

Abstract
The characterization of protein binding processes — with all of the key conformational changes — has been a grand challenge in the field of biophysics. Here, we have used the weighted ensemble path sampling strategy to orchestrate molecular dynamics simulations, yielding atomistic views of protein–peptide binding pathways involving the MDM2 oncoprotein and an intrinsically disordered p53 peptide. A total of 182 independent, continuous binding pathways were generated, yielding a kon that is in good agreement with experiment. These pathways were generated in 15 days using 3500 cores of a supercomputer, substantially faster than would be possible with “brute force” simulations. Many of these pathways involve the anchoring of p53 residue F19 into the MDM2 binding cleft when forming the metastable encounter complex, indicating that F19 may be a kinetically important residue. Our study demonstrates that it is now practical to generate pathways and calculate rate constants for protein binding processes using atomistic simulation on typical computing resources.
Graphical Abstract
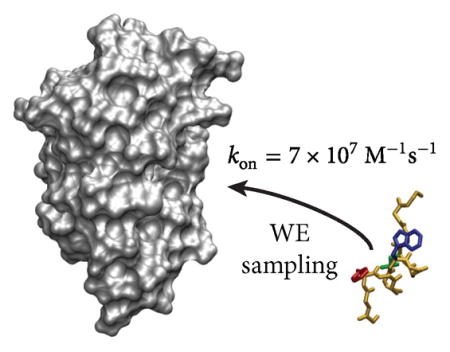
The complete characterization of protein binding processes has remained elusive to laboratory experiments due to the fleeting nature of transition and intermediate states along the pathways. Molecular dynamics (MD) simulations can, in principle, provide a “microscope” for viewing these critical biological processes in full atomic detail and high temporal resolution but are computationally demanding. While advances in computer hardware and software have led to notable successes in simulating protein binding events with small-molecule inhibitors1–4 and a phosphotetrapeptide,5 such brute force simulations (simply running the simulations long enough to capture at least one binding event) are not practical on typical computing resources.
Alternative simulation strategies have therefore been pursued extensively, and these fall roughly into two categories: methods employing discrete states using short, discontinuous trajectories and those employing continuous trajectories to connect states. Methods in the first category involve the construction of Markov state models to estimate observables such as rate constants for long-timescale processes6 and have been applied to protein–ligand binding and/or unbinding processes.7–9 Methods in the second category involve focusing computing power on the “rare”, or infrequent, functional motions rather than the stable states (e.g., protein conformational changes upon binding, and not on the unbound or bound states) without altering the underlying dynamics.10 Although “rare event” strategies have successfully generated pathways and rate constants for protein–ligand unbinding processes,11,12 such strategies (to our knowledge) have not been applied to directly simulate any protein binding processes.
One such strategy is the weighted ensemble (WE) path sampling method,13 which can generate continuous trajectories and rate constants for the rare event of interest in a rigorous manner for any type of stochastic dynamics.14 The WE strategy exhibits a set of characteristics that, taken together, set it apart from other rare events methods. First, kinetic observables can be computed from a single simulation without a Markov assumption. Second, states (e.g., unbound, intermediate, and bound states) need not be strictly defined in advance, permitting refinement of the definitions after completion of the simulation.15 Third, trajectories in WE are queried at fixed time intervals and do not need to be “caught in the act” of crossing an interface, unlike many methods (e.g., milestoning, transition interface sampling, and forward flux sampling),10 which permits facile interoperability with multiple software packages whether at the atomistic scale or beyond.16,17 Finally, our WE implementation (called WESTPA18) scales out to thousands of CPU cores and can exhibit “superlinear” performance by estimating observables with less overall computing effort than standard MD simulations.19–22
Here, we have applied the WE strategy to MD simulations of the binding process involving a p53 peptide (residues 17–29) and the MDM2 protein. Because many cancers are linked to inactivation of p53 due to overexpression of MDM2,23 there is great interest in characterizing the mechanism of the MDM2–p53 binding process in order to determine ways to disrupt the association.24 Furthermore, the MDM2–p53 complex is a classic system for studying the binding process of an intrinsically disordered peptide that folds only upon binding to its structured partner protein (in this case, adopting a helical conformation).25,26 MD simulations have provided extensive sampling of the unbound/bound states of such systems,27–29 including the p53 peptide;30–32 however, to our knowledge, no binding pathways have been reported. In this work, we generated an ensemble of pathways for the complete MDM2–p53 binding process starting from unbound states (with a separation of 30 Å), yielding converged rate constants. Consistent with previous simulation studies,33–38 our model of MDM2 is truncated (residues 25–109), lacking the flexible N-terminal lid over the binding cleft39 that is unresolved in the crystal structure.26 Our simulations were carried out using the AMBER ff99SB-ILDN force field40 and GB/SA implicit solvent41–43 with water-like viscosity at the same temperature (25 °C) and ionic strength (150 mM) as kinetics experiments of MDM2–p53 binding.44
