

Supplemental Digital Content is available in the text
Abstract
Secondary sclerosing cholangitis in critically ill patients (SSC-CIP) is an important differential diagnosis in patients presenting with cholestasis and PSC-like cholangiographic changes in endoscopic retrograde cholangiography (ERC). As a relatively newly described entity, SSC-CIP is still underdiagnosed, and the diagnosis is often delayed. The present study aims to improve the early detection of SSC-CIP and the identification of its complications.
A total of 2633 records of patients who underwent or were listed for orthotopic liver transplantation at the University Hospital Charité, Berlin, were analyzed retrospectively. The clinical presentation and outcome (mean follow-up 62.7 months) of the 16 identified SSC-CIP cases were reviewed.
Cholestasis was the first sign of SSC-CIP. GGT was the predominant enzyme of cholestasis. Hypercholesterolemia occurred in at least 75% of the patients. SSC-CIP provoked a profound weight loss (mean 18 kg) in 94% of our patients. SSC-CIP was diagnosed by ERC in all patients. The 3 different cholangiographic features detected correspond roughly to the following stages: (I) evidence of biliary casts, (II) progressive destruction of intrahepatic bile ducts, and (III) picture of pruned tree. Biliary cast formation is a hallmark of SSC-CIP and was seen in 87% of our cases. In 75% of the patients, the clinical course was complicated by cholangiosepsis, cholangitic liver abscesses, acalculous cholecystitis, or gallbladder perforation. SSC-CIP was associated with worse prognosis; transplant-free survival was ∼40 months (mean).
Because of its high rate of serious complications and unfavorable prognosis, it is imperative to diagnose SSC-CIP early and to differentiate SSC-CIP from other types of sclerosing cholangitis. Specific characteristics enable identification of SSC-CIP. Early cooperation with a transplant center and special attention to biliary complications are required after diagnosis of SSC-CIP.
INTRODUCTION
The diversity of sclerosing cholangiopathies seems greater than believed 30 years ago. The differential diagnosis includes, for example, IgG4-related sclerosing cholangitis, AIDS-related cholangiopathy, ischemic cholangiopathy, eosinophilic cholangitis, and primary sclerosing cholangitis (PSC). In recent years, there have been increasing numbers of reports on a new entity of sclerosing cholangiopathy. This type of secondary sclerosing cholangitis occurs in association with intensive-care treatment of patients with major surgery, trauma, burns, and other life-threatening events. Secondary sclerosing cholangitis in critically ill patients (SSC-CIP) is most likely caused by severe arterial hypotension/shock.1–5 SSC-CIP is a progressive cholestatic liver disease characterized by necrosis of the bile duct epithelium, which leads to the rapid destruction of the visible intrahepatic bile ducts. The disease runs a chronic, irreversible course and can only be cured by liver transplantation. Here, we report our experience with 16 SSC-CIP cases. The aim of our study was to define the characteristic clinical features of SSC-CIP.
MATERIALS AND METHODS
Here, we present clinical data on critically ill patients who developed secondary sclerosing cholangitis. All cases were recruited retrospectively by reviewing the hospital records of patients who received (2473) or were listed for orthotopic liver transplantation (OLT) at the Charité University Hospital Berlin between January 1990 and May 18, 2011. Sixteen patients with a confirmed diagnosis of SSC-CIP were identified. The Charité Department of Transplantation Surgery is a tertiary care center and most cases with SSC-CIP were referred from other hospitals. Therefore, in most patients, the initial treatment—especially intensive care treatment—was performed in outside hospitals. After listing for OLT, endoscopic management of the patients was performed at the Endoscopy Unit of Department of Gastroenterology and Hepatology/Charité, a high-volume center for biliary endoscopy. All patients had survived a prior acute life-threatening event followed by intensive care treatment. The reasons for admission to the intensive care unit (ICU) are shown in Table 1. Before admission to ICU, no patient had evidence of liver or bile duct disease and had unremarkable liver function tests. Clinical information about each patient was obtained by review of the medical records. Detailed pieces of information regarding the clinical course, laboratory parameters, ultrasound and endoscopic retrograde cholangiography (ERC) findings were collected. The diagnosis of SSC-CIP was established by ERC (16/16) and confirmed by liver histology (12/16 patients). The median follow-up was 62 months (range: 5.6–171). The onset of cholestasis was defined as an increase in the cholestatic parameters (gamma-glutamyl- transferase [GGT], alkaline phosphatase [ALP], and bilirubin) to more than twice the upper limit of normal. Data were summarized as mean and standard deviation of the mean or were presented as medians with ranges or percentages. We tested for gender differences using the 2-sided binomial probability test. Cholesterol elevation was tested for significance with the 2-sided 1-sample t test. P values of <0.05 were regarded as statistically significant. Statistical analyses were performed using Stata software, version 13.1 (Stata Corp). Transplant-free survival curves were calculated by the Kaplan–Meier method. The starting point for survival analysis was defined as the time of onset of cholestasis. Either liver transplantation or death from liver disease was counted as an event, whereas “patients alive without liver transplantation” was censored. The study was approved by the local Ethics Committee of the Charité, and all surviving patients gave their written informed consent to having their data recorded.
TABLE 1.
