

Abstract
Nox4-based NADPH oxidase is a major reactive oxygen species-generating enzyme in the vasculature, but its role in atherosclerosis remains controversial.
Objective
Our goal was to investigate the role of smooth muscle Nox4 in atherosclerosis.
Approach and results
Atherosclerosis-prone conditions (disturbed blood flow and Western diet) increased Nox4 mRNA level in smooth muscle of arteries. To address whether upregulated smooth muscle Nox4 under atherosclerosis-prone conditions was directly involved in the development of atherosclerosis, mice carrying a human Nox4 P437H dominant negative mutation (Nox4DN), specifically in smooth muscle, were generated on a FVB/N ApoE deficient genetic background to counter the effect of increased smooth muscle Nox4. Nox4DN significantly decreased aortic stiffness and atherosclerotic lesions, with no effect on blood pressure. Gene analysis indicated that soluble epoxide hydrolase 2 (sEH) was significantly downregulated in Nox4DN smooth muscle cells (SMC), at both mRNA and protein levels. Downregulation of sEH by siRNA decreased SMC proliferation and migration, and suppressed inflammation and macrophage adhesion to SMC.
Conclusions
Downregulation of smooth muscle Nox4 inhibits atherosclerosis by suppressing sEH, which, at least in part, accounts for inhibition of SMC proliferation, migration and inflammation.
Keywords: Nox4, atherosclerosis, smooth muscle, soluble epoxide hydrolase 2
1. Introduction
Atherosclerosis is the leading cause of morbidity and mortality in developed countries and occurs at lesion-prone areas of disturbed blood flow. Western diet (WD) accelerates the development of atherosclerosis. The correlation between the location of atherosclerotic lesions and regions of disturbed flow is well documented, throughout the arterial system, in experimental models of atherosclerosis (diet- and/or genetically-induced), across multiple animal species (monkeys, rabbits, pigs, and rodents) as well as in the pathophysiology of human atherosclerosis [1]. Endothelial dysfunction and leukocyte adhesion initiate the development of atherosclerotic lesions. Partial left carotid artery (LCA) ligation is an established model of acutely induced, disturbed blood flow that leads to endothelial dysfunction and atherosclerosis [2–4]. The importance of smooth muscle cells (SMC) in development of atherosclerosis remains underappreciated. When endothelium is damaged, the underlying SMC can directly stimulate leukocyte recruitment by releasing chemokines [5,6]. Recruited leukocytes become macrophages and release cytokines, which stimulate SMC migration and proliferation. Both macrophages and activated SMC uptake lipoproteins to form foam cells, the hallmark of growing atherosclerotic lesions [7,8]. In human coronary atherosclerotic plaques, a very large proportion of foam cells are SMC-derived [7]. However, the mechanisms by which SMC contribute to plaque formation are not fully elucidated [9,10].
Elevated levels of reactive oxygen species (ROS) in the vascular wall play a key role in the development of atherosclerosis. NADPH oxidases are major ROS-generating enzymes in the vasculature. Distinct from other members of the NADPH oxidase family, Nox4 is constitutively active in SMC, and its expression level determines the amount of SMC intracellular ROS, primarily hydrogen peroxide (H2O2) [11]. Our previous study in mice overexpressing a human Nox4 dominant-negative form in SMC, shows that downregulation of SMC Nox4 inhibits endothelial denudation-induced neointimal hyperplasia, by suppressing thrombospondin 1 (TSP1), indicating a critical role of SMC Nox4 in the development of neointimal lesions [12]. Several studies investigated the role of vascular NADPH oxidase’s components p47phox [13], Nox2 [14], Nox1 [15] and Nox4 [16,17] in atherosclerosis using whole body knockout on atherosclerosis-prone, ApoE deficient (ApoE−/−) mice. However, the contribution of different Nox isoforms to atherosclerosis remains far beyond conclusive. In addition, Nox4 is abundantly expressed both in SMC and endothelial cells but the relative contribution of Nox4 from each of these two cell types to atherosclerosis remains controversial. Transgenic mice overexpressing Nox4 in endothelium had decreased atherosclerotic lesions [18], while whole body Nox4 deficiency had no effect on diabetes-induced atherosclerosis [15], indicating that Nox4 contributes to the development of atherosclerosis in a cell specific manner. In human atherosclerotic coronary arteries, Nox4 is upregulated mainly during the atheroma stage of the plaque, which contains abundant α-actin-positive cells, implying that SMC Nox4 may be involved in the genesis and progression of human coronary atherosclerotic disease [19].
In the present study, we sought to explore the role of SMC Nox4 in regulating atherosclerosis and the potential molecular mechanisms involved. We found that Nox4 mRNA in smooth muscle of arteries were upregulated in atherosclerosis-prone conditions (disturbed blood flow and Western diet). To counter the effect of increased smooth muscle Nox4 under atherosclerosis-prone conditions, we generated unique transgenic mice overexpressing a human Nox4 dominant negative P437H mutant (Nox4DN) specifically in SMC [12]. We found that Nox4DN inhibits atherosclerosis in part by suppressing soluble epoxide hydrolase 2 (sEH, gene EPHX2), which inhibits SMC proliferation, migration and inflammation.
2. Material and methods
2.1. Transgenic mouse lines in FVB/N and FVB/N ApoE−/− background
Mouse lines that overexpress a human dominant negative form of Nox4 (Nox4DN P437H) in SMC (SDN) were generated in the FVB/N genetic background [12]. The mutation of proline to histidine at position 437 was confirmed by genomic DNA sequencing. Littermate non-transgenic mice (NTg) served as controls. For atherosclerosis studies, SDN were backcrossed into the FVB/N ApoE−/− background [20] to obtain SDN/ApoE−/−. Littermate ApoE−/−, without Nox4DN transgene, served as controls. Backcrossing into FVB/N ApoE−/− background did not affect Nox4DN mRNA expression levels, as confirmed by qPCR. All animal procedures was approved by the Boston University’s Institutional Animal Care and Use Committee in accordance with the provisions of the Animal Welfare Act, Public Health Service Animal Welfare Policy, and the principles of the NIH Guide for the Care and Use of Laboratory Animals, and the policies and procedures of Boston University Medical Campus. Animals were maintained in an AALAC approved Laboratory Animal Science Center staffed with licensed veterinarians.
2.2. Cell Culture
Aortic SMCs from 12 week old male SDN mice and their littermate controls were isolated and cultured as previously described [21]. SMC phenotype was confirmed by α-smooth muscle actin-positive staining. Cells from passages 3 to 10 were used.
