

Abstract
Type 1 diabetes is an autoimmune disease that afflicts millions of people worldwide. It occurs as the consequence of destruction of insulin‐producing pancreatic β‐cells triggered by genetic and environmental factors. The initiation and progression of the disease involves a complicated interaction between β‐cells and immune cells of both innate and adaptive systems. Immune cells, such as T cells, macrophages and dendritic cells, have been well documented to play crucial roles in type 1 diabetes pathogenesis. However, the particular actions of neutrophils, which are the most plentiful immune cell type and the first immune cells responding to inflammation, in the etiology of this disease might indeed be unfairly ignored. Progress over the past decades shows that neutrophils might have essential effects on the onset and perpetuation of type 1 diabetes. Neutrophil‐derived cytotoxic substances, including degranulation products, cytokines, reactive oxygen species and extracellular traps that are released during the process of neutrophil maturation or activation, could cause destruction to islet cells. In addition, these cells can initiate diabetogenic T cell response and promote type 1 diabetes development through cell–cell interactions with other immune and non‐immune cells. Furthermore, relevant antineutrophil therapies have been shown to delay and dampen the progression of insulitis and autoimmune diabetes. Here, we discuss the relationship between neutrophils and autoimmune type 1 diabetes from the aforementioned aspects to better understand the roles of these cells in the initiation and development of type 1 diabetes.
Keywords: Immune cells, Neutrophils, Type 1 diabetes
Introduction
Neutrophils, produced in the bone marrow from myeloblasts, were first discovered by Paul Ehrlich in 18791, 2. They were also called polymorphonuclear leukocytes by Elie Metchnikoff in 18933. After a 14‐day maturation in the bone marrow, neutrophils can be provisionally stored in a pond before being released into the blood4, where they circulate as dormant cells2. When activated, neutrophils are the first cells to be recruited to the locations of inflammation, and provide the first line of defense. They have traditionally been considered as short‐lived effector cells (just 8–12 h in the circulation and 1–2 days in tissues), possessing limited capacity for biosynthetic activity and releasing granules and reactive oxygen species (ROS)5, 6. However, this classical view was challenged by the development of more sensitive approaches and research tools. It has recently been shown that neutrophils have a longer circulatory life span (up to 5 days) than first suggested (Figure 1)5, 6. As an important element of the inflammatory response, neutrophils direct and guide the innate immune response by engaging in complex interactions with macrophages, natural killer cells, dendritic cells and through cross‐talk with most of the cellular effector mediators6, 7. Stimulated by type I interferons (IFNs), neutrophils can contribute to the host response towards intracellular pathogens by involving gene expression8. After activation, neutrophils can promote an innate immune response through releasing soluble pattern recognition molecules (PRMs), which have the capacity to augment phagocytosis, stimulate complement and modulate inflammation. They can also secrete a diversity of cytokines, neutrophil extracellular traps (NETs), and microorganism‐ and tissue‐damaging molecules to participate in innate resistance6. Through a combination of these cytotoxic substances, neutrophils not only eliminate inflammation, but also damage cells and tissues of the host. Additionally, neutrophils are implicated in the activation, recruitment, programming, and modulation of both innate and adaptive immune cells, and can help mediate the initiation of specific T and B cell immunity through soluble mediators or by direct cell–cell contact3, 7, 9. Accordingly, an aberrant neutrophil response can exacerbate and even initiate a variety of diseases, including autoimmune diseases, such as antineutrophil cytoplasmic antibodies‐mediated vasculitis, systemic lupus erythematosus and multiple sclerosis10, 11, 12.
Figure 1.
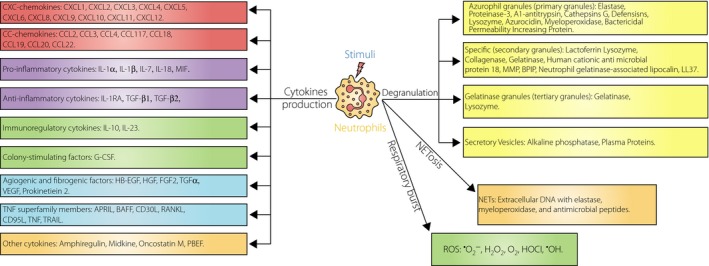
Neutrophil‐derived multiple effector molecules. During the process of maturation or on stimuli activation, neutrophils can express and/or release numerous cytotoxic substances. Three categories of granules, including azurophil granules, specific granules and gelatinase granules, are discharged during the degranulation process and are recognized as the basis content of their enzyme. Apart from these classical granules, neutrophils contain highly mobilizable secretory vesicles that serve as a reservoir primarily for plasma membrane receptors. Simultaneously with their degranulation, the initiation of nicotinamide adenine dinucleotide phosphate oxidase activity (a part of the cellular respiratory burst) in neutrophils occurs, and then various reactive oxygen species (ROS) are generated. Furthermore, neutrophils can produce numerous cytokines, which are considered to be the most critical effectors because of their vast and diverse of biological activities. In addition, neutrophils can be activated to undergo NETosis (a novel form of cell death) and extrude extracellular fibrillary networks termed neutrophil extracellular traps (NETs). APRIL, a proliferation‐inducing ligand; BAFF, B cell activating factor; BPIP, bactericidal permeability increasing protein; DNA, deoxyribonucleic acid; EGF, epidermal growth factor; G‐CSF, granulocyte colony stimulating factor; HB‐EGF, heparin binding epidermal growth factor; HGF, hepatocyte growth factor; IL‐1RA, interleukin‐1 receptor antagonist; MIF, macrophage migration inhibitory factor; PBEF, pre‐B cell colony enhancing factor; RANKL, receptor activator for nuclear factor‐κ B ligand; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; TRAIL, tumor necrosis factor‐related apoptosis inducing factor.
Over the past decades, a number of studies have implied that neutrophils are involved in the initiation and perpetuation of autoimmune diabetes as follows: (i) circulating neutrophil counts and functions were found to change in diabetes patients; (ii) they can be activated and recruited to pancreatic islets, and were found to initiate autoimmune diabetes in non‐obese diabetic (NOD) mice. Furthermore, neutrophils were observed to infiltrate the pancreas in type 1 diabetes patients; (iii) neutrophil toxic tools, such as neutrophil elastase, were proved to increase in type 1 diabetes patients and associate with the pathogenesis of β‐cell autoimmunity as well as diabetic complications; and (iv) related antineutrophil treatments were shown to dampen and reduce the development of insulitis and autoimmune diabetes. Therefore, it is logical to hypothesize that neutrophils are involved in the pathogenesis of type 1 diabetes.
Neutrophil Counts and Functions in Type 1 Diabetes
Early studies showed that type 1 diabetes patients had higher neutrophil numbers than the controls, and increased neutrophil counts were reported to correlate with an augmented risk of vascular disease (Table 1 lists the changes of neutrophil numbers)13, 14. It has been concluded that increased levels of circulating neutrophils could be caused by immoderate enlistment from the bone marrow and/or the return of marginated cells to the circulatory pond15. However, more recent studies showed that circulating neutrophil numbers decreased in patients with type 1 diabetes and healthy autoantibody‐positive subjects, which might be associated with β‐cell specific autoimmunity15, 16, 17. Reduced numbers of blood neutrophils in type 1 diabetes could be a result of abnormal neutrophil yield and maturation, peripheral consumption or damage, and tissue detainment. It was suggested that neutrophils confined in the pancreas should account for the decreased blood neutrophils16, 18. The discrepancy in alterations of neutrophil counts from different studies might result from the discoveries from different stages of diabetes or different ethnic groups. Further investigations are required to explain the variance in the changes of neutrophil counts, and a more comprehensive and longitudinal study might clarify the difference. Neutrophil functions were also reported to change at different steps in type 1 diabetes. Clinical studies and experimental data in animal models clearly showed that consistent defects in neutrophil chemotaxis19, 20, adhesion21 and microbicidal activities (Table 1)22, 23, 24. However, literature to date is contradictory in regard to other host defense functions of neutrophil including oxidative burst activity24, 25, 26, migration19, 22, 27, phagocytosis22, 23, 24,28 and apoptosis27, 29, 30. The changed neutrophil functions can be caused by upregulated expression of adhesion molecules28, downregulated receptors31, impaired calcium signaling32 and anomalous activities of adenosine triphosphate synthases on neutrophils33. Although the neutrophil apoptosis rate was significantly correlated with glycated hemoglobin (HbA1c) levels in patients with type 2 diabetes30, research has shown that HbA1c level and history of infection did not seem to affect neutrophil functions in type 1 diabetes patients28. It was reported that neutrophil functions might be closely related to β‐cell autoimmunity, as a significant decrease in neutrophil numbers can be detected in type 1 diabetes patients diagnosed within 1 year and in prediabetes, but not in type 1 diabetes patients with long duration17, 18. There is abundant evidence showing the effects of environmental factors, such as hyperglycemia and advanced glycation end‐products (AGEs) on neutrophil functions, but literature regarding the association of neutrophil dysfunction and genes is scarce. It was suggested that human leukocyte antigen‐D related‐associated genes were not related to impaired neutrophil functions, though they were closely correlated with type 1 diabetes pathogenesis.