Demographic Characteristics and Outcome of 16 Cases With SSC-CIP

RESULTS
At time of diagnosis, the patients had a mean age of 46 ± 14.6 years. All patients required intensive care treatment for a mean 39 ± 23.9 days (Table 1). The disease showed a tendency to affect men more often than women (11 men to 5 women, ratio of 2.2), but the difference was not significant (P = 0.210, 2-sided binominal probability test).
Liver Function Tests and Other Laboratory Parameters
The onset of cholestasis was seen 7 days (median) after the initial life-threatening event. Gamma-glutamyl-transferase (GGT) was the first parameter to become elevated, and alkaline phosphatase (ALP) followed within a few days. Hyperbilirubinemia could be observed after a mean of 15.5 ± 10 days (median: 13 days) and was therefore the last sign of disease detected. This characteristic sequence of GGT, ALP, and bilirubin elevation was observed in 13/16 patients (Fig. 1). Cholestasis reached a peak after 31 ± 14 days (range: 14–68 days). GGT levels peaked at ∼20 to 50 times the upper limit of normal, whereas the elevation of AP levels was less pronounced (5 to no more than 21 times upper limit of normal). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were moderately elevated (mean: 281.9 ± 212.9 U/L for AST, and 271.4 ± 165.7 U/L for ALT). Initial cholestasis was accompanied by transient creatinine elevation in half (8/16) of the patients. Data on cholesterol levels were available in 12 patients, all developed marked hypercholesterolemia (mean 535.6 ± 243.9 mg/dL; median 476.5) within the first year of disease (Table 2). Compared to the normal value of 200 mg/dL, the cholesterol value observed in our population was significantly elevated (P = 0.001). In transplanted patients, hypercholesterolemia resolved after OLT.
FIGURE 1.
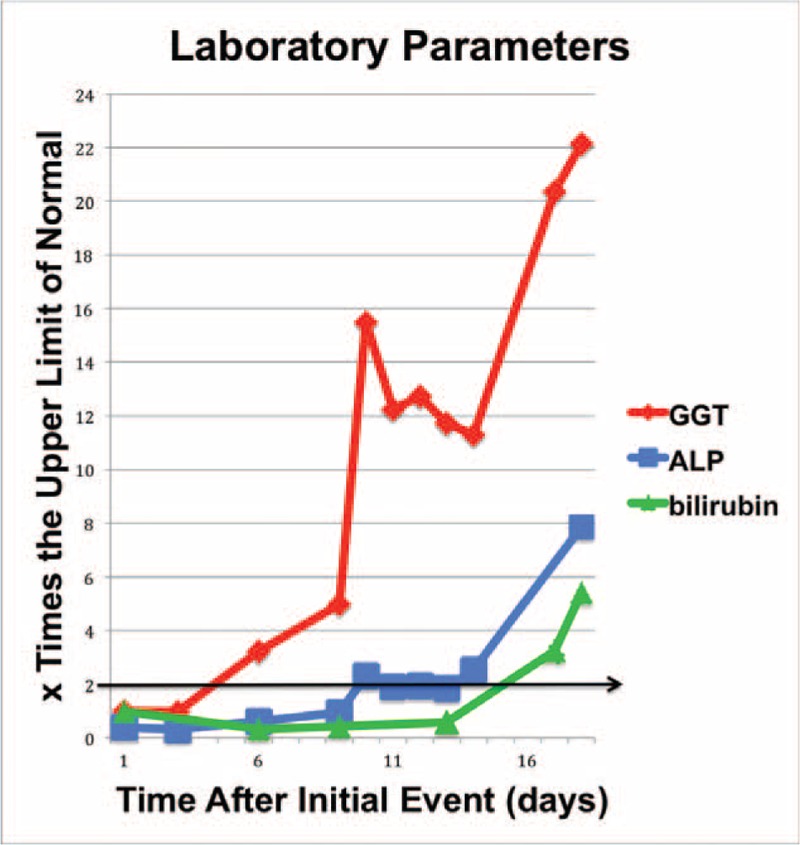
Course of cholestasis with typical sequence of GGT, ALP, and bilirubin; here, in a patient of our series with polytrauma. ALP = alkaline phosphatise, GGT = gamma-glutamyl transpeptidase.
TABLE 2.
Laboratory and Clinical Characteristics of 16 Cases With SSC-CIP
Diagnostic Studies
Ultrasound: For differential diagnosis of the cholestasis, ultrasound scans were performed in all 16 patients, starting 10.5 days (=median; mean 29.31 ± 60.05 days; range 1–247 days) after the onset of cholestasis. Dilatation of the extrahepatic bile duct was observed in 31% (5/16) of the patients. Isolated intrahepatic cholestasis was detected in another 2 patients, bringing the total number of cases with biliary system abnormalities to 7/16. Furthermore, sonographic signs of cholecystitis were observed in 8/16 patients. A total of 88% of the patients developed sonographic signs of liver cirrhosis within 6 months (mean).
