

Abstract
Parathyroid hormone (PTH) and FGF23 are the primary hormones regulating acute phosphate homeostasis. Human renal proximal tubule cells (RPTECs) were used to characterize the mechanism and signaling pathways of PTH and FGF23 on phosphate transport and the role of the PDZ protein NHERF1 in mediating PTH and FGF23 effects. RPTECs express the NPT2A phosphate transporter, αKlotho, FGFR1, FGFR3, FGFR4, and the PTH receptor. FGFR1 isoforms are formed from alternate splicing of exon 3 and of exon 8 or 9 in Ir-like loop 3. Exon 3 was absent, but mRNA containing both exons 8 and 9 is present in cytoplasm. Using an FGFR1c-specific antibody together with mass spectrometry analysis, we show that RPTECs express FGFR-β1C. The data are consistent with regulated FGFR1 splicing involving a novel cytoplasmic mechanism. PTH and FGF23 inhibited phosphate transport in a concentration-dependent manner. At maximally effective concentrations, PTH and FGF23 equivalently decreased phosphate uptake and were not additive, suggesting a shared mechanism of action. Protein kinase A or C blockade prevented PTH but not FGF23 actions. Conversely, inhibiting SGK1, blocking FGFR dimerization, or knocking down Klotho expression disrupted FGF23 actions but did not interfere with PTH effects. C-terminal FGF23(180–251) competitively and selectively blocked FGF23 action without disrupting PTH effects. However, both PTH and FGF23-sensitive phosphate transport were abolished by NHERF1 shRNA knockdown. Extended treatment with PTH or FGF23 down-regulated NPT2A without affecting NHERF1. We conclude that FGFR1c and PTHR signaling pathways converge on NHERF1 to inhibit PTH- and FGF23-sensitive phosphate transport and down-regulate NPT2A.
Keywords: alternative splicing, fibroblast growth factor receptor (FGFR), G protein-coupled receptor (GPCR), parathyroid hormone, transport, NHERF1, NPT2A, PDZ Protein, klotho
Introduction
Parathyroid hormone (PTH)2 and FGF23 display two remarkable features: 1) PTH and FGF23 exhibit parallel inhibition of renal phosphate transport mediated by NPT2A (sodium-dependent phosphate transporter-2a) but opposing actions on 1,25(OH)2-vitamin D; 2) despite being structurally and functionally distinct classes of membrane-delimited receptors, PTH and FGF receptors activate kinases that obligatorily phosphorylate NHERF1 at conserved sites required for their phosphaturic action. Phosphorus is essential for growth and maintenance of the skeleton and for generating high energy phosphate compounds. Evolutionary adaptation in humans and other terrestrial vertebrates to phosphorus-rich diets involves cell and molecular mechanisms ensuring the efficient urinary elimination of excess inorganic phosphate. The renal proximal tubule is the primary site of phosphate homeostasis and hormone-dependent phosphate transport. The NPT2A sodium-dependent phosphate cotransporter (SLC34A1) in proximal tubules is regulated by PTH and FGF23 (1, 2). PTH and FGF23 reduce phosphate uptake by sequestering and down-regulating NPT2A, thereby enhancing urinary phosphate excretion (3, 4). PTH actions are mediated by its cognate G protein-coupled PTH receptor (PTHR) (5, 6). Both PKA and PKC have been implicated in PTH-dependent inhibition of NPT2A (7–12). Using a signaling-selective form of the PTHR that activates PKA or signaling-biased PTH analogs, Jüppner and co-workers (13, 14) determined that adenylyl cyclase and PKA regulate acute PTH effects on Npt2a and phosphate transport, whereas persistent reductions of phosphate transport require PKC.
FGF23 actions on NPT2A are facilitated by receptor tyrosine kinases FGFR1 and FGFR4 with the obligate participation of the coreceptor αKlotho (15, 16). FGFR1 signaling by ERK1/2 and serum and glucocorticoid-activated kinase (SGK1) leads to inhibition of phosphate transport (17).
NPT2A harbors a canonical C-terminal PDZ recognition motif (-TRL) that binds to the PDZ scaffolding protein, Na+/H+ exchanger regulatory factor 1 (NHERF1) (18, 19). NHERF1 assembles a ternary complex with NPT2A and ezrin, thereby linking the transporter to the actin cytoskeleton. NHERF1 is essential for the inhibitory action of PTH and FGF23 on phosphate transport (12, 17, 20). NHERF1-null mice and humans harboring mutations in SLC9A3R1 exhibit pronounced phosphate wasting, nephrolithiasis, and skeletal disorders (21–23). Both PTH and FGF23 phosphorylate NHERF1 (17, 24–26). These findings imply that activation of distinct types of cell surface receptors results in phosphorylation of NHERF1, resulting in disassembly of the NPT2A-NHERF1-ezrin complex (27), internalization and down-regulation of NPT2A, and cessation of phosphate transport. Although the components of these pathways have been described, the lack of a suitable cell model has hindered a comprehensive analysis of PTH and FGF23 actions on NPT2A and cellular phosphate transport. We report here that PTH and FGF23 inhibit NPT2A-dependent phosphate transport in immortalized human renal proximal epithelial cells (RPTECs). PTH actions mediated by the PTHR involve PKA and PKC, whereas FGF23 effects proceed through FGFR1c and SGK1 and require αKlotho. Interruption of PTHR signaling inhibited PTH but not FGF23 actions on NPT2A. Conversely, disrupting FGFR1-associated proteins eradicated FGF23 but not PTH effects on phosphate transport. NHERF1 knockdown prevented both PTH and FGF23 action, indicating that PTHR and FGFR1c signaling pathways converge at the level of NHERF1.
Results and Discussion
RPTECs retain many features of proximal tubule epithelial cells, including the formation of tight junctions, PTH-stimulated cAMP accumulation, and sodium-dependent phosphate uptake (28). To extend these observations, we characterized the expression of NPT2A, NHERF1, PTHR, and FGFR and analyzed the regulation of phosphate transport by PTH and FGF23.
NPT2A concentrated in a tightly organized cluster at the apical pole of the cells, reminiscent of apical recycling endosomes described in other kidney cell types (Fig. 1A) (29, 30). NHERF1 was conspicuously expressed at the cell surface and displayed extensive colocalization with NPT2A (Fig. 1A, merge). NPT2A also localized to the primary cilium; however, NHERF1 was absent from this structure (see arrows in Fig. 1A and magnification in Fig. 1B). A similar distribution was observed using three different anti-NPT2A antibodies (data not shown). This localization contrasts with observations by Wade et al. (31), who found NPT2A and NHERF1 in microvilli of mouse proximal tubule cells. We also established that PTHR was prominently expressed at both apical and basolateral cell membranes (Fig. 1C) as described for native proximal tubules (10, 32, 33). FGFR1 was primarily expressed at the apical membrane (Fig. 1D).
FIGURE 1.
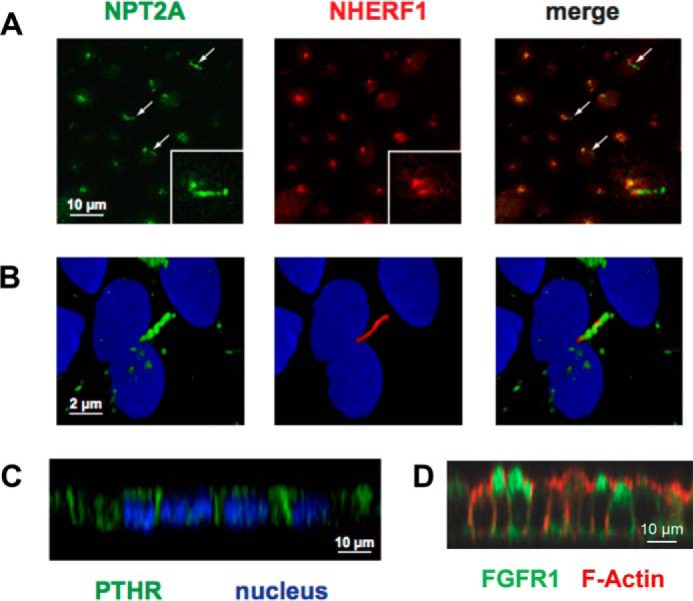
NPT2A, NHERF1, PTHR, and FGFR1 expression in RPTECs. A, distribution of NPT2A (green), NHERF1 (red), and nuclei (blue) in filter-grown RPTECs. Single optical sections from the apical region of the cell are shown. Arrows, NPT2A association with cilia. B, magnified views of a single cell. C, x-z plane depicting PTHR (green) and nuclear (blue) staining of RPTECs grown on coverslips. D, FGFR1 (green) and F-actin (red) staining of RPTECs grown on filters. The x-z plane is shown.
