1 deletion. The combination of IKZF1 deletion and p210 BCR-ABL1 (p<0.0001), high white blood cell count (p=0.021), and minimal residual disease (p=0.013) were associated with worse disease-free survival.
Keywords: Lymphoid leukemia, Prognostication, Drug resistance, Transcription factor changes
INTRODUCTION
The Philadelphia chromosome encodes a constitutively activated BCR-ABL1 tyrosine kinase, the pathogenic lesion in chronic myeloid leukemia (CML) and a subset of acute lymphoblastic leukemia (ALL). Philadelphia chromosome-positive (Ph+) ALL comprises approximately 30% of adult ALL and its incidence increases with age. Though outcomes for adults with Ph+ ALL have improved dramatically with the use of tyrosine kinase inhibitors (TKIs), treatment failure and relapse remain a major concern. Recent genomic studies have identified oncogenic lesions that cooperate with BCR-ABL1 to induce ALL as well as specific ABL1 kinase domain mutations that confer resistance to TKI therapy. Further analyses characterizing these genetic aberrations and their relationship with clinical outcomes and other established biological variables are of high priority.
The most frequent genetic alteration in Ph+ ALL is deletion of IKZF1, followed by high rates of co-deletion of CDKN2A and PAX5 [1]. IKZF1 encodes the transcription factor Ikaros, a zinc-finger nuclear protein required for normal lymphoid development. Deletion of IKZF1 results in expression of non-DNA-binding (dominant negative) isoforms of Ikaros, which appear to collaborate with BCR-ABL1 in the pathogenesis of ALL and blastic transformation of CML [1–3]. IKZF1 deletions have been shown to have a negative prognostic impact in Ph+ ALL, conferring a shorter disease-free survival even in the era of combined TKI and chemotherapy [4–5] and following allogeneic hematopoietic cell transplantation (HCT) [6].
Ph+ ALL patients who develop resistance to TKI therapy with imatinib are often found to have clones with point mutations in the ABL1 kinase domain [7]. Numerous mutations have been identified, most of which impair imatinib binding [8]. Whether these mutations are present prior to TKI therapy or emerge during treatment is uncertain. One study found that no mutations were detectable at diagnosis in 12 patients who had mutations at relapse using a quantitative pyrosequencing assay [9]. Two more recent reports, however, have shown that through utilization of more sensitive detection methods, mutations can be identified at diagnosis or before the onset of TKI therapy [10–11].
In this study, we analyzed samples collected prospectively from patients enrolled on Cancer and Leukemia Group B (CALGB) 10001, a previously reported phase II study of imatinib plus sequential chemotherapy followed by allogeneic (allo) or autologous (auto) HCT in adults with newly diagnosed Ph+ ALL [12]. One objective of this analysis was to determine the prevalence of ABL1 kinase domain mutations in patients who relapsed and to monitor the kinetics of the resistant clones. A second objective was to correlate genetic alterations of IKZF1, CDKN2A/B, and PAX5 with clinical outcomes and with other biological variables, including BCR-ABL1 fusion transcript type (p190 versus p210), presence of ABL1 kinase domain mutations, white blood cell (WBC) count at diagnosis, and minimal residual disease (MRD) status. To our knowledge, this study represents the first comprehensive correlative analysis of molecular aberrations, other biological variables, and clinical outcomes in adults with Ph+ ALL.
MATERIALS AND METHODS
Patients
Patients were eligible for CALGB 10001 if they were between 15 and 60 years of age, had an unequivocal diagnosis of t(9;22) or BCR-ABL1 ALL, had achieved either partial or complete remission following one course of induction chemotherapy with a four- or five-drug regimen, and had received imatinib for no more than six weeks before study enrollment. All patients provided written informed consent. The study received Institutional Review Board approval from each participating institution. (Clinical trial registration numbers: NCT00039377, NCT00899223)
Between April 15, 2002 and April 30, 2010, 58 patients were enrolled on CALGB 10001; one was ineligible. Patients were treated with imatinib and sequential chemotherapy followed by either auto-HCT (n=19) or allo-HCT (n=15) or combination chemotherapy plus imatinib. Maintenance therapy with imatinib was recommended until minimal residual disease was no longer detected by quantitative real-time PCR (q-PCR) for twelve months or until relapse. The clinical trial results have been published and demonstrate similar outcomes for auto-HCT and allo-HCT, including statistically similar disease-free survival (DFS) (median, 3.5 versus 4.1 years) and overall survival (OS) (median, 6.0 years versus not reached) [12].
