

Abstract
We have engineered an integration-deficient lentiviral vector, LV305, to deliver the tumor antigen NY-ESO-1 to human dendritic cells in vivo through pseudotyping with a modified Sindbis virus envelop protein. Mice immunized once with LV305 developed strong, dose-dependent, multifunctional, and cytotoxic NY-ESO-1-specific cluster of differentiation 8 (CD8) T cells within 14 days post-immunization and could be boosted with LV305 at least twice to recall peak-level CD8 T-cell responses. Immunization with LV305 protected mice against tumor growth in an NY-ESO-1-expressing CT26 lung metastasis model, with the protective effect abrogated upon depletion of CD8 T cells. Adoptive transfer of CD8 T cells, alone or together with CD4 T cells or natural killer cells, from LV305-immunized donor mice to tumor-bearing recipient mice conferred significant protection against metastatic tumor growth. Biodistribution of injected LV305 in mice was limited to the site of injection and the draining lymph node, and injected LV305 exhibited minimal excretion. Mice injected with LV305 developed little to no adverse effects, as evaluated by toxicology studies adherent to good laboratory practices. Taken together, these data support the development of LV305 as a clinical candidate for treatment against tumors expressing NY-ESO-1.
Introduction
Lentiviral vectors (LVs) are proven tools for delivering nucleic acid payloads into cells through efficient transduction of dividing and nondividing cells and have been shown to stimulate potent and long-lasting antigen-specific cluster of differentiation 8 (CD8) T-cell immune responses.1–5 To minimize the risk of insertional mutations, several methods are routinely used, such as inactivating the vector integrase and/or mutating the long terminal repeat.6,7 Our lentiviral vector platform contains two independent elements to reduce its integration rate: (i) the D64V integrase mutation within the gag/pol gene; and (ii) the deletion of the 3′-poly purine tract within the vector genome.8,9 Integration-deficient LVs (IDLVs) have been shown to induce long-term gene expression in vivo, leading to activation of cellular and humoral immune responses and generation of effective anti-tumor therapy in preclinical models.10,11 We have demonstrated that when administered to mice, IDLVs induced a similar magnitude of antigen-specific CD8 T-cell response and anti-tumor efficacy as its integrating counterpart (T. Albershardt, unpublished data). Immunization with IDLVs is therefore considered an attractive alternative to immunization with integrating LVs.
The main goal of immunization against tumors is to elicit effector T cells that are capable of eradicating malignant cells. Because dendritic cells (DCs) are professional antigen-presenting cells and potent activators of T cells,12,13 LV305 was engineered to selectively deliver the antigen-encoding NY-ESO-1 gene to human DCs in vivo, via targeting of the C-type lectin receptor DC-SIGN (CD209) on immature DCs. LV305 utilizes our previously described lentiviral vector platform—ZVex™—that targets human DCs through pseudotyping with highly mannosylated Sindbis virus envelop protein variant, SinVar1 (also referred to as E1001).9 Transduced DCs translate the gene, process and present NY-ESO-1 antigen peptides to naive CD8 T cells via major histocompatibility complex class I, leading to the generation of tumor-specific cytotoxic T cells. ZVex has been previously demonstrated in mice to generate robust CD8 T-cell responses against expressed antigens that could be boosted to protect against viral and tumor challenges.14
Given that (i) self-reactive, high-avidity T cells are eliminated from the T-cell repertoire and (ii) the remaining pool of lower avidity T cells for self-antigens may not be sufficiently activated to mount an effective anti-tumor immunity, cancer/testis antigens are considered one of the most promising groups of tumor targets, as they are generally tumor specific and shared among various cancer types. NY-ESO-1 is the prototypical cancer/testis antigen that has been targeted in various clinical trials.15,16 Expression of NY-ESO-1 is normally restricted to germ cells but not in somatic tissue. However, expression of NY-ESO-1 is frequently dysregulated and found in malignant cells.17,18 For example, NY-ESO-1 is seen in ~25–33% of all melanoma, lung, esophageal, liver, gastric, prostate, ovarian, or bladder cancers and >90% in certain subtypes of sarcoma. Although its function remains unknown, NY-ESO-1 has been found to be strongly immunogenic in some cancer patients.19,20 Therefore, immunotherapeutic approaches directed against NY-ESO-1 have the potential to be broadly applicable to many different cancer indications, while inducing minimal adverse effects to normal tissue. Because NY-ESO-1 is an intracellular antigen, immunotherapeutic strategies against NY-ESO-1 have largely focused on the generation of effector cells, either through in vivo immunization or ex vivo expansion. Immunization with HLA-A2-restricted NY-ESO-1 peptides or recombinant NY-ESO-1 protein in various formulations has shown some success in clinical trials, from the generation of NY-ESO-1-specific cellular and humoral responses to the demonstration of tumor regression or stabilization of disease in patients.21–24 Most notably, adoptive transfer of CD8 T cells expressing recombinant T-cell receptor specific for NY-ESO-1 has recently demonstrated that T cell-mediated control of NY-ESO-1-expressing tumors is feasible in human clinical settings.25,26
In this study, we evaluated the immunogenicity and therapeutic efficacy of LV305 in preclinical mouse models and demonstrated that immunization with LV305 generated a robust CD8 T cell-dependent anti-tumor protection. Our pharmacokinetic and toxicology studies showed limited biodistribution and excretion of the injected vector in mice and minimal adverse toxicity events in mice injected with LV305. These results successfully supported the on-going investigation of LV305 in a phase 1 clinical trial in cancer patients with tumors expressing NY-ESO-1 (NCT02122861).
Results
Identification of H-2d-restricted CD8 and CD4 T-cell epitopes of human NY-ESO-1
NY-ESO-1 is a human cancer-testis antigen that is not endogenously expressed in mice. Prior to assessing immunogenicity and anti-tumor efficacy of LV305 in mice, a number of mouse strains were first evaluated for their ability to develop CD8 and CD4 T cell responses to NY-ESO-1. Recognition of NY-ESO-1 epitopes by MHC haplotypes H-2b (C57BL/6), H-2d (BALB/c), and H-2b/d (B6D2F1 hybrids from female C57BL/6 and male DBA/2 cross) of mice was assessed by epitope mapping in vitro using splenocytes harvested from mice immunized with LV305 or recombinant NY-ESO-1 protein formulated in GLA-SE (Figure 1). For the initial screening, splenocytes from immunized mice were stimulated with 42 overlapping NY-ESO-1 peptides, divided into 14 pools, with each pool containing 3 peptides. Immune responses were measured by intracellular cytokine staining for interferon-γ, interleukin-2, and tumor necrosis factor in CD8 or CD4 positive T-cell populations followed by flow cytometry analysis. C57BL/6 mice immunized with LV305 did not develop detectable levels of NY-ESO-1-specific CD8 T-cell response but generated robust levels of NY-ESO-1-specific CD4 T-cell response to Peptide Pool 8. These response levels were higher than observed in splenocytes from immunized BALB/c and B6D2F1 mice (Figure 1a). BALB/c and B6D2F1 mice generated robust NY-ESO-1-specific CD8 and CD4 T-cell responses of similar magnitude. Our findings suggest that both BALB/c and B6D2F1 mouse strains are suitable models to assess CD8 and CD4 T-cell responses induced by immunizations against NY-ESO-1. We subsequently mapped NY-ESO-181–88 as the H-2d-restricted CD8 T-cell epitope (Figure 1b), consistent with previously published data.27 No H-2b-restricted CD8 T-cell epitope were found (data not shown). We also identified a novel H-2d/b-restricted CD4 T-cell epitope within peptide NY-ESO-190–107. Based on these findings, subsequent immunogenicity and therapeutic studies were done in BALB/c (H-2d) mice.