2.3. Real time quantitative PCR (qPCR) [21]
Total cellular RNA was isolated from SMCs cultured in 0.2% fetal bovine serum (FBS) Dulbecco’s Modified Eagle Medium (DMEM) or from common carotid arteries using standard procedures, and retro-transcribed to cDNA. Real time qPCR was performed with the following gene specific primers (Nox4: Forward primer-5′TGGCCAACGAAGGGGTTAAA3′, Reverse primer-5′ACACAATCCTAGGCCCAACA3′; EPHX2: Forward primer-5′CAGGAGGACACAGACACCATA3′, Reverse primer-5′TCTCAGGTAGATTGGCTCCAC3′) using SYBR-Green-based detection, or TAQMAN gene specific primers (Applied Biosystems), according to the following cycling conditions: denaturation, annealing, and extension at 95 °C, 57 °C and 72 °C for 10 s, 30 s, and 10 s, respectively, for 40 cycles. β-actin was used as internal control.
2.4. Immuno-blotting
SMC were cultured in 0.2% FBS DMEM overnight before being lysed. Proteins were subjected to SDS-PAGE and immunoblotted with specific antibodies against Nox4 (provided by Dr. Ajah Shah), phospho-p65 NFκB (Cell Signaling Technologies, CST), p65 NFκB (CST), IκBα (CST), vascular cell adhesion molecule-1 (VCAM1, Santa Cruz), sEH (Santa Cruz), TSP1 (Invitrogen); GAPDH (CST) acts as loading control. Proteins were visualized with an ECL system (Amersham Biosciences). Densitometry analysis was performed using NIH Image J software, and protein expression levels were normalized to GAPDH.
2.5. SMC proliferation assay
SMC were seeded in 96-well plate at a density of 5x103 cells per well in DMEM supplemented with 0.2% FBS overnight. Cell proliferation was stimulated by medium supplemented with 10% FBS, and 0.2% FBS medium was used as control. Culture medium with high or low serum was changed daily. A tetrazolium-based non-radioactive proliferation assay kit (Quick Cell Proliferation Assay Kit II, BioVision) was used to determine the cell number according to the manufacturer’s protocol.
2.6. Wounded monolayer migration assay [22,23]
Briefly, 106 SMC/well in 12-well plates were seeded in 0.2% FBS DMEM overnight until confluent then scratch wounds were applied to the SMC monolayer with a pipette tip. Immediately after scratching, the cells were treated with 10% FBS DMEM to stimulate cell migration. Photographs were taken at 0 h and 6 h at three fixed locations along the scratch with a light microscope and analyzed using NIH Image J software.
2.7. Small interference RNA (siRNA) transfection [21]
Using routine methods, 80% confluent SMC were transfected with EPHX2 or TSP1 siRNA or scrambled control siRNA (Applied Biosystems, 60 nmol/L) for 48 h according to the manufacturer’s protocol (Lipofectamine 2000, Invitrogen).
2.8. Adenoviral transfection in NTg SMCs [21]
Using routine methods, 80% confluent SMC were infected with an adenovirus to overexpress wild type human Nox4 [24] or an empty adenovirus control, Nox4 siRNA [25] or control siRNA at 50 MOI/cell in DMEM without serum or antibiotics for 6 h before adding DMEM containing 10% FBS for 3 days.
2.9. Application of sEH inhibitor 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) [26]
TPPU (0.1μM) was dissolved in DMSO and added to SMC in DMEM for two days before proliferation assay, migration assay and NFκB activity assay.
2.10. NFκB promoter luciferase activity assay [27]
SMC were cultured in 24-well plates with 0.2% FBS DMEM overnight, and then co-infected with firefly NFκB promoter luciferase adenovirus and pre-packaged renilla luciferase adenovirus (Vector Biolabs) for 48 h. PBS or tumor necrosis factor alpha (TNFα,10 ng/ml) was added overnight before measuring NFκB activity. Briefly, cells were washed and lysed in lysis buffer as per manufacturer’s recommendations. The ratio of firefly to renilla luciferase luminescence was calculated as indicative of NFκB promoter activity. In some experiments, SMC were transfected with EPHX2 siRNA, Nox4 siRNA or control siRNA for 48 h before measuring NFκB promoter activity.
2.11. Bone marrow-derived macrophage (BMDM) adhesion assay [27]
Bone marrow-derived mononuclear cells were isolated [28] from NTg mice and cultured in high glucose DMEM supplemented with 10% FBS and 20% L929-conditioned medium for five days, when mononuclear cells became macrophage. For macrophage adhesion assay, SMC were plated in 12-well plates, at a density of 106 cells/well, in 0.2% FBS DMEM overnight until confluent. BMDMs (5×105 cells/well) were added to SMC and incubated for 1 h, then the media was removed and SMCs washed 3 times with PBS to remove all unbound macrophages. Four images of macrophage adhesion to SMCs in each well were taken and the number of bound macrophage was counted per image area. In some experiments, SMCs were transfected with EPHX2 siRNA or TSP1 siRNA or control siRNA for 48 h before macrophage adhesion assay.
2.12. Partial left common carotid artery (LCA) ligation [3]
Anesthesia was induced in 10-week old mice, by intraperitoneal injection of xylazine (10 mg/kg) and ketamine (80 mg/kg) mixture. Mice underwent partial LCA ligation to rapidly induce disturbed blood flow. Briefly, three of the four caudal branches of the LCA (left external carotid, internal carotid, and occipital artery) were ligated with a 6-0 silk suture, while the superior thyroid artery was left intact. The right carotid artery (RCA) underwent similar manipulation but without ligation and used as a sham control. Mice were sacrificed 2 days or 2 weeks after surgery, for RNA isolation or tissue collection, respectively.
2.13. Tissue preparation and immunohistochemistry analysis [12]
Tissues were collected as described previously [12]. For each animal, 6 identical 7 μm cross-sections at 250–300μm intervals were prepared, starting at the bifurcation of the common carotid artery. The mean cross-sectional area of neotimal and medial areas were determined by using NIH Image J software. The medial area was calculated as the area encircled by the external elastic lamina minus the area encircled by the internal elastic lamina. The intima to media ratio was quantified by dividing the neointimal area by the medial area. For smooth muscle staining and macrophage staining, smooth muscle alpha actin antibody (SMA, Sigma) and Mac2 antibody (Santa Cruz) were used, according to the procedure of the Vectastain ABC kit (Vector).
2.14. Western diet (WD) induced atherosclerotic lesions [20]
Ten weeks old SDN/ApoE−/− and ApoE−/− were fed a WD (Teklad Adjusted Calories 88137; 21% wt/wt fat, 0.15% wt/wt cholesterol, 19.5% wt/wt casein, and no sodium cholate) to accelerate the formation of atherosclerotic lesions. After 12 weeks on WD, mice underwent partial LCA ligation to rapidly induce disturbed blood flow. After surgery, mice continued on WD feeding for another 2 weeks before being sacrificed to collect tissues.
2.15. Medial RNA isolation from common carotid arteries and aorta [3]
Two days after partial LCA ligation, mice were sacrificed and both LCA and RCA were isolated and carefully cleaned of periadventitial fat. The carotid lumen was quickly flushed with 150 μl of QIAzol lysis reagent (QIAGEN) using a 29-gauge insulin syringe to remove endothelium, and the leftover carotid was used to prepare RNA from media and adventitia. For mice under normal diet (ND) or WD for 1 month, whole aorta was quickly flushed with 300 μl of QIAzol lysis reagent to remove endothelium, and the leftover was used to prepare RNA from media and adventitia.