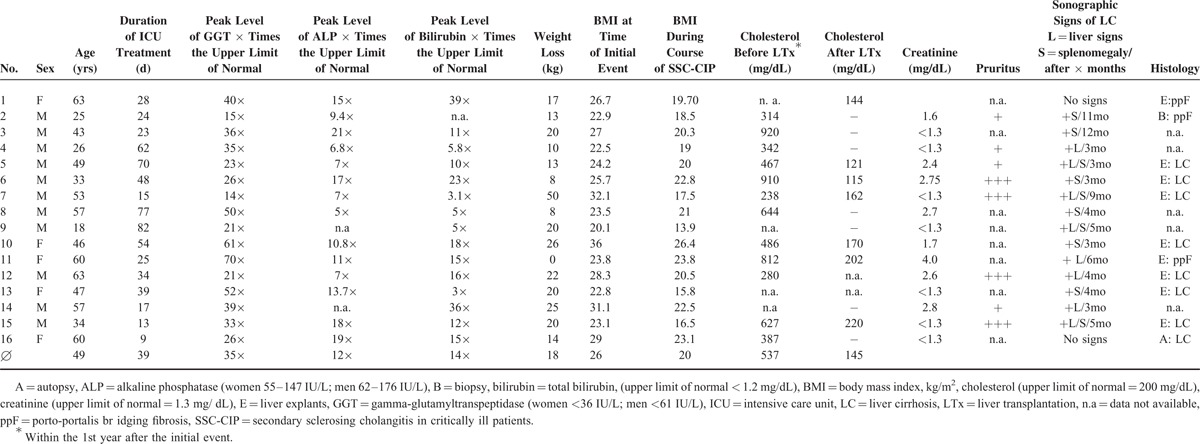
Endoscopic retrograde cholangiography (ERC): In all patients, the diagnosis of SSC-CIP was based on typical cholangiographic findings. The mean delay from first biochemical signs of cholestasis until the time of diagnosis was 85.7 ± 80.7 days (range: 13–271 days, median: 54 days). Totally 81% of the patients had only intrahepatic involvement, whereas 19% had evidence of intra- and extrahepatic bile duct involvement. However the extrahepatic ductal changes were minor. Three different cholangiographic features (I-III) of SSC-CIP were identified (Fig. 2).
FIGURE 2.
Cholangiographic findings of SCC-CIP. SSC-CIP = secondary sclerosing cholangitis in critically ill patients.
I. Early Stage: Biliary Cast Formation
The early stage of disease was characterized by the presence of multiple ribbon-like intraductal filling defects in the biliary tract. These filling defects were consistent with biliary casts and were found in 87% (14/16) of our patients in the first year after the initial injury. The shortest and longest time until detection was 13 and 265 days after the onset of cholestasis. In 3 patients, no signs of biliary cast formation were detected during the first ERC session but were observed later on the third or fourth ERC session. In 7/14 patients, biliary casts were found in multiple consecutive ERC sessions.
II. Intermediate Stage: Progressive Destruction of the Intrahepatic Bile Ducts
The further course of the disease was characterized by rapidly progressive intrahepatic bile duct destruction beyond the second bifurcation of the intrahepatic bile ducts. The destruction of the intrahepatic bile ducts due to epithelial necrosis manifests at ERC as contour defects and irregular wall contours.
III. Late Stage: Obliteration of the Intrahepatic Bile Ducts/Picture of Pruned Tree
As a result of the progressive destruction, the intrahepatic bile duct branches were eventually obliterated, leaving only a rudimentary central biliary system (see also Fig. 3). In ERC, contrast enhancement of the bile ducts occurred only below the second bifurcation. In the later stage, the picture was that of a pruned tree. At least one side of the biliary system was obliterated, but mostly both sides were affected. The median time to the appearance of this constellation was 86.5 days after the onset of cholestasis (mean: 133 ± 127 days).
FIGURE 3.
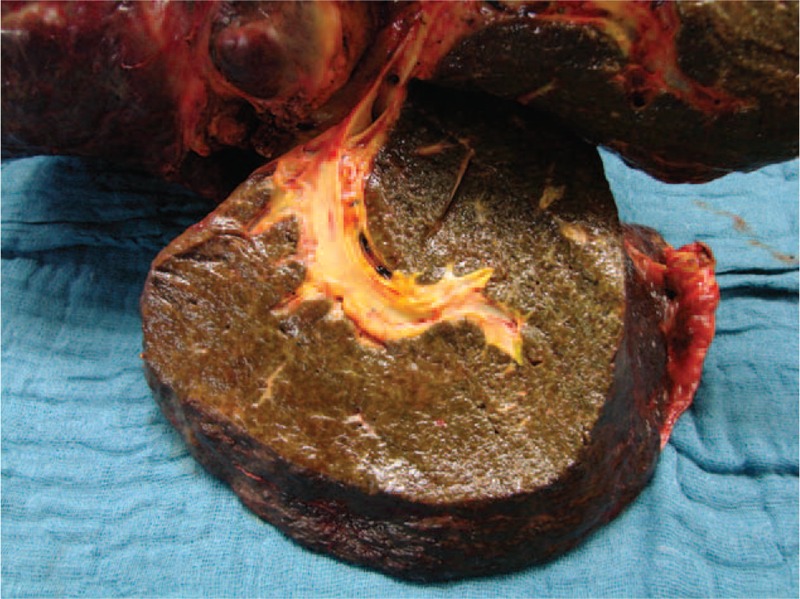
Liver explant from a patient in our series, showing advanced destruction of intrahepatic bile ducts.