RPTECs express mRNA transcripts for FGFR1, FGFR3, and FGFR4 mRNA (Fig. 2A). These results are compatible with recent findings that FGFR1, FGFR3, and FGFR4 are expressed in murine proximal tubules (34, 35). This finding is consistent with studies using recombinant extracellular Klotho domain and FGFR-Fc fusion constructs of FGFR1b and FGFR1c that showed that FGF23 binds only to FGFR1c (36). Based on these and other findings (37, 38) FGFR1, with its cofactor αKlotho, is considered the cognate FGF23 receptor regulating phosphate transport.
FIGURE 2.

FGFR mRNA and protein expression in RPTECs. A, representative 3% agarose gel of FGFR expression in RPTECs determined by RT-PCR (Table 2). 200- and 100-bp markers are shown in the left lane. B, schematic representations of the FGFR1 gene and protein structure. The FGFR-β1c splice variant harbors two extracellular immunoglobulin-like domains (II and III) with alternatively spliced exon 9 in loop III, a transmembrane domain (TM), and two intracellular tyrosine kinase domains (TK). C, FGFR1-β splice variant in a characteristic 2% agarose gel with ladder in the left lane. The 500-bp PCR product is consistent with the β-splice variant lacking exon 3. D, FGFR1b/c splice variants. Lane 1, FGFR1b; lane 2, FGFR1c; lane 3, mRNA containing both exons 8 and 9. Lane 3 PCR was performed using the forward primer for FGFR1b and the reverse primer for FGFR1c (Table 1). An illustrative 2% agarose gel is shown with a 100-bp ladder in the left lane. The PCR product in lane 2 is consistent with the exon 9 FGFR1c splice variant. Lane 3 shows an mRNA species containing both exons 8 and 9. E, FGFR1c is the primary species of FGFR1 detected by immunoblotting. FGFR1 and FGFR1c in RPTECs were detected by non-selective and isotype-selective antisera, respectively, as described under “Experimental Procedures.” The results are illustrative of n = 3 independent experiments. F, MS/MS spectrum for the identified specific peptide 355SDFHSQMAVHKLAK368 from FGFR1c. The peak heights show the relative abundances of the corresponding fragmentation ions, with the annotation of the identified matched N terminus-containing b ions in blue and the C terminus-containing y ions in red. Charge state: +3, observed m/z: 538.93969, theoretical m/z: 538.94008, precursor mass error: −0.72, Xcorr: 0.719.
FGFR1, along with FGFR2 and FGFR4, exhibit isotype variation, ligand specificity, and regulation that are determined by extensive alternative splicing (39, 40). The primary sites of FGFR1 splicing include Ig-like loops I and III (41, 42). The pattern of FGFR1 gene splicing and corresponding protein structures are shown in Fig. 2B. The α or β isoforms of FGFR are generated by inclusion or exclusion of exon 3, which encodes the first Ig-like loop (43). RT-PCR experiments using exon-spanning primers (Table 1) disclosed the absence of exon 3 (Fig. 2C), hence indicating that FGFR1 is the β form, FGFR-β1, and lacks the Ig-I loop.
TABLE 1.
FGFR1 exon splicing primers

Further FGFR variation arises from alternative splicing in Ig-like loop III. The N-terminal portion of loop III is encoded by exon 7. The C-terminal half of loop III is the extracellular ligand-binding domain of the receptor (44) and is generated by alternate inclusion of either exon 8, which then forms FGFR1b, or exon 9 to form FGFR1c. Regulatory RNA-binding proteins determine the splice events for FGFR1b and FGFR1c isoforms (42, 45). FGFR1b is considered the epithelial form of the receptor, whereas FGFR1c is accepted as expressed by mesenchymal cells (41). Thus, it would be reasonable to suppose that epithelial proximal tubule cells would express FGFR1b despite FGFR1c being accepted as the canonical FGF23 receptor. We designed exon-spanning primers (Table 1) to delineate the FGFR1 isoform in RPTECs. Using forward primers spanning the exon 7/8 boundary (FGFR1b) or the exon 7/9 boundary (FGFR1c) and reverse primers that span the exon 10/8 boundary (FGFR1b) or the exon 10/9 boundary (FGFR1c), we determined that RPTECs use exon 9 and thus express FGFR1c (Fig. 2D).
Strikingly, using the FGFR1b forward primer with the FGFR1c reverse primer, mRNA containing both exons 8 and 9 was robustly displayed in the cytoplasm (Fig. 2D). As noted above, exon 3, however, was undetectable (Fig. 2B). This suggests that splicing of exon 3 for the FGFR1α/β receptor isoform occurs in the nucleus by a canonical process, whereas exon splicing leading to tissue-specific expression of FGFR1b/c proceeds in the cytoplasm. Such noncanonical post-transcriptional mRNA processing is emerging as a cell-specific regulatory step that determines the translated splice variant (46).
Using a polyclonal FGFR1 antibody that does not distinguish between alternatively spliced FGFR1 isoforms, RPTECs express a single protein band of 72 kDa (Fig. 2E). A band of similar size was detected using an FGFR1c-specific antibody (47), confirming that FGFR1c is the only species of FGFR1 protein expressed by RPTECs. We validated this finding by mass spectrometry, where we detected a unique tryptic peptide consistent with FGFR1c (Fig. 2F). Protein containing both exons 8 and 9 was not found. Given that exons 8 and 9 each encode the mutually exclusive C-terminal portion of loop IIIc, it is unlikely that a protein harboring both domains exists or would fold correctly. Thus, RPTECs express FGFR-β1c.
Taken together, these results establish that RPTECs express NPT2A, NHERF1, PTHR, and FGFR-β1c, the proteins involved in hormone-regulated phosphate transport. Based on these considerations, we proceeded to characterize the actions of PTH and FGF23 on phosphate transport. PTH and FGF23 inhibited phosphate uptake in a concentration-dependent manner with an EC50 of 30 and 51 pm, respectively (p < 0.02) (Fig. 3A). At maximally effective concentrations of 100 nm, FGF23 and PTH similarly inhibited phosphate uptake by 40 ± 9 and 43 ± 7%, respectively (Fig. 3B). The combined addition of FGF23 and PTH modestly augmented the inhibition of phosphate uptake by an additional 20% (Fig. 3B). The non-additive response of PTH and FGF23 probably reflects a common final mechanism of action.
FIGURE 3.

PTH and FGF23 inhibit phosphate transport. A, RPTECs were treated for 2 h with 100 nm PTH(1–34) or FGF23. Phosphate uptake was measured for 10 min, as detailed under “Experimental Procedures.” Data represent the mean ± S.E. (error bars) of n = 6 independent experiments performed in triplicate. Data were normalized for each experiment, where phosphate uptake under control, untreated conditions, was defined as 0% inhibition. Data were fit to a sigmoidal relation, and Kd values were calculated with Prism. B, RPTECs on 12-well plates were treated for 2 h with the indicated concentrations of PTH or FGF23. Phosphate uptake was measured for 10 min, as outlined under “Experimental Procedures.” Data represent the mean ± S.E. of n = 6 independent experiments performed in triplicate. *, p < 0.05 versus control; **, p < 0.01 versus control. C, C-terminal FGF23(180–251) fragment blocks FGF23 but not PTH inhibitable phosphate uptake. Data are the mean ± S.E. of n = 4 experiments. **, p < 0.01 versus FGF23.
C-terminal FGF23(180–251) virtually abolished the actions of FGF23(28–251) on phosphate transport but had no effect on its own; it did not interfere with PTH-sensitive phosphate transport (Fig. 3C). FGF23(180–251) competes with full-length FGF23 for binding to the FGFR1-αKlotho complex and reduces renal phosphate excretion (48). Additional controls showed that neither FGF2 nor PTH(7–34) affected phosphate transport (data not shown), further demonstrating the specificity of FGF23 and PTH action on phosphate transport.
