

Abstract
Although it is well-known that excess renin angiotensin system (RAS) activity contributes to the pathophysiology of cardiac and vascular disease, tissue-based expression of RAS genes has given rise to the possibility that intracellularly produced angiotensin II (Ang II) may be a critical contributor to disease processes. An extended form of angiotensin I (Ang I), the dodecapeptide angiotensin-(1–12) [Ang-(1–12)], that generates Ang II directly from chymase, particularly in the human heart, reinforces the possibility that an alternative noncanonical renin independent pathway for Ang II formation may be important in explaining the mechanisms by which the hormone contributes to adverse cardiac and vascular remodeling. This review summarizes the work that has been done in evaluating the functional significance of Ang-(1–12) and how this substrate generated from angiotensinogen by a yet to be identified enzyme enhances knowledge about Ang II pathological actions.
Keywords: angiotensin-(1–12), angiotensin converting enzyme inhibitors; heart chymase; hypertension; heart failure
this article is part of a collection on Annual Meeting of the International Academy of Cardiovascular Sciences: North American Section. Other articles appearing in this collection, as well as a full archive of all collections, can be found online at http://ajpheart.physiology.org/.
The ability of polypeptide hormones to function both as chemical messengers and precursors for smaller peptides with higher specificity and biological activity endows multicellular organisms with the ability to regulate physiological and behavioral activities with a diverse molecular vocabulary. The expression of genes leading to the synthesis and secretion of hormones in tissues and organs by endocrinocytes (110) has expanded the conceptual understanding of hormone functions since these tissue-born agents act as a signaling mechanism for tissue homeostasis or the regulation of the cell's microcellular environment. The angiotensins, the effector hormones of the renin angiotensin system (RAS), constitutes a ubiquitous hormonal pathway modulating a variety of physiological functions, including sodium balance, body fluid volumes, cardiac output, and arterial blood pressure, and unfavorably regulates the myocardial and vascular responses to injury and inflammation. Although tissues other than the kidneys express the genes for the production of angiotensin peptides (103), it is now generally accepted that these locally produced angiotensins act upon surrounding cells that are receptive to them (paracrine signaling) or exert their actions on the originating cell itself (autocrine signaling) (112). An even newer concept is that angiotensin peptides act intracellularly (intracrine signaling) (21, 38) either following internalization of extracellular peptides through a receptor or a transport-mediated event or through the generation of the peptide within the intracellular compartment (33, 35, 37, 41, 78, 79, 113). A comprehensive discussion of this topic is available in a collection of review articles published in the American Journal of Physiology (30, 43, 57, 80). Intracellular detection of gene transcripts for pro(renin), renin, angiotensin converting enzyme (ACE), and angiotensin converting enzyme 2 (ACE2), as well as the activity of intracellularly generated Ang II and angiotensin-(1–7) [Ang-(1–7)], indicates the presence of a functional intracrine RAS (1, 21). Because efficient uptake of proteins and peptides occurs via cell membrane receptors, the processes involved in the expression and function of intracrine angiotensins becomes an important aspect of the mechanisms by which the intracellular RAS contributes to physiology and pathology (31).
Accumulating evidence showing that Ang II intracellular effects are not inhibited by AT1 receptor blockade (11, 34) indicates the independence of the intracrine RAS from the external environment. Intracellular Ang II synthesis is augmented in rat cardiomyocytes exposed to high glucose; this effect is not prevented by the presence of the Ang II receptor blocker candesartan (118). In hamster myocytes, losartan did not block the inotropic actions of intracellular Ang II when added to the perfusion media (34). Of importance, chronic administration of losartan, lisinopril, or both drugs combined had no effect on cardiac Ang II content even though the robust antihypertensive effects of these treatments produced the expected changes in circulating angiotensins (47). Likewise, the antihypertensive effects of chronic administration of olmesartan to mRen2.Lewis transgenic hypertensive rats did not change cardiac Ang II content even though the treatment was associated with significant increases in plasma renin concentration and plasma Ang I and Ang II content (148). Additional studies supporting the existence of an independent intracrine RAS system has been reviewed recently (1, 36). Thus there is strong experimental evidence to support the view that intracrine Ang II activity may function independent of the circulating system (45, 49).
This review addresses how the recent identification of shorter forms of angiotensinogen that are expressed in human and rodent organs reinforces the importance of intracellular RAS intracrine mechanisms and the importance of intracrine signaling. New research demonstrates that noncanonical mechanisms regulate the synthesis and processing of intracellular angiotensins and brings to light a need to reconsider the acknowledged usefulness of ACE inhibitors and Ang II receptor blockers (ARBs) as the most effective approach to halt or prevent the adverse cardiac and vascular remodeling associated with increased Ang II activity.
Currently recognized pathways for RAS expression.
The process of polypeptide biotransformation, first discussed by Burbach (20), was applied by our laboratory (46) to examine the biochemical processes associated with the formation and processing of biologically active angiotensin peptides. These guiding principles of polypeptide biotransformation, and the research that followed this methodological approach, led to the recognition that the RAS was a true example of a complex hormonal mechanism in which at least three identified angiotensinogen-derived peptides - angiotensin-(1–25) [Ang-(1–25)], angiotensin-(1–12) [Ang-(1–12)], and angiotensin I (Ang I) - act as intermediate precursors for the generation of the shorter amino acid sequences of the active hormones angiotensin-(1–9) [Ang-(1–9)], Ang II, and Ang-(1–7). These three biologically active peptides, in turn, become precursor molecules for the formation of two other active angiotensins, angiotensin III (Ang III) and angiotensin IV (Ang IV). An additional level of complexity, leading to potentially further biological selectivity, involves the decarboxylation of Ang II into des[Asp1]-[Ala1]-Ang II (Ang A) (69) and, not unexpectedly, the formation of des[Asp1]-[Ala1]-Ang-(1–7) (alamandine) from either Ang A or Ang-(1–7) (81). As ligands, angiotensin hormones bind to the G protein nuclear and transmembrane receptors AT1, AT2, mas (mas-R), and a component of the family of mas-related MRG receptors (Mrg-D) (10). Several reviews have documented our current understanding of the biochemical pathways, enzymes, biologically active peptides, and receptors encompassing the RAS (3, 45, 149).
Upstream from Ang I, the diversity of substrates and enzymes contributing to the generation of Ang II and Ang-(1–7) had been relatively ignored since most research remains focused on the catalytic actions of prorenin, renin, and their action in hydrolyzing angiotensinogen into Ang I. Over the years, a number of renin-like enzymes have been reported to release Ang I from either the angiotensinogen molecule or the model substrate tetradecapeptide [Ang-(1–14)]. Cathepsin D and G, acid and neutral proteases, tonin, the serine protease esterase B, a kallikrein-like serine protease, and mouse γ-nerve growth factor (γ-NGF) are among the enzymes identified as Ang I-forming enzymes (16, 17, 23, 42, 56, 58, 59, 73, 117, 157). Tonin, an enzyme discovered by Boucher et al. (17) and present at its highest concentration in the submaxillary gland, forms Ang II directly from the tetradecapeptide substrate [Ang-(1–14)] and from Ang I (117). A neutral protease with Ang I-forming activity that was readily separated from acid proteases and both plasma and renal renin was reported by Husain et al. (65, 66) in the canine brain. Mast cell-derived cathepsin G is an Ang II-forming enzyme implicated in the evolution of atherosclerosis and aortic aneurysms (15, 68, 111, 114, 134, 151). More recently, the characterization of the (pro)renin receptor (PRR) (100) has led to the demonstration that binding of (pro)renin to the PRR augments the catalytic activity of renin (100) as well as activating proinflammatory and profibrotic signaling pathways by both Ang II-dependent and independent mechanisms (44, 63, 105). Overexpression of the human PRR in Neuro-2A cells stimulated production of radical oxygen species via both Ang II and non-Ang II mediated mechanisms (105). In addition, the microinjection of human prorenin into the paraventricular nucleus of the rat's hypothalamus stimulates splanchnic sympathetic nerve activity, whereas the increases in inducible nitric oxide synthase and cybb gene transcripts mediated by exposure to human prorenin were not blocked by losartan (63). The potential role of PRR as a potential source for non-Ang II-dependent actions in the heart remains to be investigated since the PRR is highly expressed in the heart (115).
The extended forms of angiotensin I.
Since characterization in 1957 of the first 14 amino acids of horse angiotensinogen (119), this α2-globulin member of the serpin family remains the sole known angiotensins forming substrate (93). Although high (H) and low (L) molecular weight (MW) forms of angiotensinogen have been described, the function of the HMW form of angiotensinogen remains poorly understood. According to Tewksbury and Tewksbury and Tryon (126, 128), levels of the HMW form of angiotensinogen are less than 5% that of the LMW form. When compared with the LMW form, both the Km and Vmax of the HMW form are lower at a physiological pH, and its blood concentration doubles as a result of estrogen treatment or pregnancy (127, 129). The major known function of angiotensinogen to date is to act as a substrate for the liberation of the decapeptide angiotensin I [Ang-(1–10)] through a renin-mediated cleavage of the Leu10-Val11 amino acid bond in humans and the Leu10-Leu11 bond in nonprimate species such as the rat (93). The substitution of histidine for tyrosine at residue 13 of the human angiotensinogen protein contributes to the species-specificity of the human renin angiotensinogen reaction (94). In recent years, the identification of extended molecular sequences of Ang I as potential intermediate sources of Ang II production, through nonrenin-dependent mechanisms, has revealed new paradigms regarding the biochemical mechanisms that account for the expression and action of angiotensins (3, 149).