2.16. Measurement of plasma cholesterol and triglycerides [29]
Mice were sacrificed and blood was drawn from the right atrium into EDTA-containing tubes, for lipid measurements. Plasma was prepared via centrifugation at 850 x g for 15 minutes at 4 °C and stored at −20 °C. Measurements were carried out using an Infinity Cholesterol and Infinity Triglycerides measurement kit (Thermo Scientific), based on absorbance of samples normalized to the absorbance of a known concentration of a calibrator (200 mg/dL), provided with the kit. Data are expressed as mg/dL.
2.17. Oil Red O (ORO) staining for lipids
ORO staining was used to investigate lipid deposition. For aortic root analysis, the heart was isolated along with the aortic arch after perfusion. Aortic roots were embedded in OCT compound. Sequential sections, each 5 μm thick, were cut starting at the aortic sinus, which was identified by the appearance of the semilunar valve leaflets, and extending over the next 1.2 mm over the entire aortic sinus. Frozen whole neck sections were serially sectioned (7 μm), air-dried and mounted on superfrost glass. Briefly, slides were dipped in propylene glycol to dehydrate followed by ORO (American MasterTech) for 10 min at 60 °C. Sections were differentiated in propylene glycol for 1 min and counterstained with hematoxylin and mounted in an aqueous mounting media. For aortic roots, the areas of the ORO positive lesions in the proximal aortas were quantified. The mean cross-sectional area of ORO positive lesions and the total area were determined by using NIH Image J software.
2.18. In vivo measurement of aortic pulse wave velocity (PWV) [30]
PWV was measured 12 weeks after ND or WD using a high-resolution Doppler ultrasound instrument (VEVO770, FujiFilm) [30]. Briefly, mice were anesthetized with 2% isoflurane and mounted on a heated (37 °C) platform to monitor electrocardiogram (ECG), heart rate (HR), respiratory rate, and to eliminate movement artifacts. Hair was removed from the abdomen with a depilatory agent (Nair). Isoflurane was reduced to 0.8% with slight adjustments in order to maintain a HR of approximately 450 bpm. A b-mode cross-section image of the abdominal aorta was used to locate the point where the renal vein crosses over the aorta and identified as proximal point. The scan head was then turned longitudinally so that blood flow measurements could be made at the proximal point and about 8 mm distally (distal point). Flow wave Doppler measurements were taken at 30 MHz with a pulse-repetition frequency of 40 kHz at a depth of 6 mm. There was no difference in the HR between the proximal and distal point measurements. In offline analysis of the images, the time (ms) from the peak of the ECG R wave to the foot of the flow waves at both the proximal and distal locations were measured, in at least 5 replicates for each location per mouse. The difference between proximal and distal points arrival time yields the transit time (TT). PWV (mm/ms) was calculated from TT (Δt) and the distance between the measurement sites (Δd).
2.19. Invasive blood pressure measurements on anesthetized mice in FVB/N ApoE−/− background
Mice were anesthetized with 2% isofluorane/air inhalation and maintained under anesthesia (0.5–1% isofluorane/air; HR: 450–500 bpm; respiration rate ~130 bpm) during the procedure. Mice were in the recumbent position on a heated platform (37°C) and with paws in contact with pad electrodes for continuous ECG recording. After gentle hair removal in the neck area, a solid-state high-fidelity pressure catheter (Mikro-tip catheter transducers, SPR-1000, Millar Instruments, Houston, TX, USA) was inserted in the left carotid artery and advanced into the aortic arch. 20 ms of stable recordings were acquired and analyzed post-acquisition with an acquisition and analysis workstation (NIHem, Cardiovascular Engineering, Norwood, MA, USA) with 1 kHz bandwidth and 5 kHz sampling rate. NIHem software (v. 4.99) automatically signal-averages 20 ms of pressure waveform recordings and computes mean arterial pressure (MAP).
2.20. Radiotelemetry blood pressure measurements in FVB/N background
HR, systolic blood pressure (SBP), MAP and diastolic blood pressure (DBP) were measured in conscious, freely moving mice using radiotelemeters (Data Sciences International), as we previously described [32]. Daily recordings (1 every 4 min, 360 recordings/mouse per day) started after complete recovery from surgery (1–2 weeks) and were carried on continuously for 4 days (baseline). Angiotension II (Ang II, 1mg/kg body weight/day) was then administered via osmotic minipumps (model 1007D, Alzet, Durect Corporation, Cupertino, CA, USA) for the following 7 days. The average of 360 daily recordings for each mouse was calculated before and after Ang II.
2.21. Statistical analysis
All data are presented as mean ± standard error. Student t-test was used to analyze data from experimental and control groups. One-way or two-way ANOVA was used to compare data from 3 groups with one or two treatments, followed by repeated measures and post hoc analysis with Bonferroni multiple comparison test. For samples analyzed in separate experiments, all data were normalized to its control (NTg or control siRNA) in each individual experiment and expressed as fold change relative to control. For their statistical analysis, each data was taken log transformation and Wilcoxon signed-rank test was used to get a probability value. A probability value of <0.05 is considered statistically significant.
3. Results
3.1. Disturbed blood flow and Western diet (WD) upregulate smooth muscle Nox4 and induce atherosclerosis
Atherosclerosis occurs preferentially in arterial branches and curved areas with disturbed blood flow; WD accelerates the development of atherosclerosis. We first performed a partial LCA ligation in FVB/N ApoE −/− mice, fed normal diet (ND), to induce disturbed blood flow patterns. We found that disturbed blood flow dramatically upregulates smooth muscle Nox4 mRNA levels 2 days after ligation (Figure 1 A), and neointimal formation with lipid accumulation 2 weeks after ligation, compared with unligated RCA (Figure 1 B). WD feeding for 1 month significantly increased smooth muscle Nox4 mRNA levels (Figure 1 A), and further accelerated disturbed blood flow-induced neointimal formation and lipid accumulation compared with ND (Figure 1 B). These results imply that upregulation of smooth muscle Nox4 may contribute to the development of atherosclerosis.
Figure 1.

Both disturbed blood flow and Western diet upregulate smooth muscle Nox4 and induce atherosclerosis in FVB/N ApoE−/− mice. A. Smooth muscle Nox4 mRNA levels. Left: partial ligated left common carotid artery (LCA) vs. unligated right common carotid artery (RCA) 2 days after ligation. *p < 0.05 vs. unligated artery, n = 4. Right: Smooth muscle Nox4 mRNA levels of denuded aorta from mice fed normal diet (ND) or Western diet (WD) for 1 month. *p < 0.05 vs. ND, n = 6–7. B. Representative pictures (20 X) of Oil Red O staining of unligated RCA and ligated LCA 2 weeks after partial LCA ligation in mice fed ND or WD for 3 months.