PTHR and FGFR1c mediate the phosphaturic actions of PTH and FGF23, respectively (5, 49, 50). Both receptors are expressed on RPTECs (Fig. 2). The PTHR signals primarily through Gs and Gq, leading to downstream activation of PKA and PKC, which are stimulated in parallel following occupancy of proximal tubule PTHR (51). FGFR1 dimerizes upon activation, thereby stimulating downstream kinases associated with Ras/ERK and SGK1 (17, 52). To define the mechanism of PTH and FGF23 action on phosphate transport in RPTECs, we first characterized the signaling pathways responsible for their effects by employing a panel of pharmacological protein kinase inhibitors. The PKA inhibitor H89 and PKC inhibitor bis-indolylmaleimide I (Bis-I) blocked the actions of PTH (Fig. 4A) but had no effect on FGF23-sensitive phosphate uptake (Fig. 4B). In contrast, the SGK1 inhibitor GSK650394 blocked the actions of FGF23 (Fig. 4B) but not PTH (Fig. 4A). These results are consistent with observations that acute NPT2A down-regulation by PTH involves cAMP and PKA (14). The findings are also compatible with a recent study showing that acute PTH action involves PKA (14), whereas FGF23 effects are mediated by SGK1 (17). The ERK1/2 inhibitor PD-98059 abolished the action of FGF23 and inhibited PTH-sensitive uptake by 50% (Fig. 4, A and B). PTH activation of ERK occurs downstream of PKC activation (53, 54). This may explain the partial inhibition of PTH-sensitive phosphate transport by PD98059.
FIGURE 4.

Signaling pathways mediating PTH and FGF23 effects on phosphate transport. RPTECs were treated for 2 h with 100 nm PTH(1–34) (A) or FGF23 (B) in the presence or absence of the specified inhibitors: BisI (PKC), GSK-650394 (SGK-1), H89 (PKA), and PD (PD-98059, ERK1/2). Inhibitors were used at 1 μm except for GSK, where 10 μm was employed. Phosphate uptake was measured as before. Data represent the mean ± S.E. (error bars) of n = 6 experiments. *, p < 0.05; **, p < 0.01 versus PTH or FGF23.
The finding that RPTECs express FGFR1c (Fig. 2, E and F) implies that knockdown of this isoform with an siRNA targeted to exon 9 should inhibit FGF23-sensitive phosphate transport. siRNA specific to either exon 8 (si8, FGFR1b) or exon 9 (si9, FGFR1c) was used to knock down FGFR1 expression. A sample immunoblot showing FGFR1c abundance after transfection of RPTECs with siRNA is shown in Fig. 5A. si8 and si9 reduced FGFR1c protein expression by 82 and 89%, respectively (Fig. 5B). Detection with a polyclonal anti-FGFR1 antibody yielded similar results (data not shown). Unexpectedly, knocking down exon 8 (si8) was almost as effective as knocking down exon 9 (si9) at reducing FGFR1c protein expression. si8 reduced FGF23-dependent phosphate transport by 40%, and si9 eliminated FGF23 modulation (Fig. 5C). These data are consistent with FGFR1c being the primary mediator of the phosphaturic effects of FGF23. Furthermore, the ability of si8 to interfere with FGFR1c expression supports the supposition that the mRNA emerging from the nucleus contains both exons 8 and 9, as shown in Fig. 2C, and that regulated splicing in the cytoplasm determines the translation and expressed form of FGFR1.
FIGURE 5.
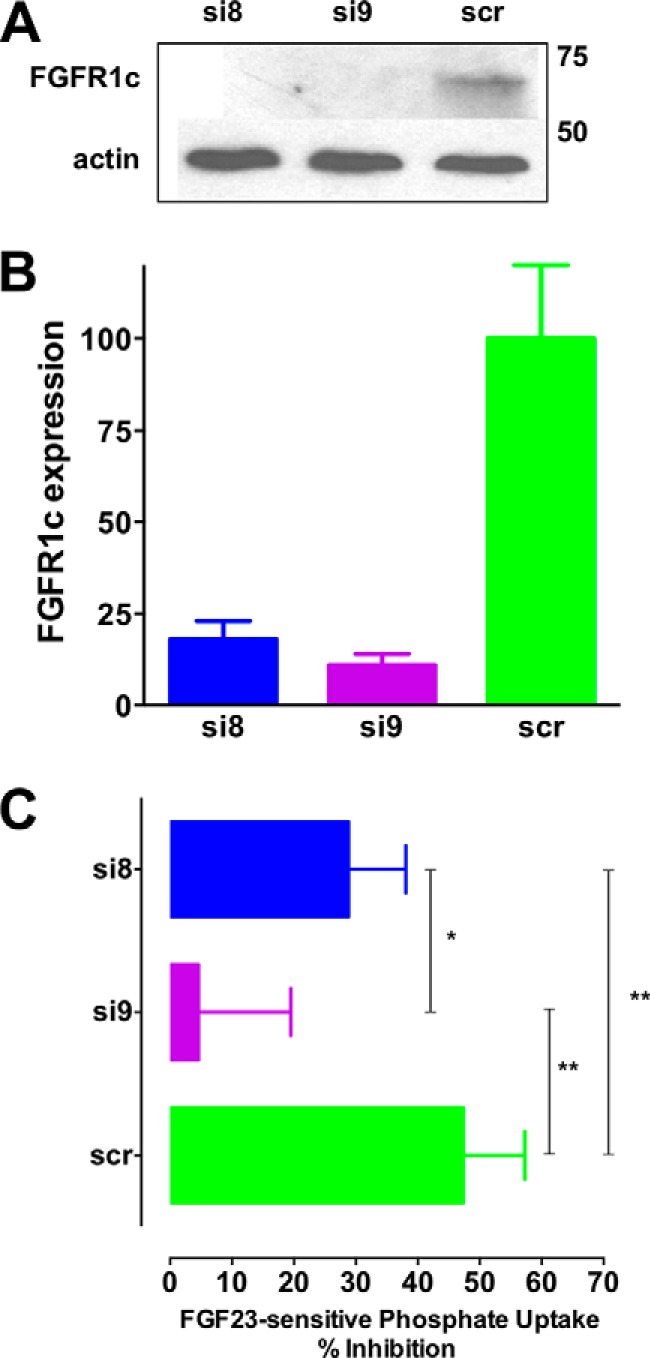
Effect of FGFR1 exon 9 and exon 8 knockdown on FGF23-regulated phosphate transport. A, FGFR1 knockdown by siRNA for exon 8 (si8), exon 9 (si9), or a scrambled control (scr) was assessed by immunoblotting using antibody that specifically recognizes FGFR1c. A representative result is presented. B, quantification of si8 and si9 knockdown of FGFR1c. Data represent the mean ± S.E. (error bars) of n = 4 experiments. C, siRNA effects on FGF23-sensitive phosphate transport. Data represent the mean ± S.E. of n = 4 experiments. *, p < 0.05; **, p < 0.01.
FGF23 effects mediated by FGFR1 require the presence of the coreceptor αKlotho (41). We confirmed that RPTECs express αKlotho (Fig. 6A). We then assessed the involvement of αKlotho as an FGFR1 cofactor to support FGF23 effects on phosphate transport. We envisioned that disrupting αKlotho would interfere with FGF23 actions but not those of PTH. Stable formation of the FGF23-FGFR1-αKlotho complex and FGFR dimerization is regulated by sulfated glycosaminoglycans (4). Sodium chlorate inhibits proteoglycan sulfation by removing cell surface glycosaminoglycans without affecting protein synthesis or other posttranslational modifications (55). Overnight treatment of RPTECs with 50 mm NaClO3 suppressed FGF23 but not PTH effects on phosphate transport in RPTECs (Fig. 6B). These data are consistent with the constitutive and sufficient expression of αKlotho by RPTECs and its requirement for FGF23-mediated signaling and NPT2A-dependent phosphate transport and with previous work showing that NaClO3 reversibly interferes with FGF23 action (4).
FIGURE 6.
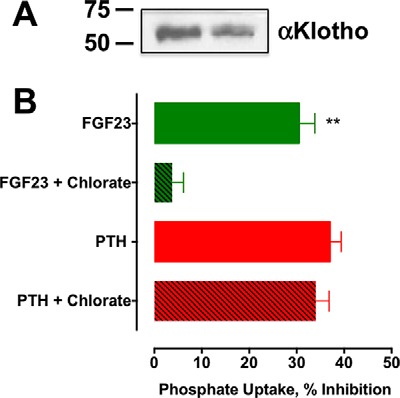
αKlotho expression and function. A, αKlotho is expressed by RPTECs. Shown is an immunoblot of 20 μg of protein lysate duplicate wells probed with rabbit polyclonal anti-Klotho antibody. Molecular mass markers representing 50 and 75 kDa are shown. B, chlorate treatment of RPTECs interfered with FGF23 but not PTH inhibition of phosphate transport. RPTECs were treated overnight with 50 mm NaClO4, followed by a 2-h treatment with 100 nm PTH(1–34) or FGF23, as indicated. Phosphate uptake was measured as described under “Experimental Procedures.” Data represent the mean ± S.E. (error bars) of n = 4 independent experiments. Data were normalized for each experiment, where phosphate uptake under control, untreated conditions was defined as 0% inhibition. **, p < 0.01 versus FGF23 alone.