Figure 1.

Epitope mapping of H-2d-restricted CD8 and CD4 epitopes of NY-ESO-1. To epitope map CD8 T-cell responses, (a) female BALB/c, C57BL/6, or B6D2F1 mice or (b) female BALB/c mice (n = 5 per group) were injected with 5 × 109 vector genomes of LV305, SC, in the tail base. To epitope map CD4 T-cell responses, mice (n = 5 per group) were injected with two doses of 5 µg recombinant NY-ESO-1 protein formulated with 5 µg GLA in 2% oil-in-water stable emulsion 21 days apart. Splenic T-cell responses were measured by ICS 12 or 7 days post-last immunization for CD8 or CD4 T-cell responses, respectively. “Pool 7” contains peptides NYESO173–87, NYESO177–91, and NYESO181–95; “Pool 8” contains peptides NYESO185–99, NYESO189–103, and NYESO193–107. Error bars represent mean ± SEM. CD, cluster of differentiation; ICS, intracellular cytokine staining; IL, interleukin; INF, interferon; SC, subcutaneous; TNF, tumor necrosis factor. Data are representative of at least three independent experiments.
LV305 induces NY-ESO-1-specific CD8 T cells in a route- and dose-dependent manner
To assess the effect of administration route on LV305-mediated immune responses, BALB/c mice were immunized with LV305 via the following routes: subcutaneous (SC) in the tail base, scruff of the neck, or hind footpad; intradermal; intravenous (IV) in the retro-orbital venous sinus; or intramuscular in the thigh. On Day 14 post-immunization, splenocytes were harvested to evaluate magnitude of antigen-specific CD8 T-cell responses via intracellular cytokine staining and flow cytometry analysis. Of the routes tested, tail base SC, scruff SC, intradermal, IV, and intramuscular induced relatively similar magnitude of CD8 T-cell responses, with footpad SC inducing the lowest response (see Supplementary Figure S1a). The generation of multifunctional CD8 T cells, capable of expressing more than one cytokine, was comparable among the various routes, with the exception of IV administration, which elicited CD8 T-cell response that lacked multifunctional quality. For the ease of mouse handling and optimal generation of multifunctional T cells, SC administration at the bail base was chosen as the default immunization route for LV305.
To assess the dose–response of LV305, BALB/c mice were immunized SC with LV305 at a dose range spanning 5-logs (5 × 106 to 5 × 1010 vector genomes). The magnitude of LV305-induced CD8 and CD4 T-cell responses correlated positively with the dose of LV305 administered (see Supplementary Figure S1b). A dose of 5 × 108 or 5 × 109 vector genomes was the lowest immunogenic dose required to induce significant levels of CD8 or CD4 T-cell response, respectively.
Homologous prime-boost immunizations with LV305 generates sustained level of NY-ESO-1-specific CD8 T cells and humoral immunity in mice
Homologous prime-boost immunizations can be achieved with LVs due to low pre-existing immunity against LVs in humans.28,29 To assess the potential of using LV305 for homologous prime-boost regimen, mice were immunized with LV305 twice, 21 days apart (Figure 2a,b). Immune responses were measured at various time points post-prime or post-boost to study the response kinetics. The average frequency of interferon-γ+ CD8 T cells at the peak of the prime (Day 16) was ~3% (Figure 2a). Both the primary and recall responses generated high-quality effector CD8 T cells, as evidenced by the additional expression of tumor necrosis factor and interleukin-2 (Figure 2b) and their antigen-specific cytotoxic activity, as validated in an in vitro cytotoxic T lymphocyte assay (Figure 2c). Mice primed and boosted once with LV305 also generated anti-NY-ESO-1 IgG (Figure 2d). Taken together, priming with LV305 induced NY-ESO-1-specific cellular and humoral immunity that could be efficiently recalled with homologous boosting with LV305.
Figure 2.

Mice immunized with LV305 generated NY-ESO-1-specific cellular and humoral immunity. (a,b) Female BALB/c mice (n = 5 per group) were injected with 5 × 109 vector genomes of LV305, SC, in the tail base on Days 0, 21, and 42. Splenic T-cell responses were measured by ICS at the time points listed in the figure. (c) Effector cells (E) isolated from LV305- or mock-immunized female BALB/c mice (n = 5 per group) were incubated with PKH26-labeled target cells (T) at E:T ratios detailed in the figure for 4 hours in 37 °C, 5% CO2 to assess CTL activity. Target cells were subsequently stained with LIVE/ DEAD Fixable Near-IR prior to flow cytometry analysis for determination of target cell % death. (d) Female BALB/c mice (n = 10 per group) were injected with 5 × 109 vector genomes of LV305, SC, in the tail base on Days 0 and 21. Total anti-NY-ESO-1 IgG in sera were measured at the time points detailed in the figure by endpoint ELISA. Error bars represent mean ± SEM (a–c) or geometric mean ± 95% confidence interval (d). CD, cluster of differentiation; CTL, cytotoxic T lymphocyte; ICS, intracellular cytokine staining; IL, interleukin; INF, interferon; SC, subcutaneous; TNF, tumor necrosis factor. Data are representative of at least two independent experiments.
Mice immunized with LV305 are protected from tumor challenge
Having determined that the BALB/c (H-2d) mouse was an appropriate model to evaluate LV305-induced T-cell responses, a tumor model was developed using the same mouse strain to evaluate LV305-induced anti-tumor efficacy. We generated a novel murine tumor cell line (which we named, “CIN”) by genetically modifying the parental CT26 colon cancer cells to express human NY-ESO-1. CIN cells were plated at near single-cell density to screen clones for uniform expression level of NY-ESO-1. CIN clones expressing high levels of NY-ESO-1 (e.g., clone 35) were completely rejected when inoculated in mice, whereas clones expressing lower levels of NY-ESO-1 grew (e.g., clone 23) (data not shown). Clone 23 (CIN.23) was chosen for subsequent studies based on its uniform expression level of NY-ESO-1 (see Supplementary Figure S2) and its ability to generate tumor nodules in lungs of BALB/c mice via IV inoculation.
BALB/c mice were inoculated with CIN.23 cells intravenously into the tail vein and 3 days later immunized with high (5.0 × 109 vector genomes), medium (1.5 × 109 vector genomes), or low (5.0 × 108 vector genomes) doses of LV305 (Figure 3). Lungs and spleens were harvested 18 days post-tumor inoculation followed by enumeration of tumor nodules (Figure 3a,b) and evaluation of splenic NY-ESO-1-specific CD8 T-cell responses (Figure 3c). Nonimmunized or mock-immunized mice developed an average of 130–150 lung nodules, whereas LV305-immunized mice developed significantly fewer lung nodules in a dose-dependent manner (Figure 3a,b). Mice treated with high-dose LV305 developed an average of fewer than 20 tumor nodules, which is more than a sixfold decrease in tumor nodule formation compared with mock-immunized mice. This dose-dependent therapeutic response correlated with the magnitude of NY-ESO-1-specific CD8 T cells generated (Figure 3c).
Figure 3.

Mice immunized with LV305 were protected from tumor challenge in a dose-dependent manner. On Day 0, female BALB/c mice (n = 10 per group) were injected with 1.5 × 105 CIN.23 cells, IV. On Day 3, tumor-bearing mice were injected with control vector or 5.0 × 108 (Lo), 1.5 × 109 (Med), or 5.0 × 109 (Hi) vector genomes of LV305, SC, in the tail base. (a,b) On Day 18, tumor nodules in lungs were enumerated. (c) On Day 18, splenic T-cell responses were measured by ICS. Error bars represent mean ± SEM. CD, cluster of differentiation; ICS, intracellular cytokine staining; IL, interleukin; INF, interferon; IV, intravenous; SC, subcutaneous; TNF, tumor necrosis factor ***P < 0.0000001, **P < 0.00001, *P < 0.0001. Data are representative of at least three independent experiments.