Liver histology: Liver histology data were available in 11/16 cases, either by liver biopsy, autopsy, or by histological findings from the explanted liver. The liver biopsy specimens were obtained 22 to 351 days after the onset of cholestasis (mean: 133 ± 124 days, median: 102 days). All biopsy specimens showed histopathological changes, which were located mainly in the portal tracts. Abnormalities such as portal tract dilatation, ductular reaction, and portal tract infiltration were described. Fibrosis of portal tracts was detected in 5 of 6 liver biopsies. Cirrhosis was found in 7/9 of the explanted livers and portal fibrosis was detected in 2/9 patients with early transplantation on day 189 and 237 (Table 2). In the explanted livers, the histological evidence of necrosis of the larger bile ducts in the hilar region is particularly worth mentioning (Supplement 1).
Weight Loss
Severe weight loss (mean: 18 ± 11 kg; median: 18.5 kg) occurred in the first year after the initial injury in 15/16 patients. The amount of weight loss was not associated with the length of ICU stay and occurred, in some cases, despite a short period of intensive care treatment (Table 2).
Complications of SSC-CIP
Biliary complications occurred in 87.5% of our patients. Recurrent cholangitic episodes/flare-ups were documented in 12/14 patients (no data were available for the remaining 2 patients). In 3/16 patients liver abscess formation was observed. Severe cholangiosepsis was seen in 3/16 (19%) patients (Table 3, Fig. 4).
TABLE 3.
Initial Event, Complications, Follow-Up, and Outcome of 16 Cases With SSC-CIP
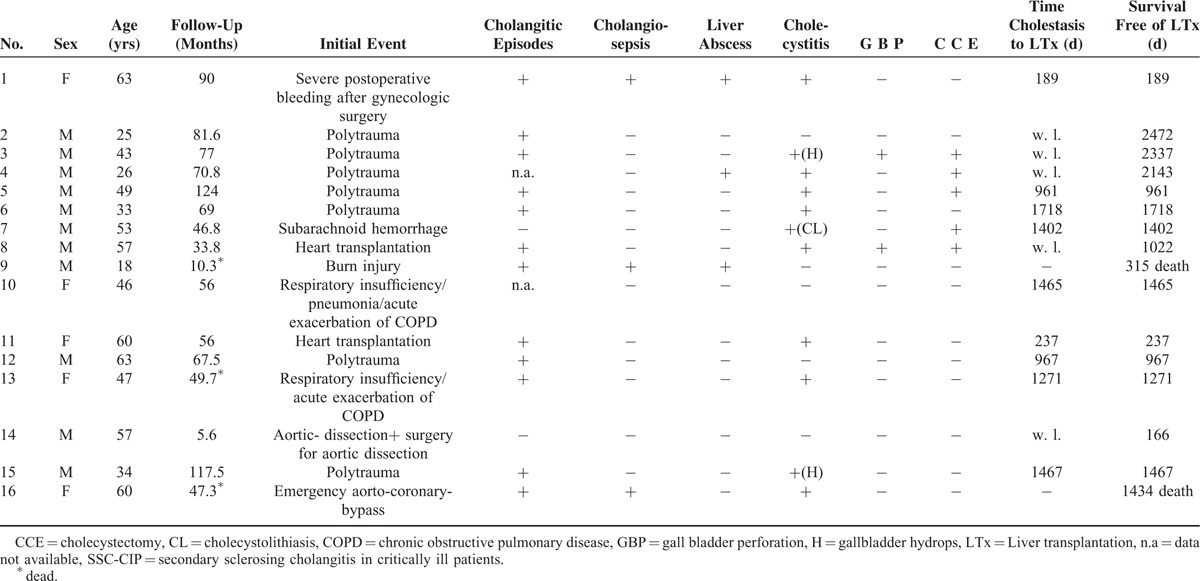
FIGURE 4.
Cholangiography (via percutaneous biliary drainage) of a patient of our series with aburn injury, showing multiple peribiliary liver abscesses. The patient died of unmanageable cholangiosepsis while on the waiting list.
Involvement of the gallbladder: Immediately following cholestasis onset, one-half of our patients had sonographic signs of acute (predominantly acalculous) cholecystitis. Within the 1st year after initial injury acute cholecystitis occurred in 69% (11/16) of the patients. Of these, 45% (5/11) required emergency cholecystectomy. The cholecystitis described was mostly an acalculous cholecystitis (10/11). Gallbladder perforation evolved in 2 of these patients—in 1 patient 24 days after the onset of cholestasis. The perforation rate was thus 18% (2/11).
Outcome, Progression, Listing, Need for Transplantation
The data relating to outcome and transplantation are shown in Tables 1 and 3 and Figure 5. After a short course of disease, clinical signs and symptoms of liver cirrhosis appeared within a few months. Most of the patients in our series (12/16) had to be placed on the waiting list for liver transplant within 1 year after the onset of cholestasis. The mean time to listing was 322 ± 257 days (median: 225 days). The mean time from the onset of cholestasis until transplantation was 35 months (median: 42 months). The earliest transplantation was performed 189 days after the onset of cholestasis in a 63-year-old woman who developed cholangiosepsis and multiple organ failure.
FIGURE 5.