3.2. Overexpression of human Nox4DN in SMCs attenuates Western diet-induced atherosclerotic lesions and arterial stiffness
To address whether upregulated SMC Nox4 is directly involved in the development of atherosclerotic lesions, we generated transgenic mice that overexpress a human Nox4 dominant negative P437H mutant (Nox4DN), specifically in SMCs (SDN) [12], to counter the effect of increased SMC Nox4 observed under atherosclerosis-prone conditions (disturbed flow and WD). We previously showed that this human Nox4DN construct effectively works as a dominant negative for murine Nox4 1) by suppressing endogenous mouse Nox4, and 2) by competing with endogenous mouse Nox4 for binding to p22phox, thus forming a non-functional Nox4-based NADPH oxidase complex [12]. Compared with non-transgenic littermate controls (NTg), SDN have lessened neointimal hyperplasia after common carotid artery endothelial denudation, and have decreased SMC proliferation and migration rates, via TSP1 suppression [12].
To study the effects of Nox4DN on atherosclerosis, SDN mice were backcrossed into the FVB/N ApoE−/− genetic background [20]. Breeding ApoE−/− and SDN/ApoE−/− produced a total of 546 healthy pups at a ratio close to 1:1 (280 ApoE−/− vs. 266 SDN/ApoE−/−), suggesting a normal embryonic development. There was no difference in body weights between ApoE−/− and SDN/ApoE−/− at different ages (male mice at 2 months old: ApoE−/−: 25.0 ± 0.6 vs. SDN/ApoE−/−: 25.2 ± 0.6 g, n = 17–18; male mice at 3.5 months old: ApoE−/−: 32.7 ± 2.0 vs. SDN/ApoE−/−: 33.0 ± 1.3 g, n = 13–14), excluding any apparent developmental growth abnormalities in SDN/ApoE−/− mice.
In comparison with ApoE−/− on C57BL/6 genetic background, obvious atherosclerotic lesions, in the aortic root, take much longer to develop in FVB/N ApoE−/− mice (up to 11 months). To accelerate the formation of atherosclerotic lesions in the aortic root, we fed mice a WD for 14 weeks [20] and, during the last 2 weeks of diet, we performed a partial LCA ligation [3]. WD did not increase body weight or total triglycerides in either ApoE−/− or SDN/ApoE−/− compared with their ND controls of the same age (6 months old). However, WD equally increased total cholesterol levels in both groups (Table 1). Importantly, lipid deposition in the aortic root, detected by ORO staining, was decreased in SDN/ApoE−/− compared with ApoE−/− (Figure 2 A).
Table 1.
Plasma total cholesterol and triglycerides levels
| ApoE ND | SDN ApoE ND | ApoE WD | SDN ApoE WD | |
|---|---|---|---|---|
| Body weight (g) | 36.5±1.8 | 39.2±1.7 | 43.9±2.8 | 42.5±2.1 |
| TG (mg/dl) | 377.7±49.5 | 388.2±47.3 | 390.6±38.8 | 400.8±43.0 |
| Cholesterol (mg/dl) | 488.5±38.2 | 495.6±46.4 | 1131.9±109.1* | 1225.5±155.2* |
Note:
p < 0.05 Western diet (WD) vs. normal diet (ND), ND, n = 10; WD, n = 16–18.
Figure 2.

Overexpression of human Nox4DN in smooth muscle attenuates arterial stiffness and Western diet induced atherosclerotic lesions. A. Representative picture of Oil Red O staining of aortic root lesions in mice fed Western diet (20 X). Quantification in graph. *p < 0.05 vs. ApoE−/−, n = 8–12. B. Pulse wave velocity (PWV). *p < 0.05 vs. ApoE−/−, n = 6–9. C. Downregulation of smooth muscle Nox4 has no effect on mean arterial pressure on anesthetized mice in FVB/N ApoE−/− background fed ND. n = 7–9. D. Downregulation of smooth muscle Nox4 has no effect on either baseline or angiotension II (Ang II)-mediated blood pressure measured by radiotelemetry in FVB/N background. n = 5 in each group. SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure.
Pulse wave velocity (PWV) is inversely proportional to aortic distensibility and directly correlates with aortic stiffness [33]. As arterial stiffness is an early biomarker of atherosclerosis [34], we measured PWV by high-resolution Doppler ultrasound along the aorta. Compared with age-matched non-ApoE−/− FVB/N on ND, ApoE−/− had increased PWV (non-ApoE−/−: 2.7 ± 0.3 vs. ApoE−/−:4.1 ± 0.5, p < 0.05, n = 6–10). WD itself slightly increased PWV in ApoE−/− but not in SDN/ApoE−/−, which had lower PWV than ApoE−/−, either on ND or WD (Figure 2 B).
To test whether a decrease in aortic stiffness in SDN/ApoE−/− was due to an effect of smooth muscle Nox4DN on blood pressure, as PWV is dependent on distending pressure, we measured MAP in anesthetized mice. There was no significant difference in MAP between SDN/ApoE−/− and ApoE−/− fed ND (Figure 2 C) or WD (ApoE−/−: 97.3 ± 2.6 vs. SDN/ApoE−/−:99.9 ± 6.5, n = 3). We also measured blood pressure by radiotelemetry, in conscious, freely moving mice. As shown in Figure 2 D, systolic, diastolic and mean arterial pressures were not different between SDN and NTg, either at baseline or after Ang II. Our data indicate that downregulation of SMC Nox4 does not affect blood pressure in our experimental conditions. Therefore, we conclude that the decrease in arterial stiffness in SDN/ApoE−/− is not due to a decrease in blood pressure but, rather, it reflects a decrease in atherosclerotic lesion formation on the aortic wall.
When we performed a partial LCA ligation to induce disturbed blood flow in ApoE−/−, we observed the formation of lipid-rich lesions, localized both in the neointima and media, even in mice fed ND, although to a much lesser extent than WD-fed mice (Figure 1 B), indicating that this model of disturbed blood flow dramatically accelerates atherosclerosis in lesion-prone areas, even without WD. In mice on WD, partial LCA ligation induced advanced atherosclerotic lesions assessed in the ligated carotid, with abundant lipid deposition, which were dramatically decreased in SDN/ApoE−/− mice (Figure 3 A). SDN/ApoE−/−, had an equally increased medial area but a significantly lower intimal area and intima to media ratio compared to ApoE−/− controls (Figure 3 B).
Figure 3.