Table 1.
Neutrophil counts and functions in autoimmune type 1 diabetes
| Neutrophil counts and functions | Alteration | Reference |
|---|---|---|
| Counts | ||
| Neutrophil counts | Upregulated | 13,14 |
| Downregulated | 15, 16, 17 | |
| Functions | ||
| Chemotaxis | No change | 19,20 |
| Adhesion | Downregulated | 21 |
| Microbicidal activities | Downregulated | 22, 23, 24 |
| Oxidative burst activity | Downregulated | 24,25 |
| Upregulated | 26 | |
| Migration | Upregulated | 19 |
| Downregulated | 22,27 | |
| Phagocytosis | No change | 23 |
| Downregulated | 22, 24, 28 | |
| Apoptosis | Upregulated | 27, 29, 30 |
Neutrophil‐Derived Effector Molecules and Type 1 Diabetes
Adhesion Molecules
Adhesion molecules, existing as soluble and membrane forms, not only play an essential role during neutrophil migration from the bloodstream into target tissue34, but are also involved in the regulation of the immune system35. In autoimmune diabetes, the migration of immune cells from blood vessels into the pancreatic islet is important to exert their potential36. Various studies have shown an involvement of two families of adhesion molecules including selectins and integrins in the development of type 1 diabetes. The selectin family consists of P‐selectin (CD62P, GMP‐140), L‐selectin (CD62L) and E‐selectin (CD62E; Table 2 summarizes the neutrophil‐related adhesion molecules). L‐selectin is predominantly expressed on the surface of various immune cells37. It is rapidly effluxed after the activation of neutrophil38. Soluble L‐selectin (sL‐selectin) was found to alter in patients with type 1 diabetes, as well as subjects in the preclinical stage of the disease39. Early studies suggested that L‐selectin could be one of the new risk markers for type 1 diabetes development in humans and animal models40. Subsequent studies further suggested that elevated levels of sL‐selectin are associated with high titers of insulinoma‐associated protein 2 antibody in children with type 1 diabetes41 and siblings of diabetic children34. Therefore, augmented sL‐selectin expression in patients can manifest an active destructive insulitis procedure34, 41. Furthermore, increased levels of sL‐selectin were also reported to correlate with seroconversion to autoantibody positivity, proposing that the activation of leukocytes coincides with the occurrence of β‐cell autoimmunity42. However, from the prospective aspect (10 and 2 years, respectively), it was shown that early‐onset of β‐cell autoimmunity cannot be displayed entirely by elevated concentrations of circulating adhesion molecules42, 43. No differences were discovered in the integrated concentrations of sL‐selectin associating with distinct autoantibody specificities and titers.
Table 2.
Neutrophil‐related adhesion molecules
| Type | Expressed cells | Ligand |
|---|---|---|
| Slectins | ||
| P‐selectin (CD62P) | Endotheliocyte | PGSL |
| L‐selectin (CD62L) | Neutrophils | Unknown |
| E‐selectin (CD62E) | Endotheliocyte | Unknown |
| Integrins | ||
| VLA‐4 (CD49d) | Neutrophils | VCAM‐1 (CD106) |
| LFA‐1 (CD11a) | Neutrophils | ICAM‐1 (CD54) |
| Mac‐1 (CD11b) | Neutrophils | ICAM‐1 (CD54) |
| p150 (CD11c) | Neutrophils | Unknown |
| CD29 | Unknown | Unknown |
ICAM‐1, intercellular adhesion molecule 1; LFA‐1, leucocyte function antigen‐1; Mac‐1, macrophage‐1; PGSL, P‐selectin glycoprotein ligand; VCAM‐1, vascular adhesion molecule‐1; VLA‐4, very late antigen 4.
Integrins, heterodimers with an α chain and a common β chain, are composed of β1‐integrins and β2‐integrins. The three β2‐integrin complexes comprise CD11a (leucocyte function antigen‐1 [LFA]), CD11b (macrophage‐1 [MAC‐1]) and CD11c (p150,95). LFA is expressed on neutrophil membranes, whereas MAC‐1 glycoprotein complex is stored in secondary granules4. LFA‐1 was shown to be involved in the initiation of insulitis in humans and type 1 diabetes animal models44, 45. Treatment with anti‐LFA‐1 monoclonal antibodies can delay the spontaneous onset of the disease in NOD mice46, 47, and has a strong preventive effect on the progression of the disease48, 49. The mechanism of prevention appears to result from suppressing effector cells to home to the pancreas. It is well known that LFA‐1 can be expressed on neutrophil membranes as well as lymphocytes, monocytes and natural killer cells. To our knowledge, the expression of LFA‐1 on neutrophils in type 1 diabetes has not been reported. However, a variety of studies have reported the expression of LFA‐1 on monocytes. An early study found that LFA‐1 alpha chain expression decreased and the percentage of LFA‐1 β chain‐positive monocytes was normal in newly diagnosed patients with type 1 diabetes50. While another study showed that a higher level of LFA‐1 on mononuclear cells was observed in overt diabetics in comparison with the controls51, there was a positive interrelation between LFA‐1 expression and islet cell autoantibodies (ICA) titer, indicating that LFA‐1 plays an important role in type 1 diabetes pathogenesis51. MAC‐1, another member of the β2‐integrins, is considered to be associated with vascular event rates, because it is prothrombotic and can mediate leukocytes vascular infiltration52. In type 1 diabetes patients, neutrophils showed higher expression of MAC‐1 receptors that are independent of duration of diabetes and HbA1c53. Increased expression of MAC‐1 can be caused by acute hyperglycemia52, and might result from impaired neutrophil actin polymerization54. It was observed that elevated expression of MAC‐1 could contribute to the pathogenesis of diabetic complications by enhancing adhesion between neutrophils and endothelial cells54, 55.
Degranulation Products
Neutrophils are sophisticated cells that communicate with their environment by dislodging and releasing various kinds of granules and vesicles1, such as azurophil granules, specific granules, gelatinase granules and secretory vesicles (Figure 1)1, 56, 57. Normally, a variety of proteases, formed during the process of neutrophil maturation, are stored intracellularly in granules, and might be liberated into the extracellular space after neutrophil activation and degranulation58. It has been suggested that neutrophil proteases show activity in membrane‐bound form and soluble form, the latter of which acts extracellularly in plasma and tissues59. Among these proteases, neutrophil elastase and myeloperoxidase, found in the azurophil granules, are considered as relatively neutrophil‐specific. Plasma lactoferrin, a member of the specific granules, is thought to derive principally from neutrophils, and is markedly associated with circulating neutrophil numbers, although it can be excreted by other diversified cells60.