Binding Pathways and Kinetics
Our WE simulation of the MDM2–p53 binding process resulted in a thorough exploration of the space around the MDM2 protein by the p53 peptide (Figure S1, Supporting Information) and generated a total of 182 independent, continuous binding trajectories in 15 days using up to 3500 cores at a time on the XSEDE Stampede supercomputer (aggregate simulation time of ~120 μs). The simulation was initiated from 1554 pre-equilibrated unbound states in which the binding partners were separated by 30 Å at random orientations, drawing from a diverse ensemble of unbound p53 peptide conformations (Figure S2). Although this ensemble included some nonhelical conformations, the peptide exhibited a high propensity for helical conformations, which is consistent with results from explicit solvent simulations30,31 as well as those from NMR31,45 and UV resonance Raman spectroscopy.30
On the basis of our simulations, binding involves a two-step process in which diffusive collisions of p53 and MDM2 form a metastable “encounter complex” intermediate, followed by rearrangement of this encounter complex to the bound state (Figure 1). Two thirds (~80 μs) of our ~120 μs of aggregate dynamics was involved in binding pathways, with ~10% of the diffusional collisions between the p53 peptide and MDM2 protein being productive (i.e., eventually resulting in the native complex); the remaining ~90% of collisions either occurred in the wrong orientation or did not occur at the binding site and further did not lead to binding on the time scale of our simulation. Thus, all of the binding pathways that were generated involved unbound states in which the binding interfaces of the partners were oriented toward each other. Of future interest are improvements to WE strategies for generating a greater diversity of binding pathways, including more optimal schemes for replication and combination of trajectory walkers along the selected progress coordinate.
Figure 1.

Representative, continuous MDM2–p53 binding pathway obtained from WE simulation, superimposed on the probability distribution over the WE progress coordinate: binding RMSD vs minimum MDM2–p53 separation (upper left) and preorganization RMSD of the p53 peptide vs binding RMSD (lower left). The preorganization RMSD reflects the similarity of the p53 peptide to its conformation in the minimized crystal structure and was calculated as the heavy-atom RMSD of the three hydrophobic anchor residues (F19, W23, and L26) of p53 after alignment on all heavy atoms of p53. At the beginning of the WE simulation (1), the p53 peptide (yellow) is separated from MDM2 (gray) by 30 Å. After diffusing to an encounter complex (2), p53 residue W23 (blue) buries in its native position (3) while F19 (red) is buried in a non-native position. The p53 peptide then rearranges to the bound state (4), with F19 (red), W23 (blue), and L26 (green) buried in their native, bound-state positions. This trajectory is also illustrated in Movie S1.
Because our WE setup (e.g., progress coordinate, scheme for combining trajectories, etc.) was intended to enhance the sampling of association events, we did not observe a sufficient number of dissociation events to directly compute rate constants for the unbinding direction and therefore focused solely on characterizing the kinetics in the binding direction (Table 1). Our computed association rate constant kon [(7 ± 4) × 107 M−1 s−1; here and throughout, uncertainties represent 95% confidence intervals] is in good agreement with experiment (9.2 × 106 M−1 s−1).44 The somewhat faster computed kon may be due to the fact that the MDM2 protein in our simulation is truncated such that the binding cleft is constitutively open in contrast to the 10% open population in full-length MDM2,46 which was used for the stopped-flow kinetics experiment.44 Nonetheless, the 8-fold difference in the computed vs experimental kon amounts to a difference of only 1.2 kcal/mol in the overall free energy barrier to association. Given that the computed rate constant for the formation of the encounter complex k1 [(1.1 ± 0.2) × 108 M−1 s−1] is approximately equal to the computed kon [(7 ± 4) × 107 M−1 s−1] (see Table 1), the kinetics of the binding process must be close to the limiting case where the dissociation of the encounter complex is much slower than its rearrangement to the bound state; that is, assuming a mechanism of
Table 1.