Recent work shows that unliganded FGFR1 is able to form homodimers or heterodimerize with αKlotho. These receptor complexes undergo conformational changes upon ligand occupancy (56). To establish directly its cofactor role in FGF23-sensitive phosphate transport, we knocked down αKlotho in RPTECs and determined its effect on FGF23 and PTH action on phosphate uptake. Three different siRNAs were screened. siRNA3 decreased αKlotho expression by 80% (Fig. 7A), and this was associated with the most pronounced disruption with the inhibition of phosphate uptake by FGF23 (Fig. 7B). The converse was also true insofar as siRNA1, which failed to reduce αKlotho expression, had no discernable inhibitory action on FGF23-sensitive phosphate transport. Thus, the extent of αKlotho knockdown corresponded to the degree of suppression of FGF23-sensitive phosphate transport. As predicted, αKlotho knockdown did not interfere with PTH inhibition of phosphate uptake (Fig. 7B), underscoring the distinct signaling and cofactor requirements for FGF23 and PTH actions. The results also imply that αKlotho acts specifically and autonomously on FGF23.
FIGURE 7.
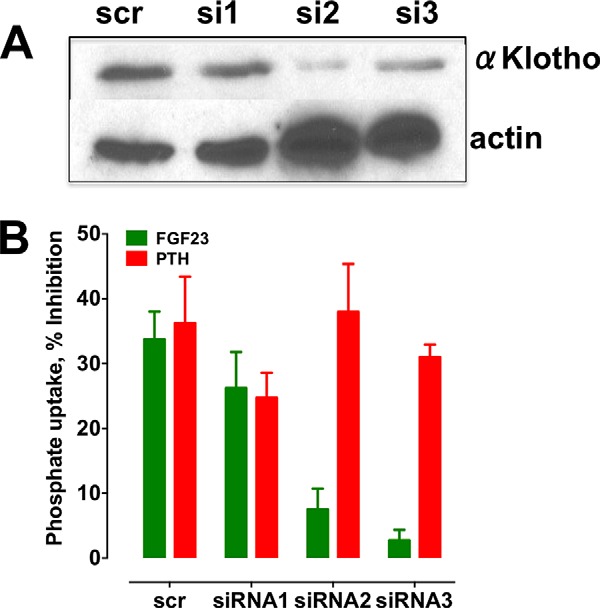
siRNA αKlotho knockdown blocks FGF23- but not PTH-inhibitable phosphate transport. αKlotho expression was knocked down in RPTECs using siRNA as described under “Experimental Procedures.” A, knockdown of αKlotho was assessed by immunoblotting. A representative experiment is depicted. The data for three siRNA duplexes plus a scrambled control (scr) are shown. B, the effects of αKlotho knockdown on PTH- and FGF23-dependent phosphate transport were measured. Data represent the mean ± S.E. (error bars) of n = 5 experiments. Data were normalized for each experiment, where phosphate uptake under control, untreated conditions was defined as 0% inhibition. **, p < 0.01; ***, p < 0.001 versus scrambled.
Contrary to these findings, a recent study suggested that exogenous recombinant αKlotho binds PTH, thereby interfering with PTH binding and signaling (57). Although we can not speak to the effects of exogenous Klotho on PTH action on phosphate transport, blocking FGFR dimerization or down-regulating αKlotho selectively impaired FGF23 actions without interfering with the inhibitory effect of PTH on phosphate transport. An alternative explanation for the apparent inhibitory effect of Klotho on PTH action may be ascribed to signaling cross-talk between GPCRs and receptor tyrosine kinases that arises from stimulation of ERK1/2 and endocytosis of unliganded receptor (58).
PTH and FGF23 acting through their respective GPCR and receptor tyrosine kinase, stimulate distinct signaling pathways, but both require NHERF1 to inhibit phosphate transport (12, 17). This raised the hypothesis that the signaling events initiated at PTHR and FGFR1 converge on NHERF1 to facilitate endocytosis and inhibit NPT2A-dependent phosphate transport. According to this view, NHERF1 knockdown should disrupt the actions of both FGF23 and PTH. Consistent with this prediction, shNHERF1 (shN1) reduced NHERF1 expression by 80% (Fig. 8A) and concomitantly virtually abolished both PTH and FGF23-inhibitable phosphate transport (Fig. 8B). These findings support the idea that although the actions of FGF23 and PTH stem from different classes of membrane receptors, their effects intersect at the level of NHERF1. This is compatible with the requirement for NHERF1 to mediate the inhibitory actions of both PTH and FGF23 on NPT2A (12, 17, 20). Phosphorylation of NHERF1 results in disassembly of the NPT2A-NHERF1-ezrin complex (27), internalization and down-regulation of NPT2A, and cessation of phosphate transport.
FIGURE 8.
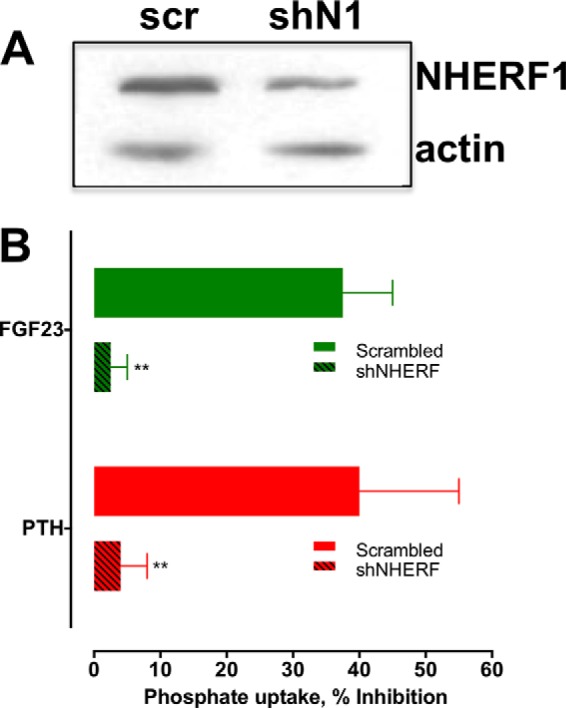
shRNA NHERF1 knockdown inhibits FGF23- and PTH-regulated phosphate transport. NHERF1 expression was knocked down in RPTECs by shRNA transfection as described under “Experimental Procedures.” A, knockdown of NHERF1 was assessed by immunoblotting. A representative experiment is depicted. The data for NHERF1 shRNA and a scrambled control (scr) are shown. B, effects of NHERF1 knockdown on PTH- and FGF23-dependent phosphate transport were assessed. Data represent the mean ± S.E. (error bars) of n = 4 experiments. Data were normalized for each experiment, where phosphate uptake under control, untreated conditions was defined as 0% inhibition. **, p < 0.01 versus FGF23 or PTH.
NHERF1 harbors 38 Ser and Thr residues. Identifying the particular residues phosphorylated following activation of PTHR and FGFR1 in a native cell model will be essential to understand the structural determinants and sites of PTH- and FGF23-dependent NHERF1 post-translational modifications.
Treatment with PTH or FGF23 for 2 h down-regulates NPT2A in RPTECs (Fig. 9A). After PTH or FGF23 exposure, NPT2A was less concentrated at the apical pole of the cell, with a significant amount now present in the cytoplasm (Fig. 9B). In contrast, NHERF1 remained associated with the apical membranes and showed diminished colocalization with NPT2A. A phenomenologically similar process has been described for PTH action on PTHR trafficking, where PTHR is endocytosed while NHERF1 remains at the cell surface (59).
FIGURE 9.
