

In skeletal muscle of mice lacking caveolin-2 (Cav2−/−), microvascular diameters and vasomotor responses to norepinephrine, ACh, and rhythmic contractions were not different from wild-type. However, rapid onset vasodilation to tetanic contraction was diminished by approximately half, while active force increased by ∼40%, revealing novel physiological roles for Cav2 in skeletal muscle.
Keywords: arteriole, blood flow, exercise, feed artery, microcirculation
Abstract
Caveolin-2 (Cav2) is a major protein component of caveolae in membranes of vascular smooth muscle and endothelium, yet its absence alters the ultrastructure of skeletal muscle fibers. To gain insight into Cav2 function in skeletal muscle, we tested the hypothesis that genetic deletion of Cav2 would alter microvascular reactivity and depress contractile function of skeletal muscle in vivo. In the left gluteus maximus muscle (GM) of anesthetized Cav2−/− and wild-type (WT) male mice (age, 6 mo), microvascular responses to physiological agonists and to GM contractions were studied at 34°C. For feed arteries (FA), first- (1A), second- (2A) and third-order (3A) arterioles, respective mean diameters at rest (45, 35, 25, 12 μm) and during maximal dilation (65, 55, 45, 30 μm) were similar between groups. Cumulative dilations to ACh (10−9 to 10−5 M) and constrictions to norepinephrine (10−9 to 10−5 M) were also similar between groups, as were steady-state dilations during rhythmic twitch contractions (2 and 4 Hz; 30 s). For single tetanic contractions (100 Hz; 100, 250, and 500 ms), rapid onset vasodilation (ROV) increased with contraction duration throughout networks in GM of both groups but was reduced by nearly half in Cav2−/− mice compared with WT mice (P < 0.05). Nevertheless, maximal force during tetanic contraction was ∼40% greater in GM of Cav2−/− vs. WT mice (152 ± 14 vs. 110 ± 3 mN per square millimeter, respectively; P < 0.05). Thus, while structural and functional properties of resistance networks are well maintained in the GM of Cav2−/− mice, diminished ROV with greater force production reveals novel physiological roles for Cav2 in skeletal muscle.
NEW AND NOTEWORTHY
In skeletal muscle of mice lacking caveolin-2 (Cav2−/−), microvascular diameters and vasomotor responses to norepinephrine, ACh, and rhythmic contractions were not different from wild-type. However, rapid onset vasodilation to tetanic contraction was diminished by approximately half, while active force increased by ∼40%, revealing novel physiological roles for Cav2 in skeletal muscle.
caveolae are specialized regions of plasma membranes that appear as flask-shaped invaginations 50–100 nm in diameter formed by the polymerization of caveolin with complementary proteins involved in signaling (27, 35, 38, 53). Of the three caveolin isoforms, caveolin-1 (Cav1) and caveolin-2 (Cav2) are coexpressed in multiple cell types, including vascular endothelium (38, 46). In contrast, the expression of caveolin-3 (Cav3) is specific for muscle cells, including smooth, cardiac, and skeletal muscle cells (16, 38, 42, 47). When Cav1 or Cav3 are present, respective isoforms form homo-oligomers in caveolae irrespective of Cav2 (38, 40, 53). However, for Cav2 to localize within caveolae requires hetero-oligomerization with Cav1 because caveolae do not form in the absence of Cav1, while Cav2 is retained within the Golgi complex (27, 28, 38, 46, 54). Caveolin proteins are integral to the anchoring and regulation of nitric oxide synthase (NOS). In skeletal muscle, binding of Cav3 to neuronal NOS (nNOS) suppresses NO production (52), while in endothelial cells (ECs) the binding of Cav1 to endothelial NOS (eNOS) performs a similar role (18, 25). Indeed, eNOS activity is elevated constitutively in the absence of Cav1, resulting in diminished vasomotor tone (27, 35, 38, 39). Whereas integral roles have been identified for Cav1 in the vasculature and Cav3 in skeletal muscle, less is known about the physiological role of Cav2. Only 38% of the amino acid sequence between Cav1 and Cav2 is identical, suggesting different functional roles for respective protein isoforms (29, 46). In mice, genetic deletion of Cav2 resulted in hypercellularity of the lung parenchyma, where thickened alveolar septa was associated with exercise intolerance attributed to impaired pulmonary gas exchange (37). In the gastrocnemius muscle, abnormalities following Cav2 deletion included dilated sarcoplasmic reticulum (SR) and intracellular aggregates of microtubules and mitochondria (40). While these latter findings suggest a role for Cav2 in skeletal muscle function, it is unknown whether Cav2 may contribute to the regulation of muscle blood flow or to the contractile function of muscle fibers.
The gluteus maximus muscle (GM) is a powerful hip extensor and integral to mammalian locomotion. Its superficial location and vascular supply are well suited for studying its microcirculation using intravital microscopy (2, 23, 44). With respect to the control of muscle blood flow, feed arteries (FA) enter the muscle and give rise to first-order arterioles (1A); these proximal branches of the resistance network govern the volume of flow entering the muscle (41). The 1A branches into second- (2A) and then third- (3A) order arterioles that govern the distribution of blood flow within the muscle (41). Respective branches are surrounded by sympathetic nerve fibers that produce vasoconstriction by releasing norepinephrine (NE) onto α-adrenergic receptors (αARs) of smooth muscle cells (SMCs) (15, 31). In contrast, ECs contribute to vasodilation by relaxing SMCs via hyperpolarization and the release of nitric oxide as evoked by ACh (3, 41). With ECs and SMCs working in concert to regulate tissue blood flow (8, 41), this study investigated whether Cav2 may be integral to the underlying signaling events. Therefore, we tested the hypothesis that genetic deletion of Cav2 attenuates vasomotor responses in microvascular resistance networks. The magnitude of vasodilation and increase in muscle blood flow correlate with exercise intensity and active force production (1, 26, 33). Because the time course and signaling events that mediate vasodilation can vary with the intensity and duration of muscle contraction (6, 8, 32, 50), a key goal of this study was to determine whether absence of Cav2 would affect vasodilation during moderate-twitch contractions or in response to a brief tetanic contraction. While rhythmic twitch contractions of the GM approximate scurrying around, brief tetanic contractions approximate jumping out of harm's way. In light of Cav2 deletion resulting in impaired swimming performance (37) and ultrastructural abnormalities within muscle fibers (40), a complementary goal of these experiments was to determine whether Cav2 was integral to contractile function. Thus, we tested the hypothesis that Cav2 deletion would attenuate active force production in skeletal muscle.
METHODS
Animal Care and Use
All procedures were approved by the Animal Care and Use Committee of the University of Missouri. Cav2−/− mice were generated using C57BL/6N background with the assistance of the University of California, Davis, Mouse Biology Program (Davis, CA). Thereby, the entire exon 2 and a 5′-portion of exon 3 of the Cav2 gene were deleted using sequence replacement strategy as described previously (29). Mice were bred and housed in the animal care facilities of the University of Missouri while maintained at ∼24°C on a 12:12-h light-dark cycle with food and water provided ad libitum. In accord with earlier findings of phenotypic changes in male but not female mice (23, 40), males were used for our criterion experiments and studied at ∼6 mo of age. To avoid the possibility of an order effect, Cav2−/− and wild-type (WT) littermates were studied on alternate days. Body mass was not different between Cav2−/− (30.8 ± 0.6 g; n = 16) and WT (29.2 ± 0.5 g; n = 16) groups. Three sets of functional experiments were performed, each using separate groups of mice. As detailed below, the first set evaluated microvascular reactivity to ACh and NE. The second set evaluated functional vasodilation to GM contractions and the third set evaluated force production during contractile activity. The first two sets of experiments utilized intravital microscopy to image the microcirculation, which was obviated in the third set due to clamping the muscle for force measurements. In each set of criterion experiments, five or six mice were studied per group.