Neutrophil elastase is capable of degrading most of the extracellular matrix proteins and plasma proteins61. In addition, neutrophil elastase can regulate inflammation by splitting different agents, such as cytokines, chemokines and cell surface receptors62, 63. Plasma and total neutrophil elastase were reported to rise substantially in type 1 diabetes patients when compared with the control13, 14. High concentrations of plasma neutrophil elastase can also be considered as a marker of the development of complications, such as diabetic angiopathy and coronary artery disease, because it might contribute to the progression of vascular disease64, 65, 66. Furthermore, the previous work in our laboratory discovered that levels of circulating neutrophil elastase released from activated neutrophils was positively associated with the counts and titers of the autoantibodies against β‐cell‐specific antigens, which suggested that neutrophil activation and elevated proteases activities might play an anetiogenic role in the process of β‐cell autoimmunity. Therefore, serum neutrophil elastase could act as a susceptible biomarker for the prediction and early diagnosis of type 1 diabetes17. Although some studies showed that poor short‐term glycemic and metabolic control in type 2 diabetes patients were correlated with higher elastase concentration in plasma and neutrophils59, 64, it has been shown in several studies that increased plasma neutrophil elastase level in type 1 diabetes patients was not related to age, leucocyte count, HbA1c, plasma glucose or duration of diabetes13, 14. Myeloperoxidase (MPO), a powerful oxidative medium, is located mainly in neutrophil primary granules and constitutes approximately 5% of the total neutrophil protein56. MPO is relevant to oxidative stress, because it catalyzes ROS formation that can facilitate atherogenesis and alter lipid as well as proteins60. Activity of MPO can be inhibited in patients with diabetes, resulting in diminished phagocytic activity of neutrophils and thus increasing susceptibility to infections67. It has been shown that no significant differences were found in plasma MPO values when children and adolescents with type 1 diabetes were compared with controls. However, another study found that diabetic children had significantly higher serum concentrations of MPO than the healthy control group, which can reflect increased risk of cardiovascular diseases in type 1 diabetes patients68. Regardless of whether the MPO level is increased or decreased, there are no significant interrelations between MPO serum concentration and diabetes duration, HbA1c value, and the level of actual blood glucose68.
Cytokines
Neutrophils, either constitutively or after appropriate motivation, not only synthesize a variety of polypeptides that are directly involved in their effector functions, but can also produce numerous inflammatory modulation proteins (Figure 1)69, 70. Among these molecules, cytokines are considered to be the most critical effectors because of their vast and diverse biological activities69, 71. Cytokines are not liberated immediately after synthesis, but are deposited in temporarily intracellular ponds and are released quickly when neutrophils are activated72. Human and mouse neutrophils were reported to have a different capacity to express cytokines, especially interleukin (IL)‐6, IL‐17A, IL‐17F and IFN‐γ6. Whether activated neutrophils secrete cytokines or not depends on the state of the cells and the type of triggering stimulus. Although it is evident that neutrophils make substantially less quantities of cytokines than other immune cells, it must be stressed that neutrophils constitute most of the infiltrating cells in inflamed tissues, and might be considered as an important origin of cytokines in those tissues69. In view of the broad spectrum of biological activities exerted by cytokines, it is reasonable to infer that neutrophils have crucial effects not only on inflammatory response, but also on innate and adaptive immune responses6, 70.
Cytokines, produced locally in and around islets during islet insulitis by more than a single cell type, are shown to play considerable roles in the development of autoimmune diabetes73. It has been proposed that cytokines, such as tumor necrosis factor (TNF)‐α, IL‐1, IFN‐γ and TNF‐β, can stimulate toxic free radical production, which might initiate pancreatic β‐cell destruction74, 75, 76. Furthermore, β‐cell function can be impaired and β‐cell mass can be reduced by IL‐1 derived from macrophages77. In addition, cytokines like IFN‐γ, IL‐1 and TNF‐α can exacerbate autoimmunity by intensifying adaptive immune responses77. Increased expression of cytokines (IFN‐α, TNF‐α and IL‐1) is associated with β‐cell destructive insulitis73. In patients with recent‐onset type 1 diabetes, increased IL‐18 is reported to be related to multiple autoantibody presence78. As a result of their important roles in type 1 diabetes, serum cytokines are promising candidates as additional markers and for monitoring interventions beyond autoantibodies to predict type 1 diabetes. However, many problems and conflicting observations exist – the major difficulty is the absence of a normal range for the majority of cytokines. Furthermore, the concept of physiological disparities, and technical alterations in assay methodologies and assay perturbations cannot be neglected. Another obstacle is the lack of specificity to diseases, even though certain cytokines have normal reference ranges and the levels in plasma can be measured by current technologies. Finally, cytokines concentrations are dynamic, especially under the condition of infections, allergies and medications79. Although cytokines have diverse biological activities and neutrophils can release a variety of cytokines, the role of cytokines derived from neutrophils in the pathogenesis and development of autoimmune diabetes is still unclear.
ROS
ROS, including superoxide anions, hydroxyl radicals and hydrogen peroxide, are produced as a consequence of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, which is a part of the cellular respiratory burst56, 80. Neutrophils are rich sources of toxic oxygen species (Figure 1)81, and the initiation of NADPH oxidase activity in neutrophils occurs almost simultaneously with their degranulation, with a delay of approximately 20 s82. Increased production of ROS from neutrophils has been reported in type 1 diabetes patients and rat models26, 83. Elevated expression of these toxic substances not only initiate pancreatic β‐cell destruction, but also predispose diabetic patients to infection by impairing anti‐oxidant defense in neutrophils84, 85, 86. The enhancement of ROS production in type 1 diabetes is suggested to result from glycosylation and glyco‐oxidation, lipid peroxidation, and activation of platelets and neutrophils81. Although AGEs possess no capacity to enhance respiratory burst alone, they appear to be capable of increasing neutrophil ROS production by upregulating NADPH oxidase and by priming neutrophils87. However, other investigators found a reduction in respiratory burst responses, and it has also been documented that neutrophil NADPH oxidase activity in vitro was impaired and superoxide production was reduced in diabetic patients24, 88. High‐glucose levels rapidly decrease ROS from stimulated neutrophils, possibly by suppressing glucose‐6‐phosphate dehydrogenase80. The contradictory results of ROS levels in these studies might be derived from patients with different metabolic states and different disease durations81. Various studies show that ROS are generated from not only neutrophils, but also macrophages, mesangial cells and glomerular epithelial cells89. It has been shown that ROS mediated by macrophages can infiltrate islets and damage islet β‐cells, directly resulting in autoimmune diabetes in NOD mice90. However, the role of ROS derived from neutrophils in the pathology of type 1 diabetes is not clear at this stage.
Neutrophil Extracellular Traps
Apart from producing classical effector molecules, such as proteases, cytokines and ROS, neutrophils might undergo NETosis (a novel kind of cell death procedure almost differentiated from both apoptosis and necrosis) and form fibrillary extracellular networks known as NETs in response to a number of stimuli6. NETs comprise nuclear constituents decorated by granular proteins and short peptidoglycan recognition protein, and are beneficial for antimicrobial processes. In addition, NETs are associated with autoimmunity, because they secrete self molecules extracellularly3. Dysregulated NET formation and NETosis were reported to be involved in a number of autoimmune diseases, such as type 1 diabetes, small vessel vasculitis and systemic lupus erythematosus10, 11, 12. Several studies have clearly proved that increased NETosis can induce autoimmunity and accelerate the occurrence of vascular disease in systemic lupus erythematosus patients91. However, literature regarding the role of NETs in autoimmune diabetes development is relatively scarce. In young NOD mice92, NET formation was observed in pancreatic islets as early as 2 weeks after birth. In autoimmune type 1 diabetes patients, NET formation was found to be elevated and closely associated with increased circulating neutrophil elastase levels, suggesting that it might play a key role in the initiation of β‐cell autoimmunity17. However, conflicting reports showed that neutrophils from diabetic patients (the diabetes type was not mentioned) released NETs at a lower level than that of healthy subjects, because a high‐glucose condition might impair and delay neutrophil NET formation93, 94. Therefore, expression of neutrophil NETs possessing antimicrobial property was reduced, providing a partial explanation for elevated susceptibility of diabetes mellitus patients to infections93, 94.
Cell–Cell Interactions in Type 1 Diabetes
Neutrophils and Other Immune Cells
Apart from using a set of membrane and intracellular molecules to respond to their local environment signals and to modify their phenotype, neutrophils engage in complex bidirectional interactions with most other types of immune cells, and shape their activation, maturation and effector functions directly or indirectly, depending on the context95. They instruct other immune cells through secreting cytokines, granules and ROS. They can also participate in the communication networks through cell–cell contact96. By interplaying with other cells, neutrophils are representatively the predominant immune cells responding to inflammatory response and exacerbating inflammation97. In autoimmune type 1 diabetes, interactions between pancreatic β‐cells and immune cells including neutrophils, as well as other immune cells, play significant roles in the progression of the disease. Recently, Diana et al.92 found that physiological β‐cell death can induce recruitment and activation of neutrophils, B‐1a cells, and plasmacytoid dendritic cells in young NOD mice. The cross‐talk between these innate immune cells was found to take place in the pancreas, and was required for the initiation of type 1 diabetes. They also found another novel cross‐talk between macrophages and β‐cells in the pancreas, which was responsible for neutrophil infiltration in the pancreas during the initiation phase of autoimmune diabetes (Figure 2)98.