Rate Constants and 95% Confidence Intervals for MDM2–p53 Association
| process | rate constant | value |
|---|---|---|
| unbound state → encounter complex | k1 | (1.1 ± 0.2) × 108 M−1 s−1 |
| encounter complex → bound state | k2 | (4 ± 2) × 105 s−1 |
| unbound state → bound state | kon | (7 ± 4) × 107 M−1 s−1 |
| (1) |
it must be the case that k−1 ≪ k2, such that the steady-state expression for the overall association rate constant kon = k1k2/(k−1 + k2) reduces to kon ≈ k1.47 [Our WE simulation yields a k2 of (4 ± 2) × 105 s−1.] Thus, we predict that the experimentally measured kon for the p53 peptide and truncated MDM2 protein would be predominated by the diffusion-controlled formation of the encounter complex. As expected, the rate constant k1 for this initial step (108 M−1 s−1) is lower than the Smoluchowski limit of ~5 × 109 M−1 s−1 due to orientational constraints.48
A caveat of all rare event strategies, including WE, is that the most probable pathways may be missed. For WE and related approaches, relevant barriers to be surmounted could be orthogonal to the progress coordinate(s) used to focus the sampling. In principle, however, one needs to focus on only the sampling along the slow, uncorrelated coordinates, thereby capturing other faster, correlated coordinates. In our simulations, the progress coordinate used to drive WE sampling explicitly included only the slow motions of the p53 peptide (folding and binding), and thus only MDM2 dynamics on time scales faster than tens of microseconds (the aggregate time of our simulation) were directly sampled in our simulation. Thus, MDM2, which undergoes large-scale conformational changes upon binding,36 was largely preorganized for binding, and any effects of long-timescale MDM2 dynamics (including inter-conversion between alternate unbound conformations) on the rate of association are absent from this simulation. Nonetheless, the estimated kon based on our simulation is in reasonable agreement with experiment. We also note that our progress coordinate was based on an experimentally determined bound pose, and though we refined the definition of the bound state based on our simulation (see the Methods section), exploration of binding in systems without a known bound pose would require a less specific progress coordinate, likely leading to increased computational expense. This expense might be mitigated by recently developed WE strategies such as WExplore,49 which can generate a diverse set of binding/unbinding metastable intermediate states at a reasonable computational cost.11 An implementation of WExplore has been implemented as a WESTPA-compatible plugin.
Role of p53 Residue F19
To further characterize the encounter complex, a probability distribution was generated as a function of the heavy-atom RMSD of three key “anchor” residues of the p53 peptide from its MDM2-bound crystallographic pose after alignment of (a) MDM2 and (b) itself (Figure 1). The anchor residues—F19, W23, and L26—are the residues of the p53 peptide that become the most buried upon binding MDM2. Such deeply buried residues have been proposed to function as anchors that smooth out the binding process by avoiding kinetically costly structural rearrangements.50 In our simulations, ~90% of the encounter complex conformations feature at least partial (>45%) burial of F19, with 25% of the conformations involving complete (>95%) burial, suggesting that F19 may be a kinetically important residue for MDM2–p53 binding. Consistent with the potential kinetic importance of F19, simulations by others have found that the F19A mutant p53 peptide is unable to anchor into the binding cleft; instead, the peptide slides along the MDM2 surface in a nonspecific manner.51 Although F19 has been identified by thermodynamic experiments to be crucial for binding,52 no kinetics experiments have yet been reported.
