

Abstract
Inflammation of the central nervous system is an important but poorly understood part of neurological disease. After acute brain injury or infection there is a complex inflammatory response that involves activation of microglia and astrocytes and increased production of cytokines, chemokines, acute phase proteins, and complement factors. Antibodies and T lymphocytes may be involved in the response as well. In neurodegenerative disease, where injury is more subtle but consistent, the inflammatory response is continuous. The purpose of this prolonged response is unclear, but it is likely that some of its components are beneficial and others are harmful. Animal models of neurological disease can be used to dissect the specific role of individual mediators of the inflammatory response and assess their potential benefit. To illustrate this approach, we discuss how mutant mice expressing different levels of the cytokine transforming growth factor β-1 (TGF-β1), a major modulator of inflammation, produce important neuroinflammatory phenotypes. We then demonstrate how crosses of TGF-β1 mutant mice with mouse models of Alzheimer's disease (AD) produced important new information on the role of inflammation in AD and on the expression of different neuropathological phenotypes that characterize this disease.
Inflammatory profile of TGF-β1 mutant mice
TGF-β1 was initially described for its ability to transform normal rat kidney cells [1]. Since then, it has been shown to also promote cell survival or induce apoptosis, stimulate cell proliferation or induce differentiation, and initiate or resolve inflammation. Its differential effects depend on the cell type involved, the cell's environment, and the duration and amount of TGF-β1 production. TGF-β receptors are found on most cell types and their activation affects the expression of a few hundred genes [2-4]. The molecular aspects of TGF-β signalling are extensively studied and we refer to several excellent reviews [2,3]. In the normal CNS, all three TGF-β isoforms and their receptors are expressed within neurons, astrocytes, and microglia, and TGF-β1 can modulate cellular responses in these cells as well as in vascular and meningeal cells [5,6]. TGF-β1 is the most abundant and best studied TGF-β isoform and an important component of the brain's response to injury. It is consistently increased after various forms of brain insults and in neurodegenerative diseases (Table 1). Still, we understand very little about the purpose and consequences of increased TGF-β1 expression to brain function.
Table 1.
TGF-β1 is elevated acutely after injury to the brain and chronically in neurodegenerative disease.
| Injury/Insult or Disease | Species | Location/Cell type | Timing | Reference |
| Degenerative Disease | ||||
| Alzheimer's Disease | Human | Entorhinal cortex and superior temporal gyrus mRNA; Brain microvessel, Senile plaques, neurofibrillary tangles, CSF protein | Chronic | [48, 80, 82-84] |
| Vascular Dementia | Human | CSF protein | Chronic | [84] |
| Parkinson's Disease | Human | CSF dopaminergic striatal brain regions, protein | Chronic | [85, 86] |
| Amyotrophic Lateral Sclerosis | Human | CSF and serum protein | Chronic | [87] [88] |
| Diabetic Neuropathy | Human | Plasma protein | Chronic | [89] |
| Acute Insult | ||||
| Transient ischemia | Rat | Hippocampus/cerebellar protein; Hippocampal mRNA; Microglial mRNA and protein | 20 min-12 weeks | [90-92] [93, 94] |
| Permanent ischemia | Human; rat; Baboon | Increased mRNA in ischemic and penumbral areas | 1–15 days | [95-98] |
| Subarachnoid hemorrhage | Human | CSF protein | 1–19 days | [99] |
| Posthemorrhagic Hydrocephalus | Human | CSF protein | 1–14 days | [12, 13, 100] |
| Spinal cord injury | Human | Spinal cord mRNA | 1–48 h | [101] |
| Triethyltin exposure | Mouse | Cortical mRNA | 6 hrs | [102] |
| Excitotoxic lesion (NMDA) | Rat | Gray matter surrounding the lesion | [103] | |
| Status epilepticus | Rat | Hippocampal | 1–3 weeks | [104] |
| Kainic acid or deafferentation-induced neurodegeneration | Rat | Reactive microglia, mRNA | [105, 106] | |
| Spinal cord Contusion | Rat | Spinal cord mRNA | 0.25–10 days | [107] |
| Penetrating brain Injury | Rat | Perilesional activated glia, meningeal cells, choroid plexus mRNA and protein | 1–14 days | [108, 109] |
| Experimentally induced glaucoma | Monkey | Optic nerve head protein | Chronic | [110] |
| Autoimmune Disease | ||||
| Multiple Sclerosis | Human | CSF protein; Mononuclear cells from blood and CSF, mRNA; Serum protein during relapses; Peri-lesional hypertrophic astrocytes, protein | Chronic | [111-114] |
| Chronic relapsing experimental autoimmune encephalitis | Mouse | Spinal cord mRNA | Chronic | [115] |
| Experimental Autoimmune Encephalitis | Rat | Spinal cord T-cell, monocyte, and microglia mRNA | Acute | [116] |
| Guillan-Barré Syndrome | Human | Serum and circulating monocyte protein | Plateau phase | [117, 118] |
| Experimental Autoimmune Neuritis | Rat | Macrophage, microglia, meningeal cells, and T-cell infiltrates | Acute | [116, 119] |
| Infection | ||||
| CMV encephalitis | Human/mouse | Astrocyte mRNA | 5-13d after infection | [120] |
| ME7 scrapie model | Mouse | Brain mRNA | [121] | |
| Bacterial Meningitis | Rat Human | CSF cellular mRNA CSF protein Brain mRNA, CSF protein | Acute | [122-124] |
| Brain Abscess | Human | Peri-abscess and abscess extracellular matrix protein | Chronic | [125] |
To study the role of TGF-β1 in the CNS we overproduced bioactive peptide under the control of glial fibrillary acidic protein (GFAP) regulatory sequences in astrocytes of two independent lines of transgenic mice (herein called TGF-β1 mice) [7]. We also analyzed brains of mice that are TGF-β1 deficient or knockout [8]. C57BL/6 mice lacking TGF-β1 have defects in vasculogenesis and angiogenesis leading to early embryonic lethality [9,10], but mice on the NIH genetic background survive up to 3–4 weeks of age before they succumb to an autoimmune wasting syndrome [11]. The effects of TGF-β1 expression and age on the expression of inflammatory phenotypes in these mice are graphically represented in what we call a "phenogram" (Figure 1).
Figure 1.

Phenograms of TGF-β1 and hAPP/TGF-β1 mice. Underexpression and knockout of TGF-β1 results in neurodegeneration. Overexpression of TGF-β1 in astrocytes produces phenotypes that are altered by the addition of a transgene expressing mutant human amyloid. TGF-β1-induced astrogliosis and microgliosis aid in clearing amyloid, and TGF-β1-induced vascular fibrosis traps amyloid in blood vessel walls, producing amyloid angiopathy.
