

Abstract
Plakins are large multi-domain proteins that interconnect cytoskeletal structures. Plectin is a prototypical plakin that tethers intermediate filaments to membrane-associated complexes. Most plakins contain a plakin domain formed by up to nine spectrin repeats (SR1–SR9) and an SH3 domain. The plakin domains of plectin and other plakins harbor binding sites for junctional proteins. We have combined x-ray crystallography with small angle x-ray scattering (SAXS) to elucidate the structure of the plakin domain of plectin, extending our previous analysis of the SR1 to SR5 region. Two crystal structures of the SR5-SR6 region allowed us to characterize its uniquely wide inter-repeat conformational variability. We also report the crystal structures of the SR7-SR8 region, refined to 1.8 Å, and the SR7–SR9 at lower resolution. The SR7–SR9 region, which is conserved in all other plakin domains, forms a rigid segment stabilized by uniquely extensive inter-repeat contacts mediated by unusually long helices in SR8 and SR9. Using SAXS we show that in solution the SR3–SR6 and SR7–SR9 regions are rod-like segments and that SR3–SR9 of plectin has an extended shape with a small central kink. Other plakins, such as bullous pemphigoid antigen 1 and microtubule and actin cross-linking factor 1, are likely to have similar extended plakin domains. In contrast, desmoplakin has a two-segment structure with a central flexible hinge. The continuous versus segmented structures of the plakin domains of plectin and desmoplakin give insight into how different plakins might respond to tension and transmit mechanical signals.
Keywords: cytoskeleton, intermediate filament, protein structure, small-angle X-ray scattering (SAXS), X-ray crystallography, desmoplakin, plakin, plectin, spectrin repeat
Introduction
Plakins are a family of very large proteins that interconnect and organize the intermediate filaments (IF),4 microtubules, and microfilaments of the cytoskeleton and tether them to membrane-associated structures (1, 2). So far, seven plakins have been described in mammals; these are plectin, bullous pemphigoid antigen 1 (BPAG1), desmoplakin, microtubule and actin cross-linking factor 1 (MACF1, also known as ACF7), envoplakin, periplakin, and epiplakin. Invertebrates have a more reduced plakin repertoire; for example Caenorhabditis elegans and Drosophila melanogaster each have a single plakin gene encoding VAB-10 and Shot (also known as Short Stop or kakapo), respectively. Most of the plakin genes produce multiple isoforms that increase the structural and functional versatility of these proteins.
Plectin is expressed in a large variety of cell types in which it acts as a highly polyvalent cytolinker that contributes to cell adhesion and the organization of the cytoskeleton. Plectin cross-links IFs to microtubules and actin filaments and mediates the attachment of IFs to cell-cell and cell-matrix junctional complexes such as hemidesmosomes, desmosomes, Z-lines, and focal contacts. Plectin also connects IFs to organelles such as the nucleus and mitochondria (3). Defects in the PLEC gene cause various forms of the blistering disease epidermolysis bullosa simplex (EBS), which may occur only with skin fragility, as found in EBS Ogna type, or may be associated with muscular dystrophy or pyloric atresia (4). These diseases highlight the important role of plectin in the homeostasis of tissues subjected to mechanical stress, such as skin and muscle.
Plectin (∼500 kDa) has a mosaic structure built up of multiple discrete domains organized in three major segments, which is prototypical of other plakins. The N-terminal region contains an actin-binding domain, formed by two calponin homology domains, followed by a plakin domain (Fig. 1A). The actin-binding domain binds to integrin α6β4 (5, 6), nesprin-3α (7), F-actin (8, 9), and dystrophin (10). A central rod domain (∼1250 amino acids) is responsible for homodimerization via coiled-coil interactions. Most of the rod domain is absent in a natural rod-less splice variant that retains the function of the full-length protein (11). Finally, the C-terminal region contains six plakin repeat domains and mediates binding to IFs (12).
FIGURE 1.