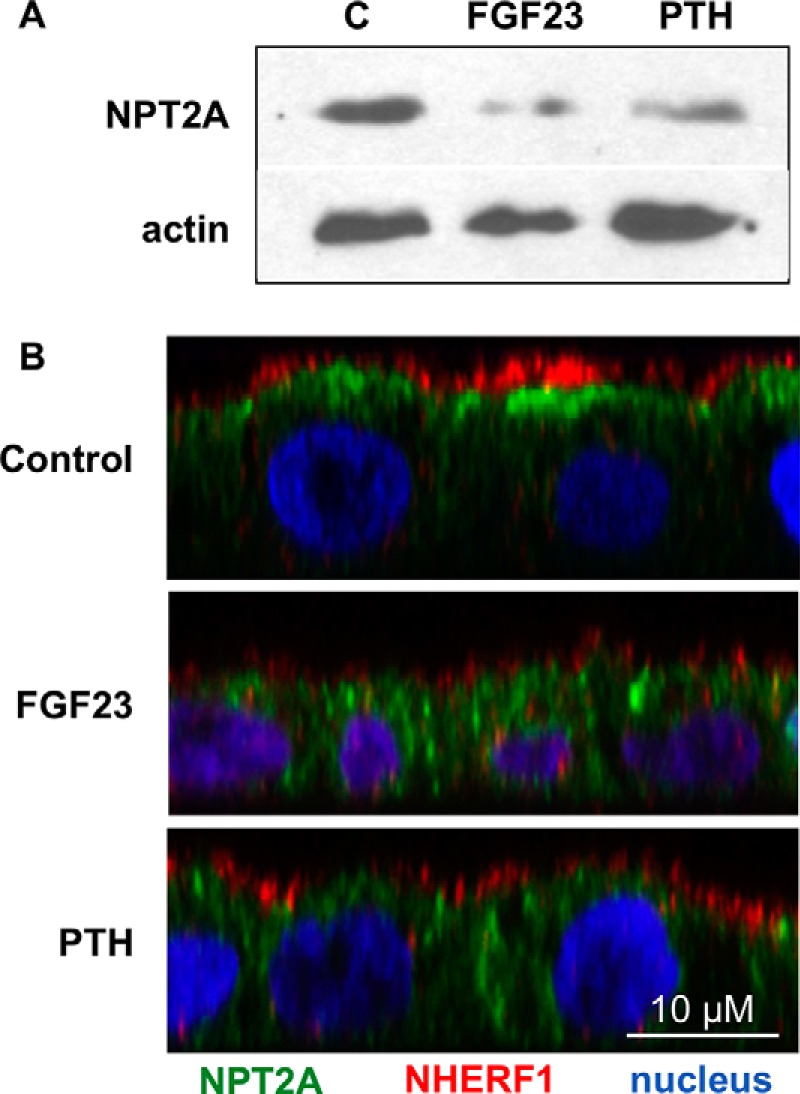
PTH and FGF23 down-regulation of NPT2A. A, NPT2A protein levels in RPTECs after a 2-h treatment with PTH or FGF23. A representative experiment is presented. B, immunofluorescence experiments depicting the change in localization of NPT2A in response to PTH or FGF23 treatment. RPTECs were left untreated (control) or treated for 2 h with 100 nm PTH or FGF23. Green, NPT2A; red, NHERF1; blue, nuclei. The x-z plane is depicted.
Studies of FGF23 action on phosphate transport using opossum kidney cells have proven controversial due to variable results (4, 50, 60, 61). The discrepancies among these reports and the work here may originate from the multiple available opossum kidney cell subtypes (62) as well as the uncertainty regarding FGFR1 and its proteoglycan requirement (63, 64). The present findings underscore the robust fidelity of RPTECs in recapitulating the hormone sensitivity and phosphate transport phenotype of native proximal tubules. RPTECs constitutively express all of the known components that regulate the renal phosphate transport mechanism, including PTHR and FGFR1c, along with its regulatory respective cofactor, and trans-acting elements that respond to PTH and FGF23. Thus, RPTECs should prove valuable for further and detailed studies of the mechanism of FGF23 and PTH action in cells from a eutherian mammal.
In summary, the present findings establish that PTH and FGF23 inhibit phosphate uptake through distinct mechanisms that impinge on a final common effector. Ligand binding of the PTHR stimulates PKA, PKC, and ERK1/2. FGF23, working through FGFR1c and αKlotho, stimulates SGK1 and ERK. The two signaling cascades converge at NHERF1. The present studies demonstrate that αKlotho is expressed in RPTECs, acting in a cis manner that is required for FGF23 but not PTH action. Also, we clarify that FGFR-β1c is the splice variant expressed by proximal tubule cells that, together with αKlotho, serves as the FGF23 receptor. We also present data consistent with the view that the final splicing decision for FGFR1c expression involves a novel cytoplasmic mechanism that may be a model for other growth factor receptors.
Experimental Procedures
Peptides
Human [Nle8,18,Tyr34]PTH(1–34) was purchased from Bachem (H9110). Recombinant human R179Q-FGF23(25–251) (referred to henceforth as FGF23), which is resistant to furin cleavage and inactivation, was obtained from R&D Systems (2604-FG-025). The C-terminal FGF23(180–251) fragment was a generous gift from Dr. Moosa Mohammadi (New York University School of Medicine).
Inhibitors
The PKA inhibitor H-89 (B1427), SGK1 inhibitor GSK-650394 (SML0773), PKC inhibitor Bis-I (B6292), and the ERK1/2 inhibitor PD98059 (P215) were purchased from Sigma. Sodium chlorate (catalog no. 403016) and other routine reagents were from Sigma.
Antibodies
Rabbit polyclonal anti-NHERF1 (ab3452), monoclonal anti-NHERF1 (Abcam ab31111), rabbit polyclonal anti-FGFR1 (ab10646), and rabbit polyclonal anti-Klotho (ab98111) antisera were purchased from Abcam. Monoclonal anti-FGFR1c (47) was acquired under license from MedImmune.
Cell Lines and Cell Culture
Human renal proximal tubule epithelial cells immortalized with hTERT (RPTECs) (11) were obtained from ATCC under license from Geron Corp. They were cultured in defined medium (DMEM/F-12 (Mediatech, 10-090-CV) supplemented with 5 pm triiodo-l-thyronine, 10 ng/ml recombinant human epidermal growth factor, 25 ng/ml prostaglandin E1, 3.5 μg/ml ascorbic acid, 1 mg/ml insulin, 0.55 mg/ml transferrin, 0.5 μg/ml sodium selenite, 25 ng/ml hydrocortisone) plus 1% penicillin and streptomycin and 0.1 mg/ml G418.
Phosphate Uptake
RPTECs were seeded on 12-well plates. When the cells reached confluence (2–3 days after passaging), they were treated with 100 nm PTH(1–34) or FGF23 in cell culture medium. After 2 h, the hormone-supplemented medium was aspirated, and the wells were washed three times with 1 ml of sodium-replete wash buffer (140 mm NaCl, 4.8 mm KCl, 1.2 mm MgSO4, 0.1 mm KH2PO4, 10 mm HEPES, pH 7.4). The cells were incubated with 1 μCi of [32P]orthophosphate (PerkinElmer Life Sciences, NEX053) in 1 ml of sodium-replete wash buffer for 10 min. Phosphate uptake was terminated by placing the plate on ice and rinsing the cells three times with sodium-free wash buffer (140 mm N-methyl-d-glucamine, 4.8 mm KCl, 1.2 mm MgSO4, 0.1 mm KH2PO4, 10 mm HEPES, pH 7.4). The cells in each well were extracted overnight at 4 °C using 500 μl of 1% Triton X-100 (Sigma). A 250-μl aliquot was counted in a Beckmann Coulter LS6500 scintillation counter. Data were normalized to phosphate uptake under control conditions defined as 100%.
Immunoblotting
Immunoblotting was performed as described (12). RPTECs were lysed with 1% Nonidet P-40 (50 mm Tris, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40) supplemented with protease inhibitor mixture I (Calbiochem). Lysis was performed for 15 min on ice. Solubilized materials were resolved on 10% SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore) using the semidry method (Bio-Rad). Membranes were blocked overnight at 4 °C with 5% nonfat dried milk in Tris-buffered saline plus Tween 20 (TBST) and incubated with the indicated antibodies (polyclonal anti-NHERF1 at 1:3000, polyclonal anti-Klotho at 1:3000, polyclonal anti-FGFR1 at 1:3000, monoclonal anti-FGFR1c at 1:500) overnight at 4 °C. The membranes were washed four times for 10 min in TBST and then incubated with goat anti-rabbit IgG (for anti-NHERF, anti-Klotho, and anti-FGFR1 primary antibodies) or goat anti-human IgG (for anti-FGFR1c primary antibody) conjugated to horseradish peroxidase at a 1:5000 dilution for 1 h at room temperature. Membranes were washed four times for 10 min in TBST. Protein bands were detected by Luminol-based enhanced chemiluminescence (EMD Millipore WBKLS0500).