TGF-β1 overexpression at high levels results in hydrocephalus
Hydrocephalus and brain fibrosis are common sequelae after whole brain inflammation due to bacterial meningitis, subarachnoid hemorrhage, or severe traumatic brain injury. High CSF levels of TGF-β1 in patients with subarachnoid hemorrhage confer an increased risk of developing chronic hydrocephalus [12,13]. TGF-β1 injected into the lateral ventricles produces hydrocephalus in mice [14]. TGF-β1 mice with high levels of expression in astrocytes had persistent communicating hydrocephalus at birth, enlargement of cerebral hemispheres, and thinning of the overlying cerebral cortex [7,15]. Additional stimulation of the injury-responsive GFAP-TGF-β1 transgene in adult low-expressor mice by CNS stab lesions leads to the development of mild hydrocephalus. These results indicate that hydrocephalus is directly related to TGF-β1 expression and not due to other developmental abnormalities [7]. Histological analysis shows decreased stratification of neuronal cell layers and leukomalacia-like areas. Given the extensive fibrosis of the meninges in TGF-β1 mice hydrocephalus may be a result of decreased CSF outflow through fibrotic arachnoid villi.
Indeed, TGF-β1 plays a key role in fibrosis in the lung [16,17] and the kidney [18]. It induces the production of a large number of extracellular matrix proteins, proteases and their inhibitors [7,19] and it may be the excess production of extracellular matrix proteins by TGF-β1 that results in hydrocephalus. The amount of TGF-β1 produced in response to injury may vary among individuals and be determined by genetic polymorphisms in the TGFB1 gene. Polymorphisms that lead to higher levels of TGF-β1 production in various assays were associated with increased risk of fibrosis in transplant recipients [20] and accelerated decline in lung function in patients with cystic fibrosis [21].
TGF-β1 overexpression causes extensive cerebrovascular fibrosis
Studies in TGF-β1 knockout mice demonstrated an essential role for TGF-β1 in vasculogenesis and angiogenesis during development [10] and other studies implicated TGF-β1 also in maintaining vascular integrity in adults [22,23]. Two TGF-β receptors on endothelial cells, endoglin and ALK1, mediate at least part of this effect and mutations in these receptors cause genetic disorders of the vasculature [24-26]. While low levels of TGF-β1 are necessary for endothelial cell proliferation and angiogenesis, higher levels result in increased synthesis of basement membrane proteins and differentiation [27-29]. Our results in TGF-β1 overexpressing mice are consistent with these findings and implicate chronically elevated TGF-β1 levels directly in cerebrovascular fibrosis.
TGF-β1 mice demonstrated an age- and dose-dependent formation of thioflavin S-positive perivascular amyloid deposits and degeneration of vascular cells [30]. The amyloid deposits had an appearance similar to those found in brains of AD cases with concomitant cerebral amyloid angiopathy (CAA). However, Aβ, the proteolytic fragment of human amyloid precursor protein (hAPP) that accumulates in AD, was at best a minor component of the deposits in TGF-β1 mice. Analysis of the progression of the cerebrovascular changes in these mice showed a significant accumulation of basement membrane proteins perlecan and fibronectin in microvessels of 3–4-month-old TGF-β1 mice. This change in the vasculature preceded the formation of thioflavin S positive amyloid and was accompanied by a thickening of the cortical capillary basement membranes [30]. Vascular fibrosis occurs in hypertension, in which TGF-β1 is elevated in serum of patients [31], as well as in AD and vascular dementia, both of which also are associated with increases in TGF-β1 (Table 1). We envision a scenario where TGF-β1 induces extensive production and accumulation of extracellular matrix proteins in the vascular wall resulting in the formation of β-pleated sheets, typically referred to as amyloid.
The deposition of amyloid in cerebral blood vessel walls is the cause of CAA. It is a common vascular abnormality in AD, where the amyloid contains large amounts of Aβ, but it can occur in the nondemented elderly as well [32,33]. CAA is a major cause of normotensive intracerebral haemorrhage [34]. It is also characterized by degeneration of cerebrovascular cell and thickening of the vascular basement membrane [35-38].
TGF-β1 overproduction results in astrogliosis
Activation of astrocytes or astrogliosis is a prominent component of the inflammatory response and an indicator of injury in the brain. These astrocytes produce a large array of inflammatory mediators, growth and neuroprotective factors, and they are involved in phagocytosis [39-41]. Again, while some of these effects are clearly beneficial, extensive astrogliosis may be detrimental and can result in the formation of "glial scars" that prevent axonal sprouting [42]. In TGF-β1 homozygous mice, GFAP and TGF-β1 immunoreactivities were strongly elevated around cerebral blood vessels, and activated astrocytes showed a characteristic perivascular arrangement, a pattern often observed in chronic hydrocephalus in humans and other animals [43]. TGF-β1 mice with moderate or low levels of TGF-β1 overexpression had a less pronounced astrogliosis but GFAP expression was consistently increased [7]. Indeed, TGF-β1 directly increases GFAP transcription in cultured astrocytes [44].
Either the absence or overproduction of TGF-β1 causes microgliosis
Activated microglia are also a typical part of the brain's inflammatory response. Although TGF-β1 is generally considered an anti-inflammatory cytokine, it has been associated with recruitment of monocytes to the site of injury at the beginning of the immune response [45]. Similarly, TGF-β1 has been implicated in the activation and recruitment of microglia and monocytes in HIV encephalitis [46]. Local expression of TGF-β1 in astrocytes also renders transgenic mice more susceptible to experimental autoimmune encephalomyelitis (EAE) [47], a rodent model of multiple sclerosis. When challenged with spinal cord homogenate, TGF-β1 mice show increased infiltration of monocyte/macrophage cells and increased expression of major histocompatibility complex (MHC) class II proteins. These mice also develop a more severe clinical phenotype and earlier onset of disease than nontransgenic littermate controls [47]. TGF-β1 has also been shown to induce expression of the proinflammatory cytokines tumor necrosis factor (TNF)-α and IL-1β when added to brain vascular endothelial cells [48]. Finally, TGF-β1 mice demonstrate an age-related microgliosis that is most prominent in the hippocampus (Figure 2). Preliminary studies suggest that this microgliosis is associated with reduced neurogenesis (Buckwalter and Wyss-Coray unpublished data).
Figure 2.
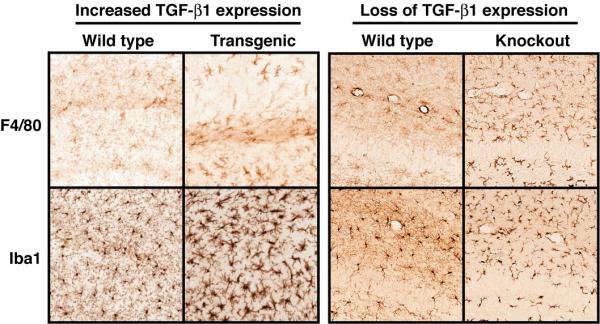
Microgliosis results from both increased and decreased levels of TGF-β1. 30-month-old TGF-β1 mice (left panel) demonstrate increased staining for F4/80, a microglial marker, in the hippocampus. A stain for Iba1, which is present in all microglia and monocytes, reveals that microglia in TGF-β1 mice are more numerous and have more cytoplasm and shorter processes than microglia in an age-matched littermates. TGF-β1 knockout mice (right panel) demonstrate dramatically increased staining with F4/80 in all brain regions and Iba1staining reveals an activated microglial morphology that is less dramatic than that seen with TGF-β1 overexpression.