Figure 2.
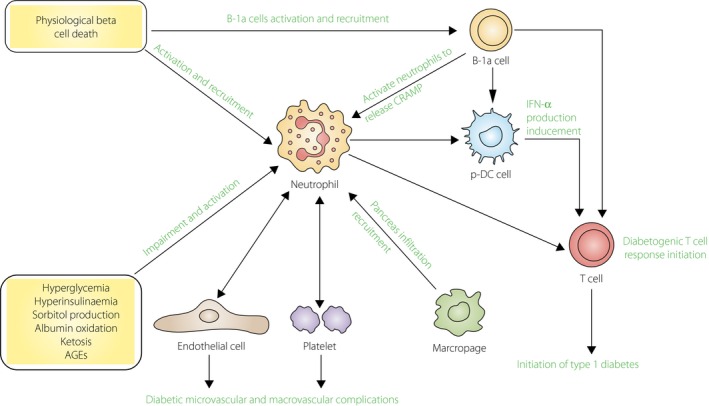
Neutrophils and type 1 diabetes. The mechanism of the initiation and pathogenesis of type 1 diabetes still remains unclear. Physiological β‐cell death was considered as an essential trigger in the development of the disease, which can recruit and activate immune cells, particularly neutrophils, to infiltrate in pancreatic islets. In the pancreas, neutrophils can release cathelicidin‐related antimicrobial peptide (CRAMP), the process of which is activated by deoxyribonucleic acid (DNA)‐specific immunoglobulin G secreted from B‐1a cells. These immunoglobulin G and CRAMP peptide complex, together with β‐cell debris like self‐deoxyribonucleic acid, can induce plasmacytoid dendritic cells to produce interferon‐α. The aforementioned cross‐talk between these immune cells is required to induce diabetogenic T‐cell response and then leads to the initiation of type 1 diabetes. Additionally, interaction between neutrophils and other non‐immune cells, such as platelets in the blood or endothelial cells on the blood vessels, is supposed to play essential roles in diabetic microvascular and macrovascular complications. Conversely, neutrophils can be activated and impaired by metabolic changes in type 1 diabetes patients. AGEs, advanced glycation end‐products; IFN, interferon.
There is considerable evidence that T cells play an essential role in the development of type 1 diabetes in both animal models and humans. In the early stage of diabetes, CD8+ T lymphocytes are considered as the most affluent immune cell type infiltrated in the pancreas during the occurrence of insulitis99. They can drive adaptive immune responses and damage pancreatic β‐cells by major histocompatibility complex class I molecules‐regulated pathogenic cytotoxicity, and both CD4+ and CD8+ T cells can secrete cytokines that could contribute to β‐cell apoptosis directly or indirectly100. In NOD mice, neutrophil trafficking into the pancreatic islets is mainly regulated by CXCR2 ligands, and neutrophils are recruited by chemokines CXCL1 and CXCL2 produced by β‐cells and macrophages98. CXCR2 blockade in early stages can lead to a dramatic reduction in specific CD8+ T cells within the islets, and then can inhibit diabetogenic T‐cell response, as well as the following development of diabetes98. Recently, decreased expressions of Cxcr1 messenger ribonucleic acid were found in both neutrophils and CD4+ T lymphocytes isolated from NOD mice when compared with diabetes‐resistant mice. Low Cxcr1 expression in NOD mice could contribute to the pathogenesis of autoimmune diabetes101. In addition, IL‐17+ β‐cell‐specific autoreactive CD4+ T cells can be observed in the circulation of type 1 diabetes patients at diagnosis102. This pro‐inflammatory IL‐17 circumstance in the pancreas can attract neutrophils to infiltrate the pancreatic tissue18. It seems that both neutrophils and T cells might contribute to the pathogenesis of type 1 diabetes with a variable degree of synergism. Therefore, interactions between neutrophils and T cells in the development of type 1 diabetes deserve us to focus on.
Neutrophils and Endothelial Cells
Interaction between neutrophils and endothelial cells is one of the earliest events in the inflammatory process and normally occurs in post‐capillary venules38. Adhesion of neutrophils to endothelial cells facilitates neutrophils to enter to the pancreatic islet and causes damage to it. However, most of the research about neutrophil–endothelial cell adhesion in diabetes have focused on the development of atherosclerosis‐mediated diseases, such as diabetic microvascular and macrovascular complications103, 104, and studies about neutrophil–endothelial cell adhesion in type 1 diabetes pathogenesis are relatively scarce. Several endothelial adhesion molecules, such as intercellular adhesion molecule‐1 (ICAM‐1), CD62E, CD62P and vascular adhesion molecule‐1 (VCAM‐1), can attract circulating neutrophils then bind with neutrophil adhesion molecules103, 105, 106. It has been shown that ICAM‐1, which is the best characterized cell surface adhesion molecule, is implicated in the pathogenesis of type 1 diabetes by being involved in the extravasation of leukocytes from the circulation into the inflamed pancreas. ICAM‐1 expression on vascular endothelial cells was reported to increase in the pancreatic islets of the NOD mouse48, 107, as well as in individuals with new‐onset type 1 diabetes108. Serum levels of circulating ICAM‐1 were also significantly increased in recent‐onset type 1 diabetes patients and their first‐degree relatives when compared with healthy controls34, 35. Among these subjects, soluble ICAM‐1 levels in prediabetics with positive autoantibodies were relatively higher than in patients with clinical diabetes, and were positively correlated with ICA and glutamicacid decarboxylase autoantibody (GADA) values63. These aforementioned advances, together with research reporting that neutrophils were found to infiltrate in the pancreatic islets, suggest that neutrophil–endothelial cell interactions are undoubtedly involved in the accumulation of inflammatory neutrophils into the pancreas and the destruction of pancreatic islets, but the mechanism that controls the process remains unclear.
Neutrophils and Platelets
Platelets and leukocytes, the latter of which consist of neutrophils, have been proven to regulate and influence each other's function by platelet–leukocyte contact and releasing soluble effector mediators109. Activated platelets promoted neutrophil activation and recruitment through expressing selectins, inflammatory cytokines, and chemokines110. Conversely, apoptotic and activated leukocytes can promote platelet recruitment and attachment111, 112. Adhesion of activated platelets to leukocytes was reported to be implicated in the development of thrombotic occurrence, and apoptotic leukocytes can induce a prothrombotic course113. It was found that type 1 diabetes patients showed increased blood platelet–leukocyte aggregation and cross‐talk in their blood114. Promotion of leukocyte–platelet interaction in the disease is probably caused by enhanced plasma elastase levels, which can induce platelet activation115 and increase production of important soluble mediators, such as platelet‐activating factor and superoxide anion109, 116, 117. AGE‐BSA, a model substance for AGEs, can augment platelet–neutrophil aggregation by inducing neutrophils apoptosis and enhancing Mac‐1expression113. As the interplay between leukocytes and platelets connects inflammation with thrombosis and might facilitate vascular obstruction as well as tissue ischemia118, 119, elevated circulating platelet–leukocyte aggregation and cross‐talk in type 1 diabetes individuals might contribute to platelet hyperactivity and the development of microvascular complications117. It was also found that an increase of platelet–neutrophil aggregation in patients with diabetes might be one of the factors leading to serious cardiovascular disease120.
Antineutrophil Therapy
The previous studies showed that neutrophils play important roles in the progression of autoimmune type 1 diabetes, raising the possibility that neutrophils might be a candidate for therapeutic interventions for the disease. The processes of neutrophil activation, binding to the endothelium, transendothelial migration, emigration into the pancreatic islet and release of cytotoxic products, are all potential targets towards which pharmacological therapy can be achieved. Therapies directed against neutrophil‐mediated injury in diabetes include direct inhibition of neutrophil recruitment and anti‐adhesion therapy.