LV305-induced anti-tumor response is CD8 T cell dependent
To elucidate the role of various lymphocyte populations in LV305-induced anti-tumor response, we evaluated anti-tumor efficacy of LV305 in mice treated with depletion antibodies against CD8, CD4, or asialo-GM1 (ASGM, expressed predominantly by natural killer (NK) cells) (Figure 4). When CD8 T cells were depleted from BALB/c mice prior to immunization, LV305-mediated anti-tumor response was abrogated, confirming CD8 T cells as required effectors for anti-tumor control (Figure 4a). CD4 T-cell depletion modestly reversed LV305-mediated anti-tumor response (Figure 4a) and compromised the generation of NY-ESO-1-specific CD8 T cells (Figure 4b). This observation is consistent with the rationale that CD4 T cell help is needed to induce optimal CD8 T cell responses and further demonstrates CD8 T cells as critical anti-tumor effectors. Depletion with the anti-ASGM antibody also abrogated LV305-induced tumor control and even resulted in a slight increase in tumor burden (Figure 4a). Interestingly, anti-ASGM antibody treatment significantly suppressed the development of NY-ESO-1-specific CD8 T-cell response (Figure 4b), again demonstrating that CD8 T cells are required for effective anti-tumor control.
Figure 4.

CD8 depletion abrogated LV305-induced anti-tumor protection in mice. On Day 0, female BALB/c mice (n = 5 per group) depleted of CD8 T cells, CD4 T cells, or natural killer (NK) cells were injected with 1.5 × 105 CIN.23 cells, IV. On Day 3, tumor-bearing mice were injected with control vector or 5 × 109 vector genomes of LV305, SC, in the tail base. On Day 18, (a) tumor nodules in lungs were enumerated, and (b) splenic T-cell responses were measured by ICS. Bars represent mean; error bars represent mean ± SEM. CD, cluster of differentiation; ICS, intracellular cytokine staining; IL, interleukin; INF, interferon; IV, intravenous; SC, subcutaneous; TNF, tumor necrosis factor; **P < 0.005, *P < 0.05. Data are representative of two independent experiments.
To further assess the role of these lymphocyte subsets in LV305-mediated tumor control, adoptive transfer experiments were conducted where lymphocytes from LV305-immunized mice were isolated and administered to tumor-challenged, nonimmunized mice. Consistent with the findings above, transfer of CD8 T cells from LV305-immunized mice to CIN.23 tumor-bearing mice conferred significant, albeit incomplete, protection against tumor nodule growth in the lungs (Figure 5 and Supplementary Figure S3a). Adoptive transfer of CD4 T cells from LV305-immunized mice also conferred significant anti-tumor protection, compared with the mock-immunized mice. Day 14 post-transfer, these recipient mice developed NY-ESO-1-specific CD8 T cells at a greater magnitude than mice having received the direct transfer of CD8 T cells from LV305-immunized mice (see Supplementary Figure S3b). This finding paralleled that of the depletion studies, demonstrating the stimulatory role of CD4 T cell help in generating optimal CD8 T-cell response. Adoptive transfer of NK cells also resulted in reduced lung nodule development. This effect was also observed when the donor mice had not been immunized with LV305, consistent with NK cell-mediated anti-tumor activity being antigen independent (see Supplementary Figure S3a). In addition, transfer of both NK cells and CD8 T cells into the same tumor-bearing recipient mice reduced tumor nodule growth to a significantly larger extent than transferring any lymphocyte population alone (Figure. 5). Lastly, cotransferring all three populations (CD8 T cells, CD4 T cells, and NK cells) conferred the best anti-tumor response.
Figure 5.
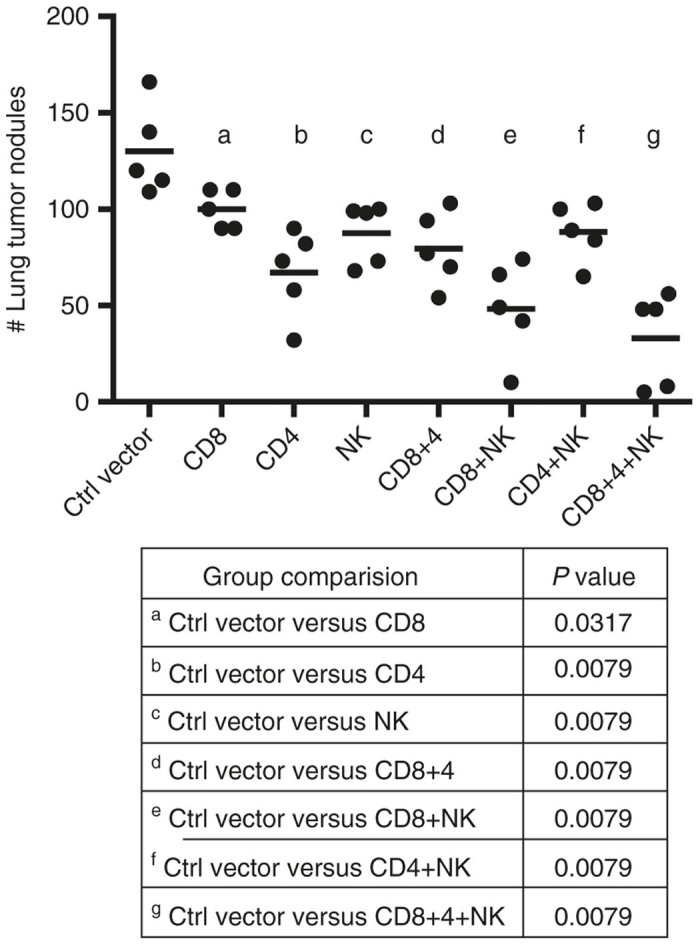
Adoptive transfer of CD8 T cells from LV305-immunized mice decreased lung tumor nodule formation in nonimmunized tumor-bearing recipient mice. On Day 0, female BALB/c mice (n = 5 per group) were injected with 1.5 × 105 CIN.23 cells, IV. On Day 3, tumor-bearing mice were injected, IV, with 1.0 × 106 cells (as detailed in the figure) isolated from LV305- or mock-immunized donor mice. On Day 18, tumor nodules in lungs were enumerated. Bars represent mean. CD, cluster of differentiation; IV, intravenous; NK, natural killer cells. Data are representative of two independent experiments.
Taken together, the above findings suggest that (i) CD8 T cells are required to mediate LV305-induced anti-tumor response; and (ii) CD4 T cells provide significant help in generating effective anti-tumor CD8 T-cell response.
Favorable pharmacokinetic and toxicology results in mice support the use of LV305 in phase 1 clinical trials
To evaluate the tropism and clearance kinetics of LV305 following multiple vector administrations, male and female BALB/c mice received four sequential immunizations, 14 days apart, of LV305 under good laboratory practices conditions (Figure 6). At various time points post-final LV305 immunization, mice were killed, and whole blood and 13 other tissues were harvested and analyzed for the presence of reverse-transcribed vector genomes (vector DNA) by qPCR. At Day 1 post-final immunization, vector DNA was detectable in LV305-injected mice at the site of injection (SOI, mean of 970 copies/1,000 ng of tissue DNA in 10 out of 10 mice) and in the draining inguinal lymph node (mean of 30 copies/1,000 ng of tissue DNA in 6 out of 10 lymph nodes). Consistent with the integration-deficient nature of LV305, vector DNA signal gradually decreased with time to Day 49 post-final immunization at the SOI to a mean of 75 copies of vector DNA/1,000 ng of tissue DNA in 9 out of 10 mice. In the draining inguinal lymph node, the vector DNA signal peaked at Day 4 post-final immunization before gradually decreasing to below the lower limit of detection. Sporadic low signal was observed in several organs that were minimally positive on Day 1 post-final immunization in fewer than 3 out of 10 mice and became negative thereafter. These signals likely represent DCs or other antigen-presenting cells transduced with the vector and migrating to organ sites.