Of the patients enrolled on CALGB 10001, 42 also registered to a companion Leukemia Tissue Bank protocol (CALGB 9665), and thus were evaluable for additional genetic mutations associated with B-lineage ALL (Figure 1). The clinical characteristics of the patients enrolled on the two protocols are statistically similar, as detailed in Table I. Median follow up for this analysis was 6.5 years.
Figure 1.

CONSORT diagrams of clinical trial CALGB 10001 and companion Leukemia Tissue Bank protocol CALGB 9665.
Mutation Analysis
Samples were examined for BCR-ABL1 expression by quantitative RT-PCR (q-PCR) as previously reported [13]. Available pretreatment and relapse samples were tested for BCR-ABL1 kinase domain mutations using a direct sequencing approach. The primer sequences were the same as those used by Soverini and colleagues [14]. For patients with BCR-ABL1 mutations detected at relapse, sequential pre-relapse samples were analyzed using the mutation-specific q-PCR method described by Gruber and colleagues [15]. The sensitivities of the assays were shown to be 1% for T315I and E255V and 0.1% for Y253H and E255K. Analysis was performed in a CLIA approved laboratory at the University of Chicago.
For the IKZF1, CDKN2A/B, and PAX5 analysis, samples were genotyped using Affymetrix SNP 6.0 microarrays according to the manufacturer’s instructions. CEL files were generated using GeneChip Command Console Software. SNP cells were generated using Genotyping Console (Affymetrix) and the Birdseed v2 algorithm with default parameters with at least 50 arrays in each analysis. Array normalization and copy number inference were performed according to a previously published workflow [1, 16]. Normalized data were viewed in dChip [17] and regions with abnormal copy number identified computationally by circular binary segmentation (CBS) [18] and analyzed as previously described [1, 16]. Analysis was performed at St. Jude Children’s Research Hospital laboratories.
Statistical Methods
Survival functions for DFS and OS were estimated using the Kaplan-Meier method [19]. DFS was defined as the time from date of complete or partial remission (CR or PR) to relapse or death, whichever came first. OS was calculated as time from study registration to death from any cause. Patients failing to reach an endpoint were censored as of their last date of evaluation. Because patients on the 10001 trial were expected to undergo either allo-HCT or auto-HCT, they were not censored for transplant in these analyses. The differences between survival distributions with respect to biological factors of interest were evaluated using the log-rank test. Hazard ratios and their 95% confidence intervals were estimated by univariable and multi-variable Cox regression models [20] that included BCR-ABL1 fusion transcript type (p190 versus p210), IKZF1, CDKN2A/B, and PAX5 deletion, WBC count at diagnosis, MRD status, age, and gender. Pairwise associations between biological factors were assessed using Fisher’s Exact Tests. Statistical analyses were performed by CALGB (Alliance) statisticians using SAS 9.2 and R 2.15.0 [21] from the study database frozen on April 22, 2014.
RESULTS
Prevalence and Clinical Impact of ABL1 Kinase Domain Mutations
Relapse samples were available for evaluation of ABL1 kinase domain mutations from 20 of the 22 patients who relapsed. Using direct sequencing, a kinase domain mutation was present at relapse in 13 out of 20 patients (65%). Four different mutations were detected: Y253H (n=4), E255K (n=4), E255V (n=2), and T315I (n=3). All of these mutations are located at the binding site between imatinib and the Abl kinase and are known to confer resistance to imatinib. None of these mutations were detected at diagnosis using direct sequencing.
Quantitative PCR assays were then performed on sequential pre-relapse samples using mutation-specific primers in order to monitor the kinetics of resistant clones. With this more sensitive method, a resistant clone was detectable prior to relapse in eight of the 13 patients and, in five of these cases, also at the time of diagnosis (Figure 2) [22]. In most cases, the kinetics of the resistant clone mirrored the overall BCR-ABL1 population, yet the resistant clone increased proportionally over time and became the dominant BCR-ABL1 clone by the time of relapse.
Figure 2.

Summary of q-PCR results showing the time point at which the resistant clone was first detected in each of the 13 patients with a kinase domain mutation present at relapse.