Recruitment of neutrophils from the circulation into the pancreas is necessary for these cells to infiltrate in the pancreatic islet and bring about their effects. It was reported that macrophages and β‐cells recruit CXCR2‐expressing neutrophils by producing chemokines CXCL1 and CXCL298. Blockade of neutrophil recruitment by CXCR2 antagonist can depress diabetogenic T‐cell response and dampen the subsequent progression of autoimmune diabetes. As aforementioned, neutrophil adhesion to endothelial venules within pancreatic sections is an important early step in the inflammatory response, and is considered as one of the essential steps in the initiation of autoimmune diabetes. Therefore, blocking different adhesion molecules, such as selectins and integrins expressed on neutrophils by specific antagonists, might be an effective approach to reduce the migration of neutrophils to the inflamed pancreas, thus inhibiting the development of insulitis. Antibodies of anti‐L‐selectin and anti‐VLA‐4 were reported to delay the appearance of insulitis by inhibiting leukocyte adhesion to the inflamed blood vessels and interpreting the recruitment of leukocyte to the islets47. Administration of anti‐α4‐integrin and/or anti‐LFA‐1 antibodies has been found to lead to an inhibition of β‐cells destruction, and protect against spontaneous and adoptively transferred diabetes in NOD mice46, 49, 121, 122. Combination treatment with the two monoclonal antibodies might have a longer delayed influence in the initiation of diabetes46. Furthermore, long‐term inhibition of both LFA‐1 and α4‐integrin not only prevents diabetes during treatment, but also has a persistent resistance to the disease long after the interference has ceased, suggesting that adhesion molecule inhibition can reduce islet insulitis by affecting effector cell functions through stimulation of suppressor cells involved in immunoregulation46. In addition, a short‐term blockade of the LFA‐1/ICAM‐1 pathway at critical periods was shown to induce a unique peripheral tolerance against β‐cell Ag(s) at an early age in NOD mice, resulting in complete protection from autoimmune diabetes123. Therefore, adhesion molecules, such as L‐selectin, VLA‐4 and LFA‐1, play significant roles in the development of type 1 diabetes, and the disease can be prevented by blocking these adhesion pathways. However, it is known that adhesion molecules are expressed not only on neutrophils, but also on lymphocytes, monocytes and natural killer cells124. Studies have shown that anti‐LFA‐1 treatment can ameliorate neutrophil‐mediated injury by reducing the adhesion, recruitment, accumulation and infiltration of neutrophils into tissues125, 126, 127, 128. In addition, the activation of neutrophil respiratory burst can also be restricted by anti‐LFA‐1 interference129. Therefore, further exhaustive studies are required to better understand the preventive effects of neutrophil‐specific anti‐adhesion therapies in the progression of spontaneous diabetes.
Concluding Remarks
Progress over the past decades shows that neutrophils play essential roles in the onset and progression of autoimmune type 1 diabetes. Recently, exciting discoveries have provided new insights into the actions of neutrophils in the initiation of the disease, which found that neutrophils can induce type 1 diabetes through infiltrating into the pancreatic islets and interplaying with other immune cells. Unfortunately, only a few studies have specifically concentrated on the relationship between neutrophils and diabetes, though neutrophils are known to the most abundant cell type in the circulation and the first cells recruited to the site of inflammation. Detailed studies are required to elucidate how neutrophils are activated and recruited to cause damage to pancreatic islets through neutrophil‐derived toxic substances or interactions with other cells. A better understanding of the role of neutrophils in type 1 diabetes pathogenesis will provide additional information for early diagnosis, therapy and even for the prevention of the disease. Although many challenges still remain in exploring the correlation between neutrophils and autoimmune type 1 diabetes, it is an encouraging and interesting research topic that deserves to be focused on.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This work is supported by the National Key Technology R&D Program (2013BAI09B12) and the Fundamental Research Funds for the Central Universities of Central South University (2014zzts085). We thank Professor Qianjin Lu and Dr Peilin Zheng for their guidance and advice.
J Diabetes Investig 2016; 7: 652–663
References
- 1. Borregaard N, Lollike K, Kjeldsen L, et al Human neutrophil granules and secretory vesicles. Eur J Haematol 1993; 51: 187–198. [DOI] [PubMed] [Google Scholar]
- 2. Borregaard N. Neutrophils, from marrow to microbes. Immunity 2010; 33: 657–670. [DOI] [PubMed] [Google Scholar]
- 3. Amulic B, Cazalet C, Hayes GL, et al Neutrophil function: from mechanisms to disease. Annu Rev Immunol 2012; 30: 459–489. [DOI] [PubMed] [Google Scholar]
- 4. Brown KA, Brain SD, Pearson JD, et al Neutrophils in development of multiple organ failure in sepsis. Lancet 2006; 368: 157–169. [DOI] [PubMed] [Google Scholar]
- 5. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol 2014; 9: 181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mantovani A, Cassatella MA, Costantini C, et al Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 2011; 11: 519–531. [DOI] [PubMed] [Google Scholar]
- 7. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6: 173–182. [DOI] [PubMed] [Google Scholar]
- 8. Berry MP, Graham CM, McNab FW, et al An interferon‐inducible neutrophil‐driven blood transcriptional signature in human tuberculosis. Nature 2010; 466: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med 2013; 210: 1283–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kessenbrock K, Krumbholz M, Schonermarck U, et al Netting neutrophils in autoimmune small‐vessel vasculitis. Nat Med 2009; 15: 623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Villanueva E, Yalavarthi S, Berthier CC, et al Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 2011; 187: 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Naegele M, Tillack K, Reinhardt S, et al Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol 2012; 242: 60–71. [DOI] [PubMed] [Google Scholar]
- 13. Collier A, Jackson M, Bell D, et al Neutrophil activation detected by increased neutrophil elastase activity in type 1 (insulin‐dependent) diabetes mellitus. Diabetes Res 1989; 10: 135–138. [PubMed] [Google Scholar]
- 14. Jackson MH, Collier A, Nicoll JJ, et al Neutrophil count and activation in vascular disease. Scott Med J 1992; 37: 41–43. [DOI] [PubMed] [Google Scholar]
- 15. Valle A, Giamporcaro GM, Scavini M, et al Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013; 62: 2072–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harsunen MH, Puff R, D'Orlando O, et al Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm Metab Res 2013; 45: 467–470. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y, Xiao Y, Zhong L, et al Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with beta‐cell autoimmunity in patients with type 1 diabetes. Diabetes 2014; 63: 4239–4248. [DOI] [PubMed] [Google Scholar]
- 18. Battaglia M. Neutrophils and type 1 autoimmune diabetes. Curr Opin Hematol 2014; 21: 8–15. [DOI] [PubMed] [Google Scholar]
- 19. Debczynski W, Pietruska Z. [Chemotaxis and spontaneous migration of neutrophil leukocytes from patients with diabetes]. Pol Tyg Lek 1994; 49: 11–13. [PubMed] [Google Scholar]