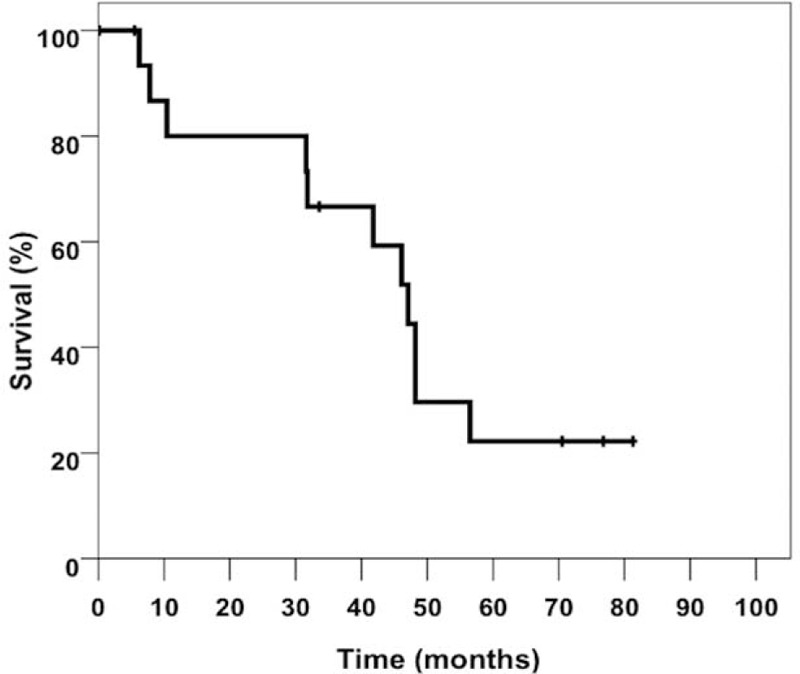
Transplant-free survival (Kaplan–Meier) of 16 cases with SSC-CIP. SSC-CIP = secondary sclerosing cholangitis in critically ill patients.
DISCUSSION
If ERC demonstrates PSC-like cholangiographic findings, all types of sclerosing cholangiopathy must be considered in the differential diagnosis. This includes secondary sclerosing cholangitis in critically ill patients (SSC-CIP). With mortality rates of up to 50%, SSC-CIP represents a challenge to gastroenterologists and intensive care specialists.5 Currently, SSC-CIP is still widely underdiagnosed. Early detection of the disease and its complications is crucial to improving the poor prognosis.
In the initial stages of the disease, SSC-CIP must be recognized as a cause of liver failure in ICU patients. In the later stages, SSC-CIP must be differentiated from other types of sclerosing cholangiopathy. This differentiation is necessary with regard to the selection and timing of liver transplantation as SSC-CIP progresses much more rapidly than other cholangiopathies. To enable clinical differentiation, characteristic features of SSC-CIP must be identified.
As its name indicates critical illness is an obligatory precondition for SSC-CIP.2–5 All of our patients had a history of intensive care treatment for a life-threatening event. Although this event often necessitates lengthy intensive care treatment, long-term treatment (>7 days) is not required for the destruction of the bile ducts, as previously believed. Rather, it seems that bile duct damage is a very early event that manifests within the first 5 to 7 days, as evidenced by the early increase of cholestatic enzymes in our series.
There is a trend that the disease affects men more often than women. In addition, SSC-CIP showed a predilection for surgical patients, especially those in whom extensive tissue destruction occurs, such as patients with polytrauma or those undergoing major surgery. The first detectable sign of SSC-CIP was an increase in the liver enzymes—as our data confirm. SSC-CIP was characterized by a cholestatic pattern. In our patients, GGT was the predominant enzyme, which increased earlier and peaked more markedly than ALP. Hyperbilirubinemia occurred in the later stages. Although not specific, these laboratory findings may be helpful in distinguishing SSC-CIP from other causes of elevated liver enzymes in ICU patients, for example, from septicaemia-associated cholestasis, in which a hyperbilirubinemia predominates.6–8
Our data show that SSC-CIP is associated with markedly elevated serum cholesterol. Elevated levels of cholesterol are common in primary biliary cirrhosis (PBC), but occur less frequently in PSC.9,10 In SSC-CIP, the combination of elevated serum-cholesterol and pruritus may reflect disturbed bile acid metabolism.
Unintentional weight loss appears to be a typical feature of SSC-CIP and affected 94% of our patients. This contrasts with reports of weight loss in only 35% to 40% of PSC patients.11 Losing a mean 18 kg, our patients experienced a level of weight loss otherwise seen only in wasting diseases. The reason for this weight loss remains unclear. Conceivably, it might have been associated with the liberation of cytokines in the presence of chronic inflammation of the bile ducts. This is a well-known mechanism in the case of cancer cachexia. In cancer patients, inflammatory mediators such as TNF-alpha can induce changes in lipid metabolism that result in increased lipolysis, hyper-cholesterolemia, and a reduction of total fat mass.12
Diagnosis
Cholangiography is mandatory for the diagnosis of SSC-CIP. In clinical practice, the decision to perform endoscopic retrograde cholangiography (ERC) in ICU patients with cholestasis is often based on ultrasound findings of dilated bile ducts. However, this approach delayed the diagnosis in our series since only a few patients had dilated extrahepatic bile ducts. Therefore, despite of the severe and striking initial course of SSC-CIP, it took 3 months (mean) to establish the final diagnosis. Typical ERC findings were biliary casts, destruction of intrahepatic bile ducts, or finally the picture of a “pruned tree.” These 3 different ERC findings occurred consecutively and might reflect distinct stages in the development of the disease. However, overlapping stages have also been observed. Our findings show that SSC-CIP is mainly a disease of the visible intrahepatic bile ducts—the extrahepatic bile ducts were only marginally affected. Biliary cast formation may be considered pathognomonic for SSC-CIP, as suggested by the high proportion of patients with biliary casts in our series. The casts completely fill the bile ducts and may distend them. In all patients initially presenting sonographically distended extrahepatic bile ducts, the first ERC's revealed biliary casts. The occurrence of biliary casts in association with either PSC or IgG4 cholangiopathy has not been described in the literature.