Immunolocalization
RPTECs were grown to confluence in a 10-cm dish, trypsinized, washed with sterile PBS, and centrifuged at 600 × g, and the cell pellet was resuspended in 16 ml of medium. Polycarbonate Transwell filters (12-mm diameter; Corning Costar 3401) were coated with a 0.2-ml solution of type IV collagen (5 mg of Sigma type VI collagen dissolved in an aqueous solution of 0.2% (v/v) glacial acetic acid), diluted 1:10 in 10 mm sodium carbonate, pH 9.0, buffer. Following a 60-min incubation at room temperature, the collagen solution was aspirated, and the Transwell filters were rinsed with PBS and allowed to dry in a cell culture hood. A 0.5-ml aliquot of the cell suspension was placed in the upper compartment of the coated Transwell filters, whereas 1.2 ml was added to the lower chamber. The cells were incubated at 37 °C in a cell culture incubator, equilibrated with 95% O2, 5% CO2 for 7–10 days. The culture medium was changed every 3 days by aspirating the two chambers and adding 0.5 ml of medium to the upper chamber medium and 1.2 ml of medium to the lower chamber. Cells cultured on Transwell filters were treated with PTH or FGF23 on both basolateral and apical surfaces, as indicated, and the medium was aspirated and replaced with 100 mm sodium cacodylate buffer, pH 7.4, containing 4% (v/v) paraformaldehyde for 30 min at room temperature. The fixative was aspirated and replaced with quench solution (20 mm glycine, pH 8.0, and 75 mm NH4Cl dissolved in PBS). The filters were subsequently placed in block solution (0.1% (v/v) saponin, 1% (v/v) teleost fish skin gelatin, dissolved in PBS) containing 5% (v/v) horse serum. The cells were incubated overnight at 4 °C with mouse monoclonal anti-NHERF1 antibody (diluted 1:30 in block solution) and one of three primary anti-Npt2a antibodies (diluted 1:200 in block solution): rabbit anti-Npt2a antibody (a gift from Dr. Mark Knepper, National Institutes of Health) or goat anti-Npt2a (Santa Cruz, sc-33928). After three washes with blocking solution, the cells were incubated for 1 h at room temperature with a mixture of ToPro3 (diluted 1:1000; Life Technologies) minimal cross-reacting donkey anti-rabbit (or goat) IgG secondary antibody conjugated with Alexa488 (diluted 1:200) and with donkey anti-mouse IgG secondary antibody conjugated with CY3 (diluted 1:200). Following three washes with blocking solution and then PBS, the filters were postfixed with 4% (v/v) paraformaldehyde (in cacodylate buffer) for 10 min, rinsed with PBS, removed from the holders, and mounted on glass slides using Slowfade mounting medium (ThermoFisher). Confocal images of NPT2A localization were obtained using a Leica CW-STED confocal microscope (in normal confocal mode; Leica Microsystems Inc., Buffalo Grove, IL), outfitted with a ×63 glycerol objective (numerical aperture = 1.3) and low noise hybrid detectors. The captured images were contrast-corrected using Volocity (PerkinElmer Life Sciences), exported as TIFF files, and assembled in Adobe Illustrator.
PTHR localization was performed in an analogous fashion on RPTECs grown on coverslips as described (27) using an antibody raised against the C terminus of the human PTHR (65).
siRNA Knockdown
siRNA for αKlotho knockdown was purchased from Integrated DNA Technologies (Coralville, IA). HuSH shRNA for NHERF1 knockdown was obtained from Origene (TR309271). Dicer substrate short interfering RNAs (Dsi-RNAs) for knockdown of FGFR1b (exon 8) and FGFR1c (exon 9) were designed using the IDTDsi-RNA tool. The Dsi-RNAs for FGFR1b were rGrGrArGrUrUrArArUrArCrCrArCrCrGrArCrArArArGrAGA (sense) and rUrCrUrCrUrUrUrGrUrCrGrGrUrGrGrUrArUrUrArArCrUrCrCrArG (antisense). The Dsi-RNAs for FGFR1c were rGrUrGrGrUrArCrCrArArGrArArGrArGrUrGrArCrUrUrCCA (sense) and rUrGrGrArArGrUrCrArCrUrCrUrUrCrUrUrGrGrUrArCrCrArCrUrC (antisense). siRNA, Dsi-RNA, and shRNA were transfected into RPTECs using Lipojet (100468, SignaGen Laboratories, Gaithersburg, MD). Cells were transfected on 60-mm dishes. 48 h after transfection, the cells were trypsinized and passaged onto 12-well plates for phosphate uptake assays. Protein lysates were extracted from a 100-μl aliquot from these cells to assess by immunoblotting the extent of knockdown.
RT-PCR
Total RNA was isolated using TRIzol (Invitrogen). First strand cDNA synthesis was completed using the Accuscript high fidelity first strand cDNA synthesis kit (Agilent, 200820). Amplification was accomplished using Phusion HF Polymerase (New England Biolabs, M0350). Exon-spanning primers (Table 1) were designed to fingerprint the specific FGFR1 subtype (α/β) and alternatively spliced isoforms present in RPTECs. For FGFR1b, we used a forward primer that spans the exon 7/8 boundary and a reverse primer that spans the exon 10/8 boundary. For FGFR1c, we employed a forward primer that spans the exon 7/9 boundary and a reverse primer that spans the exon 10/9 boundary. Primers were purchased from Integrated DNA Technologies. A second series of previously published primer sequences (Table 2) (66) was used to identify FGFR isoforms expressed in RPTECs. PCRs were performed on a PCR Sprint thermal cycler (Hybaid) with a protocol consisting of 95 °C for 5 min, followed by 40 cycles at 95 °C for 10 s; 61 °C for 10 s, 72 °C for 30 s; 72 °C for 10 min and hold at 4 °C. The PCR was analyzed on a 2% agarose gel, and the relevant bands were excised, purified, and sequenced.
TABLE 2.
FGFR Primers
| Gene | Orientation | Primer pairs | Accession number | Product size |
|---|---|---|---|---|
| bp | ||||
| FGFR1 | 5′ → 3′ | CTCGGGACAGACTGGTCTTAGG | NM_023110.2 | 150 |
| 3′ → 5′ | CGTCCGACTTCAACATCTTCAC | |||
| FGFR3 | 5′ → 3′ | GACCGAGGACAACGTGATGA | NM_000142.4 | 160 |
| 3′ → 5′ | TGAGTGTAGACTCGGTCAAACAAG | |||
| FGFR4 | 5′ → 3′ | CCCTCGAATAGGCACAGTTACC | NM_002011.4 | 120 |
| 3′ → 5′ | AGCGGAACTTGACGGTGTTC |
Mass Spectrometry
RPTEs were grown to confluence on three 15-cm dishes. On ice, the cells were washed three times with 5 ml of cold PBS. The cells were then scraped in 5 ml of PBS and transferred to a 50-ml centrifuge tube. The cells were pelleted at 200 × g at 4 °C for 4 min in a Beckman Coulter AllegraX-12R centrifuge. The supernatant was discarded, and the pellet was resuspended in a low salt buffer containing 10 mm HEPES, pH 7.5, 1.5 mm MgCl2, 10 mm KCl, and protease inhibitors. The sample was sonicated in a Fisher FB120 sonicator for 1 min (8 s on, 16 s off, 30% power) and centrifuged for 30 min at 13,000 rpm at 4 °C in an Eppendorf 5415R centrifuge. The resulting pellet was resuspended in 5 ml of high salt buffer to wash the cell membrane (4.25 ml of low salt buffer supplemented with 0.75 ml of 5 m NaCl) and centrifuged again for 30 min at 13,000 rpm at 4 °C. The pellet was resuspended in 1 ml of low salt buffer and sonicated on ice for 8 s at 30% power. The sample was then centrifuged at 2000 rpm for 10 min at 4 °C to remove unbroken cells and free nuclei. After another high speed centrifugation, the sample was divided into pellet and supernatant fractions. The pellet was resuspended in 100 μl of Laemmli buffer and resolved on an 8% SDS-polyacrylamide gel. In parallel, a small amount of pellet and supernatant fractions was examined by Western blotting to ensure that the protein of interest was in the pellet sample. The protein in cut gel pieces was reduced by 10 mm DTT and alkylated by 55 mm iodoacetamide, and in-gel digestion by trypsin was performed at 37 °C overnight. The digested peptides were resuspended in 0.1% formic acid and subjected to LC-MS/MS analysis using the LTQ Orbitrap Velos mass spectrometer (Thermo Scientific).
Statistics
Statistical analysis was performed using GraphPad Prism version 6. Single comparisons between groups were analyzed by analysis of variance with post hoc testing using the Bonferonni multiple comparisons test, whereas repeated measures were analyzed using a paired t test. Differences between three or more groups were analyzed by two-way analysis of variance and post hoc testing using Tukey's multiple comparisons test. Differences greater than p < 0.05 were assumed to be significant.
Author Contributions
P. A. F., W. B. S., O. A. W., and G. L. A. designed the study, analyzed results, and wrote the paper. W. B. S., K. X., Q. Z., G. W. R., L. I. G., and Y. R. performed the described experiments.
Acknowledgments
We are grateful to Dr. Moosa Mohammadi (New York University School of Medicine) for generously sharing reagents and to Dr. Iris Lindberg (University of Maryland) for vital guidance on preparing FGF23. MedImmune supplied FGFR1c antiserum, and we are grateful for their interest and support.