- 20. Valerius NH, Eff C, Hansen NE, et al Neutrophil and lymphocyte function in patients with diabetes mellitus. Acta Med Scand 1982; 211: 463–467. [DOI] [PubMed] [Google Scholar]
- 21. Kelly MK, Brown JM, Thong YH. Neutrophil and monocyte adherence in diabetes mellitus, alcoholic cirrhosis, uraemia and elderly patients. Int Arch Allergy Appl Immunol 1985; 78: 132–138. [DOI] [PubMed] [Google Scholar]
- 22. Cutler CW, Eke P, Arnold RR, et al Defective neutrophil function in an insulin‐dependent diabetes mellitus patients. A case report. J Periodontol 1991; 62: 394–401. [DOI] [PubMed] [Google Scholar]
- 23. Wilson RM, Reeves WG. Neutrophil phagocytosis and killing in insulin‐dependent diabetes. Clin Exp Immunol 1986; 63: 478–484. [PMC free article] [PubMed] [Google Scholar]
- 24. Marhoffer W, Stein M, Schleinkofer L, et al Evidence of ex vivo and in vitro impaired neutrophil oxidative burst and phagocytic capacity in type 1 diabetes mellitus. Diabetes Res Clin Pract 1993; 19: 183–188. [DOI] [PubMed] [Google Scholar]
- 25. Thomson GA, Fisher BM, Gemmell CG, et al Attenuated neutrophil respiratory burst following acute hypoglycaemia in diabetic patients and normal subjects. Acta Diabetol 1997; 34: 253–256. [DOI] [PubMed] [Google Scholar]
- 26. Wierusz‐Wysocka B, Wysocki H, Siekierka H, et al Evidence of polymorphonuclear neutrophils (PMN) activation in patients with insulin‐dependent diabetes mellitus. J Leukoc Biol 1987; 42: 519–523. [DOI] [PubMed] [Google Scholar]
- 27. Chanchamroen S, Kewcharoenwong C, Susaengrat W, et al Human polymorphonuclear neutrophil responses to Burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun 2009; 77: 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delamaire M, Maugendre D, Moreno M, et al [Exploration of the various steps of polymorphonuclear neutrophil function in diabetic patients]. J Mal Vasc 1995; 20: 107–112. [PubMed] [Google Scholar]
- 29. Tennenberg SD, Finkenauer R, Dwivedi A. Absence of lipopolysaccharide‐induced inhibition of neutrophil apoptosis in patients with diabetes. Arch Surg 1999; 134: 1229–1233; discussion 1233–1234. [DOI] [PubMed] [Google Scholar]
- 30. Sudo C, Ogawara H, Saleh AW, et al Clinical significance of neutrophil apoptosis in peripheral blood of patients with type 2 diabetes mellitus. Lab Hematol 2007; 13: 108–112. [DOI] [PubMed] [Google Scholar]
- 31. Spiller F, Carlos D, Souto FO, et al alpha1‐Acid glycoprotein decreases neutrophil migration and increases susceptibility to sepsis in diabetic mice. Diabetes 2012; 61: 1584–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Advani A, Marshall SM, Thomas TH. Impaired neutrophil store‐mediated calcium entry in Type 2 diabetes. Eur J Clin Invest 2004; 34: 43–49. [DOI] [PubMed] [Google Scholar]
- 33. Nath N, Biswas S, Bhelwa A, et al Plausible involvement of a diabetic serum factor in neutrophil membrane pathology. Indian J Biochem Biophys 1993; 30: 244–246. [PubMed] [Google Scholar]
- 34. Toivonen A, Kulmala P, Savola K, et al Soluble adhesion molecules in preclinical type 1 diabetes. The Childhood Diabetes in Finland Study Group. Pediatr Res 2001; 49: 24–29. [DOI] [PubMed] [Google Scholar]
- 35. Martin S. Soluble adhesion molecules in type 1 diabetes mellitus. Horm Metab Res 1997; 29: 639–642. [DOI] [PubMed] [Google Scholar]
- 36. Nicoletti F, Conget I, Di MM, et al Serum concentrations of the interferon‐gamma‐inducible chemokine IP‐10/CXCL10 are augmented in both newly diagnosed Type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 2002; 45: 1107–1110. [DOI] [PubMed] [Google Scholar]
- 37. Kretowski A, Kinalska I. L‐selectin gene T668C mutation in type 1 diabetes patients and their first degree relatives. Immunol Lett 2000; 74: 225–228. [DOI] [PubMed] [Google Scholar]
- 38. Vinten‐Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res 2004; 61: 481–497. [DOI] [PubMed] [Google Scholar]
- 39. Lampeter ER, Kishimoto TK, Rothlein R, et al Elevated levels of circulating adhesion molecules in IDDM patients and in subjects at risk for IDDM. Diabetes 1992; 41: 1668–1671. [DOI] [PubMed] [Google Scholar]
- 40. Yang XD, Michie SA, Mebius RE, et al The role of cell adhesion molecules in the development of IDDM: implications for pathogenesis and therapy. Diabetes 1996; 45: 705–710. [DOI] [PubMed] [Google Scholar]
- 41. Toivonen A, Kulmala P, Rahko J, et al Soluble adhesion molecules in Finnish schoolchildren with signs of preclinical type 1 diabetes. Diabetes Metab Res Rev 2004; 20: 48–54. [DOI] [PubMed] [Google Scholar]
- 42. Toivonen AM, Kimpimaki T, Kupila A, et al Soluble adhesion molecules in young children with signs of beta‐cell autoimmunity–prospective follow‐up from birth. Diabetes Metab Res Rev 2006; 22: 176–183. [DOI] [PubMed] [Google Scholar]
- 43. Toivonen AM, Kulmala P, Savola K, et al Soluble adhesion molecules in pre‐clinical Type 1 diabetes: a prospective study. Diabetologia 2003; 46: 492–495. [DOI] [PubMed] [Google Scholar]
- 44. Linn T, Strate C, Federlin K, et al Intercellular adhesion molecule‐1 (ICAM‐1) expression in the islets of the non‐obese diabetic and low‐dose streptozocin‐treated mouse. Histochemistry 1994; 102: 317–321. [DOI] [PubMed] [Google Scholar]
- 45. Somoza N, Vargas F, Roura‐Mir C, et al Pancreas in recent onset insulin‐dependent diabetes mellitus. Changes in HLA, adhesion molecules and autoantigens, restricted T cell receptor V beta usage, and cytokine profile. J Immunol 1994; 153: 1360–1377. [PubMed] [Google Scholar]
- 46. Ninova D, Dean PG, Stegall MD. Immunomodulation through inhibition of multiple adhesion molecules generates resistance to autoimmune diabetes in NOD mice. J Autoimmun 2004; 23: 201–209. [DOI] [PubMed] [Google Scholar]
- 47. Yang XD, Karin N, Tisch R, et al Inhibition of insulitis and prevention of diabetes in nonobese diabetic mice by blocking L‐selectin and very late antigen 4 adhesion receptors. Proc Natl Acad Sci USA 1993; 90: 10494–10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hasegawa Y, Yokono K, Taki T, et al Prevention of autoimmune insulin‐dependent diabetes in non‐obese diabetic mice by anti‐LFA‐1 and anti‐ICAM‐1 mAb. Int Immunol 1994; 6: 831–838. [DOI] [PubMed] [Google Scholar]
- 49. Scheynius A, Camp RL, Pure E. Reduced contact sensitivity reactions in mice treated with monoclonal antibodies to leukocyte function‐associated molecule‐1 and intercellular adhesion molecule‐1. J Immunol 1993; 150: 655–663. [PubMed] [Google Scholar]
- 50. Martin S, Rothe H, Tschope D, et al Decreased expression of adhesion molecules on monocytes in recent onset IDDM. Immunology 1991; 73: 123–125. [PMC free article] [PubMed] [Google Scholar]
- 51. Mysliwiec J, Kretowski A, Kinalski M, et al CD11a expression and soluble ICAM‐1 levels in peripheral blood in high‐risk and overt type 1 diabetes subjects. Immunol Lett 1999; 70: 69–72. [DOI] [PubMed] [Google Scholar]
- 52. Sampson MJ, Davies IR, Brown JC, et al Monocyte and neutrophil adhesion molecule expression during acute hyperglycemia and after antioxidant treatment in type 2 diabetes and control patients. Arterioscler Thromb Vasc Biol 2002; 22: 1187–1193. [DOI] [PubMed] [Google Scholar]
- 53. Grykiel K, Zozulinska D, Kostrzewa A, et al [Evaluation of expression of polymorphonuclear neutrophil surface receptors in patients with type 1 diabetes]. Pol Arch Med Wewn 2001; 105: 377–381. [PubMed] [Google Scholar]
- 54. Advani A, Marshall SM, Thomas TH. Impaired neutrophil actin assembly causes persistent CD11b expression and reduced primary granule exocytosis in Type II diabetes. Diabetologia 2002; 45: 719–727. [DOI] [PubMed] [Google Scholar]
- 55. Chello M, Mastroroberto P, Cirillo F, et al Neutrophil‐endothelial cells modulation in diabetic patients undergoing coronary artery bypass grafting. Eur J Cardiothorac Surg 1998; 14: 373–379. [DOI] [PubMed] [Google Scholar]