Consistent with TGF-β1's anti-inflammatory role, knockout mice showed a striking microgliosis in the neocortex and hippocampus at P1 and even more so at P21 (Figure 2). Interestingly, no concomitant increase in astrocyte activation was observed in TGF-β1 knockout mice [8]. As mentioned above, overexpression of TGF-β1 using adenovirus led to decreased production of the inflammatory chemokines MCP-1 and Mip-1α after transient cerebral ischemia [49]. These effects of TGF-β1 on the recruitment and activation of microglial cells and inflammatory responses in the CNS in general may be of importance not only for classical immune-mediated CNS diseases such as MS and HIV encephalitis, but also for other CNS diseases with an involvement of microglia and inflammatory responses, notably AD (see below).
Increased TGF-β1 is neuroprotective and decreased TGF-β1 leads to neurodegeneration
TGF-β1 has been demonstrated to protect neurons against various toxins and injurious agents in cell culture and in vivo (reviewed in [5,50]). For example, intracarotid infusion of TGF-β1 in rabbits reduces cerebral infarct size when given at the time of ischemia [51]. Rat studies also showed that TGF-β1 protects hippocampal neurons from death when given intrahippocampally or intraventricularly one hour prior to transient global ischemia [52]. Mice infected with adenovirus that overexpressed TGF-β1 five days prior to transient ischemia also had smaller infarctions than control animals [49]. Astroglial overexpression of TGF-β1 in transgenic mice protects against neurodegeneration induced with the acute neurotoxin kainic acid or associated with chronic lack of apolipoprotein E expression [8]. Boche and coworkers also demonstrated that TGF-β1 protects neurons from excitotoxic death [53]. In contrast, TGF-β1 knockout mice display signs of spontaneous neuronal death, with prominent clusters of TUNEL-positive cells in different parts of the brain including the neocortex, caudate putamen and cerebellum [8]. Besides the increase in TUNEL-positive neurons, unmanipulated 3-week-old TGF-β1 knockout mice also have significantly fewer synaptophysin positive synapses in the neocortex and hippocampus compared to wildtype littermate controls, and increased susceptibility to kainic acid-induced neurotoxicity.
It is not clear how TGF-β1 protects neurons, but several mechanisms have been postulated. For example, TGF-β1 decreases Bad, a pro-apoptotic member of the Bcl-2 family, and contributes to the phosphorylation, and thus inactivation, of Bad by activation of the Erk/MAP kinase pathway [54]. On the other hand, TGF-β1 increases production of the anti-apoptotic protein Bcl-2 [55]. TGF-β1 has also been shown to synergize with neurotrophins and/or be necessary for at least some of the effects of a number of important growth factors for neurons, including neurotrophins, fibroblast growth factor-2, and glial cell-line derived neurotrophic factor (reviewed in [50,56]). In addition, TGF-β1 increases laminin expression [7] and is necessary for normal laminin protein levels in the brain [8]. Laminin is thought to provide critical support for neuronal differentiation and survival and may be important for learning and memory [57,58]. It is also possible that TGF-β1 decreases inflammation in the infarction area, attenuating secondary neuronal damage [49].
In addition to its effects on neuronal maintenance and survival, TGF-βs and the TGF-β signalling pathway have recently been implicated in the regulation of synaptic growth and function (reviewed in [59]). Synaptic overgrowth is caused by abnormal TGF-β signalling in Drosophila with mutations in genes encoding for the late endosomal gene spinster whereas the inhibitory Smad protein Dad, and mutations in TGF-β receptors can prevent this phenotype [60]. TGF-β receptors and dSmad2 are also required for neuronal remodelling in the Drosophila brain [61]. In Aplysia, sensory neurons express a type II TGF-β receptor and recombinant human TGF-β1 induces phosphorylation and redistribution of the presynaptic protein synapsin and modulates synaptic function [62,63]. Thus, TGF-β signals may be important in modulating synaptic strength and numbers in mammals as well.
Modulation of the neuroinflammatory profile in Alzheimer's models
Neuroinflammation is a prominent characteristic of neurodegenerative diseases like AD and is likely to encompass beneficial and detrimental effects [39]. Thus, inflammatory processes may attempt to clear dying cells or aggregated proteins, initiate repair processes, but also contribute to cell death and degeneration. The neuroinflammatory profile of AD as observed by a neuropathologist does not allow him to draw conclusions about mechanisms, sequence of action, or cause and effect of any of the mediators involved. Therefore, inflammatory processes in the AD brain need to be studied in appropriate model systems in order to understand their roles in the disease process.
AD is characterized clinically by an age-dependent progressive cognitive decline and pathologically by the presence of protein deposits in the form of amyloid plaques and cerebrovascular Aβ deposits in the extracellular space. In addition, abnormal phosphorylation of the microtubule associated protein tau results in the formation of tangles inside neurons [64]. These protein deposits are associated with prominent neurodegeneration and neuroinflammation. There is strong evidence that abnormal production or accumulation of Aβ is a key factor in the pathogenesis of AD (reviewed in [65]) but many cofactors are likely to modulate Aβ toxicity. Transgenic mouse models overproducing familial AD-mutant hAPP reproduce important aspects of AD, including amyloid plaques, neurodegeneration, neuroinflammation, and cognitive deficits (for example [66-68]). Specific inflammatory mediators can be studied in these AD models by crossing them with mice lacking or overproducing selected inflammatory mediators. The phenogram of TGF-β1 mutant mice (Figure 1) illustrates and underlines the prominent effects this cytokine has on inflammatory processes in the brain. Altering TGF-β1 levels could therefore be expected to have prominent effects on the neuroinflammatory profile of AD.
TGF-β1 overexpression in AD mice results in CAA
Overexpression of TGF-β1 in hAPP mice resulted in a dramatic shift in the site of Aβ accumulation (Figure 3). While Aβ accumulates almost exclusively in parenchymal plaques in hAPP mice, most of the Aβ is associated with vascular structures in hAPP/TGF-β1 bigenic mice at 12–15 months of age. These vascular deposits in bigenic mice are already detectable at 2–3 months of age with human Aβ specific antibodies, whereas age-matched singly transgenic hAPP or TGF-β1 control mice have no such deposits [69]. This mechanism of vascular amyloid formation may be relevant for humans as well. Cortical TGF-β1 mRNA levels correlate positively with the degree of cerebrovascular amyloid deposition in AD patients, and analysis of mildly fixed cortical tissues showed that TGF-β1 immunoreactivity was elevated along cerebral blood vessels and in perivascular astrocytes [69,70]. An increase in TGF-β1 may be triggered in response to traumatic brain injury or other forms of neuronal and cellular injury. Interestingly, brain injury is considered a major environmental risk factor for AD [71], and in traumatic brain injury, blood-derived TGF-β1 stored in platelets is likely released in large amounts at the lesion site [19]. In addition, individuals with a predisposition to higher TGF-β1 production, particularly in response to injury, may be more susceptible to vascular variants of AD.