Baseline samples obtained prior to induction were available from 25 non-relapsed patients and from the seven relapsed patients with no mutations by direct sequencing. Using q-PCR, 31 of these 32 baseline samples were negative for all four kinase domain mutations. One patient was found to have a T315I mutation at diagnosis but received an allo-HCT in first CR and died of sepsis six weeks after transplant.
Among relapsed patients, the presence of a kinase domain mutation was associated with significantly shorter DFS (median, 8 versus 15.6 months; log rank p=0.0068; HR 4.5). These differences did not hold when looking at OS (log-rank p=0.53). There were no differences in outcomes between the subset of patients with a resistant clone detectable in pre-relapse samples and those with a resistant clone detected only at relapse.
Prevalence and Clinical Impact of Deletions in IKZF1, CDKN2A/B, PAX5
Alterations in IKZF1, CDKN2A/B, and PAX5 were assessed in 28 pre-treatment samples and in five samples from relapse. Prior to treatment, 22 patients (79%) had an IKZF1 deletion, 12 (43%) had a CDKN2A/B deletion, and 7 (25%) had a PAX5 deletion (Figure 3). Of the 22 patients who had an IKZF1 deletion, 13 had a deletion in IKZF1 only, four had deletions in both IKZF1 and CDKN2A/B, and five had deletions in IKZF1, CDKN2A/B, and PAX5. Of the six patients without an IKZF1 deletion, two had deletions in both CDKN2A/B and PAX5, one had deletion in CDKN2A/B only, and three had no aberrations. All seven patients with a PAX5 deletion also had a CDKN2A/B deletion, and seven of the 12 patients with a CDKN2A/B deletion also had a PAX5 deletion. The association between CDKN2A/B and PAX5 deletions was significant (p=0.0007).
Figure 3.
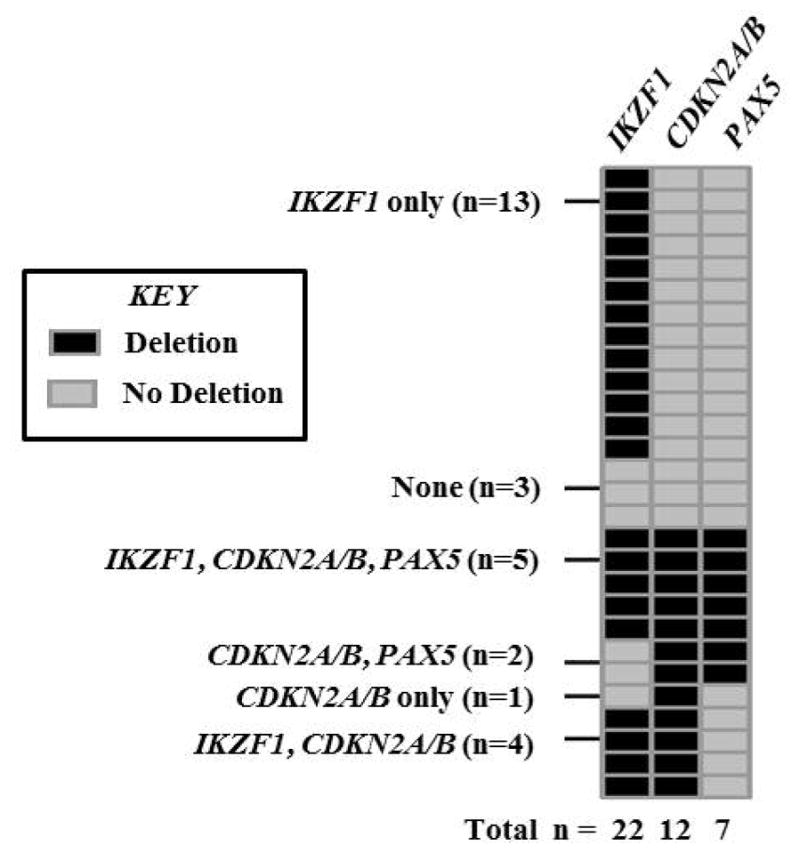
Prevalence of deletions in IKZF1, CDKN2A/B, and PAX5 in available pre-treatment samples (n=28).