This work was supported by National Institutes of Health Grants DK069998 and DK105811 (to P. A. F.), DK101484 and DK100357 (to O. A. W.), and P30 DK079307 (to G. A. and O. A. W.) and the Imaging Core of the Pittsburgh Center for Kidney Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTH
- parathyroid hormone
- PTHR
- PTH receptor
- Bis-I
- bis-indolylmaleimide I
- FGFR
- fibroblast growth factor receptor
- RPTEC
- telomerase-immortalized human renal proximal tubule cell
- Nle
- norleucine
- Dsi-RNA
- Dicer substrate siRNA.
References
- 1. Bergwitz C., and Jüppner H. (2010) Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 61, 91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gattineni J. (2014) Inherited disorders of calcium and phosphate metabolism. Curr. Opin. Pediatr. 26, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biber J., Malmström K., Reshkin S., and Murer H. (1990) Phosphate transport in established renal epithelial cell lines. Methods Enzymol. 191, 494–505 [DOI] [PubMed] [Google Scholar]
- 4. Yu X., Ibrahimi O. A., Goetz R., Zhang F., Davis S. I., Garringer H. J., Linhardt R. J., Ornitz D. M., Mohammadi M., and White K. E. (2005) Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology 146, 4647–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinman E. J., and Lederer E. D. (2012) PTH-mediated inhibition of the renal transport of phosphate. Exp. Cell Res. 318, 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Murer H., Forster I., Hernando N., and Biber J. (2008) Proximal tubular handling of phosphate. in Seldin and Giebisch's The Kidney Physiology and Pathophysiology (Alpern R. J., and Hebert S. C., eds) pp. 1979–1987, Elsevier, Burlington, MA [Google Scholar]
- 7. Cole J. A., Eber S. L., Poelling R. E., Thorne P. K., and Forte L. R. (1987) A dual mechanism for regulation of kidney phosphate transport by parathyroid hormone. Am. J. Physiol. 253, E221–E227 [DOI] [PubMed] [Google Scholar]
- 8. Lederer E. D., Sohi S. S., Mathiesen J. M., and Klein J. B. (1998) Regulation of expression of type II sodium-phosphate cotransporters by protein kinases A and C. Am. J. Physiol. 275, F270–F277 [DOI] [PubMed] [Google Scholar]
- 9. Pfister M. F., Forgo J., Ziegler U., Biber J., and Murer H. (1999) cAMP-dependent and -independent downregulation of type II Na-Pi cotransporters by PTH. Am. J. Physiol. 276, F720–F725 [DOI] [PubMed] [Google Scholar]
- 10. Traebert M., Völkl H., Biber J., Murer H., and Kaissling B. (2000) Luminal and contraluminal action of 1–34 and 3–34 PTH peptides on renal type IIa Na-Pi cotransporter. Am. J. Physiol. Renal Physiol. 278, F792–F798 [DOI] [PubMed] [Google Scholar]
- 11. Capuano P., Bacic D., Roos M., Gisler S. M., Stange G., Biber J., Kaissling B., Weinman E. J., Shenolikar S., Wagner C. A., and Murer H. (2007) Defective coupling of apical PTH receptors to phospholipase C prevents internalization of the Na+-phosphate cotransporter NaPi-IIa in Nherf1-deficient mice. Am. J. Physiol. Cell Physiol. 292, C927–C934 [DOI] [PubMed] [Google Scholar]
- 12. Cunningham R., Xiaofei E., Steplock D., Shenolikar S., and Weinman E. J. (2005) Defective PTH regulation of sodium-dependent phosphate transport in NHERF-1−/− renal proximal tubule cells and wild-type cells adapted to low phosphate media. Am. J. Physiol. Renal Physiol. 289, F933–F938 [DOI] [PubMed] [Google Scholar]
- 13. Guo J., Song L., Liu M., Segawa H., Miyamoto K., Bringhurst F. R., Kronenberg H. M., and Jüppner H. (2013) Activation of a non-cAMP/PKA signaling pathway downstream of the PTH/PTHrP receptor is essential for a sustained hypophosphatemic response to PTH infusion in male mice. Endocrinology 154, 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagai S., Okazaki M., Segawa H., Bergwitz C., Dean T., Potts J. T. Jr., Mahon M. J., Gardella T. J., and Jüppner H. (2011) Acute down-regulation of sodium-dependent phosphate transporter NPT2a involves predominantly the cAMP/PKA pathway as revealed by signaling-selective parathyroid hormone analogs. J. Biol. Chem. 286, 1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu M. C., Shi M., Zhang J., Pastor J., Nakatani T., Lanske B., Razzaque M. S., Rosenblatt K. P., Baum M. G., Kuro-o M., and Moe O. W. (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 24, 3438–3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gattineni J., Bates C., Twombley K., Dwarakanath V., Robinson M. L., Goetz R., Mohammadi M., and Baum M. (2009) FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am. J. Physiol. Renal Physiol. 297, F282–F291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrukhova O., Zeitz U., Goetz R., Mohammadi M., Lanske B., and Erben R. G. (2012) FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 51, 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hernando N., Déliot N., Gisler S. M., Lederer E., Weinman E. J., Biber J., and Murer H. (2002) PDZ-domain interactions and apical expression of type IIa Na/Pi cotransporters. Proc. Natl. Acad. Sci. U.S.A. 99, 11957–11962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mamonova T., Kurnikova M., and Friedman P. A. (2012) Structural basis for NHERF1 PDZ domain binding affinity. Biochemistry 51, 3110–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weinman E. J., Steplock D., Shenolikar S., and Biswas R. (2011) Fibroblast growth factor-23-mediated inhibition of renal phosphate transport in mice requires sodium-hydrogen exchanger regulatory factor-1 (NHERF-1) and synergizes with parathyroid hormone. J. Biol. Chem. 286, 37216–37221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shenolikar S., Voltz J. W., Minkoff C. M., Wade J. B., and Weinman E. J. (2002) Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc. Natl. Acad. Sci. U.S.A. 99, 11470–11475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morales F. C., Takahashi Y., Kreimann E. L., and Georgescu M. M. (2004) Ezrin-radixin-moesin (ERM)-binding phosphoprotein 50 organizes ERM proteins at the apical membrane of polarized epithelia. Proc. Natl. Acad. Sci. U.S.A. 101, 17705–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karim Z., Gérard B., Bakouh N., Alili R., Leroy C., Beck L., Silve C., Planelles G., Urena-Torres P., Grandchamp B., Friedlander G., and Prié D. (2008) NHERF1 mutations and responsiveness of renal parathyroid hormone. N. Engl. J. Med. 359, 1128–1135 [DOI] [PubMed] [Google Scholar]
- 24. Weinman E. J., and Lederer E. D. (2012) NHERF-1 and the regulation of renal phosphate reabsoption: a tale of three hormones. Am. J. Physiol. Renal Physiol. 303, F321–F327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weinman E. J., Steplock D., Zhang Y., Biswas R., Bloch R. J., and Shenolikar S. (2010) Cooperativity between the phosphorylation of Thr95 and Ser77 of NHERF-1 in the hormonal regulation of renal phosphate transport. J. Biol. Chem. 285, 25134–25138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Potter B. A., Ihrke G., Bruns J. R., Weixel K. M., and Weisz O. A. (2004) Specific N-glycans direct apical delivery of transmembrane, but not soluble or glycosylphosphatidylinositol-anchored forms of endolyn in Madin-Darby canine kidney cells. Mol. Biol. Cell 15, 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang B., Means C. K., Yang Y., Mamonova T., Bisello A., Altschuler D. L., Scott J. D., and Friedman P. A. (2012) Ezrin-anchored PKA coordinates phosphorylation-dependent disassembly of a NHERF1 ternary complex to regulate hormone-sensitive phosphate transport. J. Biol. Chem. 287, 24148–24163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wieser M., Stadler G., Jennings P., Streubel B., Pfaller W., Ambros P., Riedl C., Katinger H., Grillari J., and Grillari-Voglauer R. (2008) hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. Am. J. Physiol. Renal Physiol. 295, F1365–F1375 [DOI] [PubMed] [Google Scholar]
- 29. Wang E., Brown P. S., Aroeti B., Chapin S. J., Mostov K. E., and Dunn K. W. (2000) Apical and basolateral endocytic pathways of MDCK cells meet in acidic common endosomes distinct from a nearly-neutral apical recycling endosome. Traffic 1, 480–493 [DOI] [PubMed] [Google Scholar]