Figure 3.
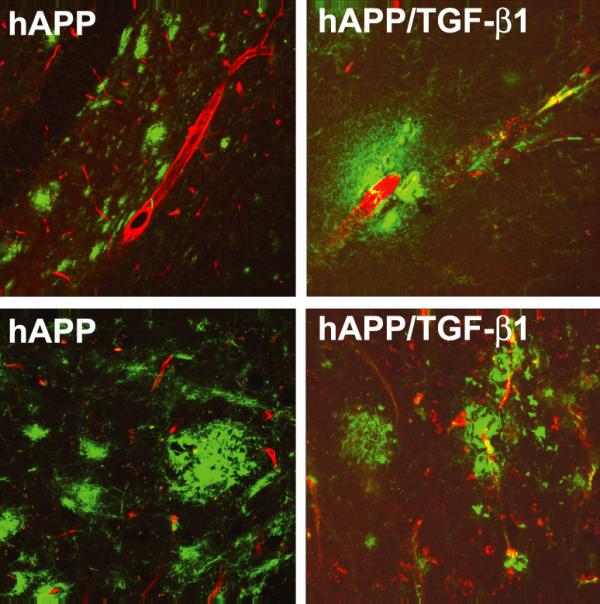
TGF-β1 overexpression in hAPP mice leads to CAA and reduces total brain amyloid. hAPP mice demonstrate amyloid plaques that are predominantly parenchymal (left panels), while bigenic hAPP/TGF-β1 mice (right panels) display fewer parenchymal amyloid plaques and have Aβ deposits localized to blood vessel walls (Aβ, green; Glut-1, red).
How does TGF-β1 cause such a dramatic change in the site of Aβ deposition? As alluded to above, TGF-β1 induces the production of many extracellular matrix proteins in the vascular basement membrane. Proteins including laminin, fibronectin, and heparan sulfate proteoglycans (HSPG) such as perlecan, have been implicated in amyloidosis (reviewed in [72,73]. In particular, glucosaminoglycan side chains of HSPGs can precipitate Aβ injected into the brain [74]. It is therefore likely that TGF-β1-induced basement membrane accumulation and fibrosis precipitates the accumulation of Aβ. In several different cell culture systems, TGF-β1 can also directly induce the expression of the APP gene [75-77]. There is currently one drug, made by Neurochem (Montreal, Quebec, Canada), which has completed phase clinical II trials that reduces amyloid deposition in transgenic mouse models by interfering with the interaction between Aβ and glucosaminoglycans.
TGF-β1-induced gliosis and amyloid clearance
Besides the accumulation of Aβ in the vasculature, bigenic hAPP/TGF-β1 mice have a 75% reduction in parenchymal amyloid plaques and overall levels of Aβ are 60–70% lower than in singly transgenic hAPP littermate controls [69]. Similar to singly-transgenic TGF-β1 mice, increased astroglial TGF-β1 production in aged bigenic mice causes extensive microglial and astroglial activation in the hippocampus and cortex [69]. Both cell types are phagocytic and we demonstrated that activation of cultured microglia with TGF-β1 results in increased degradation of Aβ [69]. In addition, primary adult astrocytes phagocytose Aβ bound to plastic or in brain sections from hAPP mice [40]. Thus, while fibrosis due to overexpression of TGF-β1 probably directs the deposition of Aβ to vascular walls, TGF-β1-activated microglia and/or astrocytes can degrade Aβ and lower brain concentration of Aβ overall (Figure 3). In AD patients, Aβ accumulation in parenchymal plaques seems to correlate inversely with Aβ in cerebral blood vessels [69,78,79] and it is tempting to speculate that TGF-β1 is involved in this process.
TGF-β1-induced neuroprotection
Given the large number of studies demonstrating that TGF-β1 is neuroprotective, it is reasonable to assume that one of the roles of TGF-β1 is to keep neurons alive in the brains of patients with Alzheimer's Disease. Indeed, expression of TGF-β1 in the superior temporal gyrus of AD brains correlates inversely with neurofibrillary tangle counts but is increased only in the late stages of disease [80]. hAPP/TGF-β1 mice have fewer dystrophic neurites than hAPP controls but this is likely confounded by the decrease in amyloid deposition in these mice [69]. Notably, TGF-β1 overproduction results not only in an overall decrease in Aβ accumulation in hAPP/TGF-β1 brains but also in a relative decrease in Aβ1–42 out of the total Aβ pool. The relative amount of Aβ1–42 appears to be a good measure for the relative toxicity and propensity of Aβ to aggregate. Chronic TGF-β1 production is likely to have different effects than acutely induced TGF-β1 and this could also have detrimental effects on neuronal survival. For example, vascular fibrosis could cause ischemia and make areas with high TGF-β1 levels more susceptible to neuronal death. Interestingly, old TGF-β1 mice have decreased blood flow to the hippocampus that correlates inversely with the thioflavin-S positive vascular deposits in this region [81]. Better tools will be necessary to separate direct neuroprotective from indirect effects of TGF-β1 in vivo.
Conclusions
Neuroinflammation occurs consistently in neurological diseases but its role is unclear. We demonstrate here that the analysis of inflammatory phenotypes in TGF-β1 mice has been helpful in understanding human disease. First, highly elevated cerebral TGF-β1 production is clearly associated with hydrocephalus in mice and humans. Interfering with local production of TGF-β1 may therefore be of potential therapeutic value in the management of hydrocephalus. Second, studying chronic overproduction of TGF-β1 in a mouse model for AD revealed that TGF-β1 has a key role in the development of CAA and also reduces amyloid deposition in the parenchyma. This highlights the utility of such models in dissecting opposing effects of inflammatory mediators in neurological diseases. Therapeutic approaches blocking the effect of TGF-β1 on the vasculature or promoting TGF-β1's effect in the brain parenchyma can be pursued based on these results. In fact, a drug that interferes with the accumulation of Aβ in the basement membrane has now completed phase II clinical trials.
Despite the progress made in understanding the role of TGF-β1 and many other factors in inflammation, many questions remain. Animal models such as Drosophila might be useful to study simple aspects of inflammation such as phagocytosis, but more complex inflammatory pathways are absent in flies and need to be studied in mammals. Drosophila could also be used to study the direct effects of cytokines on neurons and glial cells [59]. New genomic and proteomic approaches will be helpful in expanding our understanding of neuroinflammation in animal models to more complex levels. This will also require mathematical modelling systems as well as powerful database tools. Importantly, the inflammatory phenotypes generated in animal models need to be linked to functional outcome measures because these are the only measures that matter for a patient with neurological disease.