In their searching for additional bioactive angiotensin family peptides, Nagata and collaborators (98) at Miyazaki University in Japan reported in 2006 the isolation of an extended form of Ang I composed of the first 12 amino acids of angiotensinogen from the rat small intestine. The peptide, termed proangiotensin-12, was present in multiple tissues with its highest concentrations in the rat intestine (Fig. 1). Interest in this peptide as a functional Ang II-forming substrate was strengthened by the demonstration that proangiotensin 12 constricted aortic strips and, when infused intravenously, raised blood pressure in Wistar rats (98). The vasoconstrictor responses to proangiotensin 12 could be abolished by either captopril or the Ang II type I receptor blocker candesartan (98). To maintain consistency with the accepted nomenclature, proangiotensin 12 was renamed Ang-(1–12) (70).
Fig. 1.

Tissue content of angiotensin-(1–12) in Wistar rats as reported by Nagata et al. (98). Values are means ± SE.
The discovery of Ang-(1–12) and the studies conducted on it to date provide an alternate explanation as to how tissues, independently from the circulation, may avail from shorter angiotensinogen-derived peptides to give rise to Ang II and Ang-(1–7) within the immediate surrounding extracellular microenvironment or the cell itself. This idea gained support by the demonstration that both immunoreactive Ang-(1–12) expression and the left ventricular content of the dodecapeptide were markedly increased in adult spontaneously hypertensive rats (SHRs) than in normotensive Wistar Kyoto (WKY) rat controls (70). The Ang-(1–12) immunoreactive products were primarily localized within the cytoplasm of myocytes, although some positive staining was observed in the medial layer and surrounding adventitia of intracoronary arteries (70). As related by Jessup et al. (70), clusters of Ang-(1–12) immunoreactivity were primarily visualized in the endocardium of the right and left ventricles of WKY rats. This patchy Ang-(1–12) expression in the normotensive rat contrasted sharply with the more diffuse distribution of Ang-(1–12) immunoreactive staining throughout the right and left ventricular myocardium of SHRs (70). In keeping with these histological observations, direct Ang-(1–12) assays revealed higher left ventricular Ang-(1–12) tissue concentrations in the SHR than in the control WKY strain (70). The higher Ang-(1–12) content in SHRs was associated with higher tissue concentrations of Ang I and Ang II (Fig. 2). The authors inferred that Ang-(1–12) may be a cardiac precursor for angiotensin peptide formation based on the minimal presence of angiotensinogen immunoreactivity staining in the hearts of both WKY rats and SHRs (70). Although these findings should not be interpreted as excluding intracellular angiotensinogen as a contributing source of Ang-(1–12), the available literature suggest that a significant fraction of the angiotensinogen found in the heart is derived from the circulation (32, 85, 103). These data suggest that Ang-(1–12) may be a critical precursor for the formation of cardiac angiotensins, although the intracellular origin of the propeptide needs to be investigated further.
Fig. 2.
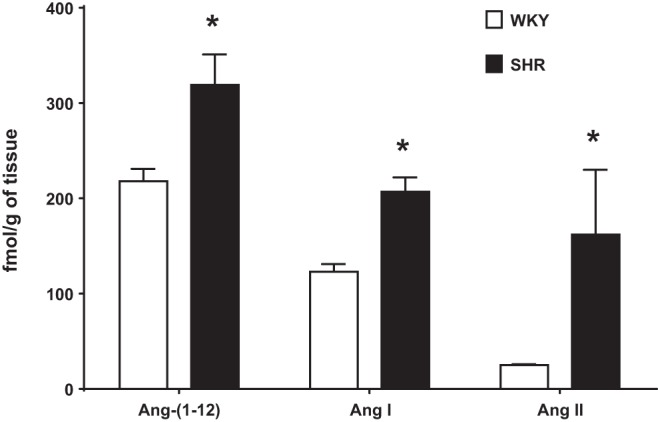
Left ventricular tissue content of angiotensin-(1–12) [Ang-(1–12)], angiotensin I (Ang I), and angiotensin II (Ang II) is higher in adult male spontaneously hypertensive rats (SHRs) than in Wistar-Kyoto (WKY) control rats. Data are reported by our laboratory and published elsewhere (70). *P < 0.05.
The characterization of Ang-(1–12) as an important source for production of angiotensins has been derived from a series of studies in which the peptide was administered both systemically and to isolated organs. Extending the initial observations of Nagata et al. (98), Ang-(1–12) elicited coronary artery vasoconstriction and impaired recovery from global ischemia as reflected by a marked increase in creatine kinase and troponin I release from the isolated perfused heart of Sprague Dawley (SD) rats subjected to ischemia reperfusion injury (108). An additional report showed that vasoconstrictor responses to Ang-(1–12) administration could be elicited in both muscular and conduit arteries of SD rats, although no responses were observed in femoral and renal arteries (109). The mechanism that accounts for the progressive reduction of vasoconstrictor responses of the isolated vessels further away from the heart remains to be fully investigated (109). Additional studies confirming the activity of Ang-(1–12) as an Ang II-forming substrate included the characterization of a modulatory action of the locally applied substrate on the central regulation of baroreflexes (9, 29) and the stimulation of neuronal circuits in both the rostral (7) and ventrolateral medulla (71). The demonstration of an antihypertensive effect of Ang-(1–12) immunoneutralization in the brain of transgenic hypertensive rats expressing the Ren-2 gene provided the first functional evidence for a role of Ang-(1–12) as a source of Ang II actions (67). In these studies, blockade of the biological response through the administration of ARBs confirmed that that the substrate was converted to Ang II before having an effect (6, 7, 28, 29, 71).
Ang-(1–12) activity as an endogenous Ang II source was investigated in several studies, including an assessment of the chronic effects of continuous infusion of the dodecapeptide in normotensive Wistar rats (77), an investigation of changes in cardiac and systemic RAS components in WKY rats 48 h post-bilateral nephrectomy (48), and an examination of the effects of stimulation of endogenous renin activity by salt depletion (97) or administration of RAS inhibitors (96). Overall, these studies confirmed that Ang-(1–12) functions as an endogenous Ang II-forming substrate and showed that the substrate is regulated independently from the circulating RAS. This conclusion is based on the increased cardiac Ang-(1–12) concentrations in anephric rats with depleted circulating renin and Ang II (48); the absence of changes in Ang-(1–12) concentrations in kidneys, small intestine, cardiac ventricles, and brain of Wistar rats fed a low-salt diet (97); and the decrease in tissue but not blood Ang-(1–12) of WKY rats and SHRs exposed to 1 wk of treatment with either the ARB losartan or the ACE inhibitor imidapril (96). The fall in cardiac and renal Ang-(1–12) content induced by a 7-day treatment with RAS inhibitors was interpreted by the authors to reflect a fall in cardiac and renal angiotensinogen gene expression (96). Ang-(1–12) functional responses appear to be primarily mediated through its conversion to Ang II (24). In this elegantly conducted study, Chan et al. (24) showed that Ang-(1–12) augmented intracellular Ca++ mobilization and phosphorylated ERK in COS-7 and Chinese Hamster Ovary cells transiently transfected with AT1 receptor cDNA at concentrations 40- to 100-fold higher than Ang II. The authors attributed the need of higher Ang-(1–12) doses to the absence of Ang-(1–12)-processing enzymes in the cells transfected with the AT1 receptor. Further studies showed that losartan but not the AT2 receptor antagonist PD123319 suppressed the Ang-(1–12) effects (24). The demonstration that Ang-(1–12)-mediated Ca++ mobilization failed to occur in Chinese Hamster Ovary cells expressing the AT2 receptor might be important in terms of differentiating responses by the two peptides (24).
It remained to be established whether tissue Ang-(1–12) is overexpressed in diseased states associated with activation of the RAS. The initial observation of augmented Ang-(1–12) expression and concentration in the heart of SHRs (70) was extended through the demonstration of increased immunohistochemical Ang-(1–12) and Ang II expression in the kidney of [mRen2]27 transgenic hypertensive rats that were suppressed by a 3-wk administration of a low-dose of a mineralocorticoid receptor (MR) antagonist (155). Because MR blockade augments Ang-(1–7) generation through ACE2 facilitation of Ang II metabolism (72), the findings in [mRen2]27 transgenic hypertensive rats implicate different pathways for Ang-(1–12) processing in tissues. Consistent with this interpretation, Westwood and Chappell (154) showed that the primary route for Ang-(1–12) metabolism in renal cortical membranes of congenic mRen2.Lewis hypertensive rats involves the formation of Ang-(1–7) and Ang-(1–4) by neprilysin.
A recently completed study affirms the importance of this alternate pathway for Ang II formation in tissues. Past studies had reported that transgenic expression of the human angiotensinogen gene in SD rats [TGR(hAGT)L1623] did not result in hypertension or cardiovascular pathology because rat renin does not cleave human angiotensinogen (54). However, a more recent study demonstrated that these transgenic rats are hypertensive and display structural and functional indexes of cardiac hypertrophy and systolic dysfunction (49). Robust human Ang-(1–12) immunofluorescence within myocytes of TGR(hAGT)L1623 rats was associated with a fourfold increase in left ventricular cardiac Ang II content (49). Because rat renin has a very low affinity for human angiotensinogen, these new studies underscored the functional importance of alternate enzymatic processes that can lead to Ang II pathology through a nonrenin-dependent pathway.