Figure 6.

Biodistribution of injected LV305 in mice was limited to the site-of-injection and the draining lymph node. Female (n = 5 per group) and male (n = 5 per group) BALB/c mice were injected with 5 × 108 vector genomes of LV305 or vehicle on Days 1, 15, 29, and 43 subcutaneously at the tail base. At the designated time points (days post-final LV305 immunization), tissues listed in the figure were harvested. The presence of reverse-transcribed vector genomes (vector DNA) was measured by qPCR using NY-ESO-1-specific primes and probe. SC, subcutaneous; LN, lymph node; LOQ, limit of quantitation. Data are representative of two independent experiments.
To evaluate vector excretion (or shedding), presence of vector RNA at the SOI and various body fluids from mice immunized with LV305 were measured by quantitative reverse transcription-polymerase chain reaction (Table 1). LV305 was administered intradermally, the administration route of choice for the on-going LV305 phase 1 clinical trial. At 1, 4, and 24 hours post-immunization, samples were collected from the SOI, oral wash fluid, urine, and feces. Vector RNA was detectable by qPCR only at the SOI at 1 hour post-immunization in two out of three mice and were below the assay’s lower limit of detection in all mice by 4 hours. Vector RNA was below the assay’s lower limit of detection in oral wash fluid, urine, or feces at all time points measured. These results demonstrate that vector RNA excretion was limited to the SOI and was cleared within 4 hours post-immunization. The signal observed at the SOI at the 1 hour time point may be attributed to (i) partial leakage of the injected bolus dose from the SOI out to the skin through the channel introduced by the needle, (ii) residual deposition of small amounts of the dose onto the skin occurring at the time of injection, perhaps through droplets originating from the syringe needle, or (iii) direct exudation through the epidermis.
Table 1. Excretion of injected LV305 in mice was limited to the site-of-injection and fell below the limit of detection within 4 hours post-injection.
| Time post-injection | Sample source | Mouse number |
Injected material |
% Recovery | |
|---|---|---|---|---|---|
| Formulation vehicle | LV305 | ||||
| 1 hour | Site of injection | 1 | BLOD | 2.84 × 107 | 0.28% |
| 2 | BLOD | 1.29 × 107 | 0.13% | ||
| 3 | BLOD | BLOD | BLOD | ||
| 4 hours | Site of injection | 1 | BLOD | BLOD | BLOD |
| 2 | BLOD | BLOD | BLOD | ||
| 3 | BLOD | BLOD | BLOD | ||
| Urine | 1 | BLOD | BLOD | BLOD | |
| 2 | BLOD | BLOD | BLOD | ||
| 3 | BLOD | BLOD | BLOD | ||
| Oral | 1 | BLOD | BLOD | BLOD | |
| 2 | BLOD | BLOD | BLOD | ||
| 3 | BLOD | BLOD | BLOD | ||
| Feces | 1 | BLOD | BLOD | BLOD | |
| 2 | BLOD | BLOD | BLOD | ||
| 3 | BLOD | BLOD | BLOD | ||
| 24 hours | Site of injection | 1 | BLOD | BLOD | BLOD |
| 2 | BLOD | BLOD | BLOD | ||
| 3 | BLOD | BLOD | BLOD | ||
Female BALB/c mice (n = 3 per group) received two adjacent intradermal injections in the lower back of a total of 1 × 1010 vector genomes. Samples were taken from the site of injection, the oral cavity, urine, and feces, at 1, 4, and 24 hours post-immunization and assayed for the presence of vector RNA by qPCR. The LOD was 5.3 copies per qPCR reaction or 7.8 × 104 vector genomes per collected sample, respectively. % Recovery was determined using the following formula: (Recovered genomes)/(Injected genomes) × 100%. Samples spiked with LV305 were found to contain vector RNA demonstrating that the sample matrix factors did not introduce inhibitory interference with the qPCR assay (not shown). Data are representative of two independent experiments.
BLOD, below the assay’s lower limit of detection; LOD, limit of detection.
Under GLP conditions, a repeat-dose toxicity study was carried out in BALB/c mice to (i) evaluate the potential local and systemic toxicity of LV305 when administered via SC injection (four sequential immunizations, 2 weeks apart) and to (ii) evaluate the reversibility, persistence, or delayed occurrence of any effects of LV305 after a 7-week recovery period. Assessment of toxicity was based on mortality, clinical observations, body weight, food consumption, dermal irritation scoring, anatomic pathology, and clinical pathology. All animals survived to their scheduled necropsies. LV305-related clinical signs were limited to unkempt appearance at both dosage levels evaluated (5 × 107 and 5 × 108 vector genomes) in males throughout the study. There were no LV305-related changes in body weights, body weight changes, or food consumption. Very slight edema and/or very slight erythema were noted in males that received 5 × 108 vector genomes (Days 2 or 16). Only one female that received 5 × 108 vector genomes developed very slight erythema (Day 30). There were no LV305-related effects among hematology, coagulation, or clinical chemistry parameters. There were also no LV305-related macroscopic findings and no treatment-related differences in absolute or relative organ weights at the terminal or recovery necropsies. LV305-related microscopic findings were limited to the inguinal lymph node (follicular lymphoid hyperplasia) and the SOI (subacute/chronic inflammation) at both of the tested doses. Follicular lymphoid hyperplasia was attributed to lymphoid stimulation secondary to injection of LV305. Generally, findings at the inguinal lymph node and inflammation at the SOI were increased at one or more necropsy time points (Days 2, 16, 30, and 44) with on-going resolution at the recovery necropsy (Day 92). Overall, none of the clinical observations, dermal changes, or microscopic findings was considered to be adverse. The duration and frequency of dosing and the findings in this GLP toxicity study in mice were reported in regulatory submissions to the US Food and Drug Administration for support of the now-approved and on-going phase 1 clinical trial (NCT02122861).
Discussion
In this report, we demonstrated that immunization with LV305, a DC-targeting, integration-deficient lentiviral vector encoding NY-ESO-1, induced a robust CD8 T cell-dependent anti-tumor response against tumor cells expressing NY-ESO-1 and exhibited favorable pharmacokinetic and toxicology profiles in mice.