We identified evidence of clonal evolution from pre-treatment to relapse. Of the five patients who were assessed for these deletions at relapse, one had an IKZF1 deletion at relapse that was not detected prior to treatment, one had IKZF1 and PAX5 deletion at relapse that was not detected prior to treatment, and one had a CDKN2A/B deletion at relapse that was not detected prior to treatment. Conversely, two patients with a PAX5 deletion prior to treatment no longer showed signs of the deletion at relapse.
In univariable analysis, median DFS in the 22 patients with IKZF1 deletions at diagnosis was only 13.2 months versus 53.1 months in the six patients with wild type IKZF1, although this difference did not reach statistical significance (p=0.18). There was no statistically significant survival difference between patients with IKZF1 deletion and those without (p=0.58). Furthermore, there were no differences in outcomes between those with deletions in IKZF1 alone (n=13) compared to those with all three deletions (n=5), or between those with no deletions (n=3) compared to those with all three deletions (n=5). Among the 22 patients with IKZF1 deletion, the presence of CDKN2A/B deletion was not associated with a significant difference in DFS (p=0.84) or OS (p=0.98).
Pairwise analyses were conducted in order to characterize the relationship between IKZF1, CDKN2A/B, and PAX5 deletions and other established biological variables. No statistically significant associations were found between these deletions and BCR-ABL1 fusion transcript type (p190 versus p210), the presence of ABL1 kinase domain mutations, WBC count at diagnosis, MRD status, age, or gender.
The Impact of BCR-ABL1 Fusion Transcript: p210 versus p190
Classification by BCR-ABL1 fusion transcript size was performed on 50 available pre-treatment samples. There were 17 patients (34%) with p210 transcripts, including B2A2 (n=11) and B3A2 (n=6), and 33 patients (66%) with p190 transcripts E1A2 (n=31) or altP190-E1A3 (n=2). In univariate analysis, patients with p210 transcripts trended toward worse DFS (Figure 4A) and OS (Figure 4B) than patients with p190 transcripts (log rank p=0.073 for DFS; p= 0.053 for OS).
Figure 4.

A) DFS and B) OS stratified by BCR-ABL1 fusion transcript. C) DFS and D) OS stratified by presence of IKZF1 deletion and BCR-ABL1 fusion transcript. E) DFS and F) OS stratified by WBC count at diagnosis.
The relationship between BCR-ABL1 fusion transcript type and IKZF1 deletion was examined. Of the 22 patients with an IKZF1 deletion, 17 (77%) had a p190 BCR-ABL1 fusion transcript, whereas the six patients with wild type IKZF1 were evenly split between p190 and p210 transcripts. The association between IKZF1 deletion and BCR-ABL1 fusion transcript type was not statistically significant (Fisher’s p=0.31).
Whereas the presence of IKZF1 deletion and p210 transcript type both trended toward worse DFS in univariable analysis, the combination of both portended significantly worse DFS in multivariable analysis (p<0.0001; Figure 4C). Controlling for fusion transcript genotype, the hazard of relapse or death for patients with IKZF1 deletion was 6.9 times that for those without. Conversely, controlling for IKZF1 status, the hazard of relapse or death for patients with a p210 transcript was 7.1 times that for those with p190 transcripts.
The Role of WBC Count at Diagnosis and MRD Status
Because patients were enrolled to CALGB 10001 after induction, WBC count at diagnosis was not available for every patient. Of the 35 patients on companion Leukemia Tissue Bank protocol CALGB 9665 with available WBC data, 18 had WBC counts greater than or equal to 20,000/ul at diagnosis (median, 51,850; range, 20,700 – 220,100) and 17 had counts below 20,000/ul (median, 7,500; range, 1,000 – 17,200). High WBC count at diagnosis was significantly associated with adverse DFS (Figure 4E; log-rank p=0.02; HR 2.5) and adverse OS, although the difference in OS was less pronounced (Figure 4F; log-rank p=0.10). There was no correlation between WBC count at diagnosis and deletions in IKZF1, CDKN2A/B, or PAX5.
Similarly, as reported previously, MRD less than or equal to 0.001 at day 120 following an auto-HCT was significantly associated with prolonged DFS and OS in the CALGB 10001 trial (p=0.01); the sample size was too small to analyze the effect of MRD on outcome among patients who underwent allo-HCT [12]. In this analysis there was no correlation between MRD status and deletions in IKZF1, CDKN2A/B, or PAX5.