Efficiency of WE Simulation
To estimate the efficiency of WE sampling relative to brute force simulation, we focus on the amount of computing time that would be required for brute force simulation to estimate the same kon and therefore the same number of independent binding events as that of our WE simulation on the same computing resource (XSEDE’s Stampede). In particular, 0.77 ms of aggregate brute force simulation time (e.g., as a single long trajectory or multiple shorter trajectories) would be required to generate 182 binding events, whereas our WE simulation required only 120 μs, yielding an efficiency of ~6× that of brute force sampling. If one is satisfied with an order-of-magnitude estimate for kon, the efficiency is ~10×, based on 58 μs of WE sampling comprised of 129 statistically independent trajectories. Furthermore, as the efficiency of WE sampling increases with the free energy barriers of the rare event process,11,21,53–55 the generation of pathways and rate constants for significantly slower processes, including unbinding processes, will become increasingly more feasible as the speed of dynamics engines continues to increase (e.g., via GPU acceleration).
In closing, we have used the WE path sampling approach13,56 to simulate the binding between an N-terminal peptide fragment of the p53 tumor suppressor with the MDM2 oncoprotein in atomic detail. Our simulation generated 182 independent, continuous binding pathways, yielding a computed kon that is consistent with experimental studies. In the absence of the N-terminal flexible lid in MDM2, MDM2–p53 binding is diffusion-controlled due to the formation of a metastable encounter complex state, which subsequently undergoes rearrangement to the native, bound state. A key feature of the encounter complex state is the anchoring of the p53 residue F19 into the binding cleft of MDM2, suggesting that F19 may be a kinetically important residue for MDM2–p53 binding. Our WE simulations provided pathways in atomic detail with rigorous rate constants for a complex biological process, namely, protein–peptide binding, in 15 days on a supercomputer, substantially more quickly than would be possible with brute force simulations. These results suggest that rare event strategies like WE could become an important part of the modern biophysics toolbox.
METHODS
Weighted Ensemble (WE) Simulation
We simulated the association between the MDM2 protein (residues 25–109) and p53 peptide (residues 17–29) using the WE strategy13 with an equilibrium reweighting procedure,56 as implemented in our open-source, high-performance WESTPA (Weighted Ensemble Simulation Toolkit with Parallelization and Analysis) software.18 In this strategy, a large number of simulations, or “walkers”, are started in parallel from the initial, unbound state and evaluated for replication or combination every τ time units (according to the standard WE algorithm13) to maintain the desired number of walkers per bin along a progress coordinate toward the target bound state. In our simulations, the initial and target states were the unbound and bound states, respectively, and a τ value of 50 ps was used. In addition, we applied a reweighting procedure56 at regular intervals of τ for the first half of the simulation to accelerate convergence in sampling. This procedure uses the local convergence of kinetics to properly redistribute weight across the entire progress coordinate space.56 As a test of simulation convergence, no equilibrium reweighting was applied in the second half of the simulation to ensure that the results remain unchanged in this part of the simulation.
A two-dimensional progress coordinate was used throughout the WE simulation, consisting of the heavy-atom RMSDs of the p53 peptide relative to its MDM2-bound crystallographic pose26 following alignment on (a) MDM2 (to monitor the extent of binding) and (b) itself (to monitor the extent of preorganization of the peptide for binding). A total of 396 iterations were performed to generate binding pathways, with a maximum trajectory length of 19.8 ns. After 200 WE iterations (about 57 μs of aggregate simulation time), both the probability distribution of progress coordinate values (Figure S3) and the kon (Figure S4) were approximately constant. All analysis was performed using the latter half of the simulation with conformations sampled every 1 ps.
State Definitions
The definition of the bound state (Figure S5A) was refined based on the probability distributions obtained from the WE simulation and confirmed by a separate control simulation that was started from the bound state (Figure S5B); the average heavy-atom RMSD of the overall bound state from the corresponding crystal structure26 was 2.5 ± 0.5 Å. Consistent with previous Brownian dynamics simulations of protein–protein association,57 the encounter complex through which all binding pathways pass was defined as a specific complex (with at least one intermolecular native contact and a certain extent of binding by the p53 anchor residues), as delineated in Figure S5B. The unbound state was defined as a p53–MDM2 separation of >20 Å. We note that the particular separation used to define the unbound state had no appreciable effect on the association kinetics (Figure S6).