In addressing the significance of Ang-(1–12) as an endogenous substrate for human tissue angiotensins, our laboratory documented the expression of the peptide in human left ventricles (5), the left atrial appendage of patients with resistant atrial fibrillation (2), and the right and left atrial tissue of subjects with left-heart disease (99). The localization of Ang-(1–12) immunoreactive products in atrial myocytes of subjects with either resistant atrial fibrillation (2) or diseases of the mitral and aortic valves, or ischemic heart disease (99), suggests that increased myocyte stretch secondary to atrial enlargement is a stimulus for the expression of the intracellular substrate. In keeping with this interpretation, Ang-(1–12) expression was significantly higher in the enlarged left atrial appendages of these subjects (99). Of importance, the increased left atrial appendage expression of Ang-(1–12) immunoreactivity was associated with increased chymase mRNA and enzymatic activity (99).
Ang-(1–12) and chymase.
Ang-(1–12) metabolism follows noncanonical pathways that seem to be independent of renin and involves enzymatic pathways that are both tissue and species specific. Trask et al. (131) investigated Ang-(1–12) processing in the isolated perfused heart of three control rat strains (SD, Lewis, and WKY) and two other hypertensive rat strains (congenic mRen2.Lewis and SHRs). Over a 60-min perfusion period of rat Ang-(1–12), a rapid appearance of both Ang I and Ang II demonstrated that the heart has the enzymes to process the substrate to the smaller peptides. A delayed appearance of Ang-(1–7) in the Ang-(1–12) perfused hearts suggested that it was generated from the accumulated Ang II. All rat strains showed a similar pattern of Ang-(1–12) processing into Ang I and Ang II, whereas addition of a rat renin-specific inhibitor [Ac-His-Pro-Phe-Val-Sta-Leu-Phe-NH2 (WFML-1)] (47) had no effect on the rate and magnitude of Ang-(1–12) metabolism in WKY rats and SHRs (131). To further evaluate a potential role of renin in hydrolyzing Ang-(1–12), a second study investigated the effects of excluding endogenous renin on the circulating and cardiac tissue concentrations of angiotensins (48). In bilaterally nephrectomized WKY rats, a marked fall in circulating Ang I and Ang II levels was associated with a significant increase in the cardiac content of Ang-(1–12), Ang I, and Ang II (48). The accumulation of angiotensins in the heart of anephric WKY rats was confirmed by the visualization of increased immunopositive fluorescence intensities for angiotensinogen, Ang II, and AT1 receptors and no differences in cardiac Ang-(1–12) expression between sham-operated and anephric WKY rats (48).
Although renin is not necessary for Ang-(1–12) processing, studies by our laboratory (2, 3, 5, 92) and others (19, 96, 97, 107–109, 116) demonstrated that Ang-(1–12) hydrolysis to Ang I and Ang II is achieved primarily through the proteolytic activities of ACE and chymase. Additional studies implicate the existence of alternate Ang-(1–12) processing in renal and vascular tissues, whereby neprilysin yields direct formation of Ang-(1–7) in rat renal cortical membranes (154) and carboxypeptidases A2 (cathepsin A) cleaves Ang-(1–12) into Ang I in rat mesenteric arteries (106) (Fig. 3). Although we have long advocated that tissue angiotensin peptide formation is markedly influenced by the composition of the enzymatic milieu and the proximity of the substrate to the enzymes, the lessons we have learned from the study of Ang-(1–12) metabolism suggest that processing of this intermediate substrate into the biologically active peptides is mediated primarily by ACE in the rat circulation (92, 98, 154), neprilysin in rat renal cortical membranes (154), and chymase in the human heart (45) (Fig. 3). Moniwa et al. (92) reported a direct role of ACE in forming Ang I and Ang II in the circulation of WKY rats and SHRs, a finding that agrees with the earlier studies of Nagata et al. (98) who showed that the vasoconstrictor response to Ang-(1–12) administration was prevented by captopril, whereas Prosser et al. (109) reported that captopril attenuated but did not abolish vascular constrictor responses in preconditioned isolated blood vessels. In cultured WKY neonatal cardiomyocytes, ACE and neprilysin cleaved Ang-(1–12) into smaller angiotensin fragments, whereas chymase showed hydrolytic activity in cardiac myocytes from SHRs (4). Interestingly, these studies also demonstrated an increased Ang-(1–12) uptake in cardiac myocytes from SHRs, paralleling the previous demonstration of an increased expression and content of Ang-(1–12) in the left ventricular tissue of SHRs (70). The demonstration of chymase activity as an Ang-(1–12) converting enzyme in neonatal myocytes from SHRs but not WKY rats suggests that this alternate pathway for Ang II generation may be linked to the mechanisms that lead to cardiac pathology in this genetic model of experimental hypertension (4). This hypothesis is supported by the additional finding of increased cardiac Ang II content in transgenic rats expressing the human angiotensinogen gene (49) and oophorectomized mRen2.Lewis transgenic hypertensive rats (150).
Fig. 3.

Rat and human processing of angiotensin-(1–12) as documented in the current literature. See article and references (2, 4, 5, 19, 26, 98, 106, 108, 141, 142, 154) for detailed information.
This noncanonical Ang-(1–12) metabolism pathway is different in rodents and human cardiac tissue (Fig. 3). Experiments investigating Ang-(1–12) metabolism in plasma membranes obtained from the left atria of patients undergoing cardiac surgery for the treatment of resistant atrial fibrillation (2) or from the left ventricle of normal subjects dying from vehicular motor accidents (5) found that chymase was almost entirely accountable for the biotransformation of this alternative substrate directly into Ang II. In these studies ACE and neprilysin displayed negligible hydrolytic activity (2). In plasma membranes isolated from the ventricles of SHRs, Ang-(1–12) rather than Ang I showed a higher hydrolytic processing activity by chymase in the generation of Ang II (Km, 57.1 μM and Vmax, 12.14 μM·mg−1·min−1 vs. Km, 88.66 μM and Vmax, 4.07 μM·mg−1·min−1, respectively), whereas the opposite is apparently true in human cardiac tissue (2, 4). Two factors may account for the differential affinities of chymase in Ang-(1–12) processing between rodents and humans. Differences in the amino acid sequence of Ang-(1–12) at the COOH terminus between rats and humans (Leu10-Leu11-Tyr12 in the rat versus Leu10-Val11-Ile12 in humans) may influence the enzyme active site at which chymase processes the peptide (2, 4, 45). In addition, multiple β-chymase isoforms are present in the rat, whereas humans express only α-chymase (25, 39). These species differences are of considerable importance since the predominant β-chymase species found in rats cleaves the Tyr4-Ile5 bond of Ang I rather than the Phe8-His9 of the decapeptide (122).
The same group of investigators from Miyazaki University in Japan (95) isolated another Ang II-forming peptide from the urine of healthy volunteers. This alternative Ang II-forming substrate, named Big angiotensin-25 [heretofore abbreviated as Ang-(1–25)], consists of the first 25 amino acids of angiotensinogen and is N-glycosylated on Asn14 with a cysteine linked to Cys18 (95). In their studies, human recombinant renin rapidly degraded angiotensinogen into Ang I, whereas production of Ang I from Ang-(1–25) was much slower. Nagata et al. (95) reported that the Km for Ang-(1–25) hydrolysis by renin averaged 95 μM compared with 1.2 μM for angiotensinogen. On the other hand, although chymase has no catalytic activity on human angiotensinogen (143), mast cell chymase digested Ang-(1–25) more efficiently than human recombinant renin, with Km and Vmax values averaging 9.6 μM and 129 pmol·min−1·ml−1, respectively (95). An antibody directed to the COOH terminus of human Ang-(1–25) stained a wide range of tissues with robust expression in kidney, heart, pancreas, adrenal medulla, testicular Sertoli cells, fallopian tubes, and placenta extravillous trophoblasts (95). The strong expression of Ang-(1–25) immunoreactivity in human pancreas, adrenal medulla, and placenta led the Japanese investigators to suggest that Ang-(1–25) may function as a direct source of intracellular Ang II activity. Whether Ang-(1–25) acts as a substrate for direct Ang II production or requires prior conversion into Ang-(1–12) remains to be investigated. Nevertheless, identification of Ang-(1–25) and Ang-(1–12) as extended Ang I-forms and precursors has revealed the possibility of new mechanisms that describe how organs such as the heart activate intracellular actions of angiotensins.