DISCUSSION
Here we expand upon recent studies that have identified oncogenic lesions in Ph+ ALL, such as deletions in IKZF1, CDKN2A, and PAX5, as well as mutations that confer imatinib resistance, examining the prevalence and clinical impact of these lesions in a cohort of patients enrolled on a clinical trial. As the first reported correlative analysis of many of the variables examined here, this study contributes several hypothesis-generating findings. Our findings must be interpreted with caution, however, given the low power of a small cohort to assess many variables and the exclusion of patients over 60 years old from the CALGB 10001 trial.
The ability to detect mutations that confer TKI resistance in small subclones present at diagnosis has important clinical implications. In this study, ABL1 kinase domain mutations were detectable at diagnosis or early during treatment in a majority (62%) of relapsed patients using sensitive q-PCR. All of the mutations detected were binding site mutations known to induce imatinib resistance. In these cases, relapse appeared to be driven by the emergence of an imatinib-resistant clone despite aggressive therapy with imatinib, chemotherapy, and HCT. The current Alliance for Clinical Trials in Oncology clinical trial for Ph+ ALL (CALGB/Alliance 10701) uses dasatinib, which is a considerably more potent TKI that is effective against many BCR-ABL1 mutants that are resistant to imatinib. With several second and third generation TKIs now available, sensitive q-PCR assays may be used to screen for mutations at diagnosis and during treatment in order to guide the choice and dosing of TKIs, thereby potentially avoiding the emergence of a resistant clone and relapse. Methods for rapid pre-screening are being developed, such as a multiplexed Real-Time PCR assay combining hybridization probes for four critical ABL1 kinase domain mutations simultaneously [23].
In the correlative analysis explored here, the presence of ABL1 kinase domain mutations was not statistically associated with any other concurrent pathogenic mutations, including deletions in IKZF1, CDKN2A, and PAX5, or with fusion transcript type. Similarly, others have demonstrated that reduced responsiveness to dasatinib among in vitro and in vivo models with IKZF1 alterations is not due to the acquisition of ABL1 kinase domain mutations [24].
Consistent with previous reports, a significant majority of the adults with Ph+ ALL in our study had deletions in IKZF1 (22 of 28; 79%). Similarly, others have found IKZF1 deletions in 36 of 43 (84%) adult and pediatric Ph+ ALL cases [1], 52 of 83 (63%) adult cases [4], and 80 of 106 (75%) adult cases [25]. Here 12 patients (43%) had a CDKN2A/B deletion and 7 (25%) had a PAX5 deletion, whereas Mullighan et al. detected CDKN2A deletions in 54% and PAX5 deletions in 51% of 43 adult and pediatric Ph+ ALL cases [1]. These comparisons are summarized in Table 2.
Table 2.
Comparison of IKZF1, CDKN2A/B, and PAX5 deletion rates across studies of Ph+ ALL
Others have reported the negative prognostic impact of IKZF1 deletion in adults, beginning with Martinelli et al. who found patients with IKZF1 deletion had significantly shorter DFS in the setting of combined TKI and chemotherapy compared to IKZF1 wild type (median DFS 10 versus 32 months; p=0.02) [4]. Recently IKZF1 and CDKN2A deletions were associated with significantly shorter DFS and OS despite allo-HCT [6]. Here we demonstrate a trend toward worse DFS in patients with IKZF1 deletion versus wild type despite combined TKI, chemotherapy, and HCT (median DFS 13.2 versus 53.1 months; p=0.18). This difference reached statistical significance only after controlling for BCR-ABL1 fusion transcript type.