Calculation of Rate Constants
Prior to kinetics analysis, the bound state definition was refined to encompass the corresponding free energy basin sampled by the WE simulation (Figure S5). The rate constant kij between states i and j is computed using the following15
where fij is the flux of probability carried by walkers originating in state i and arriving in state j and pi is the fraction of trajectories more recently in i than in j. C0 is the reference concentration of binding partners, calculated as
where r = 50 Å is the radius of the simulation region and NA is Avogadro’s number. (In these simulations, C0 = 3.17 mM.) The bimolecular form is used for the formation of the encounter complex (k1 in eq 1), and the unimolecular form is used for the rearrangement of the encounter complex to the bound state (k2). Normalization by pi amounts to a separation of equilibrium fluxes into multiple steady-state fluxes and is what allows us to extract rate constants corresponding to steady-state experiments from equilibrium data.58 The conditional flux fij from state i to state j is evaluated by tracing the continuous trajectories generated by the WE approach and noting when transitions from state i to state j occur; if such a transition occurs any time within iteration Ni of WE sampling, then that transition generates a contribution w/τ to the conditional flux fij(Ns) from state i to state j arriving within iteration Ns, where w is the weight of the walker at the time of the transition. These flux values may be correlated in time; therefore, uncertainties in the rate constants kij and the number of statistically independent binding events were determined using a blocked Monte Carlo bootstrapping strategy13,59 (see the Supporting Information for details). All reported uncertainties in rate constants correspond to 95% confidence intervals as determined by blocked bootstrapping.
Supplementary Material
Acknowledgments
This work was supported by NIH grant 1R01GM115805-01 to L.T.C. and D.M.Z., NSF CAREER grant MCB-0845216 to L.T.C., University of Pittsburgh Arts & Sciences and Mellon Fellowships to M.C.Z., NIH grant T32-DK061296 to J.L.A., NSF grant MCB-1119091 to D.M.Z., and NSF XSEDE allocation TG-MCB100109 to L.T.C. We thank the Office of the Provost and the Department of Chemistry at Drake University for providing computing resources to M.C.Z. We thank Ernesto Suárez, Steve Lettieri, Karl Debiec, Ali Saglam, Thomas Kiefhaber, and Michael Grabe for constructive discussions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b01502.
Detailed methods and Figures S1–S8, showing sampling results and simulations, p53 conformers, the evolution of the probability distribution of progress coordinate values, the evolution of flux into the bound state, state definitions refined from WE simulations, the dependence of rate constants on the minimum separation defining the unbound state, and autocorrelation results (PDF) Movie S1, showing a representative trajectory of p53 binding to MDM2 (AVI)
References
- 1.Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and Mechanism of Drug Binding to G-Protein-Coupled Receptors. Proc Natl Acad Sci U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shan Y, Kim ET, Eastwood MP, Dror RO, Seeliger MA, Shaw DE. How Does a Drug Molecule Find Its Target Binding Site? J Am Chem Soc. 2011;133:9181–9183. doi: 10.1021/ja202726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buch I, Harvey MJ, Giorgino T, Anderson DP, De Fabritiis G. High-Throughput All-Atom Molecular Dynamics Simulations Using Distributed Computing. J Chem Inf Model. 2010;50:397–403. doi: 10.1021/ci900455r. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Tajkhorshid E. Electrostatic Funneling of Substrate in Mitochondrial Inner Membrane Carriers. Proc Natl Acad Sci U S A. 2008;105:9598–9603. doi: 10.1073/pnas.0801786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giorgino T, Buch I, De Fabritiis G. Visualizing the Induced Binding of SH2-Phosphopeptide. J Chem Theory Comput. 2012;8:1171–1175. doi: 10.1021/ct300003f. [DOI] [PubMed] [Google Scholar]