A major hurdle in evaluating a new therapeutic platform is the selection of the appropriate preclinical model. ZVex, the lentiviral vector platform for LV305, was designed to target transduction of human DCs through its pseudotyped envelop recognizing the human DC-SIGN (CD209) receptor.9 This specificity is difficult to recapitulate in animal models. For example, while LV305 entry into human DCs requires the expression of DC-SIGN, expression of the homologue molecule in mice, SIGNR1, is not a limiting factor for successful transduction of murine antigen-presenting cells.14 Additionally, efficient transduction of human DCs by LV305 involves the packaging of Vpx to overcome the restriction factor, SAMHD1.9,30,31 As different lentiviral restriction factors have evolved in different species, Vpx is not expected to facilitate degradation of restriction factors found in nonhuman primates, mice, or other animal models. Lastly, immunogenicity of the studied human tumor antigen(s) may be different in animals, depending on endogenous expression of the antigen(s) and/or differences in epitope recognition. If the human antigen is not immunogenic in animal models, evaluation of immune responses against the human antigen is not possible. In cases where the human antigen is immunogenic in the selected animal model, there may be a lack of developed tumor models for therapeutic assessment. Given these hurdles, we have identified the BALB/c mouse as an appropriate animal model for LV305 evaluation for the following reasons: (i) identification of H-2d-restricted CD8 and CD4 epitopes for NY-ESO-1; (ii) induction of cellular and humoral immunity in the BALB/c mice through LV305 immunization; and (3) development of a murine tumor model by generating a syngeneic murine tumor cell line expressing NY-ESO-1 (CIN.23 cell line). The selection to use the CIN.23 lung metastasis model in the BALB/c mice was due to the lack of tumor models available that could be targeted by LV305. As NY-ESO-1 is a human cancer-testis antigen expressed by multiple cancer types, our proposed preclinical studies were based on the concept that cancer cells (regardless of type) expressing NY-ESO-1 should be targeted by LV305-induced NY-ESO-1-specific cytotoxic lymphocytes. In our preclinical models, it was therefore more critical that the cancer cells expressed the antigen-of-interest (NY-ESO-1) rather than the type of cancer that was modeled. Given the artificial nature of targeting a human antigen in a mouse model, the development of the CIN.23 lung metastasis model was not intended to recapitulate the disease state of a particular cancer type but to provide a proof-of-concept model showing that LV305 could induce effective anti-tumor response against cancer cells expressing NY-ESO-1.
The generation of antigen-specific CD8 T cells was critical for the anti-tumor efficacy of LV305. A single dose of LV305 significantly inhibited growth of metastatic lung nodules in a dose-dependent manner that correlated with the magnitude of CD8 T cell response. In tumor-bearing mice that were depleted of CD8 T cells, LV305-induced anti-tumor response was completely abrogated. The generation of effective anti-tumor CD8 T cells through LV305 immunization appeared to benefit from CD4 T cell help. Depletion of CD4 T cells in tumor-bearing mice resulted in an intermediate level of protection and a suboptimal CD8 T-cell response, presumably due to the absence of CD4 T-cell help. This is supported by the additional observation that nonimmunized tumor-bearing mice developed a greater magnitude of NY-ESO-1-specific CD8 T cells post-adoptive transfer of CD4 T cells than CD8 T cells from LV305-immunized mice. These findings are consistent with the critical role of CD4 T-cell help in activating CD8 T cells and generating memory CD8 T cells.32–35 Unexpectedly, treatment with the anti-ASGM antibody abrogated LV305-induced anti-tumor response to the same extent as CD8 T-cell depletion. These mice also failed to mount an above-background level of Ag-specific CD8 T-cell response. At first glance, these findings appear to suggest that NK cells may play a role in the development of anti-tumor CD8 T-cell response. However, the anti-ASGM antibody has been shown to deplete not just NK cells but also activated CD836–38 and CD436 T cells and to drastically decrease cytotoxic T lymphocyte activity,37,38 which could all account for the observed overall decrease in CD8 T-cell response and the resulting lack of anti-tumor protection. Given the nonspecific targeting of the anti-ASGM antibody, it is difficult to experimentally evaluate whether anti-ASGM-antibody-mediated inhibition of CD8 T-cell response was the subsequent effect of specific NK cell depletion or due to the antibody’s direct impact on T cells or both. NK cell depletion studies in NK1.1− mouse strains (e.g., BALB/c) are therefore not as straightforward as those in NK1.1+ mouse strains (e.g., C57BL/6), due to the lack of a NK cell-specific depletion antibody or a transgenic mouse model that would allow transient depletion of NK cells.39 We therefore recommend the development of tumor models in NK1.1+ mouse strains to better differentiate between the functional roles of CD8 T cells and NK cells. Although our depletion studies using the anti-AGSM antibody were inconclusive in regards to the role of NK cells in LV305-induced anti-tumor response, our adoptive transfer studies provided additional clues that highlight the complexity of the interplay between subsets of immune cells. The presumed innate immune functionality mediated by NK cells was clearly demonstrated by achieving anti-tumor efficacy through the adoptive transfer of NK cells from mock-immunized mice. Additionally, mice that received cotransfers of CD8 T cells and NK cells from LV305-immunized mice developed significantly fewer lung nodules than mice that received CD8 and CD4 T cells. Taken together, LV305-induced anti-tumor efficacy appears to be mediated by both innate and adaptive immune responses: The vector backbone alone (regardless of the encoded antigen-of-interest) activates a nonantigen-specific NK cell response, and the vector-encoded NY-ESO-1 leads to the generation of tumor antigen-specific cytotoxic T lymphocytes.
Unlike other vector systems (e.g., adenoviral or pox viral vectors), LVs are attractive therapeutic delivery systems for homologous prime-boost immunizations because (i) pre-exposure to lentiviruses in humans is rare, resulting in minimal pre-existing immunity against LVs; (ii) compared with other commonly used vector technologies, LVs induce relatively weak host immunity against the vector itself;28,40 and (iii) there is minimal pre-existing immunity against SinVar1, the modified Sindbis virus envelop protein used to pseudotype LV305. Mice primed with LV305 generated antigen-specific cellular and humoral immunity that were recalled with homologous boosting with LV305. While more rapid than the primary response, the recall response from homologous boosting with LV305 was not significantly greater than the prime response, as one would observe from a viral infection. This could be due to the development of neutralizing antibodies against the vector envelop after immunization with LV305, a vector produced using the ZVex platform. We have previously shown that neutralizing antibodies were generated against the Sindbis virus-derived envelop of ZVex, especially at higher doses and repeated immunizations.14 Due to the aggressiveness of the CIN.23 tumor model, our Institutional Animal Care and Use Committee protocol limited our ability to test the anti-tumor efficacy of repeated immunizations with LV305 in tumor-bearing mice. However, in our B16F10 murine melanoma model, compared with a single-dose immunization, four sequential immunizations at 1-week intervals with ZVex expressing the murine melanoma antigen tyrosinase-related protein 1 significantly improved anti-tumor efficacy, despite the development of neutralizing antibodies (T. Albershardt, unpublished data). While homologous prime-boost with the ZVex platform may not achieve a recall response of the greatest magnitude, it can nevertheless translate to additional improvement in therapeutic and survival benefit in certain tumor models.
To our knowledge, LV305 is the first-in-class IDLV being evaluated in the clinic for cancer immunotherapy. By targeting DCs, tumor antigens are processed for direct presentation by major histocompatibility complex-I, as opposed to cross-presentation, which has been reported to be orders of magnitude less efficient in inducing CD8 T-cell responses.41 Immunization with LV305 induced a high magnitude of NY-ESO-1-specific CD8 T cells that effectively killed tumor cells expressing NY-ESO-1 to achieve anti-tumor control in mice. This is consistent with reports demonstrating that when antigen-presenting cells are transduced with IDLVs to express tumor antigens, robust antigen-specific T-cell and antibody responses can be generated to achieve protection against tumor growth.10,11 The biodistribution and excretion of injected LV305 were limited to the SOI and/or the draining lymph node, with the vectors being cleared from injected mice within 50 days post-immunization and inducing minimal or no adverse effects. The sum of the data presented here support the use of LV305 in on-going and up-coming phase 1/2 clinical trials and validate the use of our DC-targeting, integration-deficient lentiviral vector platform, ZVex, as a therapeutic strategy for the in vivo generation of tumor antigen-specific CD8 T-cell immunity.
Materials and Methods
LV305 vector
The design and production of LV305 is based on the VP02 platform previously described.9,14,42 Briefly, LV305 was produced via the transient transfection of 293T cells with five plasmids: the transfer vector that encodes the VP02 genome and NY-ESO-1, a modified gagpol transcript (RI-gagpol), accessory protein Rev from HIV-1, accessory protein Vpx from SIVmac, and the E1001 envelop glycoprotein variant of Sindbis virus.