After liver transplantation, the occurrence of biliary casts is a known phenomenon, and an etiological association with ischemic events is assumed.13,14 However, single cases of “biliary cast syndrome” in nontransplant patients have also been described.15–18 This “biliary cast syndrome” in nontransplant patients is presumably not a separate entity, but rather an early stage of SSC-CIP, as supported by some case reports.19,20 In agreement with data from orthotopic liver transplantation we found biliary casts in SSC-CIP only within the first year after the initial event.13 Despite successful initial endoscopic removal, 7 of our patients showed recurrence of the biliary casts at subsequent ERC examinations. Whether these were merely the result of a sliding down of incompletely removed intrahepatic fragments, or whether new casts were formed is not clear.
Complications of SSC-CIP
SSC-CIP carries a high risk for hepatobiliary complications. In 75% of our patients the clinical course was complicated by acute acalculous cholecystitis, gallbladder perforation, cholangitic liver abscess, or cholangiosepsis.
Involvement of the Gallbladder
It seems that the gallbladder epithelium is involved in the pathogenetic process in SSC-CIP. This is underscored by the high rate of acute cholecystitis (in the absence of a cholelithiasis) in the early stage of disease in our series. Acute acalculous cholecystitis (AAC) is a serious and potentially fatal condition. Ischemic necrosis of the gallbladder wall, often extending into the deeper layers, causes a high spontaneous perforation rate, as our data confirm. It is known that acute acalculous cholecystitis shows a predilection to occur in critically ill patients after trauma, burns, or major surgery.21–23 Because acute acalculous cholecystitis has also been observed in association with Schönlein–Henoch purpura, antiphospholipid syndrome and lupus erythematosus, an ischemic pathogenesis for AAC is supposed.24–26 A similar pathogenetic mechanism that could explain the gallbladder involvement is hypothesized for SSC-CIP. In ICU patients, an unexplained cholestasis and the simultaneous appearance of acute acalculous cholecystitis should raise the suspicion of SSC-CIP. Conversely, acute acalculous cholecystitis is not a known phenomenon in patients with PSC or IgG4-associated cholangiopathy.
Cholangitic Episodes/Cholangiosepsis/Liver Abscesses
In our patients, the clinical course of disease was complicated by recurrent episodes of bacterial cholangitis. In SSC-CIP, these episodes may be secondary events explainable by the presence of multifocal intrahepatic biliary strictures due to extensive destruction of segmental and subsegmental bile ducts. Furthermore, as a result of the duct damage peripheral bile duct branches can lose their connection to the central ducts and become “excluded” bile ducts. The elimination of a biliary obstruction is a prerequisite for successful antibiotic treatment of cholangitis. Unfortunately in SSC-CIP this drainage is frequently inadequate since peripheral, “excluded” ducts often can no longer be reconnected by endoscopic means. The remaining obstruction impairs the penetration of antibiotics into bile, limits the effectiveness of the treatment, and increases the risk of cholangiosepsis. In the absence of drainage the mortality rate of cholangitis is high, as is known from the pre-ERC era.27–30 This problem is reflected by our data, showing a high rate of cholangiosepsis, and among these patients a high fatality rate. Two of 3 patients died from cholangiosepsis despite intensive therapeutic efforts while still on the transplant waiting list. Furthermore, the stagnation of bile flow also promotes cholangitic liver abscesses—almost 19% of our patients developed this complication.
Progression/Need for Transplantation
Two aspects contributed to the short latency between the onset of cholestasis and the need for transplantation observed in our SSC-CIP cases. First, the clinical and histology data indicate that the liver fibrosis progressed unusually quickly over a period of a couple of months. This rapid and extensive fibrotic response may be explainable by the large extent of initial cholangiocyte destruction of the intrahepatic bile ducts.31 Second, on account of the risk of cholangiosepsis, transplantation is urgently needed in cases with irreversible destruction of bile ducts and recurrent severe cholangitis. Two patients of our series died of cholangiosepsis while still on the transplant waiting list; another patient with cholangiosepsis and multiple organ failure had to be transplanted 195 days after the initial event. Median liver transplantation-free survival was only 44 months (mean 40 months) in our patients, compared to 89 to 112 months in other fibrosing cholangiopathies such as PSC, and 72 months in “classical” infective SSC.32–34 In view of the more rapid disease progression, patients with SSC-CIP require closer monitoring than patients with other forms of fibrosing cholangiopathy and an early cooperation with a transplantation center should be initiated.35,36
Survival After Liver Transplantation
It was assumed earlier that patients with secondary sclerosing cholangitis had a poor outcome after liver transplantation.33,37 Our findings refute this. Eight of the 9 transplanted patients are alive and in a good state of health. Only 1 female patient, who underwent simultaneous liver and lung transplantation, died from pulmonary complications. The resulting 1-year survival rate of 85% and 3-year survival rate of 83% correspond well with the survival rates for OLT in alcoholic liver cirrhosis.