Caveolin Protein Expression
In separate mice (n = 3 per group), the left GM was dissected free and snap-frozen in liquid nitrogen. Intact tissue samples were lysed and immunoblotted, as described previously (29) with minor modifications. Briefly, each muscle was pulverized in liquid nitrogen and then immersed in RIPA lysis buffer (50 mM Tris-HCl, 1% Nonidet-P40, 0.5% deoxycholic acid, 0.1% SDS) containing protease and phosphatase inhibitors, then homogenized, sonicated, and centrifuged for 10 min at 14,000 rpm and 4°C. Insoluble material was removed, the supernatant was mixed with Laemmli SDS loading buffer and boiled. For each sample, protein concentration was measured with a Nanodrop 2000C (ThermoFisher Scientific; Waltham, MA). Immunoblotting was performed for all samples (n = 6) simultaneously. Thus, 10 μg of protein from each GM was resolved using 12% SDS-PAGE then electro-transferred onto a nitrocellulose membrane. The membrane was washed in TBS containing 0.1% Tween, blocked in 2% fish scale gelatin and incubated with primary mouse antibodies against Cav2 and Cav3 (no. 6106865 and 610420, respectively; BD Transduction Laboratories, San Jose, CA) and primary rabbit antibodies against Cav1 and GAPDH (no. SC-894 and no. SC-25778, respectively; Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight. The membrane was then incubated for 1 h at room temperature with respective secondary anti-mouse and anti-rabbit antibodies labeled with horseradish peroxidase (no. 31432 and no. 31462, respectively; Pierce Biotechnology, Rockford, IL), and developed by enhanced chemiluminescence. Densitometric values for respective immunoblots were determined using ImageJ, and the ratio of each caveolin isoform to GAPDH was calculated for each sample.
Surgical Procedures
On the day of an experiment, a mouse was anesthetized using pentobarbital sodium via an intraperitoneal injection (60 mg/kg ip), as confirmed by the lack of withdrawal to toe pinch. Throughout the experimental protocol, a supplemental dose (20 mg/kg) was given as needed. Esophageal temperature was maintained at 37°C by placing the mouse on an aluminum warming plate (11.5 cm × 5.5 cm). The GM was prepared for observation, as described previously (2, 23, 44). Briefly, hair was shaved from the surgical area, the mouse was placed on the warming plate in the prone position, and the overlaying skin and connective tissue were removed by dissection while viewing through a stereomicroscope. To maintain physiological temperature and tissue integrity, the exposed GM was superfused continuously at 3 ml/min with a bicarbonate-buffered physiological salt solution (PSS; 34°C, pH 7.4) composed of (in mM) 131.9 NaCl, 4.7 KCl, 2 CaCl2, 1.17 MgSO4, and 18 NaHCO3 equilibrated with 5% CO2-95% N2. The GM was dissected away from its origins along the lumbar fascia, sacrum, and iliac crest, while preserving its insertion onto the femur. The muscle flap was gently spread over a transparent rubber pedestal (Sylgard 184; Dow Corning, Midland, MI) to expose its vascular supply, and the edges of the muscle were secured with 8–10 stainless-steel pins (diameter: 0.2 mm) to approximate its dimensions in situ. At the end of each day's experiments the mouse was euthanized with an overdose of pentobarbital sodium (intraperitoneal injection) followed by cervical dislocation.
Intravital Microscopy
The completed GM preparation was transferred to the stage of an intravital imaging system based upon an Olympus MVX10 Stereo Zoom microscope (Center Valley, PA) and equilibrated for 30 min. During the equilibration period, branches of the resistance network supplying the inferior region of the GM were identified as follows. The inferior gluteal artery arises from the internal iliac artery, lies external to GM, and was defined as the feed artery (FA), which enters the muscle to become a first-order (1A) arteriole. Second- order (2A) arterioles originate from 1A and branch into third-order (3A) arterioles that give rise to terminal arterioles and capillaries, which were not studied here. In each GM preparation, one segment of each microvessel branch (FA, 1A, 2A, 3A) was studied, with the sequence varied across experiments. Images of respective branches were acquired through an Olympus MV PLAPO 2XC objective (numerical aperture, 0.5) onto a monochrome charge-coupled device camera (C2400, Hamamatsu Photonics, Japan) and displayed on a video monitor (PVM 122; Sony, Tokyo, Japan) at final magnification = ×900. Internal diameter (ID) for each vessel was measured as the widest distance between luminal edges at the midpoint of the segment using a video caliper calibrated with a stage micrometer (100 × 0.01 = 1 mm; Graticules, Tonbridge, Kent, UK) with spatial resolution of ∼1 μm. Signal output from the caliper was recorded at 2 KHz using a PowerLab/4SP system (ADInstruments, Colorado Springs, CO) coupled to a personal computer.
Experimental Protocols
Resting diameters, oxygen sensitivity, and maximal diameters.
During 30 min of equilibration, the resistance network was mapped and vessel branch orders were identified. Resting ID (IDrest) was then measured in each branch. Arteriolar constriction to elevated oxygen provides a sensitive index of the viability of microvascular preparations studied in vivo (14, 24, 44). For this initial test, the superfusion solution was equilibrated with 21% O2 (with 5% CO2, balance N2) for 5–10 min and ID during elevated oxygen (IDO2) was recorded for each branch. The superfusion solution was then reequilibrated with 5% CO2-95% N2 for the remainder of the experiment. At the end of each experiment, sodium nitroprusside (SNP; 10−4 M) was added to the superfusion solution and maximal ID (IDmax) of each branch was recorded.
Concentration-response relationships.
Vasoconstriction to α-adrenoreceptor (αAR) activation was evaluated by cumulative addition of 10−9 M to 10−5 M NE in 0.5 log increments; each concentration was equilibrated for 5 min before IDs of respective branches were recorded. Superfusion with control PSS was restored for 30 min while vessels recovered to IDrest. Endothelium-dependent dilation (EDD) was evaluated by cumulative addition of 10−9 M to 10−5 M ACh to the superfusion solution in the manner described for NE. Following the final concentration of ACh, superfusion with control PSS was resumed for 30 min to restore IDrest, and then the nitric oxide synthase inhibitor l-NG-nitroarginine methyl ester hydrochloride (l-NAME; 100 μM) was added to the superfusion solution, equilibrated for 15 min and the concentration-response to ACh was repeated during continued superfusion with 100 μM l-NAME. The order in which ACh and NE were evaluated was alternated across experiments with addition of l-NAME always following evaluation of ACh alone.
Functional vasodilation.
Contraction of the GM was evoked with electrical field stimulation using platinum-iridium (90%–10%) electrodes (diameter: 250 μm) positioned on each side of the muscle (2, 23). Monophasic pulses (0.1 ms) were delivered at 12 V using a stimulation isolation unit (SIU5; Grass Technologies; Quincy, MA). To evoke rapid onset vasodilation (ROV), single maximal tetanic contractions were generated at 100 Hz (2, 23) using durations of 100, 250, and 500 ms for each vessel branch. Steady-state dilations to rhythmic twitch contractions were then evaluated at 2 and 4 Hz, each for 30 s (23). Peak response ID was recorded for each contraction followed by 2–3 min recovery to restore IDrest prior to the next contraction. The order in which respective branch orders were studied was randomized across experiments.
Skeletal muscle force production.
The GM was dissected as described above and an atraumatic clamp was positioned across the entire midline oriented perpendicular to muscle fibers. The clamp was secured to a load beam (LCL-1136; Omega, Stamford, CT) coupled to a Transbridge amplifier (TBM-4; World Precision Instruments, Sarasota, FL). The load beam was calibrated (± 0.1 g) and attached to a micrometer that enabled adjustment of muscle length to the optimum (Lo, typically 9 mm) for peak twitch tension during electrical field stimulation (0.1 ms, 20 V) at 1 Hz. To define the frequency-force relationship (43), the GM was stimulated at 10–120 Hz in 20-Hz increments (each for 1 s) while active force was recorded. To evaluate muscle water content as an index of edema, the segment of muscle used for evaluating force production was cut free and blotted of excess moisture; its wet weight (Ww) was measured (± 0.01 mg) on a microbalance (XS105; Mettler Toledo, Columbia, OH). The tissue was then dried at 40°C for 3 h, and its dry weight (Wd) was measured. Muscle cross-sectional area (mm2) was calculated as: Ww/(Lo · muscle density) with muscle density = 1.06 mg · mm−3 (43). Active force produced by the GM at a given stimulation frequency was calculated as (peak force during contraction) − (resting tension) and expressed as milliNewtons per mm2 (mN/mm2).
Data Analyses and Presentation
Vasomotor function.