List of abbreviations
AβA-beta peptide
Aβ1–42 A-beta peptide containing amino acids 1–42
AD Alzheimer's disease
CAA Cerebral amyloid angiopathy
CNS Central nervous system
CSF Cerebrospinal fluid
EAE Experimental autoimmune encephalomyelitis
GFAP Glial fibrillary acidic protein
hAPP human amyloid precursor protein
HIV Human immunodeficieny virus
HSPG Heparan sulfate proteoglycan
Iba-1 Ionized calcium-binding adaptor molecule-1
Il-1β Interleukin-1β
MAP Mitogen activated protein
MCP-1 Monocyte chemoattractant protein-1
MHC Major histocompatability complex
Mip-1α Macrophage inflammatory protein-1 alpha
MS Multiple sclerosis
NIH National Institutes of Health
P1 Postnatal day 1
P21 Postnatal day 21
TGF-β Transforming growth factor-beta
TNF-α Tumor necrosis factor alpha
TUNEL Terminal deoxynucleotidyl transferase dUTP-biotin nick-end labelling
Competing interests
None declared.
Acknowledgments
Acknowledgements
This work was supported by the National Institutes of Health grant AG20603 and the Veterans Administration GRECC.
Contributor Information
Marion S Buckwalter, Email: marion.buckwalter@stanford.edu.
Tony Wyss-Coray, Email: twc@stanford.edu.
References
- Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB. New class of transforming growth factors potentiated by epidermal growth factor: isolation from non-neoplastic tissues. Proc Natl Acad Sci U S A. 1981;78:5339–5343. doi: 10.1073/pnas.78.9.5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler S, Goumans M-J, Dijke P t. Transforming growth factor beta signal transduction. J Leukoc Biol. 2002;71:731–740. [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- Yang YC, Piek E, Zavadil J, Liang D, Xie D, Heyer J, Pavlidis P, Kucherlapati R, Roberts AB, Bottinger EP. Hierarchical model of gene regulation by transforming growth factor beta. Proc Natl Acad Sci U S A. 2003;100:10269–10274. doi: 10.1073/pnas.1834070100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor–betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/S0301-0082(97)00066-X. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WST, Hampel H, Hull M, Landreth G, Lue L-F, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyama I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neuroinflammation Working Group. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)82472-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta1. Am J Pathol. 1995;147:53–67. [PMC free article] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/S0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh C-H, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn M, Ward JM, Karlson S. Transforming growth factor-beta1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MC, Martin JS, Cousins FM, Kulkarni AB, Karlsson S, Akhurst RJ. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta1 knock out mice. Development. 1995;121:1845–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- Bonyadi M, Rusholme SAB, Cousins FM, Su HC, Biron CA, Farrall M, Akhurst RJ. Mapping of a major genetic modifier of embryonic lethality in TGFbeta1 knockout mice. Nat Genet. 1997;15:207–211. doi: 10.1038/ng0297-207. [DOI] [PubMed] [Google Scholar]
- Kitazawa K, Tada T. Elevation of transforming growth factor-beta 1 level in cerebrospinal fluid of patients with communicating hydrocephalus after subarachnoid hemorrhage. Stroke. 1994;25:1400–1404. doi: 10.1161/01.str.25.7.1400. [DOI] [PubMed] [Google Scholar]
- Takizawa T, Tada T, Kitazawa K, Tanaka Y, Hongo K, Kameko M, Uemura KI. Inflammatory cytokine cascade released by leukocytes in cerebrospinal fluid after subarachnoid hemorrhage. Neurol Res. 2001;23:724–730. doi: 10.1179/016164101101199243. [DOI] [PubMed] [Google Scholar]
- Tada T, Kanaji M, Kobayashi S. Induction of communicating hydrocephalus in mice by intrathecal injection of human recombinant transforming growth factor-beta 1. J Neuroimmunol. 1994;50:153–158. doi: 10.1016/0165-5728(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Galbreath E, Kim S-J, Park K, Brenner M, Messing A. Overexpression of TGF-beta1 in the central nervous system of transgenic mice results in hydrocephalus. J Neuropathol Exp Neurol. 1995;54:339–349. doi: 10.1097/00005072-199505000-00007. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Sigurdson SL, Sheppard D, Lwebuga-Mukasa JS. Differential modulation of integrin receptors and extracellular matrix laminin by transforming growth factor-beta 1 in rat alveolar epithelial cells. Exp Cell Res. 1995;221:385–394. doi: 10.1006/excr.1995.1389. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border WA, Noble NA. TGF-beta in kidney fibrosis: A target for gene therapy. Kidney Int. 1997;51:1388–1396. doi: 10.1038/ki.1997.190. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. Transforming growth factor-beta. In: R A F Clark, editor. The Molecular and Cellular Biology of Wound Repair. Second. New York, Plenum Press; 1996. p. 275–308. [Google Scholar]
- Awad M, El-Gamel A, Hasleton P, Turner D, Sinnott P, Hutchinson I. Genotypic variation in the transforming growth factor-beta1 gene. Transplantation. 1998;66:1014–1020. doi: 10.1097/00007890-199810270-00009. [DOI] [PubMed] [Google Scholar]
- Arkwright PD, Laurie S, Super M, Pravica V, Schwarz MJ, Webb AK, Hutchinson IV. TGF-beta genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax. 2000;55:459–462. doi: 10.1136/thorax.55.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper MS. Transforming growth factor-beta: Vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/S1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Pepper MS, Vassalli J-D, Orci L, Montesano R. Biphasic effect of transforming growth factor-beta1 on in vitro angiogenesis. Exp Cell Res. 1993;204:356–363. doi: 10.1006/excr.1993.1043. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997;61:60–67. doi: 10.1086/513903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomi J, Mount RJ, Toporsian M, Paterson AD, Wallace MC, Harrison RV, Letarte M. Cerebral vascular abnormalities in a murine model of hereditary hemorrhagic telangiectasia. Stroke. 2003;34:783–789. doi: 10.1161/01.STR.0000056170.47815.37. [DOI] [PubMed] [Google Scholar]
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahue PK, Li L, Miyazono K, Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta1 signaling in the regulation of angiogensis. Proc Natl Acad Sci USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/S1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan D, Mucke L, Masliah E. Chronic overproduction of TGF-beta1 in astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthanthiran M, Li B, Song JO, Ding R, Sharma VK, Schwartz JE, August P. Transforming growth factor–beta1 hyperexpression in African-American hypertensives: A novel mediator of hypertension and/or target organ damage. Proc Natl Acad Sci USA. 2000;97:3479–3484. doi: 10.1073/pnas.050420897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- Haan J, Maat-Schieman MLC, Roos RAC. Clinical aspects of cerebral amyloid angiopathy. Dementia. 1994;5:210–213. doi: 10.1159/000106725. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: Presentations without lobar hemorrhage. Neurology. 1993;43:2073–2079. doi: 10.1212/wnl.43.10.2073. [DOI] [PubMed] [Google Scholar]