Study Limitations
The limitations of the present study are its retrospective nature and the recruitment of the patients from a transplantation center. Patients with an initially severe course who did not live long enough to be listed, as also those with a milder course and no liver transplantation, were not included in the study. Further larger clinical studies can build on and consolidate our results. Prospective studies with a control group, which makes it possible to perform complex statistics generating P values would be needed to validate our observations.
Frequency of SSC-CIP
The actual prevalence of SSC-CIP remains unknown and is also not adequately reflected in our data (due to selection bias, as mentioned above). Overall, 0.61% of all liver transplantations performed at our center were due to SSC-CIP. Similar rates have been observed, for example, in patients with hemochromatosis (0.7% of all OLT at our center). For PSC the figure was 6.2% in our center, indicating that SSC-CIP is 10 times rarer than PSC.
CONCLUSION
Despite some similarities with other sclerosing cholangiopathies, SSC-CIP shows characteristic features that allow for discrimination of SSC-CIP from other entities. In particular, the occurrence of biliary casts in patients with equivocal findings of sclerosing cholangiopathy in ERC is suggestive of SSC-CIP and excludes PSC and IgG4 cholangiopathies. Last but not least, the fatal outcome of the complications of SSC-CIP could be prevented by better recognition of the disease and its natural history.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Peter Palm, Berlin, for the graphic design in Figure 2 and H. Niggemann for statistical analysis.
Footnotes
Abbreviations: AAC = acute acalculous cholecystitis, ALP = alkaline phosphatase, ALT = alanine aminotransferase, ASAT = aspartate aminotransferase, CK = creatinine kinase, COPD = chronic obstructive pulmonary disease, ERC = endoscopic retrograde cholangiography, GGT = gamma-glutamyl transpeptidase, ICU = intensive care unit, IgG4 = Immunoglobulin G4, mg/dL = milligrams per deciliter, OLT = orthotopic liver transplantation, PBC = primary biliary cirrhosis, PSC = primary sclerosing cholangitis, SDM = standard deviation of the mean, SSC = secondary sclerosing cholangitis, SSC-CIP = secondary sclerosing cholangitis in critically ill patients, TNF-α = tumor necrosis factor alpha, U/L = units per liter.
Authors’ contributions: SL, DS, PN, WVS, AA, ES, DE, WF contributed to the study conception, participated in data analysis and interpretation, and revised the manuscript critically. SL collected and analyzed the data and prepared the manuscript. DS analyzed the data and edited the manuscript. All authors read and approved the final version of the manuscript. SL is the guarantor of this work and, as such, had full access to all of the study data and takes full responsibility for the integrity of the data.
Funding: the work presented here was done without any financial support.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Schmitt M, Kölbel CB, Müller MK, et al. Sclerosing cholangitis after burn injury. Z Gastroenterol 1997; 35:929–934. [PubMed] [Google Scholar]
- 2.Leonhardt S, Veltzke-Schlieker W, Adler A, et al. Trigger mechanisms of secondary sclerosing cholangitis in critically ill patients. Crit Care Doi: 101186/s13054-015-0861-5 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaeger C, Mayer G, Henrich R, et al. Secondary sclerosing cholangitis after long-term treatment in an intensive care unit: clinical presentation, endoscopic findings, treatment, and follow-up. Endoscopy 2006; 38:730–734. [DOI] [PubMed] [Google Scholar]
- 4.Gelbmann CM, Rümmele P, Wimmer M, et al. Ischemic-like cholangiopathy with secondary sclerosing cholangitis in critically ill patients. Am J Gastroenterol 2007; 102:1221–1229. [DOI] [PubMed] [Google Scholar]
- 5.Voigtländer T, Negm AA, Schneider AS, et al. Secondary sclerosing cholangitis in critically ill patients: model of end-stage liver disease score and renal function predict outcome. Endoscopy 2012; 44:1055–1058. [DOI] [PubMed] [Google Scholar]
- 6.Bansal V, Schuchert VD. Jaundice in the intensive care unit. Surg Clin North Am 2006; 86:1495–1502. [DOI] [PubMed] [Google Scholar]
- 7.Kortgen A, Paxian M, Werth M, et al. Prospective assessment of hepatic function and mechanisms of dysfunction in the critically ill. Shock 2009; 32:358–365. [DOI] [PubMed] [Google Scholar]
- 8.Chand N, Sanyal AJ. Sepsis-induced cholestasis. Hepatology 2007; 45:230–241. [DOI] [PubMed] [Google Scholar]
- 9.Talwalkar JA, Lindor KD. Primary biliary cirrhosis. Lancet 2003; 362:53–61. [DOI] [PubMed] [Google Scholar]
- 10.Sinakos E, Abbas G, Jorgensen RA, et al. Serum lipids in primary sclerosing cholangitis. Dig Liver Dis 2012; 44:44–48. [DOI] [PubMed] [Google Scholar]
- 11.Wiesner RH, Grambsch PM, Dickson ER, et al. Primary sclerosing cholangitis: natural history, prognostic factors and survival analysis. Hepatology 1989; 10:430–436. [DOI] [PubMed] [Google Scholar]