- 6.Chodera JD, Noé F. Markov State Models of Biomolecular Conformational Dynamics. Curr Opin Struct Biol. 2014;25:135–144. doi: 10.1016/j.sbi.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang D, Caflisch A. The Free Energy Landscape of Small Molecule Unbinding. PLoS Comput Biol. 2011;7:e1002002. doi: 10.1371/journal.pcbi.1002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doerr S, De Fabritiis G. On-The-Fly Learning and Sampling of Ligand Binding by High-Throughput Molecular Simulations. J Chem Theory Comput. 2014;10:2064–2069. doi: 10.1021/ct400919u. [DOI] [PubMed] [Google Scholar]
- 9.Plattner N, Noé F. Protein Conformational Plasticity and Complex Ligand-Binding Kinetics Explored by Atomistic Simulations and Markov Models. Nat Commun. 2015;6:7653–10. doi: 10.1038/ncomms8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zwier MC, Chong LT. Reaching Biological Timescales With All-Atom Molecular Dynamics Simulations. Curr Opin Pharmacol. 2010;10:745–752. doi: 10.1016/j.coph.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Dickson A, Lotz SD. Ligand Release Pathways Obtained With WExplore: Residence Times and Mechanisms. J Phys Chem B. 2016;120:5377–5385. doi: 10.1021/acs.jpcb.6b04012. [DOI] [PubMed] [Google Scholar]
- 12.Teo I, Mayne CG, Schulten K, Lelièvre T. Adaptive Multilevel Splitting Method for Molecular Dynamics Calculation of Benzamidine-Trypsin Dissociation Time. J Chem Theory Comput. 2016;12:2983–2989. doi: 10.1021/acs.jctc.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber GA, Kim S. Weighted-Ensemble Brownian Dynamics Simulations for Protein Association Reactions. Biophys J. 1996;70:97–110. doi: 10.1016/S0006-3495(96)79552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang BW, Jasnow D, Zuckerman DM. The “Weighted Ensemble” Path Sampling Method Is Statistically Exact for a Broad Class of Stochastic Processes and Binning Procedures. J Chem Phys. 2010;132:054107. doi: 10.1063/1.3306345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suárez E, Lettieri S, Zwier MC, Stringer CA, Subramanian SR, Chong LT, Zuckerman DM. Simultaneous Computation of Dynamical and Equilibrium Information Using a Weighted Ensemble of Trajectories. J Chem Theory Comput. 2014;10:2658–2667. doi: 10.1021/ct401065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donovan RM, Sedgewick AJ, Faeder JR, Zuckerman DM. Efficient Stochastic Simulation of Chemical Kinetics Networks Using a Weighted Ensemble of Trajectories. J Chem Phys. 2013;139:115105. doi: 10.1063/1.4821167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donovan RM, Tapia JJ, Sullivan DP, Faeder JR, Murphy RF, Dittrich M, Zuckerman DM. Unbiased Rare Event Sampling in Spatial Stochastic Systems Biology Models Using a Weighted Ensemble of Trajectories. PLoS Comput Biol. 2016;12:e1004611–25. doi: 10.1371/journal.pcbi.1004611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zwier MC, Adelman JL, Kaus JW, Pratt AJ, Wong KF, Rego NB, Suárez E, Lettieri S, Wang DW, Grabe M, et al. WESTPA: An Interoperable, Highly Scalable Software Package for Weighted Ensemble Simulation and Analysis. J Chem Theory Comput. 2015;11:800–809. doi: 10.1021/ct5010615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zwier MC, Kaus JW, Chong LT. Efficient Explicit-Solvent Molecular Dynamics Simulations of Molecular Association Kinetics: Methane/Methane, Na+/Cl−, Methane/Benzene, and K+/18-Crown-6 Ether. J Chem Theory Comput. 2011;7:1189–1197. doi: 10.1021/ct100626x. [DOI] [PubMed] [Google Scholar]