- Mancardi GL, Perdelli F, Rivano C, Leonardi A, Bugiani O. Thickening of the basement membrane of cortical capillaries in Alzheimer’s disease. Acta Neuropathol. 1980;49:79–83. doi: 10.1007/BF00692225. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Chui HC, Saperia D, Athanikar J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 1990;508:13–19. doi: 10.1016/0006-8993(90)91111-S. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Premkumar DRD, Pax AB, Cohen DL, Lieberburg I. Production and increased detection of amyloid β protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Mol Brain Res. 1996;35:58–68. doi: 10.1016/0169-328X(95)00180-Z. [DOI] [PubMed] [Google Scholar]
- Vinters HV, Secor DL, Read SL, Frazee JG, Tomiyasu U, Stanley TM, Ferreiro JA, Akers M-A. Microvasculature in brain biopsy specimens from patients with Alzheimer’s disease: An immunohistochemical and ultrastructual study. Ultrastruct Pathol. 1994;18:333–348. doi: 10.3109/01913129409023202. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease: A double-edged sword. Neuron. 2002;35:419–432. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Eddleston MP, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa S, Planchenault T, Charriere-Bertrand C, Mouchel Y, Fages C, Juliano S, Lefrancois T, Barlovatz-Meimon G, Tardy M. Astroglial permissivity for neuritic outgrowth in neuron-astrocyte cocultures depends on regulation of laminin bioavailability. Glia. 2002;37:105–113. doi: 10.1002/glia.10015. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR. Neuropathological changes caused by hydrocephalus. Neuropahtologica. 1993;85:573–585. doi: 10.1007/BF00334666. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Maher PA, Kumari VG. Regulation of astrocyte GFAP expression by TGF-beta1 and FGF-2. Glia. 1998;22:202–210. doi: 10.1002/(SICI)1098-1136(199802)22:2<202::AID-GLIA11>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788–5792. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM. Transforming growth factor beta (TGF-beta) in inflammation: A cause and a cure. J Clin Immunol. 1992;12:61–74. doi: 10.1007/BF00918135. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Borrow P, Brooker MJ, Mucke L. Astroglial overproduction of TGF-beta1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol. 1997;77:45–50. doi: 10.1016/S0165-5728(97)00049-0. [DOI] [PubMed] [Google Scholar]
- Grammas P, Ovase R. Cerebrovascular transforming growth factor-beta contributes to inflammation in the Alzheimer’s disease brain. Am J Pathol. 2002;160:1583–1587. doi: 10.1016/s0002-9440(10)61105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang L, Ye W, Che X-M, Roessler BJ, Betz AL, Yang G-Y. Reduction of inflammatory response in the mouse brain with adenoviral-mediated transforming growth factor-beta1 expression. Stroke. 2001;32:544–552. doi: 10.1161/01.str.32.2.544. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Krieglstein K. TGF-betas and their roles in the regulation of neuron survival. Adv Exp Med Biol. 2002;513:353–374. doi: 10.1007/978-1-4615-0123-7_13. [DOI] [PubMed] [Google Scholar]
- Gross CE, Bednar MM, Howard DB, Sporn MB. Transforming growth factor-beta 1 reduces infarct size after experimental cerebral ischemia in a rabbit model. Stroke. 1993;24:558–562. doi: 10.1161/01.str.24.4.558. [DOI] [PubMed] [Google Scholar]
- Henrich-Noack P, Prehn JHM, Krieglstein J. TGF-beta1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke. 1996;27:1609–1615. doi: 10.1161/01.str.27.9.1609. [DOI] [PubMed] [Google Scholar]
- Boche D, Cunningham C, Gauldie J, Perry VH. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab. 2003;23:1174–1182. doi: 10.1097/01.WCB.0000090080.64176.44. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yang G-Y, Ahlemeyer B, Pang L, Che X-M, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-beta1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci U S A. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsicker K, Krieglstein K. Co-activation of TGF-beta and cytokine signaling pathways are required for neurotropic functions. Cytokine Growth Factor Rev. 2000;11:97–102. doi: 10.1016/S1359-6101(99)00033-7. [DOI] [PubMed] [Google Scholar]
- Luckenbill-Edds L. Laminin and the mechanism of neuronal outgrowth. Brain Res Rev. 1997;23:1–27. doi: 10.1016/S0165-0173(96)00013-6. [DOI] [PubMed] [Google Scholar]
- Venstrom KA, Reichardt LF. Extracellular matrix. 2: Role of extracellular matrix molecules and their receptors in the nervous system. Faseb J. 1993;7:996–1003. doi: 10.1096/fasebj.7.11.8370483. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kim SM, Ramaswami M. Retrograde regulation in the CNS; neuron-specific interpretations of tgf-Beta signaling. Neuron. 2004;41:845–848. doi: 10.1016/S0896-6273(04)00152-7. [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/S0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Zheng X, Wang J, Haerry TE, Wu AY, Martin J, O'Connor MB, Lee CH, Lee T. TGF-beta signaling activates steroid hormone receptor expression during neuronal remodeling in the Drosophila brain. Cell. 2003;112:303–315. doi: 10.1016/S0092-8674(03)00072-2. [DOI] [PubMed] [Google Scholar]
- Zhang F, Endo S, Cleary LJ, Eskin A, Byrne JH. Role of transforming growth factor–beta in long-term synaptic facilitation in Aplysia. Science. 1997;275:1318. doi: 10.1126/science.275.5304.1318. [DOI] [PubMed] [Google Scholar]
- Chin J, Angers A, Cleary LJ, Eskin A, Byrne JH. Transforming growth factor beta1 alters synapsin distribution and modulates synaptic depression in Aplysia. J Neurosci. 2002;22:1–6. doi: 10.1523/JNEUROSCI.22-09-j0004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Hansen LA. Structural basis of the cognitive alterations in Alzheimer disease. In: R D Terry, R Katzman and K L Bick, editor. Alzheimer Disease. New York, Raven Press; 1994. p. 179–196. [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang FS, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu G, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:614–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and Alzheimer’s disease. Nature. 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano S, Stern Y, Mayeux R. Alzheimer’s disease after remote head injury: An incidence study. J Neurol Neurosurg Psychiatry. 1997;62:119–124. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisilevsky R, Fraser PE. Aβ amyloidogenesis: unique, or variation on a systemic theme? Crit Rev Biochem Mol Biol. 1997;32:361–404. doi: 10.3109/10409239709082674. [DOI] [PubMed] [Google Scholar]