Both smooth muscle cell and macrophage contribute to the attenuated disturbed blood flow induced atherosclerotic lesions by overexpression of human Nox4DN in smooth muscle. A. Common carotid artery lesions 2 weeks after partial left carotid artery ligation in mice fed Western diet. Left: Representative pictures of Oil Red O staining (20 X). Right: Quantification in graph. *p < 0.05 vs. ApoE−/−, n = 6–8. B. Common carotid artery neointimal hyperplasia. Left: Neointimal and medial areas. *p < 0.05 vs. ApoE−/−, #p < 0.05 vs. uninjured media, n = 6–7. Right: Intima to media ratio. *p < 0.05 vs. ApoE−/−, n = 6–7. C. Both SMC and macrophage contribute to atherosclerotic lesions. Left: Representative picture of macrophage (Mac2) and smooth muscle cell (SMA) staining (20 X). Middle: Summary of neotimal area, macrophage positive area and others. *p < 0.05 vs. ApoE−/−, n = 6–8. Right: The percentages of both macrophage and non-macrophage components in neointimal area. n = 6–8.
3.3. Overexpression of human Nox4DN attenuates both smooth muscle cells and macrophages in atherosclerotic lesions
To elucidate the contribution of macrophages and SMC to the formation of atherosclerotic lesions, we immunostained ligated carotid arteries after WD (same samples as in Figure 3 A) with macrophage (Mac2) and SMC (SMA) specific antibodies. As shown in Figure 3 C, both macrophages and SMCs were present in atherosclerotic lesions. Although both the lesion area and overall macrophage-positive area were significantly decreased in SDN/ApoE−/− (Figure 3 C), the percentages of both macrophage and non-macrophage (majorly SMCs) in the lesion area were not different between SDN/ApoE−/− and ApoE−/− (Figure 3 C). Taken together, our data indicate that SMC Nox4 plays an important role in the development of atherosclerosis, and that Nox4 may have differential effects on SMC migration, proliferation and recruitment of macrophages, which may all contribute to the development of atherosclerosis.
3.4. Overexpression of human Nox4DN in SMCs downregulates soluble epoxide hydrolase 2 (sEH, gene EPHX2) and suppresses inflammation
To further explore the potential mechanisms by which Nox4 regulates SMC proliferation, migration and inflammation, we isolated SMC from aortas of both SDN/ApoE−/− and ApoE−/−. However, after a few sub-culturing passages, these SMCs lost their phenotype and morphology, rendering them unfit for further cell analysis. We suspect that the ApoE−/− genetic background affects SMC in vitro properties, and/or our culture conditions were not optimal to maintain their phenotype. Accordingly, all in vitro experiments with SMC presented here, are from SMC isolated from mice on FVB/N genetic background, which grew normally in vitro.
As shown in Figure 4 A&B, we found that mRNA and protein levels of EPHX2, a gene important for SMC function, were significantly downregulated in both arteries and aortic SMC from SDN compared with NTg controls.
Figure 4.

Overexpression of human Nox4DN in smooth muscle downregulates the expression levels of soluble epoxide hydrolase 2 and suppresses inflammation. A&B: Soluble epoxide hydrolase 2 (sEH, gene EPHX2) are downregulated in aortic SMC and arteries from SDN mice. A. sEH expression in arteries. Left: mRNA levels. *p < 0.05 vs. NTg, n = 8. Right: protein levels. *p < 0.05 vs. NTg, n = 6–8. B. sEH expression in SMC. Left: mRNA levels. *p < 0.05 vs. NTg, n = 6. Right: protein levels. *p < 0.05 vs. NTg, n = 8. C. Representative Western blots of inflammation markers from aortic SMC and quantification of band intensities in graph. *p < 0.05 vs. NTg, n = 6–9. D. NFκB activity in SMC cultured in 0.2% FBS DMEM with or without TNFα (10 ng/ml) overnight. *p < 0.05 vs. NTg in the same treatment, #p < 0.05 TNFα vs. no TNFα from same cell line, n = 6. E. Representative pictures of macrophage adhesion to SMC and quantification in graph. *p < 0.05 vs. NTg, n = 6.
Because sEH inhibition is known to downregulate proinflammatory genes and decrease inflammatory cell infiltration into the vascular wall [35] and SDN/ApoE−/− had decreased macrophages in atherosclerotic lesions than controls (Figure 3 C), we assessed VCAM1 and NFκB promoter activity, as inflammatory markers, and the interaction between macrophage and SMC in cultured aortic SMC. As shown in Figure 4 C, VCAM1 protein level was significantly decreased in SMC isolated from SDN compared with NTg, but we did not detect any difference in protein levels of phospho-p65 NFκB or IκBα, two well-known components of NFκB activation. Despite phospho-p65 NFκB levels were similar in SDN and NTg, both basal and TNFα-induced NFκB promoter activity were decreased in SDN compared with NTg, as measured by a luciferase reporter assay (Figure 4 D).
The endothelium is known to play a key role in regulating leukocyte adhesion to vessel wall, however also SMC, underlining activated/denuded endothelial cells, can regulate leukocyte recruitment by releasing chemokines [5,6]. Because human Nox4DN decreased VCAM1 expression and NFκB promoter activity in SMC, we measured macrophage adhesion to SMC, isolated from SDN and NTg. As shown in Figure 4 E, macrophage adhesion to SDN SMC was decreased compared with NTg SMC. Taken together, VCAM1 downregulation, decreased NFkB promoter activity and decreased macrophage adhesion in SDN SMC are consistent with a role of SMC Nox4 in promoting vascular inflammation.
Next, we tested the effect of smooth muscle Nox4DN on proinflammatory genes in isolated common carotid arteries. As shown in Figure 5, downregulation of smooth muscle Nox4, on both FVB/N and FVB/N ApoE−/− genetic backgrounds, suppressed monocyte chemoattractant protein 1 (MCP1), intercellular adhesion molecule 1 (ICAM1) and VCAM1 compared with littermate controls, confirming that smooth muscle Nox4 regulates inflammation in intact arteries.
Figure 5.

Downregulation of smooth muscle Nox4 suppressed proinflammatory genes of carotid artery in both FVB/N and FVB/N ApoE−/− backgrounds. *p < 0.05 vs. littermate controls, n = 5–8. MCP1, monocyte chemoattractant protein 1; ICAM1, intercellular adhesion molecule 1; VCAM1, vascular cell adhesion molecule 1.
3.5. Nox4 regulates expression of sEH and inflammation in smooth muscle cells
To validate the direct effects of Nox4 on sEH and inflammation in SMC, we carried out experiments with Nox4 downregulation or overexpression in cultured SMC, isolated from NTg aortas. Similar to Nox4DN mice, Nox4 siRNA suppressed sEH and VCAM1 expression, and inhibited NFκB promoter activity in NTg SMC (Figure 6 A). Overexpression of human Nox4 generated the opposite effect, i.e. sEH and VCAM1 upregulation and increase in NFκB promoter activity in NTg SMC (Figure 6 B), further confirming a crucial role of smooth muscle Nox4 in regulating sEH expression and inflammation.
Figure 6.