Among patients with IKZF1 deletion, those with p210 transcripts had significantly shorter DFS than those with p190 transcripts (p<0.0001). Notably, Chiaretti et al. recently reported worse outcomes among p210 Ph+ ALL patients treated with a dasatinib-based protocol, associated with a lower susceptibility to TKI, lower blast clearance, and greater incidence of relapse [26]. One possible explanation for worse outcomes in patients with p210 transcripts may be that these cases actually represent lymphoid blast crisis of chronic myeloid leukemia (CML) and may, therefore, be biologically different from de novo ALL with a BCR-ABL1 fusion [27]. Looking at other recent phase II studies of adult Ph+ ALL that reported fusion transcript type, the frequency of p210 ranged from 26 to 38% (weighted mean: 31.9%), similar to our rate of 34% [28–31]. These data are consistent with the GMALL report of a large number of pre-B ALL cases (n=2498), showing that p210 transcripts in Ph+ ALL increased in relative frequency from the adolescent cohort (12.6%) to the young adults (22.5%), then began to plateau in the 35–44 year-olds at 36.8%, and remained between 33% and 36.2% from then on [32]. Genomic analysis may be useful in distinguishing between de novo Ph+ ALL with a p210 transcript and CML in lymphoid blast crisis. With this caveat, our finding represents the first report of a significant interaction between BCR-ABL1 fusion transcript type and deletion of IKZF1. Larger studies are needed to confirm these observations.
Novel strategies to target IKZF1 are being examined. In a recently reported high content screening of hundreds of FDA-approved compounds, retinoid receptor agonists were found to reverse the leukemogenic phenotype driven by IKZF1 alterations [24]. Specifically, IKZF1 alterations were associated with acquisition of stem cell-like features such as increased adhesion and self-renewal, and retinoid receptor agonists reversed this phenotype, inducing expression of wild type IKZF1 and abrogating adherent growth, reducing proliferation, and increasing sensitivity to dasatinib. These insights underscore the importance of ongoing work to further characterize the molecular pathogenesis in Ph+ ALL and translate the findings to the clinical setting.
In conclusion, the molecular characterization of Ph+ ALL has the potential to inform prognosis and therapeutics. Screening for ABL1 kinase domain mutations at diagnosis or early during treatment using mutation-specific q-PCR assays may guide the choice and dosing of TKIs, which is particularly important given their different toxicity profiles. Greater understanding of resistance patterns may also justify regulatory approval and insurance coverage of second and third generation TKIs in the frontline setting. IKZF1 deletion is very common in Ph+ ALL and associated with worse outcomes, and in this study the combination of IKZF1 deletion and p210 transcript was associated with significantly worse DFS despite treatment with a TKI, chemotherapy, and HCT. Prospective validation in a larger patient population is warranted to further characterize these variables and determine their role in guiding complex management decisions.
Table 1.
Clinical characteristics of the 57 eligible patients registered to clinical trial CALGB 10001 and the 42 patients also registered to companion Leukemia Tissue Bank protocol CALGB 9665.
| Variable | Category value | 10001 Patients n (% ) | 9665 Patients n (%) |
|---|---|---|---|
| Gender | Female | 32 (56) | 24 (57) |
| Male | 25 (44) | 18 (43) | |
| Age | Median | 44 | 42.5 |
| Range | 22 – 58 | 22 – 58 | |
| Race | American Indian or | ||
| Alaska Native | 2 (4) | 2 (5) | |
| Asian | 4 (7) | 2 (5) | |
| African American | 9 (16) | 6 (14) | |
| Caucasian | 40 (70) | 30 (71) | |
| More than one race | 1 (2) | 1 (2) | |
| Unknown | 1 (2) | 1 (2) | |
| Ethnicity | Hispanic | 4 (7) | 4 (10) |
| Non-Hispanic | 45 (79) | 32 (76) | |
| Unknown | 8 (14) | 6 (14) | |
| ECOG Performance Status | (0) Normal | 18 (32) | 13 (31) |
| (1) Ambulatory | 33 (58) | 24 (57) | |
| (2) <50% Time in Bed | 5 (9) | 4 (10) | |
| (3) >50% Time in Bed | 1 (2) | 1 (2) | |
| WBC at diagnosis | < 20,000/ul | 19 (33) | 17 (40) |
| ≥ 20,000/ul | 21 (37) | 18 (43) | |
| Missing | 17 (30) | 7 (17) | |
| BCR-ABL Fusion Transcript | p190 | 33 (58) | 28 (67) |
| p210 | 17 (30) | 12 (29) | |
| Missing | 7 (12) | 2 (5) | |
| Transplant | Allogeneic | 15 (26) | 12 (29) |
| Autologous | 19 (33) | 12 (29) | |
| None | 23 (40) | 18 (43) | |
| Response to Induction | Complete Response | 54 (95) | 39 (93) |
| Partial Response | 3 (5) | 3 (7) |
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers: 1-U10-CA180835-01 to the University of Chicago, U10CA180821 and U10CA180882 to the Alliance for Clinical Trials in Oncology, and the following grants to the legacy Cancer and Leukemia Group B (CALGB): CA31946, CA33601, CA41287, CA59518, CA101140, CA77658, and CA32291.