- Fillit H, Leveugle B. Disorders of the extracellular matrix and the pathogenesis of senile dementia of the Alzheimer’s type. Lab Invest. 1995;72:249–253. [PubMed] [Google Scholar]
- Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG. An important role of heparan sulfate proteoglycan (perlecan) in a model system for the deposition and persistence of fibrillar Aβ-amyloid in rat brain. Neuron. 1994;12:219–234. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- Monning U, Sandbrink R, Banati RB, Masters CL, Beyreuther K. Transforming growth factor beta mediates increase of mature transmembrane amyloid precursor protein in microglial cells. FEBS Lett. 1994;342:267–272. doi: 10.1016/0014-5793(94)80514-8. [DOI] [PubMed] [Google Scholar]
- Burton T, Liang B, Dibrov A, Amara F. Transforming growth factor-beta-induced transcription of the Alzheimer beta-amyloid precursor protein gene involves interaction between the CTCF-complex and Smads. Biochem Biophys Res Commun. 2002;295:713–723. doi: 10.1016/S0006-291X(02)00725-8. [DOI] [PubMed] [Google Scholar]
- Lesne S, Docagne F, Gabriel C, Liot G, Lahiri DK, Buee L, Plawinski L, Delacourte A, MacKenzie ET, Buisson A, Vivien D. Transforming growth factor-beta 1 potentiates amyloid-beta generation in astrocytes and in transgenic mice. J Biol Chem. 2003;278:18408–18418. doi: 10.1074/jbc.M300819200. [DOI] [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo Y–M, Roher AE. Cerebral amyloid angiopathy. Amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Shi J, Bailey K, Mann DM. Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer's disease. Neurosci Lett. 2003;352:137–140. doi: 10.1016/j.neulet.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Luterman JD, Haroutunian V, Yemul S, Ho L, Purohit D, Aisen PS. Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch Neurol. 2000;2000:1153–1160. doi: 10.1001/archneur.57.8.1153. [DOI] [PubMed] [Google Scholar]
- Buckwalter M, Pepper JP, Gaertner RF, Von Euw D, Lacombe P, Wyss-Coray T. Molecular and functional dissection of TGF-beta1-induced cerebrovascular abnormalities in transgenic mice. Ann N Y Acad Sci. 2002;977:87–95. doi: 10.1111/j.1749-6632.2002.tb04801.x. [DOI] [PubMed] [Google Scholar]
- Peress NS, Perillo E. Differential expression of TGF-beta 1, 2 and 3 isotypes in Alzheimer's disease: a comparative immunohistochemical study with cerebral infarction, aged human and mouse control brains. J Neuropathol Exp Neurol. 1995;54:802–811. doi: 10.1097/00005072-199511000-00007. [DOI] [PubMed] [Google Scholar]
- van der Wal EA, Gómez-Pinilla F, Cotman CW. Transforming growth factor-beta1 is in plaques in Alzheimer and Down pathologies. Neuroreport. 1993;4:69–72. doi: 10.1097/00001756-199301000-00018. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002;23:237–243. doi: 10.1016/S0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Dillon-Carter O, Tourtellotte WW, Carvey P, Freed WJ. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson's disease in ventricular cerebrospinal fluid. Exp Neurol. 1996;142:313–322. doi: 10.1006/exnr.1996.0200. [DOI] [PubMed] [Google Scholar]
- Mogi M, Harada M, Kondo T, Narabayashi H, Riederer P, Nagatsu T. Transforming growth factor-beta 1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson's disease. Neurosci Lett. 1995;193:129–132. doi: 10.1016/0304-3940(95)11686-Q. [DOI] [PubMed] [Google Scholar]
- Ilzecka J, Stelmasiak Z, Dobosz B. Transforming growth factor-Beta 1 (tgf-Beta 1) in patients with amyotrophic lateral sclerosis. Cytokine. 2002;20:239–243. doi: 10.1006/cyto.2002.2005. [DOI] [PubMed] [Google Scholar]
- Houi K, Kobayashi T, Kato S, Mochio S, Inoue K. Increased plasma TGF-beta1 in patients with amyotrophic lateral sclerosis. Acta Neurol Scand. 2002;106:299–301. doi: 10.1034/j.1600-0404.2002.01301.x. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Middelberg-Bisping K, Drewes C, Schatz H. Elevated plasma levels of transforming growth factor-beta 1 in NIDDM. Diabetes Care. 1996;19:1113–1117. doi: 10.2337/diacare.19.10.1113. [DOI] [PubMed] [Google Scholar]
- Wiessner C, Gehrmann J, Lindholm D, Topper R, Kreutzberg GW, Hossmann KA. Expression of transforming growth factor-beta 1 and interleukin-1 beta mRNA in rat brain following transient forebrain ischemia. Acta Neuropathol (Berl) 1993;86:439–446. doi: 10.1007/BF00228578. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Kiefer R, Finsen B, Diemer NH, Zimmer J, Hartung HP. Cytokines in cerebral ischemia: expression of transforming growth factor beta-1 (TGF-beta 1) mRNA in the postischemic adult rat hippocampus. Exp Neurol. 1995;131:114–123. doi: 10.1016/0014-4886(95)90013-6. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, Diemer NH, Hartung HP, Finsen B. Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia. 1998;24:437–448. doi: 10.1002/(SICI)1098-1136(199812)24:4<437::AID-GLIA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Mol Brain Res. 1996;40:1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- Martinez G, Carnazza ML, Di Giacomo C, Sorrenti V, Vanella A. Expression of bone morphogenetic protein-6 and transforming growth factor-beta1 in the rat brain after a mild and reversible ischemic damage. Brain Res. 2001;894:1–11. doi: 10.1016/S0006-8993(00)03140-1. [DOI] [PubMed] [Google Scholar]
- Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams C, Gluckman PD. Hypoxia-ischemia induces transforming growth factor beta 1 mRNA in the infant rat brain. Brain Res Mol Brain Res. 1992;13:93–101. doi: 10.1016/0169-328X(92)90048-G. [DOI] [PubMed] [Google Scholar]
- Wang X, Yue TL, White RF, Barone FC, Feuerstein GZ. Transforming growth factor-beta 1 exhibits delayed gene expression following focal cerebral ischemia. Brain Res Bull. 1995;36:607–609. doi: 10.1016/0361-9230(94)00243-T. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-beta1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27:852–857. doi: 10.1161/01.str.27.5.852. [DOI] [PubMed] [Google Scholar]
- Ali C, Docagne F, Nicole O, Lesne S, Toutain J, Young A, Chazalviel L, Divoux D, Caly M, Cabal P, Derlon JM, MacKenzie ET, Buisson A, Vivien D. Increased expression of transforming growth factor-beta after cerebral ischemia in the baboon: an endogenous marker of neuronal stress? J Cereb Blood Flow Metab. 2001;21:820–827. doi: 10.1097/00004647-200107000-00007. [DOI] [PubMed] [Google Scholar]
- Flood C, Akinwunmi J, Lagord C, Daniel M, Berry M, Jackowski A, Logan A. Transforming growth factor-beta1 in the cerebrospinal fluid of patients with subarachnoid hemorrhage: titers derived from exogenous and endogenous sources. J Cereb Blood Flow Metab. 2001;21:157–162. doi: 10.1097/00004647-200102000-00007. [DOI] [PubMed] [Google Scholar]