Spontaneous vasomotor tone (%) in each branch order was calculated as: [(IDmax − IDrest)/IDmax] × 100%. Vasoconstriction (%) to NE was calculated as [(IDrest − IDssr)/Drest] × 100%, where IDssr is the ID recorded during the steady-state response to a given agonist concentration. Vasodilation (%) to ACh was calculated as [(IDssr − IDrest)/IDrest] × 100%. To compare functional responses to GM contractions across respective vessel branch orders having different IDs, vasodilation was calculated as Vasodilator Capacity (%) = [(IDpeak − IDrest)/(IDmax − IDrest)] × 100%. Diameter change was calculated as the difference between IDrest and ID response.
Contractile function.
Active force (P) produced through a range of stimulation frequencies defines the classic frequency-force relationship (43). Respective values were normalized to peak tetanic force (i.e., P/Po × 100%) to provide an index of potential differences in the efficacy of excitation-contraction coupling and the speed of contraction/relaxation (43). Force × time integrals were calculated for each duration of tetanic contraction used to evaluate ROV. Data were analyzed with one- or two-way ANOVA and Student's t-tests (GraphPad Prism 5; GraphPad, La Jolla, CA). When significant F ratios were obtained with ANOVA, post hoc comparisons were performed using Bonferroni tests. Differences were accepted as statistically significant with P < 0.05.
RESULTS
Caveolin Protein Expression
Cav1, Cav2, and Cav3 were expressed in the GM of WT mice; normalization to GAPDH indicates that each isoform was present at similar levels (Fig. 1). For Cav2−/− mice, absence of Cav2 confirms the efficacy of genetic deletion (Fig. 1, A and B). Although the level of Cav1 appeared lower in Cav2−/− vs. WT mice (Fig. 1C), this difference was not statistically significant (P = 0.096), nor was the expression of Cav3 between respective groups (Fig. 1D).
Fig. 1.
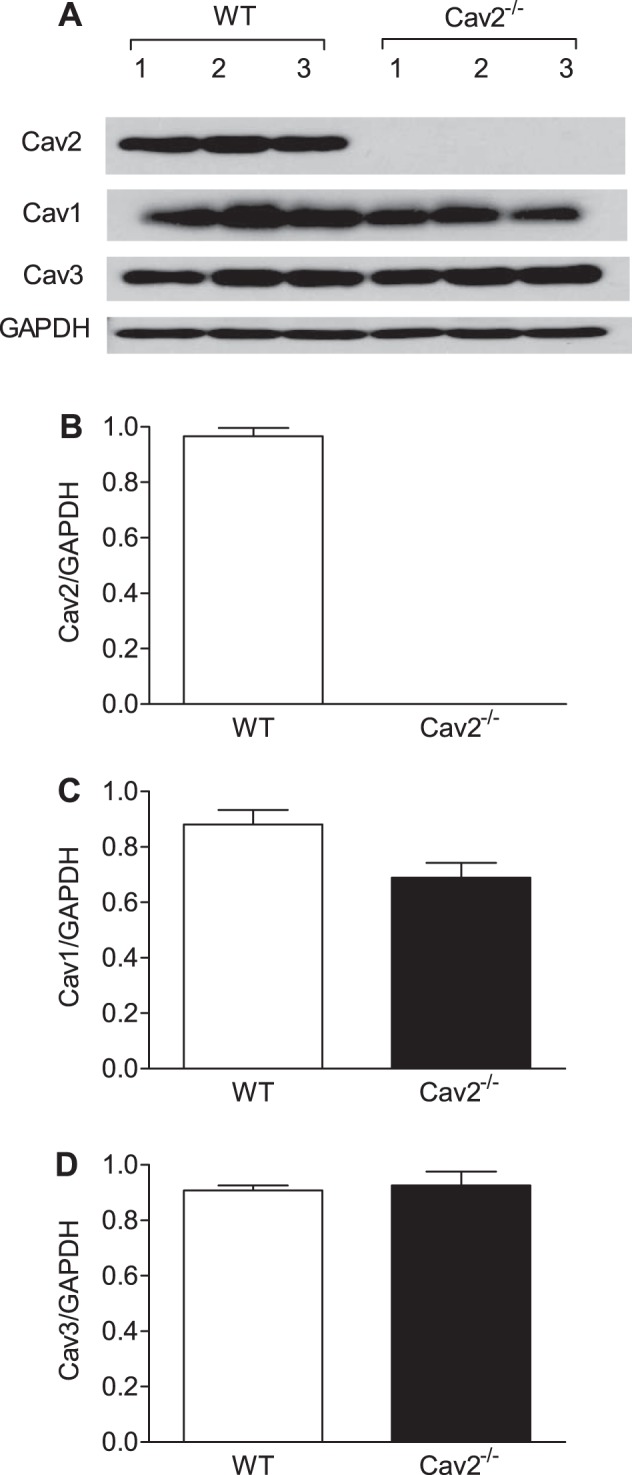
Cav2, Cav1, and Cav3 protein expression in gluteus maximus muscle (GM) of wild-type (WT) and Cav2−/− mice. A: immunoblots for Cav2, Cav1, Cav3, and GAPDH extracted from whole muscle lysates. B–D: summary data for quantitative densitometric ratios of Cav2/GAPDH, Cav1/GAPDH, and Cav3/GAPDH immunoblots are calculated as means ± SD; n = 3 per group.
Resting and Maximal Diameters, Vasomotor Tone, and O2 Response
Microvessel IDs decreased as vessel branch order increased (Fig. 2, FA > 1A > 2A > 3A; P < 0.05); however, there was no significant difference in IDrest (Fig. 2A) or IDmax (Fig. 2B) between Cav2−/− and WT mice. Thus, vasomotor tone increased with branch order (Fig. 2C; FA < 1A < 2A < 3A; P < 0.05) in a similar manner for both groups. All branch orders studied in Cav2−/− and WT mice constricted ∼3–5 μm during elevation of O2 in the superfusion solution to 21%, confirming the viability of GM preparations.
Fig. 2.
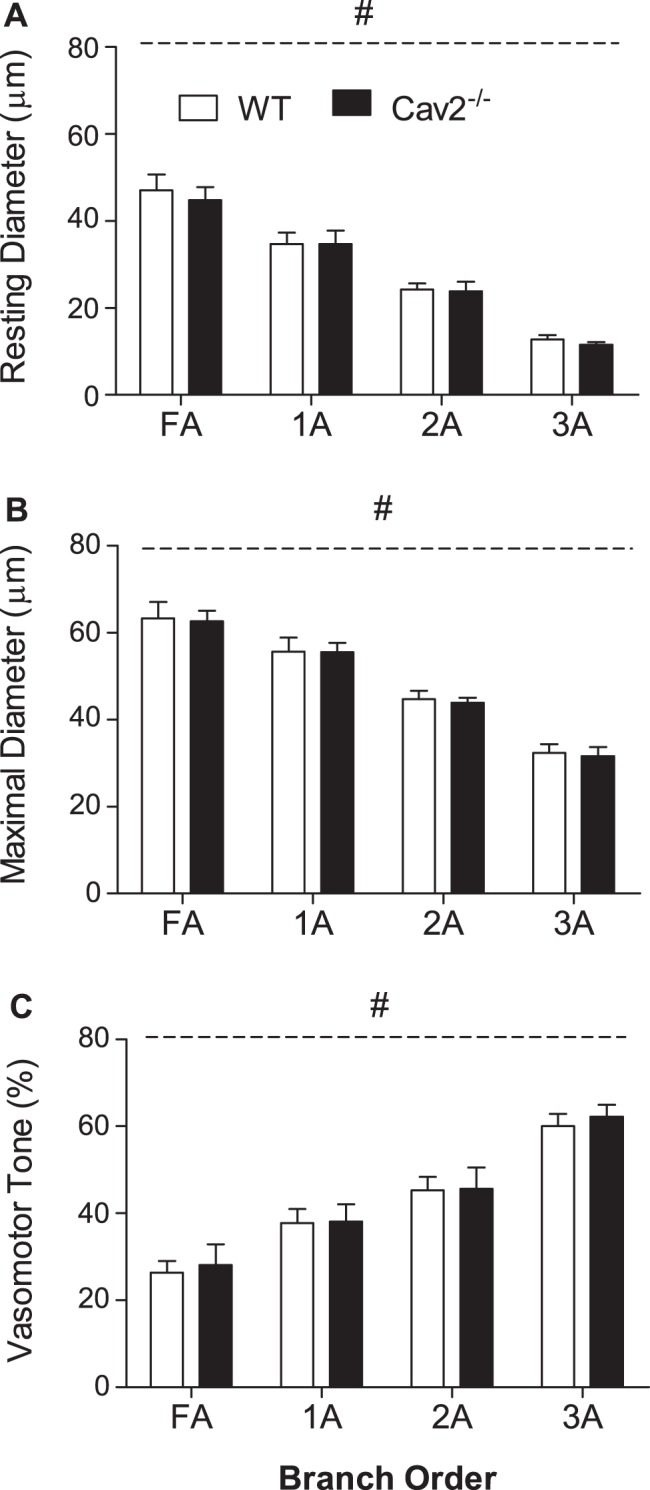
Vessel diameters and spontaneous tone in WT and Cav2−/− mice. Resting (A) and maximal (B) diameters decreased as vessel branch order increased. Vasomotor tone (C) increased with vessel branch order. Summary data are means ± SE; n = 11 per group. #P < 0.05, main effect of vessel branch order; no differences between groups.