LV305 quantitation
Genomic RNA was isolated from vector particles using the QIAamp Viral RNA Mini kit (Qiagen, Valencia, CA). To eliminate contaminating DNA, the extracted nucleic acid was digested with DNase I (Invitrogen, Grand Island, NY). Two dilutions of each DNase I-treated RNA sample were then analyzed by quantitative reverse transcription-PCR using the RNA Ultrasense One-Step Quantitative Reverse Transcription-PCR System (Invitrogen) and vector-specific primers and probe (Integrated DNA Technologies, Coralville, IA):
Forward primer: 5′-GGCAAGCAGGGAGCTAGAAC-3′
Reverse primer: 5′-GTTGTAGCTGTCCCAGTATTTGTC-3′
Probe: 5′-(FAM)-TCGCAGTTAATCCTGGCCTGTTAGA-(BHQ)-3′
The vector RNA copy number was calculated in reference to a standard curve comprised of linearized plasmid DNA containing the target sequences, diluted over a 7-log range (1 × 107 to 1 × 101 copies). The genome titer used throughout the experiments reflects the number of physical vector particles, calculated based on genomes, with each vector particle predicted to contain two single-stranded copies of genomic RNA.
Mice
Female 7- to 8-week-old C57BL/6 and BALB/c mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and housed under specific pathogen-reduced conditions in a BSL-2+ level room in the Infectious Disease Research Institute animal facility (Seattle, WA). All procedures were approved by the Infectious Disease Research Institute Institutional Animal Care and Use Committee.
Generation of syngeneic, NY-ESO-1-expressing tumor cell line, CIN.23
CT26 cells were transduced with lentiviral vector encoding NY-ESO-1. Transduced cells were serially diluted to near single-cell density per well of a 96-well plate for clonal expansion. Each clone was stained with anti-NY-ESO-1 antibody (Invitrogen, Grand Island, NY) to confirm expression of NY-ESO-1 by flow cytometry analysis. Clones expressing high, medium, or low level of NY-ESO-1 were inoculated subcutaneously into flanks of BALB/c mice for evaluation of tumor take. Clone 23 (named, CIN.23) was selected for its uniform expression of NY-ESO-1 (see Supplementary Figure S3) and reproducible induction of tumor nodule growth in lungs when inoculated intravenously in mice. CIN.23 cells proliferated faster (divided once per 0.8 day) than its parental CT26 cells (divided once per 1.1 day).
CIN.23 tumor inoculation
In adherence to the Institutional Animal Care and Use Committee protocol, we first demonstrated that IV injection of 1.5 × 105 CIN.23 cells led to the development of 100–150 tumor nodules in the lungs around 18 days post-inoculation (in contrast, 14 days for the parental CT26 cells). On the day of tumor inoculation, CIN.23 cells in logarithmic growth were resuspended in HBSS at 1.5 × 105 cells/100 µl and transported on ice to the animal facility for injections. Each mouse was placed in a conventional slotted restrainer with the tail accessible. To improve visualization of the tail vein, each tail was dabbed for a few seconds with gauze wetted with warm water. CIN.23 cells were then inoculated intravenously, via a 100-µl injection into the tail vein using a 29G 0.3 ml insulin syringe (BD Biosciences, San Jose, CA). On Day 18 post-tumor inoculation, lungs were harvested from mice, stained with India ink, and fixed in Fekete’s solution for enumeration of tumor nodules, as previously described.43
LV305 immunizations
When needed, aliquots of LV305 stored at −80 °C were thawed at room temperature and then kept on ice. Vector was diluted in cold sterile HBSS and transported to the animal facility on ice for injections. Mice were placed in a conventional slotted restrainer with the tail base accessible. Vector was administered via a 50-µl injection using a 29G 0.3 ml insulin syringe (BD Biosciences, San Jose, CA) inserted subcutaneously at the tail base, ~1 cm caudal to the anus, leading to minor but notable distension of the skin around the tail base.
Antibody depletion
Mice were administered 100 µg/dose of anti-CD8 (clone 2.43, Bio X Cell, West Lebanon, NH), anti-CD4 (clone GK1.5, Bio X Cell), anti-asialo-GM1 (Wako Chemicals USA, Richmond, VA), or the corresponding isotype control antibody, intraperitoneally, 7 days prior to tumor challenge. Depletion antibodies were continuously administered every 3–4 days until the end of study. To confirm successful depletion (>95%) of the target population, blood from mice was collected, stained for B220 (eBioscience, San Diego, CA), CD3ϵ (eBioscience), CD8 (clone H35-17.2, eBioscience), CD4 (RM4-5, eBioscience), and NKp46 (29A1.4, eBioscience), and analyzed by flow cytometry.
Adoptive transfer
CD4, CD8, or NK cells were isolated from spleens harvested from LV305- or mock-immunized mice using negative selection kits from Miltenyi Biotec (San Diego, CA), following the manufacturer’s protocol. Isolated cells were stained for B220, CD3ϵ, CD8, CD4, and NKp46, and analyzed by flow cytometry to ensure the purity was >95%. Cells were transferred to mice at 1 × 106 cells/100 µl injection, intravenously.
NY-ESO-1 peptide library for epitope mapping
A peptide library covering the complete sequence of NY-ESO-1 was purchased from Sigma-Aldrich (St. Louis, MO). The library consisted of 42 15-mer peptides overlapping by 11 amino acids. Each lyophilized peptide was reconstituted at 20 mg/ml dimethyl sulfoxide and stored at −20 °C. Peptides were pooled in a total of 14 pools, with Pool 1 containing peptides 1–3; Pool 2, peptides 4–6; and so on. These 14 peptide pools were used for the initial screening for positive CD8 and CD4 T-cell responses. Individual peptides from pools that tested positive were further screened to determine the 15-mers that contained the stimulating epitope. Minimal epitope sequences were not identified.
Intracellular cytokine staining for flow cytometry analysis
Spleens were homogenized with gentleMACS Octo Dissociator (Miltenyi Biotec, San Diego, CA), per manufacturer’s protocol. Red blood cells were lysed with 1× RBC Lysis Buffer (BioLegend, San Diego, CA), per manufacturer’s protocol. For analysis of cytokines, cells were stimulated in 96-well round-bottom plates with peptides at a concentration of 1 µg/ml per peptide in complete Roswell Park Memorial Institute medium (10% fetal bovine serum, 10 mmol/l N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid, 2 mmol/l β-mercaptoethanol, and l-glutamine) for 5 hours at 37 °C, 5% CO2 in the presence of brefeldin A (GolgiPlug, BD Biosciences, San Jose, CA). Peptides, including NY-ESO-181–88 (H-2d-restricted CD8 epitope), NY-ESO-190–107 (H-2d/b-restricted CD4 epitope), and MAGE-A3281–291 (irrelevant peptide), were manufactured at 95% purity by New England Peptide (Gardner, MA). After peptide stimulation, surface staining was carried out in flow cytometry staining buffer (phosphate-buffered saline, 1% fetal bovine serum, 2 mmol/l ethylenediaminetetraacetic acid) in the presence of FcR blocking antibody 2.4G2 (eBioscience, San Diego, CA) and LIVE/DEAD Fixable Near-IR (L/D NIR; Invitrogen, Grand Island, NY). Antibodies used for surface staining included anti-mouse CD3ϵ-PerCP/Cy5.5 (eBioscience), CD4-Alexa Fluor 700 (eBioscience), CD8-eFluor 450 (eBioscience), and B220-V500 (BD Biosciences). After surface staining, cells were washed with flow cytometry staining buffer, fixed with Cytofix (BD Biosciences), and stored overnight at 4 °C in flow cytometry staining buffer. Cells were then permeabilized with Perm/Wash buffer (BD Biosciences) containing 5% rat serum (Sigma-Aldrich, St. Louis, MO). Antibodies for intracellular staining were diluted in Perm/Wash buffer containing 5% rat serum and added to permeabilized cells. Antibodies included anti-mouse tumor necrosis factor-FITC (eBioscience), interferon-γ-PE (eBioscience), and interleukin-2-APC (eBioscience). Cells were washed with Perm/ Wash buffer, resuspended in flow cytometry staining buffer, and analyzed on a 3-laser LSRFortessa with High Throughput Sampler (BD Biosciences). Data were analyzed using FlowJo software (Tree Star, Ashland, OR). Viable CD8 T cells were gated as follows: lymphocytes (FSCint, SSClo), single cells (SSC-A = SSC-H), live (L/D NIRlo), CD3ϵ+ B220−, CD4- CD8+. Cytokine gates were based on the 99.9th percentile (<0.1% of positive events in nonstimulated cells).