Nox4 regulates expression of soluble epoxide hydrolase 2 and inflammation in smooth muscle cells. A. Downregulation of Nox4 by siRNA suppresses the expression of sEH and VCAM1, and inhibits NFκB activity in NTg SMC. Left: Representative Western blots of decreased sEH and VCAM1 by knocking down of Nox4. Middle: Quantification of band intensities in graph. Right: NFκB activity. *p < 0.05 vs. control siRNA, n = 6. B. Overexpression of wild type human Nox4 upregulates the expression levels of both sEH and VCAM1, and increases NFκB activity in NTg SMC. Left: Representative Western blots of indicated proteins in NTg SMC with overexpression of empty adenovirus (Adv) or adenovirus human Nox4 wild type (Nox4). Middle: Quantification of band intensities in graph. Right: NFκB activity. *p < 0.05 vs. Adv, n=6–7.
3.6. Downregulation of TSP1 in smooth muscle cells has no effect on sEH and inflammation
As we previously reported that TSP1 regulated by Nox4 is directly involved in SMC proliferation and migration [12], in the current study we knocked down TSP1 in SMC to test whether TSP1 is an upstream regulator of sEH and inflammation. As shown in Figure 7, downregulation of TSP1 with siRNA had no effects on the expression levels of sEH and VCAM1, nor on macrophage adhesion to SMC, compared with control siRNA, indicating that downregulation of TSP1 does not account for Nox4DN anti-inflammatory effect, despite inhibiting SMC proliferation and migration [12].
Figure 7.

Knocking down thrombospondin 1 by siRNA has no effect on the expression of soluble epoxide hydrolase 2 and inflammation. A. Representative Western blots of indicated proteins in control and TSP1 siRNA-transfected SMC. Quantification in graph. *p < 0.05 vs. control siRNA, n = 6–12. B. Macrophage adhesion to SMC. n = 6.
3.7. Downregulation of sEH inhibits SMC proliferation, migration and inflammation
To further study the effect of sEH on SMC proliferation, migration and inflammation, we used EPHX2 siRNA to downregulate sEH expression in NTg aortic SMC. As shown in Figure 8 A, downregulation of sEH decreased protein levels of TSP1 and VCAM1, but had no effect on phospho-p65 NFkB or Nox4 compared with control siRNA. In addition, downregulation of sEH by EPHX2 siRNA inhibited both basal and TNFα-induced NFκB promoter activity (Figure 8 B), serum-induced SMC proliferation and migration, and macrophage adhesion to SMC, compared with control siRNA (Figure 8 C). Therefore, EPHX2 siRNA fully mimicked the effect of Nox4DN or Nox4 siRNA on pathways involved in SMC proliferation, migration and inflammation.
Figure 8.

Downregulation of soluble epoxide hydrolase 2 inhibits cell proliferation, migration, and inflammation in NTg SMC. A–C. Downregulation of sEH by siRNA. A. Representative Western blots for indicated proteins. Quantification in graph. *p < 0.05 vs. control siRNA, n = 6–11. B. NFκB activity. *p < 0.05 vs. control siRNA in the same treatment, #p < 0.05 TNFα vs. basal, n = 6. C. Downregulation of sEH inhibits cell proliferation, migration and macrophage adhesion to SMC. Proliferation assay: *p < 0.05 vs. control siRNA, n = 11–12; migration assay: *p < 0.05 vs. control siRNA, n = 6; macrophage adhesion to SMC: *p < 0.05 vs. control siRNA, n = 6. D. sEH inhibitor TPPU inhibits serum induced SMC proliferation and migration, and NFκB activity. *p < 0.05 vs. DMSO, n = 6.
We further explored the role of sEH in SMC by using a specific sEH inhibitor, TPPU [26]. As shown in Figure 8 D, similar to sEH siRNA, sEH inhibitor TPPU inhibited serum-induced SMC proliferation, migration and NFκB promoter activity, confirming the contribution of sEH in regulating SMC function. Overall, our results demonstrate that sEH is an upstream regulator of TSP1 and NFκB and is an essential factor, regulated by SMC Nox4, in controlling SMC biological functions in atherosclerosis.
4. Discussion
The data presented here, using a smooth muscle specific Nox4 genetic knockdown, represent the first direct and in vivo evidence of an important role of SMC Nox4 in promoting atherosclerosis. First, we found that SMC Nox4 mRNA was upregulated in arteries in response to disturbed blood flow and Western diet, two experimental pro-atherosclerosis conditions, implying that there is a potential link between SMC Nox4 and the development of atherosclerosis. Secondly, consistent with our previous study [12], downregulation of SMC Nox4 in vivo inhibited the extent of atherosclerotic lesions in ligated carotids and SMC proliferation, migration and expression of pro-inflammatory pathways assesses on SMC isolated from SDN mice. Thirdly, in an attempt to elucidate the molecular mechanisms by which Nox4 regulates SMC biological functions in atherosclerotic lesions, we found that downregulation of SMC Nox4 suppresses gene and protein expression of sEH, a well-known regulator of SMC, both in vivo and in vitro. Of note, sEH appears to regulate another important SMC factor, TSP1, since knocking down sEH by siRNA suppressed TSP1 expression, but not vice versa. As summarized in Figure 9, we hypothesize that atherosclerosis-prone conditions, such as disturbed blood flow and/or western diet, upregulate smooth muscle Nox4 to promote atherosclerosis, by stimulating SMC proliferation, migration and inflammation. On the contrary, downregulation of SMC Nox4 suppresses sEH, which, in turn, inhibits atherosclerosis by inhibiting TSP1, thus decreasing SMC proliferation and migration. In addition, sEH inhibits NFkB activity and VCAM1, which mediate inflammation and macrophage adhesion, in a TSP-1-independent manner. Overall, our findings strongly implicate smooth muscle Nox4 as one of the major determinants of atherosclerosis.
Figure 9.
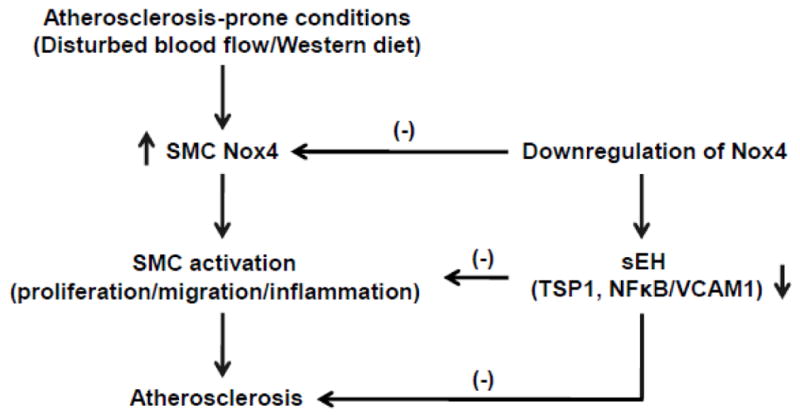
The potential mechanisms of downregulation of smooth muscle Nox4 on atherosclerosis.