- Whitelaw A, Christie S, Pople I. Transforming growth factor-beta1: a possible signal molecule for posthemorrhagic hydrocephalus? Pediatr Res. 1999;46:576–580. doi: 10.1203/00006450-199911000-00014. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Mehta PS, Bruccoleri A, Brown HW, Harry GJ. Increase in brain stem cytokine mRNA levels as an early response to chemical-induced myelin edema. J Neuroimmunol. 1998;88:154–164. doi: 10.1016/S0165-5728(98)00116-7. [DOI] [PubMed] [Google Scholar]
- Acarin L, Gonzalez B, Castellano B. Neuronal, astroglial and microglial cytokine expression after an excitotoxic lesion in the immature rat brain. Eur J Neurosci. 2000;12:3505–3520. doi: 10.1046/j.1460-9568.2000.00226.x. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA. Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci. 2000;12:2333–2344. doi: 10.1046/j.1460-9568.2000.00131.x. [DOI] [PubMed] [Google Scholar]
- Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-beta1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol. 1993;120:291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Nichols NR, Tocco G, Morgan T, Laping N, Finch CE. Transforming growth factor beta1 and fibronectin messenger RNA in rat brain: Responses to injury and cell-type localization. Neuroscience. 1993;54:893–907. doi: 10.1016/0306-4522(93)90583-2. [DOI] [PubMed] [Google Scholar]
- Semple-Rowland SL, Mahatme A, Popovich PG, Green DA, Hassler G., Jr., Stokes BT, Streit WJ. Analysis of TGF-beta 1 gene expression in contused rat spinal cord using quantitative RT-PCR. J Neurotrauma. 1995;12:1003–1014. doi: 10.1089/neu.1995.12.1003. [DOI] [PubMed] [Google Scholar]
- Logan A, Frautschy SA, Gonzalez A-M, Sporn MB, Baird A. Enhanced expression of transforming growth factor beta1 in the rat brain after a localised cerebral injury. Brain Res. 1992;587:216–225. doi: 10.1016/0006-8993(92)91000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta1 in the rat brain: Increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi T, Ueda J, Hanyu T, Abe H, Sawaguchi S. Distribution and expression of transforming growth factor-beta and platelet-derived growth factor in the normal and glaucomatous monkey optic nerve heads. Jpn J Ophthalmol. 2001;45:592–599. doi: 10.1016/S0021-5155(01)00414-2. [DOI] [PubMed] [Google Scholar]
- Link J, Sîderstrîm M, Olsson T, Hîjeberg B, Ljungdahl è, Link H. Increased transforming growth factor-beta, interleukin-4, and interferon-gamma in multiple sclerosis. Ann Neurol. 1994;36:379–386. doi: 10.1002/ana.410360309. [DOI] [PubMed] [Google Scholar]
- Rollnik JD, Sindern E, Schweppe C, Malin JP. Biologically active TGF-beta 1 is increased in cerebrospinal fluid while it is reduced in serum in multiple sclerosis patients. Acta Neurol Scand. 1997;96:101–105. doi: 10.1111/j.1600-0404.1997.tb00248.x. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, DiMarco R, Patti F, Reggio E, Nicoletti A, Zaccone P, Stivala F, Meroni PL, Reggio A. Blood levels of transforming growth factor-beta 1 (TGF-beta1) are elevated in both relapsing remitting and chronic progressive multiple sclerosis (MS) patients and are further augmented by treatment with interferon-beta 1b (IFN-beta1b) Clin Exp Immunol. 1998;113:96–99. doi: 10.1046/j.1365-2249.1998.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot CJA, Montagne L, Barten AD, Sminia P, Van Der Valk P. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta Type I and Type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J Neuropathol Exp Neurol. 1999;58:174–187. doi: 10.1097/00005072-199902000-00007. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, Bielecki B, Ransohoff RM. Chemokine upregulation follows cytokine expression in chronic relapsing experimental autoimmune encephalomyelitis. Scand J Immunol. 2003;58:81–88. doi: 10.1046/j.1365-3083.2003.01285.x. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Funa K, Schweitzer T, Jung S, Bourde O, Toyka KV, Hartung HP. Transforming growth factor-beta 1 in experimental autoimmune neuritis. Cellular localization and time course. Am J Pathol. 1996;148:211–223. [PMC free article] [PubMed] [Google Scholar]
- Sindern E, Schweppe K, Ossege LM, Malin JP. Potential role of transforming growth factor-beta 1 in terminating the immune response in patients with Guillain-Barre syndrome. J Neurol. 1996;243:264–268. doi: 10.1007/BF00868524. [DOI] [PubMed] [Google Scholar]
- Dahle C, Kvarnstrom M, Ekerfelt C, Samuelsson M, Ernerudh J. Elevated number of cells secreting transforming growth factor beta in Guillain-Barre syndrome. Apmis. 2003;111:1095–1104. doi: 10.1111/j.1600-0463.2003.apm1111204.x. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Gold R, Gehrmann J, Lindholm D, Wekerle H, Kreutzberg GW. Transforming growth factor beta expression in reactive spinal cord microglia and meningeal inflammatory cells during experimental allergic neuritis. J Neurosci Res. 1993;36:391–398. doi: 10.1002/jnr.490360405. [DOI] [PubMed] [Google Scholar]
- Kossmann T, Morganti-Kossmann MC, Orenstein JM, Britt WJ, Wahl SM, Smith PD. Cytomegalovirus production by infected astrocytes correlates with transforming growth factor-beta release. J Infect Dis. 2003;187:534–541. doi: 10.1086/373995. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Boche D, Perry VH. Transforming growth factor beta1, the dominant cytokine in murine prion disease: Influence on inflammatory cytokine synthesis and alteration of vascular extracellular matrix. Neuropathol Appl Neurobiol. 2002;28:107–119. doi: 10.1046/j.1365-2990.2002.00383.x. [DOI] [PubMed] [Google Scholar]
- Ossege LM, Voss B, Wiethege T, Sindern E, Malin JP. Detection of transforming growth factor beta 1 mRNA in cerebrospinal fluid cells of patients with meningitis by non-radioactive in situ hybridization. J Neurol. 1994;242:14–19. doi: 10.1007/BF00920569. [DOI] [PubMed] [Google Scholar]
- Diab A, Zhu J, Lindquist L, Wretlind B, Bakhiet M, Link H. Haemophilus influenzae and Streptococcus pneumoniae induce different intracerebral mRNA cytokine patterns during the course of experimental bacterial meningitis. Clin Exp Immunol. 1997;109:233–241. doi: 10.1046/j.1365-2249.1997.4441343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Chang YC, Chow NH, Wang ST. Level of transforming growth factor beta 1 is elevated in cerebrospinal fluid of children with acute bacterial meningitis. J Neurol. 1997;244:634–638. doi: 10.1007/s004150050159. [DOI] [PubMed] [Google Scholar]
- Liu HM, Yang HB, Chen RM. Expression of basic fibroblast growth factor, nerve growth factor, platelet-derived growth factor and transforming growth factor-beta in human brain abscess. Acta Neuropathol (Berl) 1994;88:143–150. doi: 10.1007/s004010050142. [DOI] [PubMed] [Google Scholar]