The predominant role of chymase in cardiac tissue Ang-(1–12) processing, particularly in human cardiac tissue, is consistent with a robust literature documenting a singular role for chymase in the production of Ang II from Ang I in humans (156). In their examination of the finding that serine proteinase inhibitors were more potent than captopril in preventing Ang II formation from Ang I in human left ventricular tissue (142), Urata and coworkers (143) purified and characterized cardiac chymase as the major enzyme in the production of Ang II in the human heart. Human chymase was found to display heterogeneous and widespread tissue distribution throughout the human body with the highest expression in alimentary tract tissue, uterus, and tonsil (144). According to Urata et al. (144), the highest levels of chymase-like enzymatic activity were found in the skin, gastrointestinal track, and uterus, whereas human ventricular tissue, the lung, and renal cortex express lower levels of chymase activity. Cardiac sources of chymase in the human heart were reported in cytosolic granules of mast and endothelial cells, as well as mesenchymal interstitial cells (140). Using specific antibodies, we showed high chymase expression in atrial myocytes of patients with diverse cardiac pathology (2, 99). In addition, expression of Ang-(1–12), chymase mRNA, and enzymatic activity were significantly higher in the left than in the right atrial appendage of human diseased hearts (99).
Other studies have documented the role of chymase as the key enzyme forming Ang II from Ang I in humans (13, 39, 40, 64, 84, 130, 136–138). Excellent reviews by Takai (121, 122) and Dell′Italia and Husain (39) provide a comprehensive analysis of chymase contribution to cardiovascular disease. A schematic diagram of chymase contribution to cardiovascular function is summarized in Fig. 4. For the specific objectives of this review article, we would point out that: (1)- chymase shows no enzymatic activity in the circulation partly because of the presence of internal serine protease inhibitors (53) and strict intracellular localization; (2)- chymase inhibition has no direct antihypertensive or vasodilator effects nor does it increase plasma renin activity (122); (3)- although secretory granules of mast cells are the recognized principal source of chymase, our recent studies identified rat cardiac fibroblasts as enriched with chymase (51, 52); and (4)- chymase is a potent intermediate of collagen deposition as it activates the precursor of TGF-β to its active form (120, 158) and promotes an increase in matrix metallopeptidase (MMP-9) activity (27, 101, 125). Fu et al. (51, 52) reported recently that chymase of fibroblast origin induced autophagic degradation of newly synthesized intracellular procollagen I in an experimental model of cardiac volume overload. Mechanisms associated with chymase-mediated cardiac Ang II production may be influenced by multiple factors other than stretch since chronic estrogen treatment attenuates the increases in cardiac Ang II, Ang-(1–7), chymase mRNA, and mast cell number induced in oophorectomized mRen2.Lewis hypertensive rats (150, 159). In addition, Li et al. (83) reported recently that chymase and tryptase induction of cardiac fibrosis may require the protease-activated receptor-2 (PAR-2).
Fig. 4.

Schematic diagram depicting multiple chymase actions including direct breakdown of laminin and fibronectin important in cell survival (82), activation of matrix metalloproteinases (MMPs) (139), TGF-β (152), stem cell factor (SCF) (86), kallikrein and chemotaxis (50), IL-6 and IL-1β (91), pre-proendothelin I (pre-proET1) (74, 124), and activation of other growth factors and enzymes in tissue injury. BK, bradykinin.
CLINICAL PERSPECTIVE
Further investigation of the noncanonical pathways through which tissue Ang II may lead to cardiac and vascular pathology is warranted given the possibility that enzymes and precursor proteins currently been characterized as existing between the primary angiotensinogen substrate and Ang I may explain why ACE inhibitors or even ARBs have limited efficacy in reversing or halting the progression of cardiovascular disease (12, 18, 132, 133). Arguments in favor of this conclusion have been advanced by us (45, 49) and others (60, 90, 156) previously. These issues were recently underscored in a report summarizing the current limitations for heart failure therapies as elaborated upon a broad representation of health care providers that included clinical and basic science investigators, regulators, and pharmaceutical industry representatives attending a meeting facilitated by the Food and Drug Administration (55). Although it may be argued that the limited effectiveness of RAS blockers may reflect the limited contribution of this system to cardiovascular disease progression, this interpretation does not agree with the overwhelming knowledge that continues to accumulate regarding the cellular and molecular mechanisms by which Ang II contributes to adverse cardiac and vascular remodeling (55). Rigorous analysis of the reduction in important clinical cardiovascular end points in hypertensive patients (18, 132, 133), ischemic heart disease (12), and heart failure (60) are in keeping with this interpretation. In a recently reported meta-analysis of the impact of ACE and ARBs in cardiovascular mortality, Brugts et al. (18) stated that “The annual incidence rate of all-cause mortality was 0.0233 in patients randomized to RAAS inhibitors versus 0.0252 in controls (hazard ratio, 0.95; 95% confidence interval, 0.91 to 0.99).” Furthermore, although ACE inhibitors reduced the combined end point of all-cause and cardiovascular mortality and myocardial infarction, the number of patients that needed to be treated (NNT) for 4.3 years to avoid one event averaged 116 for patients medicated with an ACE inhibitor and 409 for those treated with an ARB (18). A separate analysis of all-cause mortality demonstrated a minor improvement in patients medicated with ACE inhibitors [hazard ratio (HR), 0.90; 95% CI, 0.84 to 0.97] and no differences in those treated with ARBs (HR, 0.99; 95% CI, 0.94 to 1.04) (18). Another comprehensive analysis of the comparative effectiveness of ACE inhibitors and ARBs in ischemic heart disease found a relative risk (RR) reduction of 0.87 (95% CI, 0.81 to 0.94) for total mortality and a RR reduction of 0.83 (CI, 0.73 to 0.94) for nonfatal myocardial infarction with ACE inhibitors (12). Although these data show a benefit from the use of ACE inhibitors in the treatment of heart disease, the residual risk of events (1.0 minus RR) documented in these studies is close to or greater than 70%. Additional studies from well-conducted clinical trials shows that the “residual risk” for cardiovascular events is substantially greater than the risk reduction achieved by these agents (1, 12, 18, 36, 60, 75, 132, 133, 145–147).
We suggest that this discrepancy may be in part accounted for by 1) the existence of tissue-specific noncanonical mechanisms in which ACE is not the obligatory processing pathway for Ang II formation (137); and 2) the almost exclusive role of chymase, rather than ACE, as the Ang II forming enzyme in the human heart (156) and blood vessels (8). It is also possible that ACE inhibition may upregulate the alternate expression of other Ang II-forming enzymes as observed by cardiac chymase upregulation during ACE inhibition in mice (153) or increased vascular rat elastase-2 expression in enalapril-treated rats (14). As noted by Takai et al. (121, 123), a renewed effort for the discovery of better tolerated, orally active chymase inhibitors should be undertaken to effectively and specifically prevent the pathological actions of Ang II in humans.
As documented in this review, chymase is the major Ang II-forming mechanism in humans. Mast cells contain a plethora of mediators, of which the most important in tissue injury are mast cell proteases including tryptase, chymase, and dipeptidyl peptidase I (DPP I), in addition to TNF-α, and stem cell factor (SCF), which are responsible for recruitment of macrophages and neutrophils (22, 104). Targeting mast cells with mast cell stabilizers resulted in left ventricular and cardiomyocyte dysfunction in dogs with mitral regurgitation due to inherent calcium-entry blocking effects of mast cell stabilizers (102). As indicated by chymase's multiple actions (Fig. 4), cardiac chymase has been identified to contribute to left ventricular remodeling post-myocardial infarction (61, 153), atherosclerosis (8, 135), type 2 diabetes (62, 76), heart failure (87), and cardiac ischemia reperfusion injury (160).
As demonstrated in Fig. 4, blockade of chymase has implications beyond the effects on Ang II formation because of the many other direct protease actions of chymase that have major effects on tissue remodeling, especially in the breakdown of matricellular connections. Our recent studies showed that dual inhibition of ACE and chymase was significantly more effective in decreasing infarct size and left ventricular dilatation, as well as improving cardiac function, compared with ACE inhibitor or chymase inhibitor alone in hamsters 1 mo after coronary occlusion (153). These results underscore the fact that chymase inhibitors can target other enzymes in the pathophysiology of left ventricular remodeling and thus may synergize with ACE inhibition or AT1 receptor blockade in the treatment of heart failure.
Finally, our new finding of chymase within the cardiomyocyte during volume overload in the rat opens up an entirely new potential mechanism for Ang II formation and protease-mediated destructive actions within the cell that are untouched by ACE inhibitors or AT1 receptor blockers that act on the cell surface (102). The presence of the chymase-Ang-(1–12) pathway is further exemplified by the failure to achieve additional benefit when more complete blockade of the canonical pathway was obtained with the renin inhibitor aliskerin in patients with heart failure (88). The immediacy of translation of this work is underscored by an ongoing clinical trial of a new type of chymase inhibitor. BAY1142524 (Bayer Health Care) is in phase III clinic trial (NCT02452515; www.clinicaltrials.gov) to treat patients with left ventricular dysfunction after myocardial infarction. In view of the relative plateau in efficacy of neurohormonal blockade in cardiovascular disease including the improvement obtained with the combination of valsartan and the neprilysin inhibitor sacubitril (88, 89), we look forward to future applications for the addition of chymase inhibition as an adjunct to existing pharmacotherapy in cardiovascular disease.
Although further research is clearly needed to assess the importance of a nonrenin-dependent Ang II-forming pathway from Ang-(1–12) in human cardiovascular disease, the data reported thus far demonstrate the existence of this functional alternate substrate in cardiovascular tissues, its increased expression and content in the heart of hypertensive strains and the left atrial appendage of heart disease patients, and a singular role for chymase in forming Ang II directly from Ang-(1–12), particularly in human subjects.