Vasoconstriction to NE
Norepinephrine produced a concentration-dependent constriction of all vessel branch orders for both Cav2−/− and WT mice; however, there was no difference between groups (Fig. 3). In all GM preparations, 2A and 3A constricted to closure with 10−5 M NE. Thus, the ability to activate SMC contraction throughout the resistance network via αARs was unaffected by loss of Cav2.
Fig. 3.
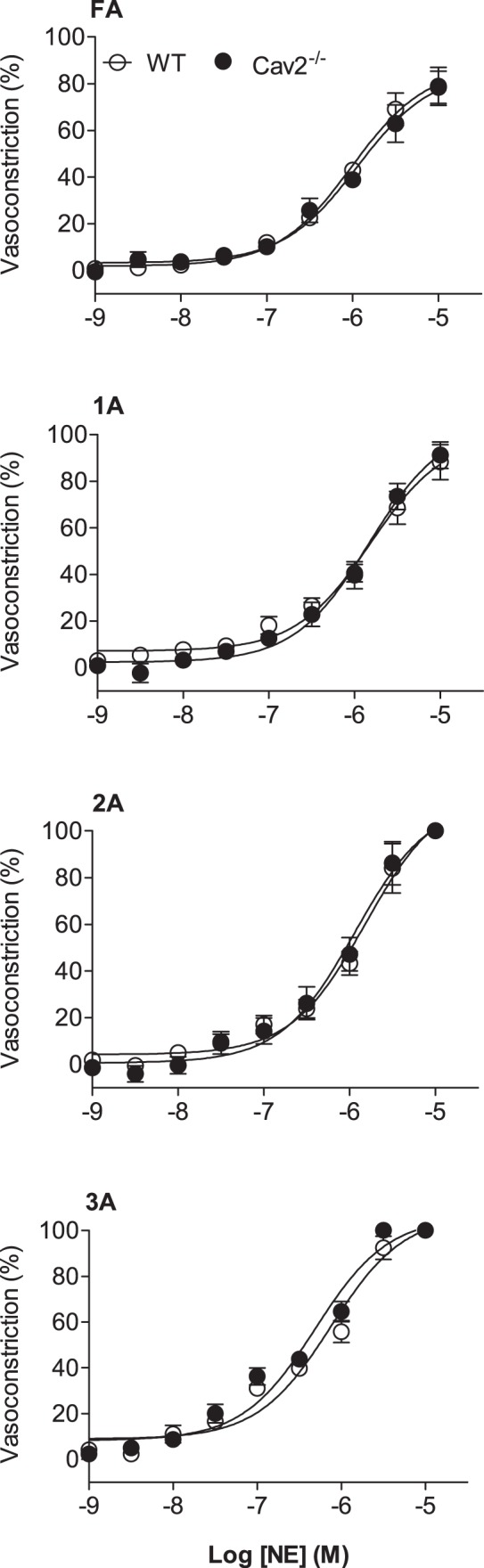
Concentration-response curves to norepinephrine (NE) of WT and Cav2−/− mice. Vasoconstriction increased with NE concentration in all vessel branch orders with no difference between groups. Summary data are means ± SE; n = 6 per group.
Vasodilation to ACh
ACh induced a concentration-dependent dilation of all vessel branch orders for both Cav2−/− and WT mice (Fig. 4). With no difference between groups, the relative response to ACh increased with branch order (P < 0.05). In the presence of l-NAME, vasodilation to ACh was reduced to a similar extent across branch orders of both groups (Fig. 4). Thus EDD was maintained throughout the resistance network in the absence of Cav2, as was the contribution of NO to SMC relaxation.
Fig. 4.
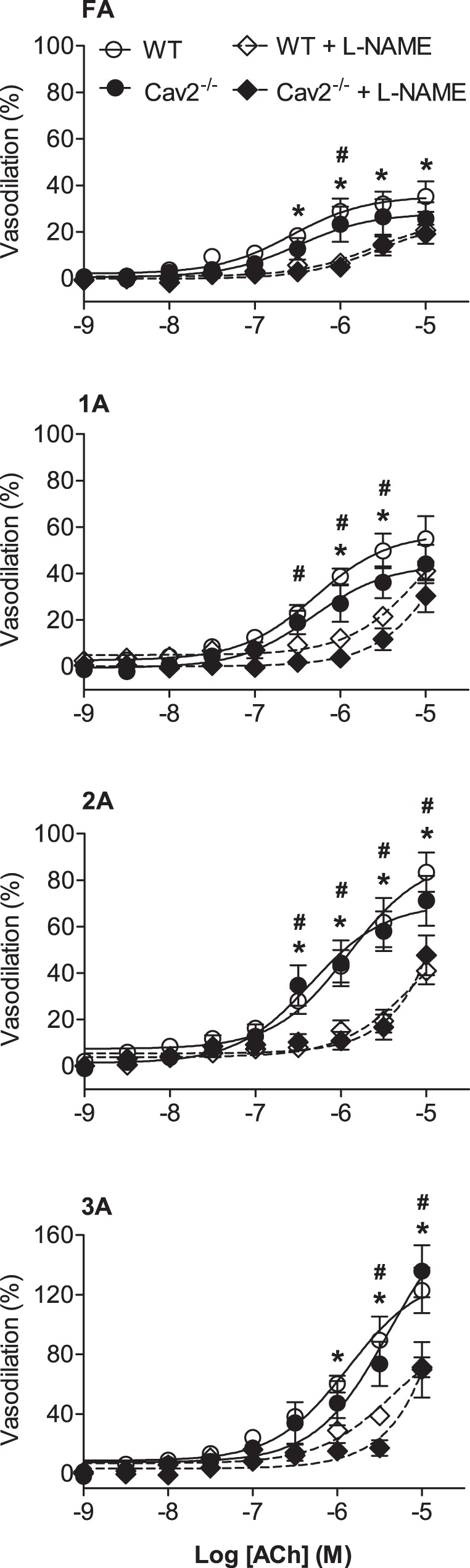
Concentration-response curves to ACh in the absence and presence of l-NG-nitroarginine methyl ester hydrochloride (l-NAME) in WT and Cav2−/− mice. Vasodilation increased with ACh concentration in all branch orders with no differences between groups. Vasodilation to ACh decreased significantly and to the same extent in the presence of l-NAME in all branch orders of both groups. Summary data are means ± SE; n = 6 per group. *P < 0.05 for effect of l-NAME in WT mice. #P < 0.05 for effect of l-NAME in Cav2−/− mice.
Functional Vasodilation to Muscle Contraction
During twitch contractions for 30 s, vasodilation typically began after several seconds and increased to a plateau within 15–20 s. These steady-state responses were significantly greater at 4 Hz vs. 2 Hz (P < 0.05) for all branch orders in Cav2−/− and WT mice (Fig. 5). However, there were no differences between groups for any branch order. In response to single tetanic contractions, ROV peaked within a few seconds, and its amplitude increased with contraction duration (P < 0.05) in all branch orders of both groups (Fig. 6). A key finding was the attenuation of ROV by approximately half across branch orders of Cav2−/− mice compared with WT mice (P < 0.05). Thus, while the ability to maintain steady-state dilations during mild rhythmic contractions was preserved, the absence of Cav2 was associated with a selective and pronounced impairment in the ability of microvascular resistance networks to undergo ROV.
Fig. 5.