- 20.Adelman JL, Dale AL, Zwier MC, Bhatt D, Chong LT, Zuckerman DM, Grabe M. Simulations of the Alternating Access Mechanism of the Sodium Symporter Mhp1. Biophys J. 2011;101:2399–2407. doi: 10.1016/j.bpj.2011.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saglam AS, Chong LT. Highly Efficient Computation Of The Basal kon Using Direct Simulation Of Protein-Protein Association With Flexible Molecular Models. J Phys Chem B. 2016;120:117–122. doi: 10.1021/acs.jpcb.5b10747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adelman JL, Grabe M. Simulating Current-Voltage Relationships for a Narrow Ion Channel Using the Weighted Ensemble Method. J Chem Theory Comput. 2015;11:1907–1918. doi: 10.1021/ct501134s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 Tumor Suppressor Gene: Clues to Cancer Etiology and Molecular Pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 24.Toledo F, Wahl GM. Regulating the p53 Pathway: In Vitro Hypotheses, in Vivo Veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 25.Joerger AC, Fersht AR. Structural Biology of the Tumor Suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 26.Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 27.Stanley N, Esteban-Martín S, De Fabritiis G. Kinetic Modulation of a Disordered Protein Domain by Phosphorylation. Nat Commun. 2014;5:5272. doi: 10.1038/ncomms6272. [DOI] [PubMed] [Google Scholar]
- 28.Knott M, Best RB. A Preformed Binding Interface in the Unbound Ensemble of an Intrinsically Disordered Protein: Evidence From Molecular Simulations. PLoS Comput Biol. 2012;8:e1002605–10. doi: 10.1371/journal.pcbi.1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ganguly D, Chen J. Modulation of the Disordered Conformational Ensembles of the p53 Transactivation Domain by Cancer-Associated Mutations. PLoS Comput Biol. 2015;11:e1004247–19. doi: 10.1371/journal.pcbi.1004247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong K, Zwier MC, Myshakina NS, Burger VM, Asher SA, Chong LT. Direct Observations of Conformational Distributions of Intrinsically Disordered p53 Peptides Using UV Raman and Explicit Solvent Simulations. J Phys Chem A. 2011;115:9520–9527. doi: 10.1021/jp112235d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittal J, Yoo TH, Georgiou G, Truskett TM. Structural Ensemble of an Intrinsically Disordered Polypeptide. J Phys Chem B. 2013;117:118–124. doi: 10.1021/jp308984e. [DOI] [PubMed] [Google Scholar]
- 32.Pantelopulos GA, Mukherjee S, Voelz VA. Microsecond Simulations of MDM2 and Its Complex With p53 Yield Insight Into Force Field Accuracy and Conformational Dynamics. Proteins: Struct, Funct Genet. 2015;83:1665–1676. doi: 10.1002/prot.24852. [DOI] [PubMed] [Google Scholar]
- 33.Espinoza-Fonseca LM, Trujillo-Ferrara JG. Conformational Changes of the p53-Binding Cleft of MDM2 Revealed by Molecular Dynamics Simulations. Biopolymers. 2006;83:365–373. doi: 10.1002/bip.20566. [DOI] [PubMed] [Google Scholar]
- 34.Eyrisch S, Helms V. Transient Pockets on Protein Surfaces Involved in Protein-Protein Interaction. J Med Chem. 2007;50:3457–3464. doi: 10.1021/jm070095g. [DOI] [PubMed] [Google Scholar]
- 35.Chen HF, Luo R. Binding Induced Folding in p53-MDM2 Complex. J Am Chem Soc. 2007;129:2930–2937. doi: 10.1021/ja0678774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dastidar SG, Lane DP, Verma CS. Multiple Peptide Conformations Give Rise to Similar Binding Affinities: Molecular Simulations of p53-MDM2. J Am Chem Soc. 2008;130:13514–13515. doi: 10.1021/ja804289g. [DOI] [PubMed] [Google Scholar]
- 37.Dastidar SG, Madhumalar A, Fuentes G, Lane DP, Verma CS. Forces Mediating Protein-Protein Interactions: A Computational Study of p53 “Approaching” MDM2. Theor Chem Acc. 2010;125:621–635. [Google Scholar]
- 38.Dastidar SG, Lane DP, Verma CS. Modulation of p53 Binding to MDM2: Computational Studies Reveal Important Roles of Tyr100. BMC Bioinf. 2009;10:S6. doi: 10.1186/1471-2105-10-S15-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy MA, Gesell JJ, Senior MM, Wyss DF. Flexible Lid to the p53-Binding Domain of Human MDM2: Implications for p53 Regulation. Proc Natl Acad Sci U S A. 2003;100:1645–1648. doi: 10.1073/pnas.0334477100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins: Struct, Funct Genet. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 42.Schaefer M, Bartels C, Karplus M. Solution Conformations and Thermodynamics of Structured Peptides: Molecular Dynamics Simulation With an Implicit Solvation Model. J Mol Biol. 1998;284:835–848. doi: 10.1006/jmbi.1998.2172. [DOI] [PubMed] [Google Scholar]