- 12.Batista ML, Jr, Peres SB, McDonald ME, et al. Adipose tissue inflammation and cancer cachexia: possible role of nuclear transcription factors. Cytokine 2012; 57:9–16. [DOI] [PubMed] [Google Scholar]
- 13.Shah JN, Haigh WG, Lee SP, et al. Biliary casts after orthotopic liver transplantation: clinical factors, treatment, biochemical analysis. Am J Gastroenterol 2003; 98:1861–1867. [DOI] [PubMed] [Google Scholar]
- 14.Gor NV, Levy RM, Ahn J, et al. Biliary cast syndrome following liver transplantation: predictive factors and clinical outcomes. Liver Transpl 2008; 14:1466–1472. [DOI] [PubMed] [Google Scholar]
- 15.Gleeson FC, Czaja AJ, Baron TH. Successful endoscopic management of biliary cast syndrome in nonliver transplant patients. J Clin Gastroenterol 2008; 42:752–755. [DOI] [PubMed] [Google Scholar]
- 16.Kusnierz K, Nowakowska-Dulawa E, Pilch-Kowalczyk J, et al. Biliary cast syndrome in a non-transplant patient with acute pancreatitis. Dig Liver Dis 2012; 44:534–535. [DOI] [PubMed] [Google Scholar]
- 17.Hosoi M, Nannya Y, Sasaki T, et al. Biliary cast syndrome and benign biliary stricture as complications of allogeneic hematopoietic stem cell transplantation. Ann Hematol 2010; 89:1287–1289. [DOI] [PubMed] [Google Scholar]
- 18.Dziurkowska-Marek A, Hartleb M, Marek TA, et al. Fatal case of biliary cast syndrome in nontransplant patient. Endoscopy 2009; 41 Suppl 2:E256. [DOI] [PubMed] [Google Scholar]
- 19.Byrne MF, Chong HI, O’Donovan D, et al. Idiopathic cholangiopathy in a biliary cast syndrome necessitating liver transplantation following head trauma. Eur J Gastroenterol Hepatol 2003; 15:415–417. [DOI] [PubMed] [Google Scholar]
- 20.Kwon ON, Cho SH, Park CK, et al. Biliary cast formation with sclerosing cholangitis in critically ill patient: case report and literature review. Korean J Radiol 2012; 13:358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ullman M, Hasselgren PO, Tveit E. Posttraumatic and postoperative acute acalculous cholecystitis. Acta Chir Scand 1984; 150:507–509. [PubMed] [Google Scholar]
- 22.Castana O, Rempelos G, Anagiotos G, et al. Acute acalculous cholecystitis—a rare complication of burn injury. Ann Burns Fire Disasters 2009; 22:48–50. [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed N. Acute acalculous cholecystitis complicating major trauma: a report of five cases. South Med J 2008; 101:1146–1149. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann JC, Cremer P, Preiss JC, et al. Gallbladder involvement of Henoch–Schönlein purpura mimicking acute acalculous cholecystitis. Digestion 2004; 70:45–48. [DOI] [PubMed] [Google Scholar]
- 25.Bando H, Kobayashi S, Matsumoto T, et al. Acute acalculous cholecystitis induced by mesenteric inflammatory veno-occlusive disease (MIVOD) in systemic lupus erythematosus. Clin Rheumatol 2003; 22:447–449. [DOI] [PubMed] [Google Scholar]
- 26.Desailloud R, Papo T, Vaneecloo S, et al. Acalculous ischemic gallbladder necrosis in the catastrophic antiphospholipid syndrome. Arthritis Rheum 1998; 41:1318–1320. [DOI] [PubMed] [Google Scholar]
- 27.Blenkharn JI, Habib N, Mok D, et al. Decreased biliary excretion of piperacillin after percutaneous relief of extrahepatic obstructive jaundice. Antimicrob Agents Chemother 1985; 6:778–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung JW, Chan RC, Cheung SW, et al. The effect of obstruction on the biliary excretion of cefoperazone and ceftazidime. J Antimicrob Chemother 1990; 25:399–406. [DOI] [PubMed] [Google Scholar]
- 29.Welch JP, Donaldson GA. The urgency of diagnosis and surgical treatment of acute suppurative cholangitis. Am J Surg 1976; 131:527–532. [DOI] [PubMed] [Google Scholar]
- 30.O’Connor MJ, Schwartz ML, McQuarrie DG, et al. Acute bacterial cholangitis: an analysis of clinical manifestation. Arch Surg 1982; 117:437–441. [DOI] [PubMed] [Google Scholar]
- 31.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology 2004; 127:1565–1577. [DOI] [PubMed] [Google Scholar]
- 32.Talwalkar JA, Lindor KD. Natural history and prognostic models in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol 2001; 15:563–575. [DOI] [PubMed] [Google Scholar]
- 33.Gossard AA, Angulo P, Lindor KD. Secondary sclerosing cholangitis: a comparison to primary sclerosing cholangitis. Am J Gastroenterol 2005; 100:1330–1333. [DOI] [PubMed] [Google Scholar]
- 34.Broomé U, Olsson R, Lööf L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996; 38:610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiesner RH, Porayko MK, Dickson ER, et al. Selection and timing of liver transplantation in primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 1992; 16:1290–1299. [PubMed] [Google Scholar]
- 36.Farges O, Malassagne B, Sebagh M, et al. Primary sclerosing cholangitis: liver transplantation or biliary surgery. Surgery 1995; 117:146–155. [DOI] [PubMed] [Google Scholar]
- 37.Kirchner GI, Scherer MN, Obed A, et al. Outcome of patients with ischemic-like cholangiopathy with secondary sclerosing cholangitis after liver transplantation. Scand J Gastroenterol 2011; 46:471–478. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.