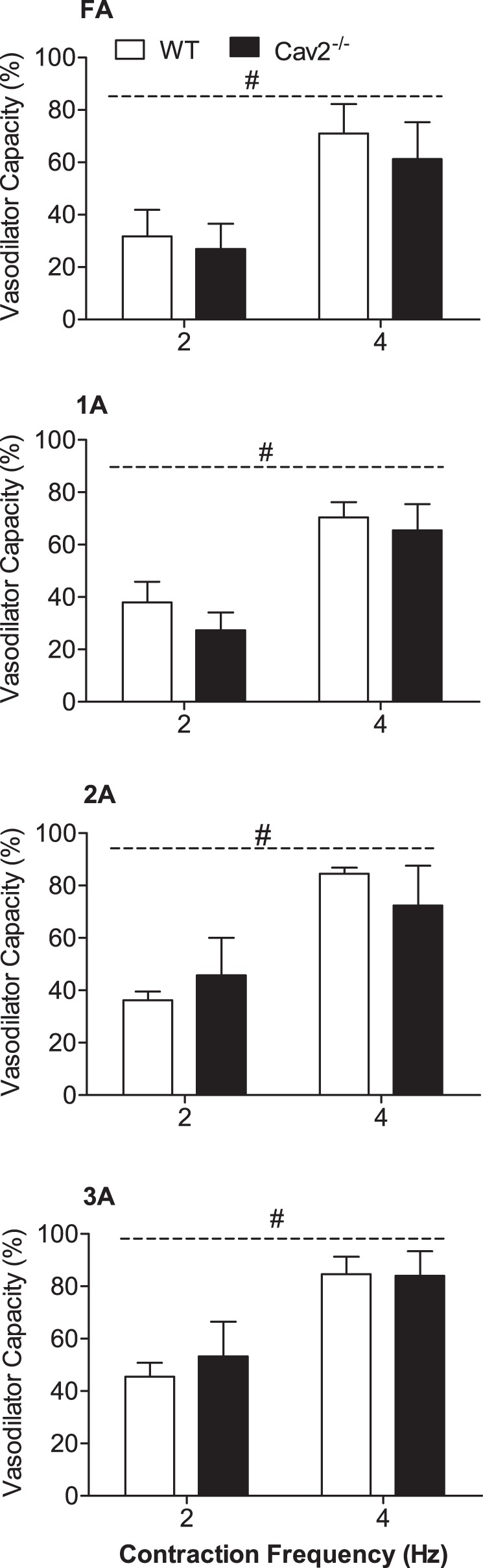
Steady-state vasodilation in response to rhythmic twitch contractions at 2 Hz and 4 Hz in WT and Cav2−/− mice. Vasodilator Capacity calculated as described in methods. Across vessel branch orders, diameter changes were 5–10 μm during 2-Hz contractions and 15–20 μm during 4-Hz contractions with no differences between WT and Cav2−/− mice. Summary data are means ± SE; n = 5 per group. #P < 0.05 for contraction duration across groups.
Fig. 6.
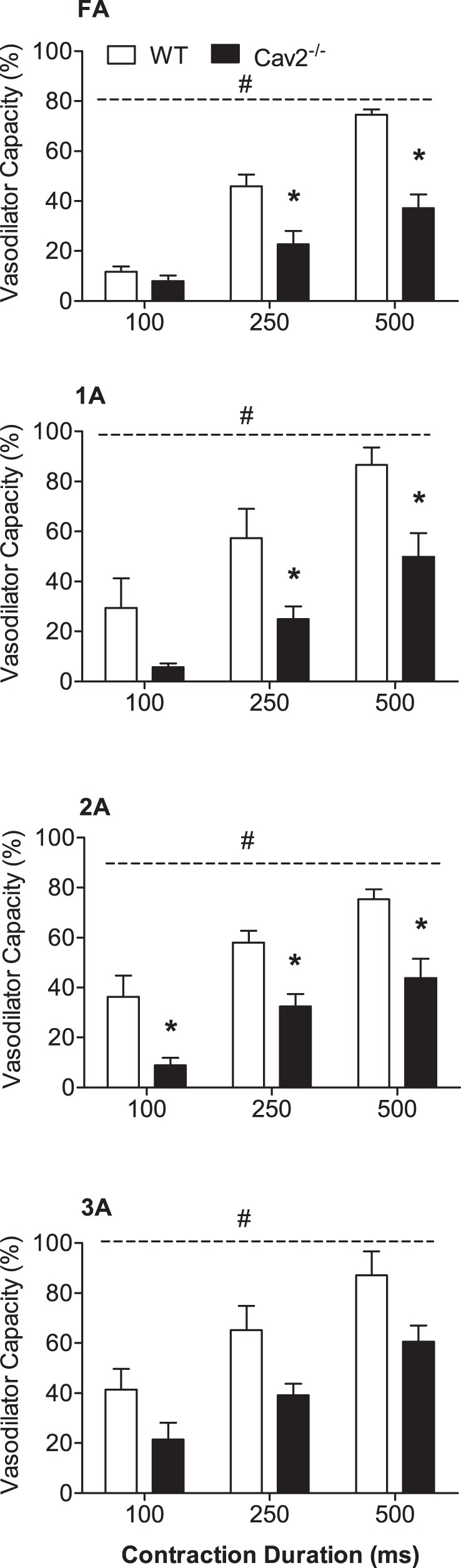
Rapid onset vasodilation (ROV) in response to single tetanic contractions in WT and Cav2−/− mice. For all vessel branch orders, ROV increased with contraction duration and was attenuated in Cav2−/− compared with WT mice. Summary data are means ± SE; n = 5 per group. *P < 0.05 for Cav2−/− vs. WT at indicated contraction duration. #P < 0.05 for groups across contraction durations. Respective diameter changes (μm) are shown in Fig. 8.
Skeletal Muscle Force Production
In response to electrical field stimulation, active force produced by the GM at 40–120 Hz was significantly greater (P < 0.05) in Cav2−/− mice compared with WT mice (Fig. 7). Normalized to muscle cross-sectional area, maximal tetanic force (Po at 100 Hz) was 152 ± 14 mN/mm2 for Cav2−/− mice compared with 110 ± 3 mN/mm2 in WT mice (P < 0.05, n = 5 per group). There were no significant differences in muscle wet weights (Ww = 48.7 ± 2.3 and 48.2 ± 3.8 mg), dry weights (Wd = 12.2 ± 0.6 and 10.9 ± 0.6 mg), water content (37.5 and 37.2 mg; 77% for both), or calculated cross-sectional area (5.1 ± 0.2 and 5.1 ± 0.4 mm2) between WT and Cav2−/− mice, respectively. Thus, the absence of Cav2 is associated with enhanced specific force production by the contractile machinery of skeletal muscle fibers. When normalized to maximal force (Po) for respective groups, frequency-force responses of Cav2−/− mice were shifted slightly up and to the left between 20 and 80 Hz compared with WT mice, and this difference was statistically significant at 60 Hz (P < 0.05). Thus, in the steepest portion of the response curve, a given stimulation frequency evoked ∼10% greater relative force in Cav2−/− mice (Fig. 7B).
Fig. 7.
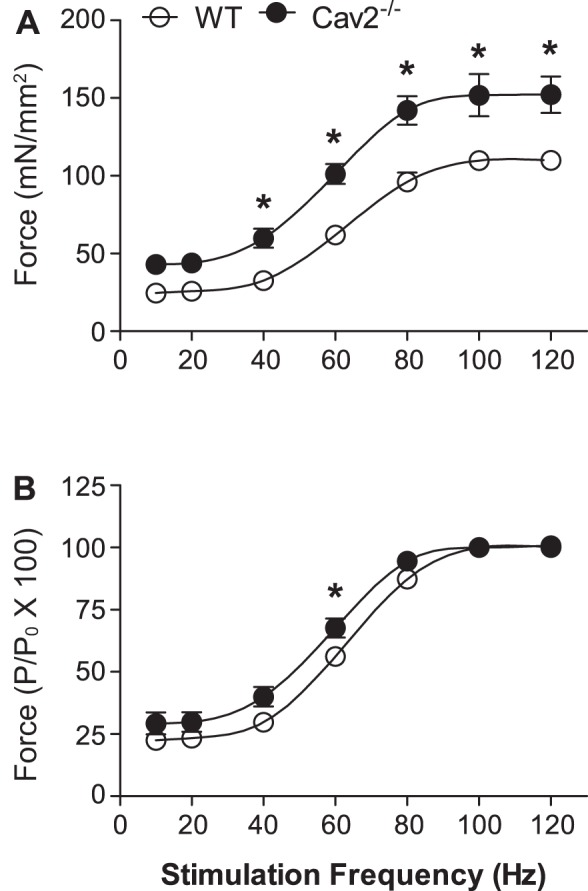
Frequency-force relationship for GM contractions in WT and Cav2−/− mice. A: specific force production was greater from 40 Hz to 120 Hz in Cav2−/− mice compared with WT mice. B: active force produced at each stimulation frequency (P) normalized to maximal force production (Po). Summary data are means ± SE; n = 5 per group. *P < 0.05 for Cav2−/− vs. WT at indicated stimulation frequencies.