- 43.Onufriev A, Bashford D, Case DA. Modification of the Generalized Born Model Suitable for Macromolecules. J Phys Chem B. 2000;104:3712–3720. [Google Scholar]
- 44.Schon O, Friedler A, Bycroft M, Freund SM, Fersht AR. Molecular Mechanism of the Interaction Between MDM2 and p53. J Mol Biol. 2002;323:491–501. doi: 10.1016/s0022-2836(02)00852-5. [DOI] [PubMed] [Google Scholar]
- 45.Zondlo SC, Lee AE, Zondlo NJ. Determinants of Specificity of MDM2 for the Activation Domains of p53 and P65: Proline27 Disrupts the MDM2-binding Motif of p53. Biochemistry. 2006;45:11945–11957. doi: 10.1021/bi060309g. [DOI] [PubMed] [Google Scholar]
- 46.Showalter SA, Bruschweiler-Li L, Johnson E, Zhang F, Brüschweiler R. Quantitative Lid Dynamics of MDM2 Reveals Differential Ligand Binding Modes of the p53-Binding Cleft. J Am Chem Soc. 2008;130:6472–6478. doi: 10.1021/ja800201j. [DOI] [PubMed] [Google Scholar]
- 47.Kiefhaber T, Bachmann A, Jensen KS. Dynamics and Mechanisms of Coupled Protein Folding and Binding Reactions. Curr Opin Struct Biol. 2012;22:21–29. doi: 10.1016/j.sbi.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 48.Schreiber G, Haran G, Zhou HX. Fundamental Aspects of Protein-Protein Association Kinetics. Chem Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dickson A, Brooks CL., III WExplore: Hierarchical Exploration of High-Dimensional Spaces Using the Weighted Ensemble Algorithm. J Phys Chem B. 2014;118:3532–3542. doi: 10.1021/jp411479c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajamani D, Thiel S, Vajda S, Camacho CJ. Anchor Residues in Protein-Protein Interactions. Proc Natl Acad Sci U S A. 2004;101:11287–11292. doi: 10.1073/pnas.0401942101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dastidar SG, Lane DP, Verma CS. Why Is F19Ap53 Unable to Bind MDM2?: Simulations Suggest Crack Propagation Modulates Binding. Cell Cycle. 2012;11:2239–2247. doi: 10.4161/cc.20333. [DOI] [PubMed] [Google Scholar]
- 52.Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, Lu WY, Lu W. Systematic Mutational Analysis of Peptide Inhibition of the p53-MDM2/Mdmx Interactions. J Mol Biol. 2010;398:200–213. doi: 10.1016/j.jmb.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rojnuckarin A, Livesay DR, Subramaniam S. Bimolecular Reaction Simulation Using Weighted Ensemble Brownian Dynamics and the University of Houston Brownian Dynamics Program. Biophys J. 2000;79:686–693. doi: 10.1016/S0006-3495(00)76327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang BW, Jasnow D, Zuckerman DM. Efficient and Verified Simulation of a Path Ensemble for Conformational Change in a United-Residue Model of Calmodulin. Proc Natl Acad Sci U S A. 2007;104:18043–18048. doi: 10.1073/pnas.0706349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adelman JL, Grabe M. Simulating Rare Events Using a Weighted Ensemble-Based String Method. J Chem Phys. 2013;138:044105. doi: 10.1063/1.4773892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhatt D, Zhang BW, Zuckerman DM. Steady-State Simulations Using Weighted Ensemble Path Sampling. J Chem Phys. 2010;133:014110. doi: 10.1063/1.3456985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gabdoulline RR, Wade RC. On the Protein-Protein Diffusional Encounter Complex. J Mol Recognit. 1999;12:226–234. doi: 10.1002/(SICI)1099-1352(199907/08)12:4<226::AID-JMR462>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 58.Bhatt D, Zuckerman DM. Beyond Microscopic Reversibility: Are Observable Nonequilibrium Processes Precisely Reversible? J Chem Theory Comput. 2011;7:2520–2527. doi: 10.1021/ct200086k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Efron BYB, Tibshirani R. Bootstrap Methods for Standard Errors, Confidence Intervals, and Other Measures of Statistical Accuracy. Stat Sci. 1986;1:54–75. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.