One of the novel findings of our study is that downregulation of SMC Nox4 suppresses sEH, which appears to be one of the major Nox4 downstream mediators of inhibition of SMC proliferation, migration and inflammation in vitro and decreased atherosclerosis in SDN/ApoE−/− mice. Cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids (EETs) are hyperpolarizing factors which modulate vascular tone. sEH catalyzes the hydrolysis of EETs and other fatty acid epoxides, thus limiting their biological actions, including modulation of inflammation, SMC migration and platelet aggregation [36,37]. sEH inhibitors or EETs inhibit SMC proliferation and migration [38,39] and the role of sEH in cardiovascular inflammation has been recently reviewed [40]. In cultured endothelial cells, EETs attenuate TNFα-induced VCAM1 expression and leukocyte adhesion, partially through inhibition of NFκB activation [41]. sEH inhibitors decrease proinflammatory factors, both circulating and in the aorta, as well as inflammatory cell infiltration into the vascular wall [35]. sEH deficient mice have significantly decreased neointima after femoral artery cuff occlusion [42], and carotid artery ligation [43], while administration of an sEH inhibitor to Ang II-treated ApoE−/− mice significantly attenuates abdominal aortic aneurysm formation and atherosclerotic lesion area [35,44]. In addition, sEH inhibition decreases blood pressure in angiotensin-dependent and deoxycorticosterone-salt hypertension models [45,46]. Although the role of sEH in neointimal hyperplasia and atherosclerosis have been studied in sEH deficient mice or mice treated with sEH inhibitors, the contribution of SMC sEH to vascular diseases is unclear. In our study, downregulation of SMC sEH by Nox4DN implies that SMC sEH, as regulated by Nox4, can play an important role in atherosclerosis and neointimal hyperplasia, but has no evident effect on blood pressure before or during Ang II administration.
The limitation of current study is that we do not know the exact mechanism by which SMC Nox4 downregulates sEH. We did not observe consistent effects of H2O2, PEG-catalase or overexpression of catalase on sEH expression in cultured SMCs (data not shown), suggesting that Nox4 may regulate sEH in H2O2-independent fashion.
The role of Nox4 in atherosclerosis remains controversial. One study using Nox4 deficient mice, on ApoE−/− genetic background, found no effect of Nox4 on atherosclerosis in a model of type I diabetes [15]. To the contrary, a recent publication using an inducible Nox4 knockout, indicates that Nox4 is protective in the progression of atherosclerotic lesions [16]. Since Nox4 is abundantly expressed in both endothelial cell and SMC, it is imperative to study the role of Nox4 in these individual cell types on atherosclerosis, which cannot be addressed using mice globally deficient in Nox4, as in previous studies. Our own unpublished data indicate that downregulating Nox4 in endothelium accelerates the formation of atherosclerosis in settings of type I diabetes, while overexpressing Nox4 in the endothelium ameliorates WD-induced atherosclerosis. These findings are in accordance with a study using transgenic mice that overexpress Nox4 in the endothelium, indicating that endothelial Nox4 is protective from atherosclerosis induced by WD [18]. Compared with the data from SDN/ApoE−/− mice presented here, indicating that SMC Nox4 promotes atherosclerosis, it is clear that Nox4 in SMC and endothelium play opposite roles thus explaining the overall lack of effect on atherosclerosis in global Nox4 null mice, which might be the result of endothelial and SMC Nox4 countering off each other’s effects [15]. Our study addresses the limitation of using whole body Nox4 ablated mice and underlines the importance of studying cell specific roles for Nox4 in cardiovascular pathologies.
In conclusion, our study indicates for the first time that SMC Nox4 plays an essential role in the development of atherosclerosis. Pro-atherosclerosis conditions, such as disturbed blood flow and Western diet, may promote atherosclerosis through upregulation of smooth muscle Nox4. By using unique transgenic mice overexpressing human Nox4DN in SMCs, we found that SMC Nox4 promotes atherosclerosis by stimulating SMC proliferation, migration and inflammation, and that sEH is one of SMC Nox4 downstream mediators. Inhibiting SMC Nox4 and/or sEH could be therapeutically beneficial to prevent atherosclerosis.
Highlights.
Atherosclerosis-prone conditions increase Nox4 in smooth muscle of arteries.
Smooth muscle Nox4 plays a detrimental role in the development of atherosclerosis.
Downregulation of smooth muscle Nox4 inhibits atherosclerosis by suppressing sEH.
Nox4 regulates SMC proliferation, migration and inflammation via sEH.
Acknowledgments
The authors thank Dr. Jan L. Breslow from Rockefeller University for FVB/N ApoE−/−mice and Dr. Ajah Shah for Nox4 antibody. This work was supported by National Natural Science Foundation of China (31571172, Tong X), Fundamental Research Funds for the Central Universities (0236015202008, Tong X), American Diabetes Association award (7-09-JF-69, Tong X), NIH R01 HL031607 (Cohen RA and Tong X), R37 HL104017, Boston University Medical Center Department of Medicine Evans Center Arterial Stiffness ARC. B.D.H and K.S.S.L. is partially supported by the NIEHS grant R01 ES002710, NIEHS Superfund Research Program grant P42 ES004699 and NIH CounterAct U54 NS079202 and K.S.S.L. has been partially supported by the NIH Pathway to Independence Award from NIH/NIEHS (1K99ES024806-01).