- 30. Apodaca G., Katz L. A., and Mostov K. E. (1994) Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. J. Cell Biol. 125, 67–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wade J. B., Liu J., Coleman R. A., Cunningham R., Steplock D. A., Lee-Kwon W., Pallone T. L., Shenolikar S., and Weinman E. J. (2003) Localization and interaction of NHERF isoforms in the renal proximal tubule of the mouse. Am. J. Physiol. Cell Physiol. 285, C1494–C1503 [DOI] [PubMed] [Google Scholar]
- 32. Amizuka N., Lee H. S., Kwan M. Y., Arazani A., Warshawsky H., Hendy G. N., Ozawa H., White J. H., and Goltzman D. (1997) Cell-specific expression of the parathyroid hormone (PTH)/PTH-related peptide receptor gene in kidney from kidney-specific and ubiquitous promoters. Endocrinology 138, 469–481 [DOI] [PubMed] [Google Scholar]
- 33. Ba J., Brown D., and Friedman P. A. (2003) CaSR regulation of PTH-inhibitable proximal tubule phosphate transport. Am. J. Physiol. Renal Physiol. 285, F1233–F1243 [DOI] [PubMed] [Google Scholar]
- 34. Gattineni J., Alphonse P., Zhang Q., Mathews N., Bates C. M., and Baum M. (2014) Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am. J. Physiol. Renal Physiol. 306, F351–F358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J. W., Chou C. L., and Knepper M. A. (2015) Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J. Am. Soc. Nephrol. 26, 2669–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Urakawa I., Yamazaki Y., Shimada T., Iijima K., Hasegawa H., Okawa K., Fujita T., Fukumoto S., and Yamashita T. (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–77417086194 [Google Scholar]
- 37. Han X., Yang J., Li L., Huang J., King G., and Quarles L. D. (2016) Conditional deletion of fgfr1 in the proximal and distal tubule identifies distinct roles in phosphate and calcium transport. PLoS One 11, e0147845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu S., Vierthaler L., Tang W., Zhou J., and Quarles L. D. (2008) FGFR3 and FGFR4 do not mediate renal effects of FGF23. J. Am. Soc. Nephrol. 19, 2342–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mistry N., Harrington W., Lasda E., Wagner E. J., and Garcia-Blanco M. A. (2003) Of urchins and men: evolution of an alternative splicing unit in fibroblast growth factor receptor genes. RNA 9, 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ornitz D. M., and Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Belov A. A., and Mohammadi M. (2013) Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a015958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gong S. G. (2014) Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 229, 1887–1895 [DOI] [PubMed] [Google Scholar]
- 43. Johnson D. E., Lu J., Chen H., Werner S., and Williams L. T. (1991) The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 11, 4627–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson D. E., and Williams L. T. (1993) Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 60, 1–41 [DOI] [PubMed] [Google Scholar]
- 45. Hovhannisyan R. H., and Carstens R. P. (2007) Heterogeneous ribonucleoprotein m is a splicing regulatory protein that can enhance or silence splicing of alternatively spliced exons. J. Biol. Chem. 282, 36265–36274 [DOI] [PubMed] [Google Scholar]
- 46. Buckley P. T., Khaladkar M., Kim J., and Eberwine J. (2014) Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip. Rev. RNA 5, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lelliott C. J., Ahnmark A., Admyre T., Ahlstedt I., Irving L., Keyes F., Patterson L., Mumphrey M. B., Bjursell M., Gorman T., Bohlooly-Y M., Buchanan A., Harrison P., Vaughan T., Berthoud H. R., and Lindén D. (2014) Monoclonal antibody targeting of fibroblast growth factor receptor 1c ameliorates obesity and glucose intolerance via central mechanisms. PLoS One 9, e112109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goetz R., Nakada Y., Hu M. C., Kurosu H., Wang L., Nakatani T., Shi M., Eliseenkova A. V., Razzaque M. S., Moe O. W., Kuro-o M., and Mohammadi M. (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. U.S.A. 107, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murer H., and Biber J. (2010) Phosphate transport in the kidney. J. Nephrol. 23, S145–S151 [PubMed] [Google Scholar]
- 50. Yamashita T., Konishi M., Miyake A., Inui K., and Itoh N. (2002) Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 277, 28265–28270 [DOI] [PubMed] [Google Scholar]
- 51. Friedman P. A., Coutermarsh B. A., Kennedy S. M., and Gesek F. A. (1996) Parathyroid hormone stimulation of calcium transport is mediated by dual signaling mechanisms involving PKA and PKC. Endocrinology 137, 13–20 [DOI] [PubMed] [Google Scholar]
- 52. Kouhara H., Hadari Y. R., Spivak-Kroizman T., Schilling J., Bar-Sagi D., Lax I., and Schlessinger J. (1997) A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89, 693–702 [DOI] [PubMed] [Google Scholar]
- 53. Sneddon W. B., Liu F., Gesek F. A., and Friedman P. A. (2000) Obligate MAP kinase activation in parathyroid hormone stimulation of calcium transport but not calcium signaling. Endocrinology 141, 4185–4193 [DOI] [PubMed] [Google Scholar]
- 54. Sneddon W. B., Yang Y., Ba J., Harinstein L. M., and Friedman P. A. (2007) Extracellular signal-regulated kinase activation by parathyroid hormone in distal tubule cells. Am. J. Physiol. Renal Physiol 292, F1028–F1034 [DOI] [PubMed] [Google Scholar]
- 55. Safaiyan F., Kolset S. O., Prydz K., Gottfridsson E., Lindahl U., and Salmivirta M. (1999) Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J. Biol. Chem. 274, 36267–36273 [DOI] [PubMed] [Google Scholar]
- 56. Comps-Agrar L., Dunshee D. R., Eaton D. L., and Sonoda J. (2015) Unliganded fibroblast growth factor receptor 1 forms density-independent dimers. J. Biol. Chem. 290, 24166–24177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takenaka T., Inoue T., Miyazaki T., Hayashi M., and Suzuki H. (2016) Xeno-klotho inhibits parathyroid hormone signaling. J. Bone Miner. Res. 31, 455–462 [DOI] [PubMed] [Google Scholar]
- 58. Paradis J. S., Ly S., Blondel-Tepaz É., Galan J. A., Beautrait A., Scott M. G., Enslen H., Marullo S., Roux P. P., and Bouvier M. (2015) Receptor sequestration in response to β-arrestin-2 phosphorylation by ERK1/2 governs steady-state levels of GPCR cell-surface expression. Proc. Natl. Acad. Sci. U.S.A. 112, E5160–E5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ardura J. A., Wang B., Watkins S. C., Vilardaga J. P., and Friedman P. A. (2011) Dynamic Na+-H+ exchanger regulatory factor-1 association and dissociation regulate PTH receptor trafficking at membrane microdomains. J. Biol. Chem. 286, 35020–35029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bowe A. E., Finnegan R., Jan de Beur S. M., Cho J., Levine M. A., Kumar R., and Schiavi S. C. (2001) FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem. Biophys. Res. Commun. 284, 977–981 [DOI] [PubMed] [Google Scholar]
- 61. Shimada T., Mizutani S., Muto T., Yoneya T., Hino R., Takeda S., Takeuchi Y., Fujita T., Fukumoto S., and Yamashita T. (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. U.S.A. 98, 6500–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cole J. A., Forte L. R., Krause W. J., and Thorne P. K. (1989) Clonal sublines that are morphologically and functionally distinct from parental OK cells. Am. J. Physiol. 256, F672–F679 [DOI] [PubMed] [Google Scholar]
- 63. Ibrahimi O. A., Zhang F., Hrstka S. C., Mohammadi M., and Linhardt R. J. (2004) Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry 43, 4724–4730 [DOI] [PubMed] [Google Scholar]
- 64. Goetz R., Beenken A., Ibrahimi O. A., Kalinina J., Olsen S. K., Eliseenkova A. V., Xu C., Neubert T. A., Zhang F., Linhardt R. J., Yu X., White K. E., Inagaki T., Kliewer S. A., Yamamoto M., et al. (2007) Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 27, 3417–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lupp A., Klenk C., Röcken C., Evert M., Mawrin C., and Schulz S. (2010) Immunohistochemical identification of the PTHR1 parathyroid hormone receptor in normal and neoplastic human tissues. Eur. J. Endocrinol. 162, 979–986 [DOI] [PubMed] [Google Scholar]
- 66. Roidl A., Foo P., Wong W., Mann C., Bechtold S., Berger H. J., Streit S., Ruhe J. E., Hart S., Ullrich A., and Ho H. K. (2010) The FGFR4 Y367C mutant is a dominant oncogene in MDA-MB453 breast cancer cells. Oncogene 29, 1543–1552 [DOI] [PubMed] [Google Scholar]