GRANTS
The studies described here were funded, in part, by Program Project Grant 2P01 HL-051952 from the National Heart, Lung, and Blood Institute of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.M.F. and L.J.D. conception and design of research; C.M.F., S.A., J.V., C.P.C., L.G., H.W., J.F.C., and L.J.D. performed experiments; C.M.F., S.A., J.V., C.P.C., L.G., H.W., J.F.C., and L.J.D. analyzed data; C.M.F., S.A., J.V., C.P.C., L.G., H.W., and L.J.D. interpreted results of experiments; C.M.F. and L.J.D. prepared figures; C.M.F. and L.G. drafted manuscript; C.M.F., S.A., J.V., C.P.C., L.G., H.W., J.F.C., and L.J.D. edited and revised manuscript; C.M.F., S.A., J.V., C.P.C., L.G., H.W., J.F.C., and L.J.D. approved final version of manuscript.
REFERENCES
- 1.Abadir PM, Walston JD, Carey RM. Subcellular characteristics of functional intracellular renin-angiotensin systems. Peptides 38: 437–445, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1–12) in human atrial tissue. PLoS One 6: e28501, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad S, Varagic J, Groban L, Dell′Italia LJ, Nagata S, Kon ND, Ferrario CM. Angiotensin-(1–12): a chymase-mediated cellular angiotensin II substrate. Curr Hypertens Rep 16: 429, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad S, Varagic J, Westwood BM, Chappell MC, Ferrario CM. Uptake and metabolism of the novel peptide angiotensin-(1–12) by neonatal cardiac myocytes. PLoS One 6: e15759, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad S, Wei CC, Tallaj J, Dell′Italia LJ, Moniwa N, Varagic J, Ferrario CM. Chymase mediates angiotensin-(1–12) metabolism in normal human hearts. J Am Soc Hypertens 7: 128–136, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arakawa H, Chitravanshi VC, Sapru HN. The hypothalamic arcuate nucleus: a new site of cardiovascular action of angiotensin-(1–12) and angiotensin II. Am J Physiol Heart Circ Physiol 300: H951–H960, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arakawa H, Kawabe K, Sapru HN. Angiotensin-(1–12) in the rostral ventrolateral medullary pressor area of the rat elicits sympathoexcitatory responses. Exp Physiol 98: 94–108, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arakawa K, Urata H. Hypothesis regarding the pathophysiological role of alternative pathways of angiotensin II formation in atherosclerosis. Hypertension 36: 638–641, 2000. [DOI] [PubMed] [Google Scholar]
- 9.Arnold AC, Isa K, Shaltout HA, Nautiyal M, Ferrario CM, Chappell MC, Diz DI. Angiotensin-(1–12) requires angiotensin converting enzyme and AT1 receptors for cardiovascular actions within the solitary tract nucleus. Am J Physiol Heart Circ Physiol 299: H763–H771, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bader M, Alenina N, Andrade-Navarro MA, Santos RA. MAS and its related G protein-coupled receptors, Mrgprs. Pharmacol Rev 66: 1080–1105, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Baker KM, Kumar R. Intracellular angiotensin II induces cell proliferation independent of AT1 receptor. Am J Physiol Cell Physiol 291: C995–C1001, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Baker WL, Coleman CI, Kluger J, Reinhart KM, Talati R, Quercia R, Phung OJ, White CM. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors or angiotensin II-receptor blockers for ischemic heart disease. Ann Intern Med 151: 861–871, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Balcells E, Meng QC, Johnson WH Jr, Oparil S, Dell′Italia LJ. Angiotensin II formation from ACE and chymase in human and animal hearts: methods and species considerations. Am J Physiol Heart Circ Physiol 273: H1769–H1774, 1997. [DOI] [PubMed] [Google Scholar]
- 14.Becari C, Teixeira FR, Oliveira EB, Salgado MC. Angiotensin-converting enzyme inhibition augments the expression of rat elastase-2, an angiotensin II-forming enzyme. Am J Physiol Heart Circ Physiol 301: H565–H570, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Belova LA. Angiotensin II-generating enzymes. Biochemistry (Mosc) 65: 1337–1345, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Berg T, Wassdal I, Sletten K. Immunohistochemical localization of rat submandibular gland esterase B (homologous to the RSKG-7 kallikrein gene) in relation to other serine proteases of the kallikrein family. J Histochem Cytochem 40: 83–92, 1992. [DOI] [PubMed] [Google Scholar]
- 17.Boucher R, Demassieux S, Garcia R, Gutkowska Y, Genest J. [The tonin-angiotensin II system]. Union Med Can 106: 502–507, 1977. [PubMed] [Google Scholar]
- 18.Brugts JJ, van VL, Akkerhuis M, Bertrand M, Fox K, Mourad JJ, Boersma E. Impact of renin-angiotensin system inhibitors on mortality and major cardiovascular endpoints in hypertension: a number-needed-to-treat analysis. Int J Cardiol 181: 425–429, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Bujak-Gizycka B, Olszanecki R, Suski M, Madek J, Stachowicz A, Korbut R. Angiotensinogen metabolism in rat aorta: robust formation of proangiotensin-12. J Physiol Pharmacol 61: 679–682, 2010. [PubMed] [Google Scholar]
- 20.Burbach JP. Action of proteolytic enzymes on lipotropins and endorphins: biosynthesis, biotransformation and fate. Pharmacol Ther 24: 321–354, 1984. [DOI] [PubMed] [Google Scholar]
- 21.Carey RM. Functional intracellular renin-angiotensin systems: potential for pathophysiology of disease. Am J Physiol Regul Integr Comp Physiol 302: R479–R481, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev 217: 141–154, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cemassieux S, Boucher R, Crise C, Genest J. Purification and characterization of tonin. Can J Biochem 54: 788–795, 1976. [DOI] [PubMed] [Google Scholar]
- 24.Chan KH, Chen YH, Zhang Y, Wong YH, Dun NJ. Angiotensin-[1–12] interacts with angiotensin type I receptors. Neuropharmacology 81: 267–273, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chandrasekharan UM, Sanker S, Glynias MJ, Karnik SS, Husain A. Angiotensin II-forming activity in a reconstructed ancestral chymase. Science 271: 502–505, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Chappell MC. Biochemical evaluation of the renin-angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol 310: H137–H152, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen LY, Li P, He Q, Jiang LQ, Cui CJ, Xu L, Liu LS. Transgenic study of the function of chymase in heart remodeling. J Hypertens 20: 2047–2055, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Chitravanshi VC, Proddutur A, Sapru HN. Cardiovascular actions of angiotensin-(1–12) in the hypothalamic paraventricular nucleus of the rat are mediated via angiotensin II. Exp Physiol 97: 1001–1017, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chitravanshi VC, Sapru HN. Cardiovascular responses elicited by a new endogenous angiotensin in the nucleus tractus solitarius of the rat. Am J Physiol Heart Circ Physiol 300: H230–H240, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol 302: R482–R493, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook JL, Zhang Z, Re RN. In vitro evidence for an intracellular site of angiotensin action. Circ Res 89: 1138–1146, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Danser AH, van Kats JP, Admiraal PJ, Derkx FH, Lamers JM, Verdouw PD, Saxena PR, Schalekamp MA. Cardiac renin and angiotensins. Uptake from plasma versus in situ synthesis. Hypertension 24: 37–48, 1994. [DOI] [PubMed] [Google Scholar]
- 33.De Mello WC. Influence of intracellular renin on heart cell communication. Hypertension 25: 1172–1177, 1995. [DOI] [PubMed] [Google Scholar]
- 34.De Mello WC. Intracellular angiotensin II regulates the inward calcium current in cardiac myocytes. Hypertension 32: 976–982, 1998. [DOI] [PubMed] [Google Scholar]
- 35.De Mello WC. Angiotensin (1–7) re-establishes impulse conduction in cardiac muscle during ischaemia-reperfusion. The role of the sodium pump. J Renin Angiotensin Aldosterone Syst 5: 203–208, 2004. [DOI] [PubMed] [Google Scholar]
- 36.De Mello WC. Intracellular angiotensin II as a regulator of muscle tone in vascular resistance vessels. Pathophysiological implications. Peptides 78: 87–90, 2016. [DOI] [PubMed] [Google Scholar]
- 37.De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension 35: 1183–1188, 2000. [DOI] [PubMed] [Google Scholar]
- 38.De Mello WC, Frohlich ED. On the local cardiac renin angiotensin system. Basic and clinical implications. Peptides 32: 1774–1779, 2011. [DOI] [PubMed] [Google Scholar]