Abbreviations
- ApoE−/−
ApoE deficient
- DBP
diastolic blood pressure
- EETs
epoxyeicosatrienoic acids
- H2O2
hydrogen peroxide
- ICAM1
intercellular adhesion molecule 1
- LCA
left common carotid artery
- MAP
mean arterial pressure
- MCP1
monocyte chemoattractant protein 1
- ND
normal diet
- NTg
non-transgenic littermate mice
- Nox4DN
Nox4 dominant negative form P437H
- ORO
Oil Red O
- PWV
pulse wave velocity
- ROS
reactive oxygen species
- SBP
systolic blood pressure
- SDN
smooth muscle human Nox4DN transgenic mice
- sEH
soluble epoxide hydrolase 2
- siRNA
small interference RNA
- SMA
smooth muscle alpha actin
- SMC
smooth muscle cells
- TNFα
tumor necrosis factor alpha
- TPPU
1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea
- TSP1
thrombospondin 1
- VCAM1
vascular cell adhesion molecule 1
- WD
Western diet
Footnotes
Disclosure
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–87. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nam D, Ni C-W, Rezvan A, Suo J, Budzyn K, Llanos A, et al. A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium. J Vis Exp. 2010 doi: 10.3791/1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–H1543. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, et al. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood. 2010;116:e66–e73. doi: 10.1182/blood-2010-04-278192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simon DI. Inflammation and Vascular Injury. Circ J. 2012;76:1811–1818. doi: 10.1253/circj.CJ-12-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–542. doi: 10.1152/physrev.00004.2012. [DOI] [PubMed] [Google Scholar]
- 7.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis Ga. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 8.Allahverdian S, Pannu PS, Francis Ga. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc Res. 2012;95:165–72. doi: 10.1093/cvr/cvs094. [DOI] [PubMed] [Google Scholar]
- 9.Ozaki MR, de Almeida EA. Evolution and involution of atherosclerosis and its relationship with vascular reactivity in hypercholesterolemic rabbits. Exp Toxicol Pathol. 2013;65:297–304. doi: 10.1016/j.etp.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Ross R. Atherosclerosis: current understanding of mechanisms and future strategies in therapy. Transplant Proc. 1993;25:2041–2043. [PubMed] [Google Scholar]
- 11.Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol. 2010;30:653–661. doi: 10.1161/ATVBAHA.108.181610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, et al. Role of smooth muscle Nox4-based NADPH oxidase in neointimal hyperplasia. J Mol Cell Cardiol. 2015;89:185–194. doi: 10.1016/j.yjmcc.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 13.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, et al. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI200111927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Judkins CP, Diep H, Broughton BRS, Mast AE, Hooker EU, Miller AA, et al. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am J Physiol Heart Circ Physiol. 2010;298:H24–H32. doi: 10.1152/ajpheart.00799.2009. [DOI] [PubMed] [Google Scholar]
- 15.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, et al. Nox1 Plays a Key Role in Diabetes Accelerated Atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 16.Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. 2015:ehv460. doi: 10.1093/eurheartj/ehv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation. 2013;127:1888–902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 18.Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE− /− mice from atherosclerotic lesions. Free Radic Biol Med. 2015;89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 20.Dansky HM, Charlton SA, Sikes JL, Heath SC, Simantov R, Levin LF, et al. Genetic background determines the extent of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1960–1968. doi: 10.1161/01.ATV.19.8.1960. [DOI] [PubMed] [Google Scholar]
- 21.Tong X, Hou X, Jourd’heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGF{beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010;107:975–983. doi: 10.1161/CIRCRESAHA.110.221242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44:361–369. doi: 10.1016/j.yjmcc.2007.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong X, Schröder K. NADPH oxidases are responsible for the failure of nitric oxide to inhibit migration of smooth muscle cells exposed to high glucose. Free Radic Biol Med. 2009;47:1578–1583. doi: 10.1016/j.freeradbiomed.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen K, Kirber MT, Xiao H, Yang Y, Keaney JF. Regulation of ROS signal transduction by NADPH oxidase 4 localization. J Cell Biol. 2008;181:1129–1139. doi: 10.1083/jcb.200709049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanborn R, Hammock BD. NIH Public Access. 2012;53:7067–7075. doi: 10.1021/jm100691c.1-Aryl-3-(1-acylpiperidin-4-yl)urea. [DOI] [Google Scholar]
- 27.Bayat H, Xu S, Pimentel D, Cohen RA, Jiang B. Activation of thromboxane receptor upregulates interleukin (IL)-1beta-induced VCAM-1 expression through JNK signaling. Arterioscler Thromb Vasc Biol. 2008;28:127–134. doi: 10.1161/ATVBAHA.107.150250. [DOI] [PubMed] [Google Scholar]
- 28.Wang HW, Liu PY, Oyama N, Rikitake Y, Kitamoto S, Gitlin J, et al. Deficiency of ROCK1 in bone marrow-derived cells protects against atherosclerosis in LDLR−/− mice. FASEB J. 2008;22:3561–3570. doi: 10.1096/fj.08-108829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuccollo A, Shi C, Mastroianni R, Maitland-Toolan KA, Weisbrod RM, Zang M, et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation. 2005;112:3001–3008. doi: 10.1016/S0749-4041(08)70346-4. [DOI] [PubMed] [Google Scholar]
- 30.Williams SB, Cusco JA, Roddy MA, Johnstone MT, Creager MA. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1996;27:567–574. doi: 10.1016/0735-1097(95)00522-6. [DOI] [PubMed] [Google Scholar]
- 31.Van Vliet BN, McGuire J, Chafe L, Leonard A, Joshi A, Montani JP. Phenotyping the level of blood pressure by telemetry in mice. Clin Exp Pharmacol Physiol. 2006;33:1007–1015. doi: 10.1111/j.1440-1681.2006.04479.x. [DOI] [PubMed] [Google Scholar]
- 32.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King Ca, et al. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62:1105–10. doi: 10.1161/HYPERTENSIONAHA.113.01744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lane HA, Smith JC, Davies JS. Noninvasive assessment of preclinical atherosclerosis. Vasc Health Risk Manag. 2006;2:19–30. doi: 10.2147/vhrm.2006.2.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamatelopoulos K, Karatzi K, Sidossis LS. Noninvasive methods for assessing early markers of atherosclerosis: the role of body composition and nutrition. Curr Opin Clin Nutr Metab Care. 2009;12:467–473. doi: 10.1097/MCO.0b013e32832f0d99. [DOI] [PubMed] [Google Scholar]
- 35.Zhang LN, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan SK, et al. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol. 2009;29:1265–1270. doi: 10.1161/ATVBAHA.109.186064. [DOI] [PubMed] [Google Scholar]
- 36.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.RES.87.11.992. [DOI] [PubMed] [Google Scholar]
- 37.Elmarakby AA. Reno-protective mechanisms of epoxyeicosatrienoic acids in cardiovascular disease. AJP Regul Integr Comp Physiol. 2012;302:R321–R330. doi: 10.1152/ajpregu.00606.2011. [DOI] [PubMed] [Google Scholar]
- 38.Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2002;99:2222–2227. doi: 10.1073/pnas.261710799. papers://89a9a5ef-0e36-4406-8469-1548b73d210c/Paper/p2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun J, Sui X, Bradbury JA, Zeldin DC, Conte MS, Liao JK. Inhibition of vascular smooth muscle cell migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ Res. 2002;90:1020–7. doi: 10.1161/01.res.0000017727.35930.33. http://www.google.com/search?client=safari&rls=en-us&q=Inhibition+of+vascular+smooth+muscle+cell+migration+by+cytochrome+p450+epoxygenase+derived+eicosanoids&ie=UTF-8&oe=UTF-8\npapers2://publication/uuid/C2905CE1-43C8-4479-90AC-1B28E47E5C79. [DOI] [PubMed] [Google Scholar]
- 40.Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol. 2010;48:331–341. doi: 10.1016/j.yjmcc.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Revermann M, Schloss M, Barbosa-Sicard E, Mieth A, Liebner S, Morisseau C, et al. Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2010;30:909–914. doi: 10.1161/ATVBAHA.110.204099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol. 2010;298:H795–H806. doi: 10.1152/ajpheart.00543.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, et al. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol. 2008;52:314–323. doi: 10.1097/FJC.0b013e318185fa3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, et al. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–F748. doi: 10.1152/ajprenal.00098.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anandan SK, Webb HK, Chen D, Wang YXJ, Aavula BR, Cases S, et al. 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg Med Chem Lett. 2011;21:983–988. doi: 10.1016/j.bmcl.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]