Authorship Contributions and Conflict of Interest Disclosures
| Author | Contributions | Disclosures |
|---|---|---|
| DeBoer, R |
|
None |
| Koval, G |
|
None |
| Mulkey, F |
|
None |
| Wetzler, M |
|
Consultant for Novartis |
| Devine, S |
|
None |
| Marcucci, G |
|
None |
| Stone, RM |
|
None |
| Larson, RA |
|
None |
| Bloomfield, CD |
|
None |
| Geyer, S |
|
None |
| Mullighan, CG |
|
None |
| Stock, W |
|
None |
Footnotes
References
- 1.Mullighan C, Miller C, Radtke I, et al. BCR-ABL1 lymphoblastic leukemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–114. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 2.Virely C, Moulin S, Cobaleda C, et al. Haploinsufficiency of the IKZF1 (IKAROS) tumor suppressor gene cooperates with BCR-ABL in a transgenic model of acute lymphoblastic leukemia. Leukemia. 2010;24(6):1200–1204. doi: 10.1038/leu.2010.63. [DOI] [PubMed] [Google Scholar]
- 3.Schjerven H, McLaughlin J, Arenzana TL, et al. Selective regulation of lymphopoiesis and leukemogenesis by individual zinc fingers of Ikaros. Nature Immunology. 2013;14:1073–1083. doi: 10.1038/ni.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martinelli G, Iacobucci I, Storlazzi C, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapase: A GIMEMA AL WB report. Journal of Clinical Oncology. 2009;27(31):5202–5207. doi: 10.1200/JCO.2008.21.6408. [DOI] [PubMed] [Google Scholar]
- 5.van der Veer A, Zaliova M, Mottadelli F, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. 2014;123(11):1691–1698. doi: 10.1182/blood-2013-06-509794. [DOI] [PubMed] [Google Scholar]
- 6.Pfeifer H, Raum K, Markovic S, et al. In adult Philadelphia chromosome positive acute lymphoblastic leukemia, the negative prognostic impact of IKZF1, CDKN2A/B and PAX5 deletions is not abrogated by allogeneic stem cell tranplantation in first complete remission. ASH Annual Meeting Abstracts. 2013;122(21):231. [Google Scholar]
- 7.Hofmann W, Komor M, Wassmann B, et al. Presence of the BCR-ABL mutation Glu255Lys prior to STI571 (imatinib) treatment in patients with Ph+ acute lymphoblastic leukemia. Blood. 2003;102:659–661. doi: 10.1182/blood-2002-06-1756. [DOI] [PubMed] [Google Scholar]
- 8.Pfeifer H, Wassmann B, Pavlova A, et al. Kinase domain mutations of BCR-ABL frequently preced imatinib-based therapy and give rise to relapse in patients with de novo Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Blood. 2007;110:727–734. doi: 10.1182/blood-2006-11-052373. [DOI] [PubMed] [Google Scholar]
- 9.Jones D, Thomas D, Yin C, et al. Kinase domain mutations in Philadelphia chromosome-positive acute lymphoblastic leukemia emerge after therapy with BCR-ABL kinase inhibitors. Cancer. 2008;113:985–994. doi: 10.1002/cncr.23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soverini S, Vitale A, Poerio A, et al. Philadelphia-positive acute lymphoblastic leukemia patients already harbor BCR-ABL kinase domain mutations at low levels at the time of diagnosis. Haematologica. 2011;96:552–557. doi: 10.3324/haematol.2010.034173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeifer H, Lange T, Wystub S, et al. Prevalence and dynamics of bcr-abl kinase domain mutations during imatinib treatment differ in patients with newly diagnosed and recurrent bcr-abl positive acute lymphoblastic leukemia. Leukemia. 2012;26:1475–1481. doi: 10.1038/leu.2012.5. [DOI] [PubMed] [Google Scholar]
- 12.Wetzler M, Watson D, Stock W, et al. Autologous transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia achieves outcomes similar to allogeneic transplantation: results of a CALGB Study 10001 (Alliance) Haematologica. 2014;99(1):111–115. doi: 10.3324/haematol.2013.085811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stock W, Yu D, Karrison T, et al. Quantitative real-time RT-PCR monitoring of BCR-ABL in chronic myelogenous leukemia shows lack of agreement in blood and bone marrow samples. Int J Oncol. 2006;28:1099–1103. [PubMed] [Google Scholar]