- 39.Dell′Italia LJ, Husain A. Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol 17: 374–379, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Dell′Italia LJ, Meng QC, Balcells E, Wei CC, Palmer R, Hageman GR, Durand J, Hankes GH, Oparil S. Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. J Clin Invest 100: 253–258, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dostal DE. The cardiac renin-angiotensin system: novel signaling mechanisms related to cardiac growth and function. Regul Pept 91: 1–11, 2000. [DOI] [PubMed] [Google Scholar]
- 42.Dzau VJ, Brenner A, Emmett N, Haber E. Identification of renin and renin-like enzymes in rat brain by a renin-specific antibody. Clin Sci (Lond) 59, Suppl 6: 45s–47s, 1980. [DOI] [PubMed] [Google Scholar]
- 43.Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol 302: R494–R509, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng Y. ANG II-independent prorenin/(pro)renin receptor signaling pathways in the central nervous system. Am J Physiol Heart Circ Physiol 309: H731–H733, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, Dell′Italia LJ. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond) 126: 461–469, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrario CM, Brosnihan KB, Diz DI, Jaiswal N, Khosla MC, Milsted A, Tallant EA. Angiotensin-(1–7): a new hormone of the angiotensin system. Hypertension 18: III126–III133, 1991. [DOI] [PubMed] [Google Scholar]
- 47.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111: 2605–2610, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Ferrario CM, Varagic J, Habibi J, Nagata S, Kato J, Chappell MC, Trask AJ, Kitamura K, Whaley-Connell A, Sowers JR. Differential regulation of angiotensin-(1–12) in plasma and cardiac tissue in response to bilateral nephrectomy. Am J Physiol Heart Circ Physiol 296: H1184–H1192, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrario CM, VonCannon J, Jiao Y, Ahmad S, Bader M, Dell′Italia LJ, Groban L, Varagic J. Cardiac angiotensin-(1–12) expression and systemic hypertension in rats expressing the human angiotensinogen gene. Am J Physiol Heart Circ Physiol 310: H995–H1002, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forteza R, Lauredo I, Abraham WM, Conner GE. Bronchial tissue kallikrein activity is regulated by hyaluronic acid binding. Am J Respir Cell Mol Biol 21: 666–674, 1999. [DOI] [PubMed] [Google Scholar]
- 51.Fu L, Wei CC, Powell PC, Bradley WE, Ahmad S, Ferrario CM, Collawn JF, Dell′Italia LJ. Increased fibroblast chymase production mediates procollagen autophagic digestion in volume overload. J Mol Cell Cardiol 92: 1–9, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fu L, Wei CC, Powell PC, Bradley WE, Collawn JF, Dell′Italia LJ. Volume overload induces autophagic degradation of procollagen in cardiac fibroblasts. J Mol Cell Cardiol 89: 241–250, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fukusen N, Kato Y, Kido H, Katunuma N. Kinetic studies on the inhibitions of mast cell chymase by natural serine protease inhibitors: indications for potential biological functions of these inhibitors. Biochem Med Metab Biol 38: 165–169, 1987. [DOI] [PubMed] [Google Scholar]
- 54.Ganten D, Wagner J, Zeh K, Bader M, Michel JB, Paul M, Zimmermann F, Ruf P, Hilgenfeldt U, Ganten U. Species specificity of renin kinetics in transgenic rats harboring the human renin and angiotensinogen genes. Proc Natl Acad Sci U S A 89: 7806–7810, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gheorghiade M, Larson CJ, Shah SJ, Greene SJ, Cleland JGF, Colucci WS, Dunnmon P, Epstein SE, Kim RJ, Parsey RV, Stockbridge N, Carr J, Dinh W, Krahn T, Kramer F, Wahlander K, Deckelbaum LI, Crandall D, Okada S, Senni M, Sikora S, Sabbah HN, Butler J. Developing new treatments for heart failure. Focus on the heart. Circ Heart Fail 9: e002727, 2016. [DOI] [PubMed] [Google Scholar]
- 56.Gutkowska J, Boucher R, Demassieux S, Garcia R, Genest J. A direct radioimmunoassay for tonin. Can J Biochem 56: 769–773, 1978. [DOI] [PubMed] [Google Scholar]
- 57.Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol 302: R518–R530, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haas E, Lewis L, Koshy TJ, Varde AU, Renerts L, Bagai RC. Angiotensin II-producing enzyme III from acidified serum of nephrectomized dogs. Am J Hypertens 2: 696–707, 1989. [DOI] [PubMed] [Google Scholar]
- 59.Hanssens M, Pijnenborg R, Keirse MJ, Vercruysse L, Verbist L, Van Assche FA. Renin-like immunoreactivity in uterus and placenta from normotensive and hypertensive pregnancies. Eur J Obstet Gynecol Reprod Biol 81: 177–184, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Heran BS, Musini VM, Bassett K, Taylor RS, Wright JM. Angiotensin receptor blockers for heart failure. Cochrane Database Syst Rev 4: CD003040, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoshino F, Urata H, Inoue Y, Saito Y, Yahiro E, Ideishi M, Arakawa K, Saku K. Chymase inhibitor improves survival in hamsters with myocardial infarction. J Cardiovasc Pharmacol 41, Suppl 1: S11–S18, 2003. [PubMed] [Google Scholar]
- 62.Huang XR, Chen WY, Truong LD, Lan HY. Chymase is upregulated in diabetic nephropathy: implications for an alternative pathway of angiotensin II-mediated diabetic renal and vascular disease. J Am Soc Nephrol 14: 1738–1747, 2003. [DOI] [PubMed] [Google Scholar]
- 63.Huber MJ, Basu R, Cecchettini C, Cuadra AE, Chen QH, Shan Z. Activation of the (pro)renin receptor in the paraventricular nucleus increases sympathetic outflow in anesthetized rats. Am J Physiol Heart Circ Physiol 309: H880–H887, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Husain A. The chymase-angiotensin system in humans. J Hypertens 11: 1155–1159, 1993. [PubMed] [Google Scholar]
- 65.Husain A, Smeby RR, Wilk D, Dzau VJ, Bumpus FM. Biochemical and immunological properties of dog brain isorenin. Endocrinology 114: 2210–2215, 1984. [DOI] [PubMed] [Google Scholar]
- 66.Husain A, Wilk D, Smeby RR, Dzau VJ, Bumpus FM. Isorenin in dog brain and other tissues. Clin Exp Hypertens A 6: 1795–1799, 1984. [DOI] [PubMed] [Google Scholar]
- 67.Isa K, Garcia-Espinosa MA, Arnold AC, Pirro NT, Tommasi EN, Ganten D, Chappell MC, Ferrario CM, Diz DI. Chronic immunoneutralization of brain angiotensin-(1–12) lowers blood pressure in transgenic (mRen2)27 hypertensive rats. Am J Physiol Regul Integr Comp Physiol 297: R111–R115, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jahanyar J, Youker KA, Loebe M, Assad-Kottner C, Koerner MM, Torre-Amione G, Noon GP. Mast cell-derived cathepsin g: a possible role in the adverse remodeling of the failing human heart. J Surg Res 140: 199–203, 2007. [DOI] [PubMed] [Google Scholar]
- 69.Jankowski V, Toelle M, van der Giet M, Jankowski J. Angiotensin A, an ANG-II like peptide stimulating the AT2 receptor: PP.24461. J Hypertens 28: e1–e651, 2010.21928474 [Google Scholar]
- 70.Jessup JA, Trask AJ, Chappell MC, Nagata S, Kato J, Kitamura K, Ferrario CM. Localization of the novel angiotensin peptide, Angiotensin-(1–12), in heart and kidney of hypertensive and normotensive rats. Am J Physiol Heart Circ Physiol 294: H2614–H2618, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawabe T, Kawabe K, Sapru HN. Cardiovascular effect of angiotensin-(1–12) in the caudal ventrolateral medullary depressor area of the rat. Am J Physiol Heart Circ Physiol 306: H438–H449, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keidar S, Gamliel-Lazarovich A, Kaplan M, Pavlotzky E, Hamoud S, Hayek T, Karry R, Abassi Z. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res 97: 946–953, 2005. [DOI] [PubMed] [Google Scholar]
- 73.Khullar M, Scicli G, Carretero OA, Scicli AG. Purification and characterization of a serine protease (esterase B) from rat submandibular glands. Biochemistry 25: 1851–1857, 1986. [DOI] [PubMed] [Google Scholar]
- 74.Kido H, Yokogoshi Y, Katunuma N. Kunitz-type protease inhibitor found in rat mast cells. Purification, properties, and amino acid sequence. J Biol Chem 263: 18104–18107, 1988. [PubMed] [Google Scholar]
- 75.Kjeldsen SE, Westheim AS, Os I. Prevention of cardiovascular events and diabetes with angiotensin-receptor blockers in hypertension: LIFE, SCOPE, and VALUE. Curr Hypertens Rep 7: 155–157, 2005. [DOI] [PubMed] [Google Scholar]
- 76.Koka V, Wang W, Huang XR, Kim-Mitsuyama S, Truong LD, Lan HY. Advanced glycation end products activate a chymase-dependent angiotensin II-generating pathway in diabetic complications. Circulation 113: 1353–1360, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Komatsu Y, Kida N, Nozaki N, Kuwasako K, Nagata S, Kitamura K, Kato J. Effects of proangiotensin-12 infused continuously over 14 days in conscious rats. Eur J Pharmacol 683: 186–189, 2012. [DOI] [PubMed] [Google Scholar]
- 78.Kumar R, Boim MA. Diversity of pathways for intracellular angiotensin II synthesis. Curr Opin Nephrol Hypertens 18: 33–39, 2009. [DOI] [PubMed] [Google Scholar]