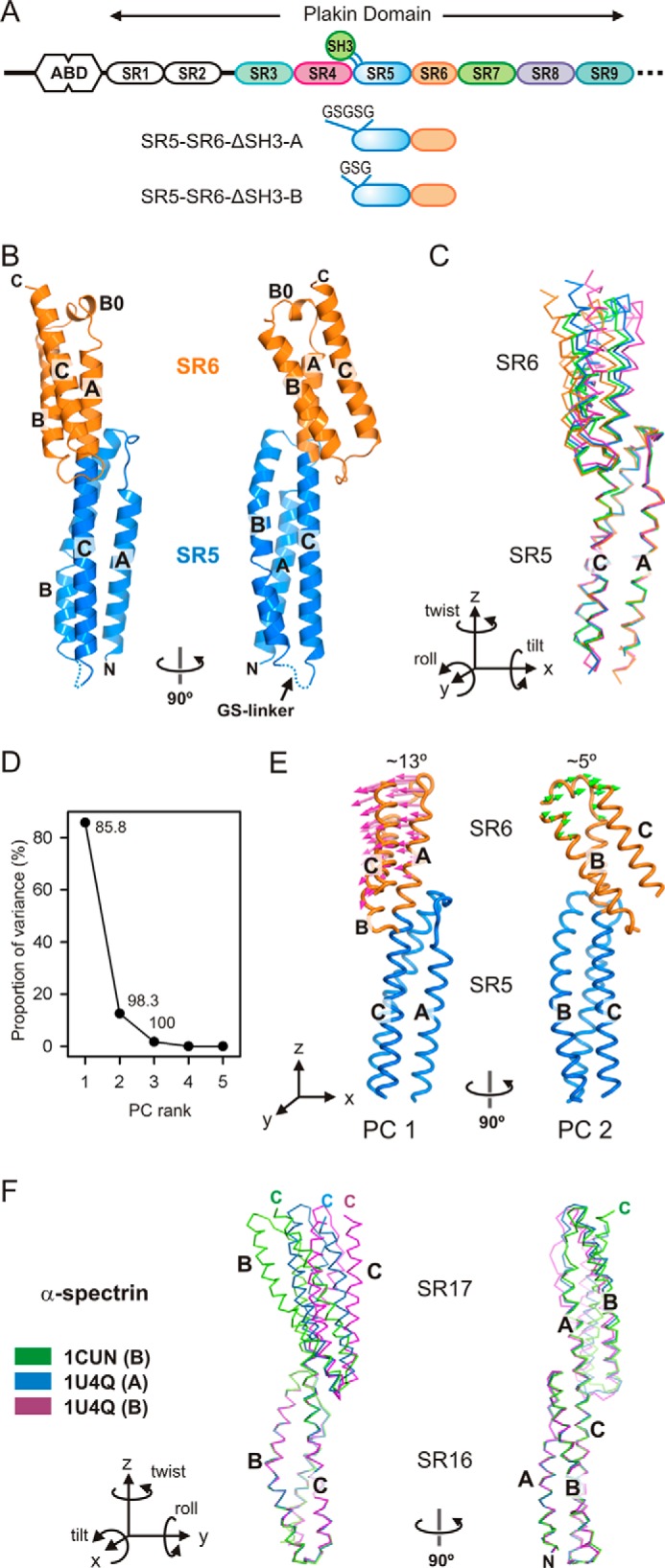
Crystal structures and conformational variability of the SR5-SR6 of plectin. A, domain organization of the N-terminal moiety of plectin and the two SR5-SR6-ΔSH3 constructs. B, orthogonal views of the ribbon representation of the SR5-SR6-ΔSH3-A structure. C, Cα trace representation of the four different molecules of SR5-SR6 present in the asymmetric units of the SR5-SR6-ΔSH3-A (blue and green) and SR5-SR6-ΔSH3-B (orange and magenta). The molecules were superimposed by aligning the SR5 domains. A diagram of the orientation of the Cartesian axes and the rotations around them is shown. D, PC analysis of domain-wide conformational differences between the four structures. The percentage of the total variance captured by each eigenvector is plotted. The numbers indicate the cumulative sum of the total variance accounted for in all of the preceding eigenvectors. E, porcupine illustration of the two main PCs (PC1 and PC2) derived from the four individual SR5-SR6 structures. F, conformational variations between structures of the SR16-SR17 region of α-spectrin. Shown is a representative Cα trace of the SR16-SR17 region of α-spectrin in different monomers of the crystal structures of the SR16-SR17 (PDB code 1CUN) and the SR15–SR17 fragments (PDB code 1U4Q). For clarity only three monomers illustrating the divergence in the orientation of the two repeats are shown. The structures were superimposed by matching the Cα atoms of the repeat SR16.
The plakin domain of plectin (∼1070 amino acids) consists of nine spectrin repeats (SR1–SR9) and a Src homology 3 (SH3) domain embedded in the SR5 (13). SRs are ∼100–110-amino acid domains that contain three amphipathic α-helices (A, B, and C) connected by short loops. The helices fold into a left-handed helical bundle that encloses a hydrophobic core formed by residues in positions a and d of the helical heptad repeats. Juxtaposed SRs are connected by helical linkers that fuse helix C of the anterior repeat with helix A of the posterior repeat; therefore, arrays of SR form rod-like bendable structures. Despite the conservation of the SR fold, there are large variations in the relative orientation of different pairs of tandem repeats. The crystal structures of several multi-repeat segments of BPAG1 (14), plectin (13, 15), and desmoplakin (16) have unveiled the details of the N-terminal part of the plakin domain. The SR1-SR2 region is connected by a predicted disordered linker to the rod-like SR3–SR6 segment. The SH3 lacks a canonical poly-Pro-binding site and makes extensive contacts with the SR4. The plakin domain of plectin participate in protein-protein interactions harboring binding sites for integrin α6β4 (17), BPAG2 (also known as BP180 or type XVII collagen) (18), β-dystroglycan (10), β-synemin (19), and the kinase Fer (20), although the specific binding sites for these proteins are not known precisely.