- 56. Segal AW. How neutrophils kill microbes. Annu Rev Immunol 2005; 23: 197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Borregaard N, Sorensen OE, Theilgaard‐Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol 2007; 28: 340–345. [DOI] [PubMed] [Google Scholar]
- 58. Witko‐Sarsat V, Rieu P, Descamps‐Latscha B, et al Neutrophils: molecules, functions and pathophysiological aspects. Lab Invest 2000; 80: 617–653. [DOI] [PubMed] [Google Scholar]
- 59. Zurawska‐Plaksej E, Piwowar A, Knapik‐Kordecka M, et al Activities of neutrophil membrane‐bound proteases in type 2 diabetic patients. Arch Med Res 2014; 45: 36–43. [DOI] [PubMed] [Google Scholar]
- 60. Vengen IT, Dale AC, Wiseth R, et al Lactoferrin is a novel predictor of fatal ischemic heart disease in diabetes mellitus type 2: long‐term follow‐up of the HUNT 1 study. Atherosclerosis 2010; 212: 614–620. [DOI] [PubMed] [Google Scholar]
- 61. Kawabata K, Hagio T, Matsuoka S. The role of neutrophil elastase in acute lung injury. Eur J Pharmacol 2002; 451: 1–10. [DOI] [PubMed] [Google Scholar]
- 62. Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc 2006; 3: 424–427. [DOI] [PubMed] [Google Scholar]
- 63. Owen CA, Campbell MA, Sannes PL, et al Cell surface‐bound elastase and cathepsin G on human neutrophils: a novel, non‐oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 1995; 131: 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Piwowar A, Knapik‐Kordecka M, Warwas M. Concentration of leukocyte elastase in plasma and polymorphonuclear neutrophil extracts in type 2 diabetes. Clin Chem Lab Med 2000; 38: 1257–1261. [DOI] [PubMed] [Google Scholar]
- 65. Greer IA, Haddad NG, Dawes J, et al Increased neutrophil activation in diabetic pregnancy and in nonpregnant diabetic women. Obstet Gynecol 1989; 74: 878–881. [PubMed] [Google Scholar]
- 66. Amaro A, Gude F, Gonzalez‐Juanatey R, et al Plasma leukocyte elastase concentration in angiographically diagnosed coronary artery disease. Eur Heart J 1995; 16: 615–622. [DOI] [PubMed] [Google Scholar]
- 67. Saeed FA, Castle GE. Neutrophil chemiluminescence during phagocytosis is inhibited by abnormally elevated levels of acetoacetate: implications for diabetic susceptibility to infections. Clin Diagn Lab Immunol 1998; 5: 740–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Heilman K, Zilmer M, Zilmer K, et al Arterial stiffness, carotid artery intima‐media thickness and plasma myeloperoxidase level in children with type 1 diabetes. Diabetes Res Clin Pract 2009; 84: 168–173. [DOI] [PubMed] [Google Scholar]
- 69. Cassatella MA. The production of cytokines by polymorphonuclear neutrophils. Immunol Today 1995; 16: 21–26. [DOI] [PubMed] [Google Scholar]
- 70. Cassatella MA. Neutrophil‐derived proteins: selling cytokines by the pound. Adv Immunol 1999; 73: 369–509. [DOI] [PubMed] [Google Scholar]
- 71. Tecchio C, Micheletti A, Cassatella MA. Neutrophil‐derived cytokines: facts beyond expression. Front Immunol 2014; 5: 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Scapini P, Bazzoni F, Cassatella MA. Regulation of B‐cell‐activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett 2008; 116: 1–6. [DOI] [PubMed] [Google Scholar]
- 73. Rabinovitch A. An update on cytokines in the pathogenesis of insulin‐dependent diabetes mellitus. Diabetes Metab Rev 1998; 14: 129–151. [DOI] [PubMed] [Google Scholar]
- 74. Nerup J, Mandrup‐Poulsen T, Helqvist S, et al On the pathogenesis of IDDM. Diabetologia 1994; 37(Suppl 2): S82–S89. [DOI] [PubMed] [Google Scholar]
- 75. Rabinovitch A, Suarez‐Pinzon WL, Strynadka K, et al Human pancreatic islet beta‐cell destruction by cytokines involves oxygen free radicals and aldehyde production. J Clin Endocrinol Metab 1996; 81: 3197–3202. [DOI] [PubMed] [Google Scholar]
- 76. Dominguez C, Ruiz E, Gussinye M, et al Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care 1998; 21: 1736–1742. [DOI] [PubMed] [Google Scholar]
- 77. Padgett LE, Broniowska KA, Hansen PA, et al The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 2013; 1281: 16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hanifi‐Moghaddam P, Schloot NC, Kappler S, et al An association of autoantibody status and serum cytokine levels in type 1 diabetes. Diabetes 2003; 52: 1137–1142. [DOI] [PubMed] [Google Scholar]
- 79. Wasserfall CH, Atkinson MA. Autoantibody markers for the diagnosis and prediction of type 1 diabetes. Autoimmun Rev 2006; 5: 424–428. [DOI] [PubMed] [Google Scholar]
- 80. Perner A, Nielsen SE, Rask‐Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med 2003; 29: 642–645. [DOI] [PubMed] [Google Scholar]
- 81. Zozulinska DA, Wierusz‐Wysocka B, Wysocki H, et al The influence of insulin‐dependent diabetes mellitus (IDDM) duration on superoxide anion and hydrogen peroxide production by polymorphonuclear neutrophils. Diabetes Res Clin Pract 1996; 33: 139–144. [DOI] [PubMed] [Google Scholar]
- 82. Segal AW, Dorling J, Coade S. Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes. Biochemical and morphological studies. J Cell Biol 1980; 85: 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rocha RE, Coelho I, Pequito DC, et al Interval training attenuates the metabolic disturbances in type 1 diabetes rat model. Arq Bras Endocrinol Metabol 2013; 57: 594–602. [DOI] [PubMed] [Google Scholar]
- 84. Uchimura K, Nagasaka A, Hayashi R, et al Changes in superoxide dismutase activities and concentrations and myeloperoxidase activities in leukocytes from patients with diabetes mellitus. J Diabetes Complications 1999; 13: 264–270. [DOI] [PubMed] [Google Scholar]
- 85. Pitozzi V, Giovannelli L, Bardini G, et al Oxidative DNA damage in peripheral blood cells in type 2 diabetes mellitus: higher vulnerability of polymorphonuclear leukocytes. Mutat Res 2003; 529: 129–133. [DOI] [PubMed] [Google Scholar]
- 86. Dincer Y, Akcay T, Ilkova H, et al DNA damage and antioxidant defense in peripheral leukocytes of patients with Type I diabetes mellitus. Mutat Res 2003; 527: 49–55. [DOI] [PubMed] [Google Scholar]
- 87. Wong RK, Pettit AI, Davies JE, et al Augmentation of the neutrophil respiratory burst through the action of advanced glycation end products: a potential contributor to vascular oxidant stress. Diabetes 2002; 51: 2846–2853. [DOI] [PubMed] [Google Scholar]
- 88. Inoue S, Lan Y, Muran J, et al Reduced hydrogen peroxide production in neutrophils from patients with diabetes. Diabetes Res Clin Pract 1996; 33: 119–127. [DOI] [PubMed] [Google Scholar]
- 89. Heinzelmann M, Mercer‐Jones MA, Passmore JC. Neutrophils and renal failure. Am J Kidney Dis 1999; 34: 384–399. [DOI] [PubMed] [Google Scholar]
- 90. Horio F, Fukuda M, Katoh H, et al Reactive oxygen intermediates in autoimmune islet cell destruction of the NOD mouse induced by peritoneal exudate cells (rich in macrophages) but not T cells. Diabetologia 1994; 37: 22–31. [DOI] [PubMed] [Google Scholar]
- 91. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol 2011; 7: 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Diana J, Simoni Y, Furio L, et al Crosstalk between neutrophils, B‐1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med 2013; 19: 65–73. [DOI] [PubMed] [Google Scholar]
- 93. Riyapa D, Buddhisa S, Korbsrisate S, et al Neutrophil extracellular traps exhibit antibacterial activity against burkholderia pseudomallei and are influenced by bacterial and host factors. Infect Immun 2012; 80: 3921–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Joshi MB, Lad A, Bharath PAS, et al High glucose modulates IL‐6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett 2013; 587: 2241–2246. [DOI] [PubMed] [Google Scholar]