- 79.Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (Lond) 123: 273–284, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kumar R, Yong QC, Thomas CM, Baker KM. Intracardiac intracellular angiotensin system in diabetes. Am J Physiol Regul Integr Comp Physiol 302: R510–R517, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, Jankowski J, Jankowski V, Sousa F, Alzamora A, Soares E, Barbosa C, Kjeldsen F, Oliveira A, Braga J, Savergnini S, Maia G, Peluso AB, Passos-Silva D, Ferreira A, Alves F, Martins A, Raizada M, Paula R, Motta-Santos D, Klempin F, Pimenta A, Alenina N, Sinisterra R, Bader M, Campagnole-Santos MJ, Santos RA. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res 112: 1104–1111, 2013. [DOI] [PubMed] [Google Scholar]
- 82.Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol 23: 238–243, 2003. [DOI] [PubMed] [Google Scholar]
- 83.Li J, Jubair S, Levick SP, Janicki JS. The autocrine role of tryptase in pressure overload-induced mast cell activation, chymase release and cardiac fibrosis. IJC Metab Endocr 10: 16–23, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li M, Liu K, Michalicek J, Angus JA, Hunt JE, Dell′Italia LJ, Feneley MP, Graham RM, Husain A. Involvement of chymase-mediated angiotensin II generation in blood pressure regulation. J Clin Invest 114: 112–120, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lindpaintner K, Jin MW, Niedermaier N, Wilhelm MJ, Ganten D. Cardiac angiotensinogen and its local activation in the isolated perfused beating heart. Circ Res 67: 564–573, 1990. [DOI] [PubMed] [Google Scholar]
- 86.Longley BJ, Tyrrell L, Ma Y, Williams DA, Halaban R, Langley K, Lu HS, Schechter NM. Chymase cleavage of stem cell factor yields a bioactive, soluble product. Proc Natl Acad Sci U S A 94: 9017–9021, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 107: 2555–2558, 2003. [DOI] [PubMed] [Google Scholar]
- 88.McMurray JJ, Krum H, Abraham WT, Dickstein K, Kober LV, Desai AS, Solomon SD, Greenlaw N, Ali MA, Chiang Y, Shao Q, Tarnesby G, Massie BM. Aliskiren, enalapril, or aliskiren and enalapril in heart failure. N Engl J Med 374: 1521–1532, 2016. [DOI] [PubMed] [Google Scholar]
- 89.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 371: 993–1004, 2014. [DOI] [PubMed] [Google Scholar]
- 90.Miura S, Ideishi M, Sakai T, Motoyama M, Kinoshita A, Sasaguri M, Tanaka H, Shindo M, Arakawa K. Angiotensin II formation by an alternative pathway during exercise in humans. J Hypertens 12: 1177–1181, 1994. [PubMed] [Google Scholar]
- 91.Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (IL-1 beta) to an active IL-1 species by human mast cell chymase. J Exp Med 174: 821–825, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moniwa N, Varagic J, Simington SW, Ahmad S, Nagata S, VonCannon JL, Ferrario CM. Primacy of angiotensin converting enzyme in angiotensin-(1–12) metabolism. Am J Physiol Heart Circ Physiol 305: H644–H650, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Morgan L, Broughton PF, Kalsheker N. Angiotensinogen: molecular biology, biochemistry and physiology. Int J Biochem Cell Biol 28: 1211–1222, 1996. [DOI] [PubMed] [Google Scholar]
- 94.Morris BJ. Human renin protein and gene structures: present and future targets for renin blockade in treatment of hypertension. J Hypertens Suppl 7: S9–S14, 1989. [DOI] [PubMed] [Google Scholar]
- 95.Nagata S, Hatakeyama K, Asami M, Tokashiki M, Hibino H, Nishiuchi Y, Kuwasako K, Kato J, Asada Y, Kitamura K. Big angiotensin-25: a novel glycosylated angiotensin-related peptide isolated from human urine. Biochem Biophys Res Commun 441: 757–762, 2013. [DOI] [PubMed] [Google Scholar]
- 96.Nagata S, Kato J, Kuwasako K, Asami M, Kitamura K. Plasma and tissue concentrations of proangiotensin-12 in rats treated with inhibitors of the renin-angiotensin system. Hypertens Res 35: 234–238, 2012. [DOI] [PubMed] [Google Scholar]
- 97.Nagata S, Kato J, Kuwasako K, Kitamura K. Plasma and tissue levels of proangiotensin-12 and components of the renin-angiotensin system (RAS) following low- or high-salt feeding in rats. Peptides 31: 889–892, 2010. [DOI] [PubMed] [Google Scholar]
- 98.Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun 350: 1026–1031, 2006. [DOI] [PubMed] [Google Scholar]
- 99.Nagata S, Varagic J, Kon ND, Wang H, Groban L, Simington SW, Ahmad S, Dell′Italia LJ, VonCannon JL, Deal D, Ferrario CM. Differential expression of the angiotensin-(1–12)/chymase axis in human atrial tissue. Ther Adv Cardiovasc Dis 9: 168–180, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 100.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest 109: 1417–1427, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Oyamada S, Bianchi C, Takai S, Chu LM, Sellke FW. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J Pharmacol Exp Ther 339: 143–151, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pat B, Killingsworth C, Chen Y, Gladden JD, Walcott G, Powell PC, Denney T, Gupta H, Desai R, Tillson M, Dillon AR, Dell′Italia LJ. Mast cell stabilization decreases cardiomyocyte and LV function in dogs with isolated mitral regurgitation. J Card Fail 16: 769–776, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paul M, Poyan MA, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 86: 747–803, 2006. [DOI] [PubMed] [Google Scholar]
- 104.Pejler G, Ronnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood 115: 4981–4990, 2010. [DOI] [PubMed] [Google Scholar]
- 105.Peng H, Li W, Seth DM, Nair AR, Francis J, Feng Y. (Pro)renin receptor mediates both angiotensin II-dependent and -independent oxidative stress in neuronal cells. PLoS One 8: e58339, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pereira HJ, Souza LL, Costa-Neto CM, Salgado MC, Oliveira EB. Carboxypeptidases A1 and A2 from the perfusate of rat mesenteric arterial bed differentially process angiotensin peptides. Peptides 33: 67–76, 2012. [DOI] [PubMed] [Google Scholar]
- 107.Pereira RM, Dos Santos RA, Teixeira MM, Leite VH, Costa LP, Costa Dias FL, Barcelos LS, Collares GB, Simoes E, Silva AC. The renin-angiotensin system in a rat model of hepatic fibrosis: evidence for a protective role of Angiotensin-(1–7). J Hepatol 46: 674–681, 2007. [DOI] [PubMed] [Google Scholar]
- 108.Prosser HC, Forster ME, Richards AM, Pemberton CJ. Cardiac chymase converts rat proAngiotensin-12 (PA12) to angiotensin II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res 82: 40–50, 2009. [DOI] [PubMed] [Google Scholar]
- 109.Prosser HC, Richards AM, Forster ME, Pemberton CJ. Regional vascular response to ProAngiotensin-12 (PA12) through the rat arterial system. Peptides 31: 1540–1545, 2010. [DOI] [PubMed] [Google Scholar]
- 110.Puzyrev AA, Ivanova VF. [“Mixed” endocrine gland cells of the duodenal epithelium in various vertebrate animals and man]. Arkh Anat Gistol Embriol 90: 48–54, 1986. [PubMed] [Google Scholar]
- 111.Ramaha A, Patston PA. Release and degradation of angiotensin I and angiotensin II from angiotensinogen by neutrophil serine proteinases. Arch Biochem Biophys 397: 77–83, 2002. [DOI] [PubMed] [Google Scholar]
- 112.Re RN. The cellular biology of angiotensin: paracrine, autocrine and intracrine actions in cardiovascular tissues. J Mol Cell Cardiol 21, Suppl 5: 63–69, 1989. [DOI] [PubMed] [Google Scholar]
- 113.Re RN. A mechanism for mineralocortcoid participation in renal disease and heart failure. J Am Soc Hypertens 9: 586–591, 2015. [DOI] [PubMed] [Google Scholar]
- 114.Rykl J, Thiemann J, Kurzawski S, Pohl T, Gobom J, Zidek W, Schluter H. Renal cathepsin G and angiotensin II generation. J Hypertens 24: 1797–1807, 2006. [DOI] [PubMed] [Google Scholar]
- 115.Saris JJ, ′t Hoen PA, Garrelds IM, Dekkers DH, den Dunnen JT, Lamers JM, Jan Danser AH. Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension 48: 564–571, 2006. [DOI] [PubMed] [Google Scholar]
- 116.Satou R, Kobori H. Regulation of a novel angiotensin II precursor, proangiotensin-12, in the tissues by blockade of the renin-angiotensin system. Hypertens Res 35: 153–154, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schiffrin EL, Garcia R, Gutkowska J, Boucher R, Genest J. Pressor effect of tonin in anephric animals. Can J Physiol Pharmacol 59: 864–871, 1981. [DOI] [PubMed] [Google Scholar]
- 118.Singh VP, Le B, Bhat VB, Baker KM, Kumar R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol 293: H939–H948, 2007. [DOI] [PubMed] [Google Scholar]
- 119.Skeggs LT Jr, Kahn JR, Lentz K, Shumway NP. The preparation, purification, and amino acid sequence of a polypeptide renin substrate. J Exp Med 106: 439–453, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taipale J, Lohi J, Saarinen J, Kovanen PT, Keski-Oja J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-beta 1 from the extracellular matrix of cultured human epithelial and endothelial cells. J Biol Chem 270: 4689–4696, 1995. [DOI] [PubMed] [Google Scholar]