The plakin domain is present in all plakins except epiplakin. The pairwise sequence identity between the SR3–SR9 region of mammalian plakins ranges from ∼21 to 53%, although some proteins have shorter forms of the plakin domain. The epithelial isoform of BPAG1 (BPAG1e, also known as BP230) lacks the SR1, and the SR1-SR2 tandem is absent in desmoplakin, periplakin, and envoplakin. In desmoplakin the SR6 is connected to the SR7 by a uniquely long linker that is sensitive to proteases (21). Similarly, in periplakin and envoplakin the SR6 is replaced by 15–20-residue-long sequences. Analysis of the plakin domains of desmoplakin, periplakin, and envoplakin in solution by small angle x-ray scattering (SAXS) has revealed various degrees of segmental flexibility caused by the unique SR6-SR7 or SR5–SR7 linkers (21).
Despite recent advances in the characterization of plakin domains, no atomic structure of the SR7–SR9 region has been described to date. Similarly, the entire structure of plakin domains that lack long SR6-SR7 linkers, such as those of plectin, BPAG1, and MACF1, remains unknown. Here we have combined x-ray crystallography with SAXS to elucidate the structure of the plakin domain of plectin, which has an extended rod-like shape. In addition, a comparative analysis of the plakin domain of desmoplakin has revealed differences in the segmental flexibility of these two proteins. Our results have implications for the properties of plakins and their contribution to the stability and mechanobiology of tissues subjected to mechanical stress.
Results and Discussion
To obtain a comprehensive description of the structure of the plakin domain of plectin, we produced recombinant multi-domain fragments that cover the SR3–SR9 region, and we combined detailed analysis of two- and three-repeat fragments by crystallography with information on the global structure of larger segments, including the complete SR3–SR9 region, obtained by SAXS.
Crystal Structures of the SR5-SR6 Region of Plectin Reveal Details of Interdomain Bending
Previously, we elucidated the crystal structures of the SR3-SR4 and SR4-SR5-SH3 regions of plectin (15). To complete the analysis of the SR3–SR6 region, we attempted unsuccessfully to crystallize the SR3–SR6 and SR4–SR6 fragments. Therefore, we focused on the SR5-SR6 region. Because the SH3 makes extensive contacts with the SR4, we created two SR5-SR6 constructs in which the SH3 domain was replaced by a five-residue (SR5-SR6-ΔSH3-A) or a three-residue (SR5-SR6-ΔSH3-B) sequence (Fig. 1A). Both proteins were crystallized, and their structures were refined against data to 2.8 and 3.0 Å, respectively. Each crystal form contains two SR5-SR6 molecules in the asymmetric unit (AU). The individual structures of the SR5 and SR6 domains are highly conserved in the four molecules. After pairwise superimposition of the four structures of the SR5 domain, the root mean square deviation (rmsd) for 99 Cα atoms ranged from 0.46 to 0.67 Å. Similarly, the four copies of SR6 were superposed with the rmsd for 77 Cα atoms between 0.44 and 0.65 Å. The SR5 exhibits a canonical SR fold, whereas the SR6 is smaller and has an additional short helix (B0) between helices A and B (Fig. 1B). These repeats of plectin are very similar to the equivalent domains in the crystal structure of the SR3–SR6 region of desmoplakin (16); individually the SR5 and SR6 domains of plectin and desmoplakin superimpose with a Cα rmsd of 1.07–1.24 Å and 0.97–1.20 Å, respectively. Thus, exchanging the SH3 domain by short Gly-Ser sequences does not disrupt the overall structure of the SR5 domain.
Helix B0 of the SR6 is reminiscent of the short helix B0 of the SR5 observed in the structure of the SR4-SR5 region of plectin (15). In the structures of the SR5-SR6-ΔSH3 fragments, the helix SR5-B0 is rearranged, being part of helix B (Fig. 2). Analogously, the region of the SR6 around helix B0, which is predicted to be close to the SR7, might adopt a different conformation in the full-length protein. Similar helical rearrangements near the loops of SRs have been observed in spectrins (22).
FIGURE 2.