In vitro cytotoxic T lymphocyte assay
Spleens from LV305- or mock-immunized mice were homogenized with gentleMACS Octo Dissociator (Miltenyi Biotec, San Diego, CA), per manufacturer’s protocol. Red blood cells were lysed with 1× RBC Lysis Buffer (BioLegend, San Diego, CA), per manufacturer’s protocol. The resulting mixed lymphocytes were then cultured for 5 days in 37 °C, 5% CO2 in completed Roswell Park Memorial Institute medium (10% fetal bovine serum, 10 mmol/l N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid, 2 mmol/l β-mercaptoethanol, and l-glutamine), supplemented with 1 µg/ml NY-ESO-181–88 stimulating peptide or MAGE-A3281–291 irrelevant peptide. Post-culture, nonadherent effector cells (E) were incubated with PKH26-labeled target cells (T, CT26 cells pulsed with 1 µg/ml NY-ESO-181–88 stimulating peptide) at E:T ratios of 0.15:1 to 100:1 for 4 hours in 37 °C, 5% CO2. Target cells were subsequently stained with LIVE/DEAD Fixable Near-IR (Invitrogen, Grand Island, NY) prior to flow cytometry analysis. Target cell % death was determined using the following formula: (Number of L/D-NIR+ PKH26+ cells)/(Number of PKH26+ cells) × 100%.
Endpoint ELISA for anti-NY-ESO-1 total IgG titer
Blood from terminally bled mice were collected in Microtainer tubes with serum separator (BD Biosciences, San Jose, CA) and centrifuged at 17,000×g for 20 minutes. Sera were transferred and stored in new microcentrifuge tubes at −20 °C until ELISA analysis. ELISA plates were coated with recombinant NY-ESO-1 (Ludwig Institute for Cancer Research, New York, NY) for 2 hours at room temperature and blocked overnight at 4 °C. Coated and blocked plates were washed three times with ddH2O. Each serum sample, with a starting dilution of 1:1,000 (prime only) or 1:4,500 (prime and boost), was serially diluted threefold until signal fell below the limit of detection. Each diluted sample was added to each well of a coated and blocked plate and incubated for 2 hours at room temperature. Goat anti-mouse IgG horseradish peroxidase conjugate at a dilution of 1:1,000 was added and incubated for 1 hour at room temperature. 3, 3’, 5, 5’-tetramethylbenzidine peroxidase substrate was then added and incubated for 15 minutes at room temperature. The colorimetric reaction was then stopped with 4 mol/l H2SO4. Plate wells were read at 450 nm by a SpectraMax M5e Microplate Reader (Molecular Devices, Sunnyvale, CA). Endpoint titer was defined as the highest dilution factor needed to achieve a mean OD value greater than the mean OD value + 2 SD from sera of nonimmunized mice.
LV305 biodistribution
Mice (5 male and 5 female BALB/c mice per group) received 5 × 108 vector genomes of LV305 or vehicle on Days 1, 15, 29, and 43 subcutaneously at the tail base (four immunizations at 2-week intervals). The presence of reverse-transcribed vector DNA was analyzed by qPCR at 1, 4, 7, 14, 28, or 49 days post-final LV305 immunization in the following tissues: SOI (tail base), bone marrow, brain, heart, liver, lung, kidney, ovaries, testes, small intestines, spleens, and draining (inguinal) and nondraining (cervical) lymph nodes. Tissues were processed in Fastprep Lysing Matrix D tubes using a Fastprep-24 homogenizer (MP Biomedicals, Santa Ana, CA) and genomic DNA was isolated from homogenates using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Eluted DNA (200 ng per sample) was analyzed by qPCR in quadruplicate using EXPRESS qPCR Supermix Universal (Life Technologies, Carlsbad, CA) and the same primers/probe set used for LV305 quantitation. All reactions were performed using the Bio-Rad CFX384 and analyzed using Bio-Rad CFX Manager software (Bio-Rad Laboratories, Hercules, CA). The vector DNA copy number was calculated in reference to a standard curve comprised of plasmid DNA containing the target sequences diluted over a 7-log range (107–101 copies).
LV305 GLP repeat-dose toxicity study
Mice (75 male and 75 female BALB/c mice per group) received 5 × 107 or 5 × 108 vector genomes of LV305 or vehicle on Days 1, 15, 29, and 43 subcutaneously at the tail base (four immunizations at 2-week intervals). Mice were then monitored for mortality, clinical observations, body weight, food consumption, dermal irritation scoring, anatomic pathology, and clinical pathology. At each of the scheduled terminal (Days 2, 16, 30, and 44) and recovery (Day 92) necropsies, organs were harvested from killed mice (15 mice/sex/group), weighed and inspected for macroscopic and microscopic changes. The dose levels and range for this toxicity study were the maximum feasible dose and volume that could be administered to mice from the mid (1 × 109 vector genomes/ml and 1 × 1010 vector genomes/ml) doses of filled clinical LV305 drug product. LV305 was produced in a manner comparable to the manufacturing process that was used to produce phase 1 clinical material. These doses were both within the biologically effective dose range of research-grade LV305 in mice established in previously completed immunogenicity, prophylactic, and therapeutic tumor model studies.
Acknowledgments
We thank the past and present Immune Design colleagues: Scott Robbins for research direction; Carlos Paya, Stephen Brady, and Julie Urvater for manuscript review; Jacob Archer and Chris Nicolai for research-grade vector production; Anshika Bajaj and Chintan Vin for vector quantitation; and Patrick Flynn for experimental assistance. All of the presented work was designed and conducted by Immune Design, except LV305 Biodistribution and LV305 GLP Repeat-Dose Toxicity studies, which were conducted in collaboration with MPI Research (Mattawan, MI).
T.C.A., D.C., A.P., M.S., J.M., and P.B. are at least one of the current employees of Immune Design, a holder of stock and/or options in Immune Design, or an inventor on an issued patent or patent application for technology discussed in this paper, which has been assigned to Immune Design.