- 14.Soverini S, Martinelli G, Amabile M, et al. Denaturing HPLC-based assay for detection of ABL mutations in chronic myeloid leukemia patients resistant to imanitib. Clin Chem. 2004;50:1205–1213. doi: 10.1373/clinchem.2004.031112. [DOI] [PubMed] [Google Scholar]
- 15.Gruber F, Lamark T, Anonli A, et al. Selecting and deselecting imatinib resistant clones: observations made by longitudinal, quantitative monitoring of mutated BCR-ABL. Leukemia. 2005;19:2159–2165. doi: 10.1038/sj.leu.2403983. [DOI] [PubMed] [Google Scholar]
- 16.Mullighan C, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 17.Lin M, Wei L, Sellers W, Liebergarb M, Wong W, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 18.Venkatraman E, Olshen A. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American Statistical Association. 1958;53:457–481. [Google Scholar]
- 20.Cox DR. Regression Models and Life-Tables. J Royal Stat Soc Series B (Methodological) 1972;34(2):187–220. [Google Scholar]
- 21.R: A language and environment for statistical computing. R Foundation for statistical Computing; Vienna, Austria: 2014. (R Core Team). http://www.R-project.org/ [Google Scholar]
- 22.Koval G, Wetzler M, Watson D, et al. Abl kinase domain mutations leading to relapse of Ph+ acute lymphoblastic leukemia (ALL) occur commonly and can be detected at initial diagnosis: molecular results from CALGB 10001. ASH Annual Meeting Abstracts. 2011;118(21):2541. [Google Scholar]
- 23.Martinez-Serra J, Gutierrez A, Marcus TF, et al. Four-channel asymmetric Real-Time PCR hybridization probe assay: A rapid pre-screening method for critical BCR-ABL kinase domain mutations. Clinical Biochemistry. 2012;45:345–351. doi: 10.1016/j.clinbiochem.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Churchman M, Low J, Qu C, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell. 2015;28:343–356. doi: 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iacobucci I, Storlazzi C, Cilloni D, et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’ Adulto Acute Leukemia Working Party (GIMEMA AL WP) Lymphoid Neoplasia. 2009;114(10):2159–2167. doi: 10.1182/blood-2008-08-173963. [DOI] [PubMed] [Google Scholar]
- 26.Chiaretti S, Vitale A, Elia L, et al. First results of the multicenter total therapy Gimema LAL 1509 protocol for de novo adult Philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia (ALL) patients. ASH Annual Meeting Abstracts. 2014;124(21):797. [Google Scholar]
- 27.Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. PNAS. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravandi F, O’Brien S, Thomas D, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116(12):2070–2077. doi: 10.1182/blood-2009-12-261586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foa R, Vitale A, Vignetti M, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118(25):6521–6528. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 30.Daver N, Thomas D, Ravandi F, et al. Final report of a phase II study if imatinib mesylate with hyper-CVAD for the front-line treatment of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica. 2015;100(5):653–661. doi: 10.3324/haematol.2014.118588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jabbour E, Kantarjian H, Ravandi F, et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a single-centre, phase 2 study. Lancet Oncology. 2015;16(15):1547–1555. doi: 10.1016/S1470-2045(15)00207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burmeister T, Schwartz S, Bartram CR, et al. Patients’ age and BCR-ABL frequency in adult B-precursor ALL: a retrospective analysis from the GMALL study group. Blood. 2008;112(3):918–919. doi: 10.1182/blood-2008-04-149286. [DOI] [PubMed] [Google Scholar]
- 33.DeBoer R, Mulkey F, Koval G, et al. Clinical impact of Abl1 kinase and IKZF1 mutations in adults with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia (ALL): molecular analysis of CALGB 10001 and 9665 (Alliance) Haematologica. 2014;99(supplement 1):258. doi: 10.3109/10428194.2016.1144881. EHA 19 Abstract Book. [DOI] [PMC free article] [PubMed] [Google Scholar]