- 95. Jaillon S, Galdiero MR, Del PD, et al Neutrophils in innate and adaptive immunity. Semin Immunopathol 2013; 35: 377–394. [DOI] [PubMed] [Google Scholar]
- 96. van Gisbergen KP, Sanchez‐Hernandez M, Geijtenbeek TB, et al Neutrophils mediate immune modulation of dendritic cells through glycosylation‐dependent interactions between Mac‐1 and DC‐SIGN. J Exp Med 2005; 201: 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Talukdar S, Oh DY, Bandyopadhyay G, et al Neutrophils mediate insulin resistance in mice fed a high‐fat diet through secreted elastase. Nat Med 2012; 18: 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Diana J, Lehuen A. Macrophages and beta‐cells are responsible for CXCR2‐mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol Med 2014; 6: 1090–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Willcox A, Richardson SJ, Bone AJ, et al Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 2009; 155: 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lehuen A, Diana J, Zaccone P, et al Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 2010; 10: 501–513. [DOI] [PubMed] [Google Scholar]
- 101. Haurogne K, Pavlovic M, Rogniaux H, et al Type 1 diabetes prone NOD mice have diminished Cxcr1 mRNA expression in polymorphonuclear neutrophils and CD4+ T lymphocytes. PLoS One 2015; 10: e0134365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Arif S, Moore F, Marks K, et al Peripheral and islet interleukin‐17 pathway activation characterizes human autoimmune diabetes and promotes cytokine‐mediated beta‐cell death. Diabetes 2011; 60: 2112–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Omi H, Okayama N, Shimizu M, et al Participation of high glucose concentrations in neutrophil adhesion and surface expression of adhesion molecules on cultured human endothelial cells: effect of antidiabetic medicines. J Diabetes Complications 2002; 16: 201–208. [DOI] [PubMed] [Google Scholar]
- 104. Okouchi M, Okayama N, Imai S, et al High insulin enhances neutrophil transendothelial migration through increasing surface expression of platelet endothelial cell adhesion molecule‐1 via activation of mitogen activated protein kinase. Diabetologia 2002; 45: 1449–1456. [DOI] [PubMed] [Google Scholar]
- 105. Rawling LD, Advani A, Marshall SM, et al Neutrophil antigen exposure is altered with age in relatives of patients with Type 2 diabetes. Diabetologia 2004; 47: 353–355. [DOI] [PubMed] [Google Scholar]
- 106. Okayama N, Omi H, Okouchi M, et al Mechanisms of inhibitory activity of the aldose reductase inhibitor, epalrestat, on high glucose‐mediated endothelial injury: neutrophil‐endothelial cell adhesion and surface expression of endothelial adhesion molecules. J Diabetes Complications 2002; 16: 321–326. [DOI] [PubMed] [Google Scholar]
- 107. Hanninen A, Salmi M, Simell O, et al Endothelial cell‐binding properties of lymphocytes infiltrated into human diabetic pancreas. Implications for pathogenesis of IDDM. Diabetes 1993; 42: 1656–1662. [DOI] [PubMed] [Google Scholar]
- 108. Itoh N, Hanafusa T, Miyazaki A, et al Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin‐dependent diabetes mellitus patients. J Clin Invest 1993; 92: 2313–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Li N, Hu H, Lindqvist M, et al Platelet‐leukocyte cross talk in whole blood. Arterioscler Thromb Vasc Biol 2000; 20: 2702–2708. [DOI] [PubMed] [Google Scholar]
- 110. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol 2011; 11: 264–274. [DOI] [PubMed] [Google Scholar]
- 111. Repka J, Nicoluzzi J, Belila R, et al Neutrophil oxidative metabolism in diabetic patients undergoing pancreas transplantation. Transplant Proc 2006; 38: 3023–3025. [DOI] [PubMed] [Google Scholar]
- 112. Hartz S, Menart B, Tschoepe D. Leukocyte apoptosis in whole blood involves platelet‐dependent coaggregation. Cytometry A 2003; 52: 117–121. [DOI] [PubMed] [Google Scholar]
- 113. Gawlowski T, Stratmann B, Stirban AO, et al AGEs and methylglyoxal induce apoptosis and expression of Mac‐1 on neutrophils resulting in platelet‐neutrophil aggregation. Thromb Res 2007; 121: 117–126. [DOI] [PubMed] [Google Scholar]
- 114. Jain SK, Krueger KS, McVie R, et al Relationship of blood thromboxane‐B2 (TxB2) with lipid peroxides and effect of vitamin E and placebo supplementation on TxB2 and lipid peroxide levels in type 1 diabetic patients. Diabetes Care 1998; 21: 1511–1516. [DOI] [PubMed] [Google Scholar]
- 115. Renesto P, Chignard M. Enhancement of cathepsin G‐induced platelet activation by leukocyte elastase: consequence for the neutrophil‐mediated platelet activation. Blood 1993; 82: 139–144. [PubMed] [Google Scholar]
- 116. Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes 1991; 40: 405–412. [DOI] [PubMed] [Google Scholar]
- 117. Hu H, Li N, Yngen M, et al Enhanced leukocyte‐platelet cross‐talk in Type 1 diabetes mellitus: relationship to microangiopathy. J Thromb Haemost 2004; 2: 58–64. [DOI] [PubMed] [Google Scholar]
- 118. Entman ML, Ballantyne CM. Association of neutrophils with platelet aggregates in unstable angina. Should we alter therapy. Circulation 1996; 94: 1206–1208. [DOI] [PubMed] [Google Scholar]
- 119. Libby P, Simon DI. Inflammation and thrombosis: the clot thickens. Circulation 2001; 103: 1718–1720. [DOI] [PubMed] [Google Scholar]
- 120. Tuttle HA, Davis‐Gorman G, Goldman S, et al Platelet‐neutrophil conjugate formation is increased in diabetic women with cardiovascular disease. Cardiovasc Diabetol 2003; 2: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Baron JL, Reich EP, Visintin I, et al The pathogenesis of adoptive murine autoimmune diabetes requires an interaction between alpha 4‐integrins and vascular cell adhesion molecule‐1. J Clin Invest 1994; 93: 1700–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Burkly LC, Jakubowski A, Hattori M. Protection against adoptive transfer of autoimmune diabetes mediated through very late antigen‐4 integrin. Diabetes 1994; 43: 529–534. [DOI] [PubMed] [Google Scholar]
- 123. Moriyama H, Yokono K, Amano K, et al Induction of tolerance in murine autoimmune diabetes by transient blockade of leukocyte function‐associated antigen‐1/intercellular adhesion molecule‐1 pathway. J Immunol 1996; 157: 3737–3743. [PubMed] [Google Scholar]
- 124. Dunne JL, Collins RG, Beaudet AL, et al Mac‐1, but not LFA‐1, uses intercellular adhesion molecule‐1 to mediate slow leukocyte rolling in TNF‐alpha‐induced inflammation. J Immunol 2003; 171: 6105–6111. [DOI] [PubMed] [Google Scholar]
- 125. Oubenaissa A, Mouas C, Bourgeois F, et al Evidence for an involvement of the neutrophil integrin lymphocyte function‐associated antigen‐1 in early failure of heart transplants. Circulation 1996; 94: II254–II259. [PubMed] [Google Scholar]
- 126. Basit A, Reutershan J, Morris MA, et al ICAM‐1 and LFA‐1 play critical roles in LPS‐induced neutrophil recruitment into the alveolar space. Am J Physiol Lung Cell Mol Physiol 2006; 291: L200–L207. [DOI] [PubMed] [Google Scholar]
- 127. Forsyth KD, Levinsky RJ. Role of the LFA‐1 adhesion glycoprotein in neutrophil adhesion to endothelium and plastic surfaces. Clin Exp Immunol 1989; 75: 265–268. [PMC free article] [PubMed] [Google Scholar]
- 128. Asaduzzaman M, Zhang S, Lavasani S, et al LFA‐1 and MAC‐1 mediate pulmonary recruitment of neutrophils and tissue damage in abdominal sepsis. Shock 2008; 30: 254–259. [DOI] [PubMed] [Google Scholar]
- 129. Berton G, Laudanna C, Sorio C, et al Generation of signals activating neutrophil functions by leukocyte integrins: LFA‐1 and gp150/95, but not CR3, are able to stimulate the respiratory burst of human neutrophils. J Cell Biol 1992; 116: 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]