References
- Dullaers, M, Van Meirvenne, S, Heirman, C, Straetman, L, Bonehill, A, Aerts, JL et al. (2006). Induction of effective therapeutic antitumor immunity by direct in vivo administration of lentiviral vectors. Gene Ther 13: 630–640. [DOI] [PubMed] [Google Scholar]
- Esslinger, C, Chapatte, L, Finke, D, Miconnet, I, Guillaume, P, Lévy, F et al. (2003). In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest 111: 1673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y, Zhang, J, Donahue, C and Falo, LD Jr. (2006). Skin-derived dendritic cells induce potent CD8(+) T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 24: 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y, Zhang, J, Mi, Z, Robbins, P and Falo, LD Jr. (2005). Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol 174: 3808–3817. [DOI] [PubMed] [Google Scholar]
- Yang, L, Yang, H, Rideout, K, Cho, T, Joo, KI, Ziegler, L et al. (2008). Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol 26: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasik, MB and McCray, PB Jr. (2010). Integrase-defective lentiviral vectors: progress and applications. Gene Ther 17: 150–157. [DOI] [PubMed] [Google Scholar]
- Apolonia, L, Waddington, SN, Fernandes, C, Ward, NJ, Bouma, G, Blundell, MP et al. (2007). Stable gene transfer to muscle using non-integrating lentiviral vectors. Mol Ther 15: 1947–1954. [DOI] [PubMed] [Google Scholar]
- Kantor, B, Bayer, M, Ma, H, Samulski, J, Li, C, McCown, T et al. (2011). Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther 19: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen, SU, Kelley-Clarke, B, Nicolai, CJ, Cassiano, LA, Nelson, LT, Slough, MM et al. (2014). Design of a novel integration-deficient lentivector technology that incorporates genetic and posttranslational elements to target human dendritic cells. Mol Ther 22: 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwacz, K, Mukherjee, S, Apolonia, L, Blundell, MP, Bouma, G, Escors, D et al. (2009). Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J Virol 83: 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B, Yang, H, Dai, B, Tai, A and Wang, P (2009). Nonintegrating lentiviral vectors can effectively deliver ovalbumin antigen for induction of antitumor immunity. Hum Gene Ther 20: 1652–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia, A and Sallusto, F (2001). Regulation of T cell immunity by dendritic cells. Cell 106: 263–266. [DOI] [PubMed] [Google Scholar]
- Mellman, I and Steinman, RM (2001). Dendritic cells: specialized and regulated antigen processing machines. Cell 106: 255–258. [DOI] [PubMed] [Google Scholar]
- Odegard, JM, Kelley-Clarke, B, Tareen, SU, Campbell, DJ, Flynn, PA, Nicolai, CJ et al. (2015). Virological and preclinical characterization of a dendritic cell targeting, integration-deficient lentiviral vector for cancer immunotherapy. J Immunother 38: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholaou, T, Ebert, L, Davis, ID, Robson, N, Klein, O, Maraskovsky, E et al. (2006). Directions in the immune targeting of cancer: lessons learned from the cancer-testis Ag NY-ESO-1. Immunol Cell Biol 84: 303–317. [DOI] [PubMed] [Google Scholar]
- Cebon, J, Knights, A, Ebert, L, Jackson, H and Chen, W (2010). Evaluation of cellular immune responses in cancer vaccine recipients: lessons from NY-ESO-1. Expert Rev Vaccines 9: 617–629. [DOI] [PubMed] [Google Scholar]
- Gnjatic, S, Nishikawa, H, Jungbluth, AA, Güre, AO, Ritter, G, Jäger, E et al. (2006). NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res 95: 1–30. [DOI] [PubMed] [Google Scholar]
- Sato, S, Noguchi, Y, Wada, H, Fujita, S, Nakamura, S, Tanaka, R et al. (2005). Quantitative real-time RT-PCR analysis of NY-ESO-1 and LAGE-1a mRNA expression in normal tissues and tumors, and correlation of the protein expression with the mRNA copy number. Int J Oncol 26: 57–63. [PubMed] [Google Scholar]
- Jäger, E, Chen, YT, Drijfhout, JW, Karbach, J, Ringhoffer, M, Jäger, D et al. (1998). Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med 187: 265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, RF, Johnston, SL, Zeng, G, Topalian, SL, Schwartzentruber, DJ and Rosenberg, SA (1998). A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol 161: 3598–3606. [PubMed] [Google Scholar]
- Adams, S, O’Neill, DW, Nonaka, D, Hardin, E, Chiriboga, L, Siu, K et al. (2008). Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J Immunol 181: 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, ID, Chen, W, Jackson, H, Parente, P, Shackleton, M, Hopkins, W et al. (2004). Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci USA 101: 10697–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger, E, Gnjatic, S, Nagata, Y, Stockert, E, Jäger, D, Karbach, J et al. (2000). Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA 97: 12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odunsi, K, Qian, F, Matsuzaki, J, Mhawech-Fauceglia, P, Andrews, C, Hoffman, EW et al. (2007). Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci USA 104: 12837–12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Morgan, RA, Feldman, SA, Yang, JC, Sherry, RM, Dudley, ME et al. (2011). Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, AP, Stadtmauer, EA, Binder-Scholl, GK, Goloubeva, O, Vogl, DT, Lacey, SF et al. (2015). NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21: 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka, D, Nishikawa, H, Noguchi, T, Wang, L, Harada, N, Sato, E et al. (2013). Establishment of animal models to analyze the kinetics and distribution of human tumor antigen-specific CD8⁺ T cells. Vaccine 31: 2110–2118. [DOI] [PubMed] [Google Scholar]
- Kootstra, NA and Verma, IM (2003). Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol 43: 413–439. [DOI] [PubMed] [Google Scholar]
- Nayak, S and Herzog, RW (2010). Progress and prospects: immune responses to viral vectors. Gene Ther 17: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrecka, K, Hao, C, Gierszewska, M, Swanson, SK, Kesik-Brodacka, M, Srivastava, S et al. (2011). Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474: 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette, N, Sobhian, B, Casartelli, N, Ringeard, M, Chable-Bessia, C, Ségéral, E et al. (2011). SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474: 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan, MJ (2004). Helping the CD8(+) T-cell response. Nat Rev Immunol 4: 595–602. [DOI] [PubMed] [Google Scholar]
- Janssen, EM, Lemmens, EE, Wolfe, T, Christen, U, von Herrath, MG and Schoenberger, SP (2003). CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421: 852–856. [DOI] [PubMed] [Google Scholar]
- Shedlock, DJ and Shen, H (2003). Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 300: 337–339. [DOI] [PubMed] [Google Scholar]
- Sun, JC and Bevan, MJ (2003). Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300: 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka, MK, Pagarigan, RR and Whitton, JL (2000). NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol 164: 2009–2015. [DOI] [PubMed] [Google Scholar]
- Stitz, L, Baenziger, J, Pircher, H, Hengartner, H and Zinkernagel, RM (1986). Effect of rabbit anti-asialo GM1 treatment in vivo or with anti-asialo GM1 plus complement in vitro on cytotoxic T cell activities. J Immunol 136: 4674–4680. [PubMed] [Google Scholar]
- Ehl, S, Nuesch, R, Tanaka, T, Myasaka, M, Hengartner, H and Zinkernagel, R (1996). A comparison of efficacy and specificity of three NK depleting antibodies. J Immunol Methods 199: 149–153. [DOI] [PubMed] [Google Scholar]
- Walzer, T, Bléry, M, Chaix, J, Fuseri, N, Chasson, L, Robbins, SH et al. (2007). Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci USA 104: 3384–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B, Tai, A and Wang, P (2011). Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunol Rev 239: 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel, RM (2014). On the role of dendritic cells versus other cells in inducing protective CD8+ T cell responses. Front Immunol 5: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareen, SU, Nicolai, CJ, Campbell, DJ, Flynn, PA, Slough, MM, Vin, CD et al. (2013). A Rev-independent gag/pol eliminates detectable psi-gag recombination in Lentiviral Vectors. Biores Open Access 2: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman, M, Hu, X and Liu, K (2010) Experimental metastasis and CTL adoptive transfer immunotherapy mouse model. J Vis Exp 45. doi: 10.3791/2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.