To illustrate key differences in phenotype between Cav2−/− and WT mice, the diameter change in respective vessel branch orders for each ROV contraction duration was plotted with respect to the corresponding force × time integral (Fig. 8). Thus, despite greater force production during each contraction, the resistance vasculature of Cav2−/− mice responded with diminished ROV.
Fig. 8.
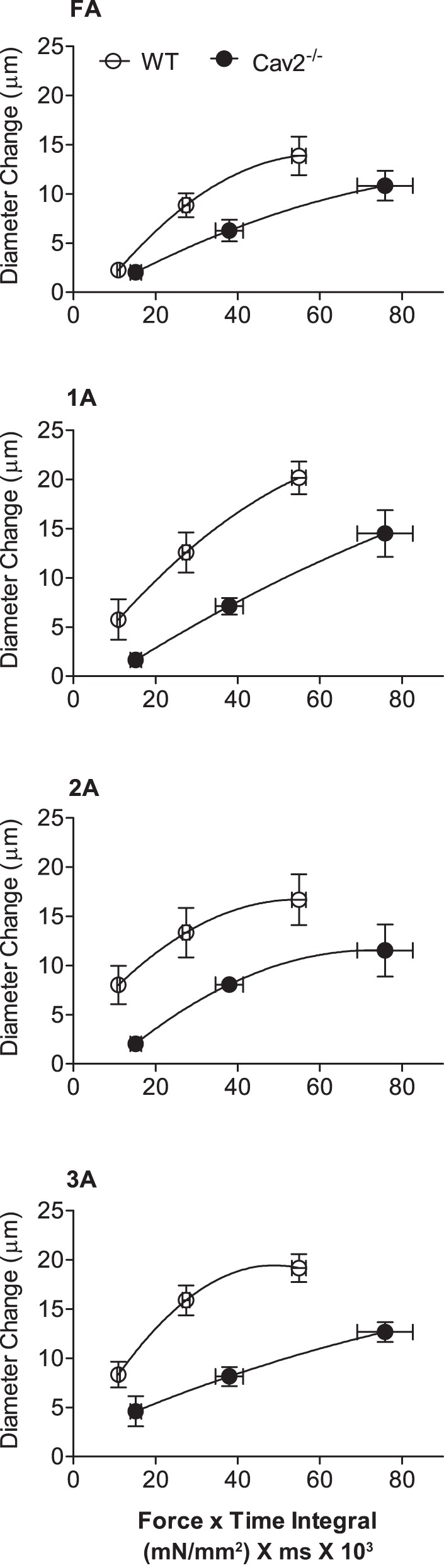
Relationships between force × time integral and diameter change in WT and Cav2−/− mice. Respective values for diameter change are from data shown in Fig. 6 at contraction durations of 100, 250, and 500 ms; force × time integral values calculated from respective Po values at 100 Hz in Fig. 7.
Additional Controls
Depressed ROV in male mice (23) and human subjects (5) with advanced age was associated with enhanced activation of αARs. Therefore, we tested whether depressed ROV responses in male Cav2−/− mice at 6 mo of age (Fig. 6) could be reversed by inhibiting αARs with a nonselective αAR antagonist. However, phentolamine (10−6 M) had no effect on ROV at any contraction duration, thereby excluding a role for αARs in modulating ROV with Cav2 deletion. In light of findings that Cav2 deletion was associated with alterations in muscle fiber morphology at 3 mo of age (40), we tested whether the increase in active force production observed at 6 mo in Cav2−/− mice was manifest at 3 mo. However, we found no difference in either the frequency-force relationship or Po (109 ± 11 and 116 ± 6 mN/mm2) between Cav2−/− and WT male mice (n = 3 per group), respectively, at 3 mo, nor were these values significantly different from WT mice at 6 mo (see above). Thus, augmentation of force production in the GM of Cav2−/− mice appears to develop between 3 and 6 mo of age. In light of alterations in muscle fiber morphology of male (but not female) Cav2−/− mice (40), we evaluated force production in Cav2−/− and WT female mice at 3 and 6 mo to test for a sex difference in muscle function. However, there was no difference in the frequency-force relationship or Po between Cav2−/− females at 3 or 6 mo (Po = 107 ± 14 and 112 ± 2 mN/mm2, respectively; n = 3 per group), nor were these values different from WT males of respective age groups. Thus, the ∼40% increase in force production observed at 6 mo in Cav2−/− mice appears to be selective for males.
DISCUSSION
The effect of Cav2 deletion has been difficult to resolve, likely because its role has appeared secondary to that of Cav1 (27, 28). While Cav2 is not expressed in mature skeletal muscle fibers, their ultrastructure is altered by Cav2 deletion (40), and it was unknown whether there are functional consequences of Cav2 deletion to the microvascular supply of skeletal muscle or to its contractile function. To explore these relationships, the present study evaluated microvascular resistance networks and isometric force production in the GM of male mice at 6 mo of age, which corresponds to ∼30 yr of age in humans (17). Our findings reveal a remarkable phenotype: while vasomotor tone, adrenergic vasoconstriction, EDD, and steady-state vasodilation during rhythmic twitch contractions remain intact, there is a profound reduction in ROV. Thus, the ability of the resistance vasculature to dilate rapidly in response to single tetanic contractions is severely compromised in the absence of Cav2. Remarkably, impaired ROV is manifest in the presence of a profound increase in the force produced by skeletal muscle fibers. The loss of Cav2 thereby dissociates the historically strong correspondence between force exerted during muscle contraction and the magnitude of functional vasodilation/hyperemia (1, 4, 9, 50).
Caveolin Protein Expression
The absence of Cav2 protein following genetic deletion (29) provides a positive control, consistent with earlier studies of Cav2−/− mice generated independently (37). The present data are also consistent with Cav2−/− offspring being viable and exhibiting postnatal growth and body mass not different from WT mice (37, 40). Although not statistically different from WT mice, the level of Cav1 protein was ∼20% lower in the GM of Cav2−/− mice (Fig. 1C). An earlier study reported that Cav1 protein was reduced by approximately half in lung and heart tissues following Cav2−/− deletion (37). However, it was subsequently found that levels of Cav1 protein in skeletal (gastrocnemius) muscle of Cav2−/− mice were not different from those in WT mice (40), which more closely agrees with the present data from the GM. The finding that levels of Cav3 protein in the GM of Cav2−/− mice was not different from those in WT mice (Fig. 1D) is consistent with earlier data for the gastrocnemius muscle (40), as well as the heart (37). Although our experiments did not evaluate the subcellular localization of respective caveolin isoforms, as done previously (37), these data collectively suggest that neither changes in Cav1 nor changes in Cav3 expression contribute to the functional differences associated with Cav2 deletion reported here.
Vessel Diameters and Reactivity
Irrespective of Cav2 deletion, vessel diameters in the GM decreased as branch order increased (FA > 1A > 2A > 3A; Fig. 1), consistent with the topology of resistance networks supplying other skeletal muscles (13, 30, 50). Thus, the morphology of microvessels studied here appears to be independent of Cav2 expression, as does their ability to constrict in response to elevated Po2. Spontaneous vasomotor tone reflects the myogenic response of vascular SMCs to transmural pressure under physiological conditions, and the increase with vessel branch order observed here is consistent with previous findings (12, 13, 44). As SMCs are the only cell type known to express all three caveolin isoforms (38, 53), the absence of Cav2 may have little effect on SMC function, particularly with Cav3 being the predominant isoform expressed in muscle cells (16, 38, 42, 47). Vasoconstriction through αAR activation reflects perivascular sympathetic nerve activity (21, 30). When αARs were stimulated with the sympathetic neurotransmitter NE, respective branches constricted (reversibly upon washout) in a concentration-dependent manner (Fig. 3), with no differences between groups. Thus, in addition to the myogenic response, respective signaling pathways for SMC activation appear well maintained in the absence of Cav2. ACh is also a neurotransmitter and has long been associated with vasodilation in skeletal muscle (22). Upon activating muscarinic receptors on ECs, ACh evokes SMC relaxation via the release of autacoids from ECs (e.g., NO) and through membrane hyperpolarization via myoendothelial coupling through gap junctions (3, 41). With Cav2 and eNOS localized to EC caveolae (46), we investigated whether Cav2 is integral to EDD of resistance networks supplying the GM. Remarkably, throughout concentration-response relationships, there were no differences in vasodilation to ACh between Cav2−/− and WT mice in any of the four branch orders. Further, inhibition of NOS with l-NAME consistently depressed vasodilation to a similar extent in all vessels of both experimental groups (Fig. 4). These findings are consistent with earlier reports that the regulation of adrenergic vasoconstriction and EDD are well maintained in the absence of Cav2, as is the integrity of EC caveolae (36, 37). The present findings are the first to illustrate that the absence of Cav2 does not alter the reactivity of microvascular resistance networks to an array of physiological stimuli in vivo.