- 121.Takai S, Jin D, Miyazaki M. New approaches to blockade of the renin-angiotensin-aldosterone system: chymase as an important target to prevent organ damage. J Pharmacol Sci 113: 301–309, 2010. [DOI] [PubMed] [Google Scholar]
- 122.Takai S, Jin D, Miyazaki M. Multiple mechanisms for the action of chymase inhibitors. J Pharmacol Sci 118: 311–316, 2012. [DOI] [PubMed] [Google Scholar]
- 123.Takai S, Jin D, Muramatsu M, Okamoto Y, Miyazaki M. Therapeutic applications of chymase inhibitors in cardiovascular diseases and fibrosis. Eur J Pharmacol 501: 1–8, 2004. [DOI] [PubMed] [Google Scholar]
- 124.Tani K, Ogushi F, Kido H, Kawano T, Kunori Y, Kamimura T, Cui P, Sone S. Chymase is a potent chemoattractant for human monocytes and neutrophils. J Leukoc Biol 67: 585–589, 2000. [DOI] [PubMed] [Google Scholar]
- 125.Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem 280: 9291–9296, 2005. [DOI] [PubMed] [Google Scholar]
- 126.Tewksbury DA. Quantitation of five forms of high molecular weight angiotensinogen from human placenta. Am J Hypertens 9: 1029–1034, 1996. [DOI] [PubMed] [Google Scholar]
- 127.Tewksbury DA, Dart RA. High molecular weight angiotensinogen levels in hypertensive pregnant women. Hypertension 4: 729–734, 1982. [DOI] [PubMed] [Google Scholar]
- 128.Tewksbury DA, Tryon ES. Immunochemical comparison of high molecular weight angiotensinogen from amniotic fluid, plasma of men, and plasma of pregnant women. Am J Hypertens 2: 411–413, 1989. [DOI] [PubMed] [Google Scholar]
- 129.Tewksbury DA, Tryon ES, Burrill RE, Dart RA. High molecular weight angiotensinogen: a pregnancy associated protein. Clin Chim Acta 158: 7–12, 1986. [DOI] [PubMed] [Google Scholar]
- 130.Tojo H, Urata H. Chymase inhibition and cardiovascular protection. Cardiovasc Drugs Ther 27: 139–143, 2013. [DOI] [PubMed] [Google Scholar]
- 131.Trask AJ, Jessup JA, Chappell MC, Ferrario CM. Angiotensin-(1–12) is an alternate substrate for angiotensin peptide production in the heart. Am J Physiol Heart Circ Physiol 294: H2242–H2247, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Turnbull F, Neal B, Ninomiya T, Algert C, Arima H, Barzi F, Bulpitt C, Chalmers J, Fagard R, Gleason A, Heritier S, Li N, Perkovic V, Woodward M, MacMahon S. Effects of different regimens to lower blood pressure on major cardiovascular events in older and younger adults: meta-analysis of randomised trials. BMJ 336: 1121–1123, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Turnbull F, Neal B, Pfeffer M, Kostis J, Algert C, Woodward M, Chalmers J, Zanchetti A, MacMahon S. Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system. J Hypertens 25: 951–958, 2007. [DOI] [PubMed] [Google Scholar]
- 134.Uehara Y, Miura S, Yahiro E, Saku K. Non-ACE pathway-induced angiotensin II production. Curr Pharm Des 19: 3054–3059, 2013. [DOI] [PubMed] [Google Scholar]
- 135.Uehara Y, Urata H, Sasaguri M, Ideishi M, Sakata N, Tashiro T, Kimura M, Arakawa K. Increased chymase activity in internal thoracic artery of patients with hypercholesterolemia. Hypertension 35: 55–60, 2000. [DOI] [PubMed] [Google Scholar]
- 136.Urata H. Pathological involvement of chymase-dependent angiotensin II formation in the development of cardiovascular disease. J Renin Angiotensin Aldosterone Syst 1: S35–S37, 2000. [DOI] [PubMed] [Google Scholar]
- 137.Urata H. [Involvement of chymase in pathogenesis of cardiovascular diseases]. Nihon Rinsho 62, Suppl 3: 147–151, 2004. [PubMed] [Google Scholar]
- 138.Urata H. [Chymase-dependent angiotensin II-formation]. Nihon Rinsho 63, Suppl 3: 60–64, 2005. [PubMed] [Google Scholar]
- 139.Urata H. Chymase and matrix metalloproteinase. Hypertens Res 30: 3–4, 2007. [DOI] [PubMed] [Google Scholar]
- 140.Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, Husain A. Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest 91: 1269–1281, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Urata H, Ganten D. Cardiac angiotensin II formation: the angiotensin-I converting enzyme and human chymase. Eur Heart J 14, Suppl I: 177–182, 1993. [PubMed] [Google Scholar]
- 142.Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Angiotensin II-forming pathways in normal and failing human hearts. Circ Res 66: 883–890, 1990. [DOI] [PubMed] [Google Scholar]
- 143.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem 265: 22348–22357, 1990. [PubMed] [Google Scholar]
- 144.Urata H, Strobel F, Ganten D. Widespread tissue distribution of human chymase. J Hypertens Suppl 12: S17–S22, 1994. [PubMed] [Google Scholar]
- 145.van der Leeuw J, Oemrawsingh RM, van der Graaf Y, Brugts JJ, Deckers JW, Bertrand M, Fox K, Ferrari R, Remme WJ, Simoons ML, Boersma E, Visseren FL. Prediction of absolute risk reduction of cardiovascular events with perindopril for individual patients with stable coronary artery disease - results from EUROPA. Int J Cardiol 182: 194–199, 2015. [DOI] [PubMed] [Google Scholar]
- 146.van Vark LC, Bertrand M, Akkerhuis KM, Brugts JJ, Fox K, Mourad JJ, Boersma E. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur Heart J 33: 2088–2097, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vanuzzo D. The epidemiological concept of residual risk. Intern Emerg Med 6, Suppl 1: 45–51, 2011. [DOI] [PubMed] [Google Scholar]
- 148.Varagic J, Ahmad S, VonCannon JL, Moniwa N, Brosnihan KB, Wysocki J, Batlle D, Ferrario CM. Predominance of AT1 blockade over mas-mediated angiotensin-(1–7) mechanisms in the regulation of blood pressure and renin-angiotensin system in mRen2.Lewis rats. Am J Hypertens 26: 583–590, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Varagic J, Trask AJ, Jessup JA, Chappell MC, Ferrario CM. New angiotensins. J Mol Med (Berl) 86: 663–671, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang H, Jessup JA, Zhao Z, Da SJ, Lin M, MacNamara LM, Ahmad S, Chappell MC, Ferrario CM, Groban L. Characterization of the cardiac renin angiotensin system in oophorectomized and estrogen-replete mRen2.Lewis rats. PLoS One 8: e76992, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang J, Sukhova GK, Liu J, Ozaki K, Lesner A, Libby P, Kovanen PT, Shi GP. Cathepsin G deficiency reduces periaortic calcium chloride injury-induced abdominal aortic aneurysms in mice. J Vasc Surg 62: 1615–1624, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang Y, Shiota N, Leskinen MJ, Lindstedt KA, Kovanen PT. Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: transforming growth factor-beta1-dependent and -independent effects. Arterioscler Thromb Vasc Biol 21: 1928–1933, 2001. [DOI] [PubMed] [Google Scholar]
- 153.Wei CC, Hase N, Inoue Y, Bradley EW, Yahiro E, Li M, Naqvi N, Powell PC, Shi K, Takahashi Y, Saku K, Urata H, Dell′Italia LJ, Husain A. Mast cell chymase limits the ca rdiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J Clin Invest 120: 1229–1239, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Westwood BM, Chappell MC. Divergent pathways for the angiotensin-(1–12) metabolism in the rat circulation and kidney. Peptides 35: 190–195, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Whaley-Connell A, Habibi J, Wei Y, Gutweiler A, Jellison J, Wiedmeyer CE, Ferrario CM, Sowers JR. Mineralocorticoid receptor antagonism attenuates glomerular filtration barrier remodeling in the transgenic Ren2 rat. Am J Physiol Renal Physiol 296: F1013–F1022, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wolny A, Clozel JP, Rein J, Mory P, Vogt P, Turino M, Kiowski W, Fischli W. Functional and biochemical analysis of angiotensin II-forming pathways in the human heart. Circ Res 80: 219–227, 1997. [DOI] [PubMed] [Google Scholar]
- 157.Xiong W, Chen LM, Chao J. Purification and characterization of a kallikrein-like T-kininogenase. J Biol Chem 265: 2822–2827, 1990. [PubMed] [Google Scholar]
- 158.Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H, Shang FJ, Niu XL, He YP, Lu XL. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol Cell Biochem 310: 159–166, 2008. [DOI] [PubMed] [Google Scholar]
- 159.Zhao Z, Wang H, Lin M, Groban L. GPR30 decreases cardiac chymase/angiotensin II by inhibiting local mast cell number. Biochem Biophys Res Commun 459: 131–136, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zheng J, Wei CC, Hase N, Shi K, Killingsworth CR, Litovsky SH, Powell PC, Kobayashi T, Ferrario CM, Rab A, Aban I, Collawn JF, Dell′Italia LJ. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS One 9: e94732, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]