Functional Vasodilation
The ability to increase skeletal muscle blood flow is integral to physical performance. While reduced swimming performance in Cav2−/− mice has been attributed to impaired gas exchange in the lung (37), there have been no studies of muscle blood flow regulation in the absence of Cav2. The kinetics of functional vasodilation and its underlying signaling events vary with the nature of contraction (6, 8, 32, 50). Therefore, we applied stimulation protocols designed to evoke either a gradual response to rhythmic twitch contractions or a rapid response to brief maximal contraction (23, 34). The increase in steady-state vasodilation from 2- to 4-Hz twitch contractions (Fig. 5) is consistent with muscle blood flow increasing with metabolic demand, work intensity, and running speed (1, 26, 33). In contrast, vasodilation occurs near-instantaneously with single brief contractions in rodents (23, 34, 51), as well as humans (9–11, 48). While several mechanisms have been proposed to initiate ROV (10, 48, 51), as well the inhibition of ROV through αAR activation (5, 23), no studies have investigated whether there may be a role for Cav2 in this regard. A key finding here is that loss of Cav2 depressed ROV by approximately half throughout the resistance network (Fig. 6). Since phentolamine had no effect on restoring ROV in Cav2−/− mice, the attenuation of ROV seen here is not via the activation of αARs, as occurs with advanced age (5, 23). Thus, the mechanism by which Cav2 affects ROV remains to be identified. Given that the absence of Cav2 had no effect on steady-state vasodilation during rhythmic contractions or on vasodilation to ACh, the present findings highlight the need for future studies to elucidate the role of Cav2 in the ability of the resistance vasculature to increase muscle blood flow at the onset of contractile activity.
Force Production During Muscle Contraction
Of the three caveolin isoforms, only Cav3 is expressed in mature skeletal muscle fibers (19, 20, 45); however, Cav2 is expressed in skeletal muscle stem (i.e., “satellite”) cells and disappears with myocyte differentiation, suggesting that Cav2 may contribute to skeletal muscle development (40). While Cav3 deletion or mutation results in muscle wasting with diminished force production (19, 20, 45), there have been no studies addressing whether Cav2 has a role in contractile function. Thus, we tested the effect of Cav2 deletion on specific force produced (i.e., per cross-sectional area) during muscle contractions. Remarkably, with no difference in tissue water content (thereby excluding any edema), specific force production was ∼40% greater in the GM of Cav2−/− mice compared with WT mice (Fig. 6). When frequency-force relationships of the GM from Cav2−/− and WT mice were normalized to respective maximum values to address potential differences in the speed of contraction and/or relaxation (43), the response curves from both groups nearly overlapped, albeit with a slight shift to the left for Cav2−/− (Fig. 6). This finding indicates that there may be a subtle shift to a slower fiber type in the absence of Cav2. In turn, adaptations may occur in the contractile proteins and/or their regulation by intracellular Ca2+, as recently reported for cardiac myocytes (7). This reasoning is supported by the finding that Cav2 deletion increased the volume of SR in fibers of the gastrocnemius muscle (40), which may thereby augment cross-bridge interactions during excitation-contraction coupling. For myocytes from patients with Rippling muscle disease and cachexia as a consequence of Cav3 depletion (20), defective excitation-contraction coupling was associated with disarray of the voltage-sensing dihydropyridine receptor (49), which governs intracellular release of Ca2+ from the SR. In light of finding no differences in force production between WT male mice at 3 or 6 mo, for female Cav2−/− mice of either genotype at 3 or 6 mo, or for male Cav2−/− mice at 3 mo, the enhanced specific force production with Cav2 deletion reported here appears to develop selectively in males between 3 and 6 mo of age. Resolving this time frame provides a reference for future studies of how Cav2 affects force production in skeletal muscle fibers.
Summary and Conclusion
The present study provides new insight into functional roles of Cav2 using genetic deletion in the mouse. Whether in humans (9–11, 48) or rodents (23, 34, 51), a consistent finding is that ROV increases with the force developed during contraction. Thus, with depressed ROV in Cav2−/− mice, it would be predicted that active force developed by the GM would also be depressed in the absence of Cav2, particularly with aberrant muscle fiber ultrastructure (40). Remarkably, specific force produced by the GM was consistently ∼40% greater in Cav2−/− mice than of WT littermate controls, such that the resistance vasculature responds with diminished ROV despite greater force production (Fig. 8). Thus while vessel diameters, myogenic tone, reactivity to oxygen and physiological agonists, as well as functional dilation during rhythmic contractions remain unchanged with Cav2 deletion, the present experiments are the first to dissociate the classic relationship between vasodilation and muscle force production. This Cav2−/− phenotype appears to be selective for males and to develop in a defined time period (3–6 mo) that corresponds to the third decade of life in humans (17). Future studies are needed to understand how manipulating the expression of caveolin isoforms affects the interaction between force production and microvascular reactivity in skeletal muscle.
GRANTS
This research was supported by National Heart, Lung and Blood Institute (NHLBI) Grant R37HL-041026 (to S. S. Segal) G. Sowa is supported by the Department of Defense Grant W81XWH-15-1-0624.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.A.F., Y.L. G.S. and S.S.S. conception and design of research; C.A.F. and Y.L. performed experiments; C.A.F., Y.L., G.S. and S.S.S. analyzed data; C.A.F., Y.L., G.S. and S.S.S. interpreted results of experiments; Y.L. bred and genotyped mice; C.A.F. and Y.L. prepared figures; C.A.F. drafted manuscript; Y.L., G.S. and S.S.S. edited and revised manuscript; C.A.F., Y.L., G.S and S.S.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Drs. Shenghua Sinkler and Camila Manrique provided valuable technical assistance.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Andersen P, Saltin B. Maximal perfusion of skeletal muscle in man. J Physiol 366: 233–249, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bearden SE, Payne GW, Chisty A, Segal SS. Arteriolar network architecture and vasomotor function with ageing in mouse gluteus maximus muscle. J Physiol 561: 535–545, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte P, Weston A. EDHF: bringing the concepts together. Trends Pharmacol Sci 23: 374–380, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Carlson RE, Kirby BS, Voyles WF, Dinenno FA. Evidence for impaired skeletal muscle contraction-induced rapid vasodilation in aging humans. Am J Physiol Heart Circ Physiol 294: H1963–H1970, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Casey DP, Joyner MJ. Influence of α-adrenergic vasoconstriction on the blunted skeletal muscle contraction-induced rapid vasodilation with aging. J Appl Physiol 113: 1201–1212, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celichowski J. Mechanisms underlying the regulation of motor unit contraction in the skeletal muscle. J Physiol Pharmacol 51: 17–33, 2000. [PubMed] [Google Scholar]
- 7.Clay SA, Domeier TL, Hanft LM, McDonald KS, Krenz M. Elevated Ca2+ transients and increased myofibrillar power generation cause cardiac hypercontractility in a model of Noonan syndrome with multiple lentigines. Am J Physiol Heart Circ Physiol 308: H1086–H1095, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clifford PS, Hellsten Y. Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97: 393–403, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Corcondilas A, Koroxenidis GT, Shepherd JT. Effect of a brief contraction of forearm muscles on forearm blood flow. J Appl Physiol 19: 142–146, 1964. [DOI] [PubMed] [Google Scholar]
- 10.Crecelius AR, Kirby BS, Luckasen GJ, Larson DG, Dinenno FA. Mechanisms of rapid vasodilation after a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305: H29–H40, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Credeur DP, Holwerda SW, Restaino RM, King PM, Crutcher KL, Laughlin MH, Padilla J, Fadel PJ. Characterizing rapid-onset vasodilation to single muscle contractions in the human leg. J Appl Physiol 118: 455–464, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis MJ. Myogenic response gradient in an arteriolar network. Am J Physiol Heart Circ Physiol 264: H2168–H2179, 1993. [DOI] [PubMed] [Google Scholar]
- 13.Dodd LR, Johnson PC. Diameter changes in arteriolar networks of contracting skeletal muscle. Am J Physiol Heart Circ Physiol 260: H662–H670, 1991. [DOI] [PubMed] [Google Scholar]
- 14.Duling BR, Berne RM. Oxygen and the local regulation of blood flow: possible significance of longitudinal gradients in arterial blood oxygen tension. Circ Res 28: Suppl 1: 65–69, 1971. [PubMed] [Google Scholar]
- 15.Faber JE. In situ analysis of α-adrenoceptors on arteriolar and venular smooth muscle in rat skeletal muscle microcirculation. Circ Res 62: 37–50, 1988. [DOI] [PubMed] [Google Scholar]
- 16.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, Michel T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J Biol Chem 271: 22,810–22,814, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Flurkey K, Currer J, Harrison D. Mouse models in aging research. In: The Mouse in Biomedical Research, edited by Fox JG, Davisson MT, Quimby FW, Barthold SW, Newcomer CE, and Smith AL. Burlington, VT: American College of Laboratory Animal Medicine (Elsevier), 2007, p. 637–672. [Google Scholar]
- 18.García-Cardeña G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the NOS caveolin binding domain in vivo. J Biol Chem 272: 25,437–25,440, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Gazzerro E, Sotgia F, Bruno C, Lisanti MP, Minetti C. Caveolinopathies: from the biology of caveolin-3 to human diseases. Eur J Hum Genet 18: 137–145, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet 9: 3047–3054, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Haug SJ, Segal SS. Sympathetic neural inhibition of conducted vasodilatation along hamster feed arteries: complementary effects of α1- and α2-adrenoreceptor activation. J Physiol 563: 541–555, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilton SM. A peripheral arterial conducting mechanism underlying dilatation of the femoral artery and concerned in functional vasodilatation in skeletal muscle. J Physiol 149: 93–111, 1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson DN, Moore AW, Segal SS. Blunting of rapid onset vasodilatation and blood flow restriction in arterioles of exercising skeletal muscle with ageing in male mice. J Physiol 588: 2269–2282, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson WF. Regional differences in mechanism of action of oxygen on hamster arterioles. Am J Physiol Heart Circ Physiol 265: H599–H603, 1993. [DOI] [PubMed] [Google Scholar]
- 25.Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide syntsynthase and caveolin-1 inhibits synthase activity. J Biol Chem 272: 18,522–18,525, 1997. [DOI] [PubMed] [Google Scholar]
- 26.Laughlin MH, Armstrong RB. Muscular blood flow distribution patterns as a function of running speed in rats. Am J Physiol Heart Circ Physiol 243: H296–H306, 1982. [DOI] [PubMed] [Google Scholar]
- 27.Le Lay S, Kurzchalia TV. Getting rid of caveolins: phenotypes of caveolin-deficient animals. Biochim Biophys Acta 1746: 322–333, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Galbiati F, Volonte D, Sargiacomo M, Engelman JA, Das K, Scherer PE, Lisanti MP. Mutational analysis of caveolin-induced vesicle formation. Expression of caveolin-1 recruits caveolin-2 to caveolae membranes. FEBS Lett 434: 127–134, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Jang S, Xie L, Sowa G. Host deficiency in caveolin-2 inhibits lung carcinoma tumor growth by impairing tumor angiogenesis. Cancer Res 74: 6452–6462, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marshall JM. The influence of the sympathetic nervous system on individual vessels of the microcirculation of skeletal muscle of the rat. J Physiol 332: 169–186, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moore AW, Jackson WF, Segal SS. Regional heterogeneity of α-adrenoreceptor subtypes in arteriolar networks of mouse skeletal muscle. J Physiol 588: 4261–4274, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murrant CL. Stimulation characteristics that determine arteriolar dilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 289: R505–R513, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Musch TI, Friedman DB, Pitetti KH, Haidet GC, Stray-Gundersen J, Mitchell JH, Ordway GA. Regional distribution of blood flow of dogs during graded dynamic exercise. J Appl Physiol 63: 2269–2277, 1987. [DOI] [PubMed] [Google Scholar]
- 34.Novielli NM, Jackson DN. Contraction-evoked vasodilation and functional hyperaemia are compromised in branching skeletal muscle arterioles of young pre-diabetic mice. Acta Physiol (Oxf) 211: 371–384, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 8: 185–194, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, Di Vizio D, Hou H Jr, Kneitz B, Lagaud G, Christ GJ, Edelmann W, Lisanti MP. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38,121–38,138, 2001. [DOI] [PubMed] [Google Scholar]
- 37.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H Jr, Christ GJ, Edelmann W, Lisanti MP. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol 22: 2329–2344, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev 54: 431–467, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1−/− knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, l-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 277: 40,091–40,098, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Schubert W., Sotgia F, Cohen AW, Capozza F, Bonuccelli G, Bruno C, Minetti C, Bonilla E, Dimauro S, Lisanti MP. Caveolin-1−/−- and caveolin-2−/−-deficient mice both display numerous skeletal muscle abnormalities, with tubular aggregate formation. Am J Pathol 170: 316–333, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Segal SS. Integration and modulation of intercellular signaling underlying blood flow control. J Vasc Res 52: 136–157, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Segal SS, Brett SE, Sessa WC. Codistribution of NOS and caveolin throughout peripheral vasculature and skeletal muscle of hamsters. Am J Physiol Heart Circ Physiol 277: H1167–H1177, 1999. [DOI] [PubMed] [Google Scholar]
- 43.Segal SS, Faulkner JA, White TP. Skeletal muscle fatigue in vitro is temperature dependent. J Appl Physiol 61: 660–665, 1986. [DOI] [PubMed] [Google Scholar]
- 44.Sinkler SY, Segal SS. Aging alters reactivity of microvascular resistance networks in mouse gluteus maximus muscle. Am J Physiol Heart Circ Physiol 307: H830–H839, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smythe GM, Eby JC, Disatnik MH, Rando TA. A caveolin-3 mutant that causes limb girdle muscular dystrophy type 1C disrupts Src localization and activity and induces apoptosis in skeletal myotubes. J Cell Sci 116: 4739–4749, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol 2: 120, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang ZSP, Okamoto T, Song K, Chu C, Kohtz DS, Nishimoto I, Lodish HF, Lisanti MP. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem 4: 2255–2261, 1996. [DOI] [PubMed] [Google Scholar]
- 48.Tschakovsky ME, Sheriff DD. Immediate exercise hyperemia: contributions of the muscle pump vs. rapid vasodilation. J Appl Physiol 97: 739–747, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Ullrich ND, Fischer D, Kornblum C, Walter MC, Niggli E, Zorzato F, Treves S. Alterations of excitation-contraction coupling and excitation coupled Ca2+ entry in human myotubes carrying CAV3 mutations linked to rippling muscle. Hum Mutat 32: 309–317, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.VanTeeffelen JW, Segal SS. Effect of motor unit recruitment on functional vasodilatation in hamster retractor muscle. J Physiol 524: 267–278, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.VanTeeffelen JW, Segal SS. Rapid dilation of arterioles with single contraction of hamster skeletal muscle. Am J Physiol Heart Circ Physiol 290: H119–H127, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Venema VJJ, Zou H, Venema R, RC. Interaction of neuronal nitric-oxide synthase with caveolin-3 in skeletal muscle. Identification of a novel caveolin scaffolding/inhibitory domain. J Biol Chem 272: 28,187–28,190, 1997. [DOI] [PubMed] [Google Scholar]
- 53.Williams TM, Lisanti MP. The Caveolin genes: from cell biology to medicine. Ann Med 36: 584–595, 2004. [DOI] [PubMed] [Google Scholar]
- 54.Woodman SE, Ashton AW, Schubert W, Lee H, Williams TM, Medina FA, Wyckoff JB, Combs TP, Lisanti MP. Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol 162: 2059–2068, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]