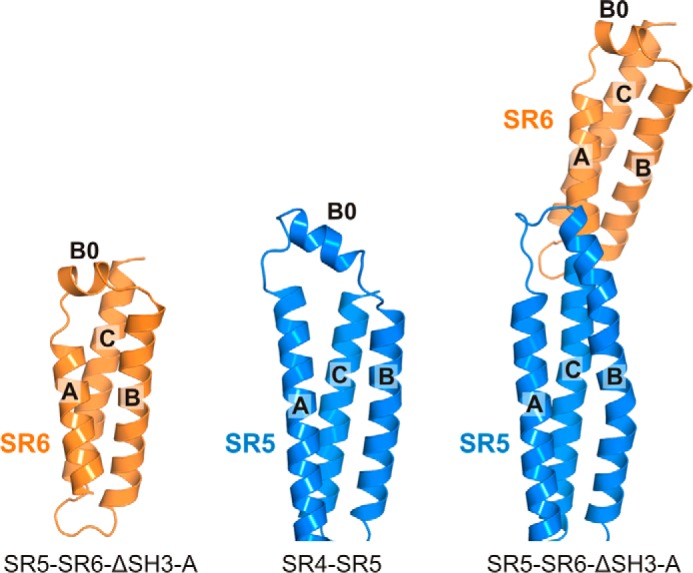
Structure of the region around helices A and B in the SR5 and SR6 region of plectin. Ribbon representation of SR6 in the structure of the SR5-SR6-ΔSH3-A fragment (left) and of SR5 in the structure of SR4-SR5 (center) and of the SR5-SR6-ΔSH3-A fragment (right). In the absence of SR6, the SR5 helices B0 and B of SR5 are separated by a non-helical segment (center). This is similar to the presence of B0 in the SR6 repeat (left); but in the construct that contains the SR5 and SR6 repeats, the region between helices A and B of SR5 is reorganized by extending helix B (right).
In contrast to the structural conservation of individual domains, superposition of the SR5 repeats revealed differences in the position of the SR6 in the four molecules (Fig. 1C). These variations are due mainly to differences in the roll (−3.3° to −19.6°) and tilt (−16.5° to −23.3°) angles; hence, they correspond to changes in inter-repeat bending (supplemental Table S1). Our crystal structures represent snapshots of the inter-repeat conformational variability. Principal component (PC) analysis revealed that the four conformers of the SR5-SR6 region are related by two major perpendicular rigid-body oscillations of ∼13° and ∼5° amplitude, which correspond approximately to roll (PC1) and tilt (PC2) rotations (Fig. 1, D and E). In both movements the beginning of helix SR6-A acts as a hinge.
The arrangement of the SR5-SR6 segment of plectin is very similar to that of desmoplakin. Yet, the conformational variations of this region are larger in plectin than in desmoplakin. The variability of the inter-repeat orientation is a major source of flexibility of SR arrays (22). Bending oscillations also occur in other segments of the plakin domain (e.g. SR3-SR4), but those of the SR5-SR6 pair show the largest variations (supplemental Table S1). Notably, the conformational changes in the SR5-SR6 region of plectin are similar to those in the SR16-SR17 region of α-spectrin (22, 23) (Fig. 1F and supplemental Table S2). In summary, the SR5-SR6 region is a preferentially bendable site of the plakin domain.
Finally, the SR6 region of one of the molecules in the AU of the SR5-SR6-ΔSH3-A crystals (protein chain B), which only makes crystal lattice contacts through the helix B0, has poorer electron density and higher average B-factors (134 Å2) than the other SRs in the crystal (59–103 Å2), suggesting that SR6 has an intrinsically relaxed structure. Collectively, our data support the assertion that SR6 is a region of substantial structural plasticity within the plakin domain.
Crystal Structure of the SR7-SR8 Region of Plectin
To elucidate the structure of the C-terminal segment of the plakin domain of plectin, initially we solved the crystal structure of the SR7-SR8 region to 1.80 Å resolution. The AU of the crystals contains two SR7-SR8 molecules. After superimposition of the individual repeats, the rmsd for the Cα atoms of SR7 and SR8 was 0.18 and 0.30 Å, respectively. A similar Cα rmsd of 0.32 Å was obtained when the full SR7-SR8 region was superimposed, revealing that the relative orientation of the two repeats is almost identical in the two monomers.
SR7 and SR8 have a canonical SR fold (Fig. 3A), yet their B helices are longer than those in other repeats of the SR1–SR6 region of the plakin domain. Specifically, helix SR8-B is unusually long (∼46 residues) and extends at the C terminus about two more turns than helix SR7-B.
FIGURE 3.

Crystal structure of SR7-SR8 of plectin. A, ribbon representation of the SR7-SR8 region in two orthogonal views. B, close-up of the SR7-SR8 interface (dotted line square in A). Residues in the AB loop of SR7 and the BC loop of SR8, which mediate inter-repeat contacts, are shown as sticks. Polar contacts between the two repeats, either directly or through solvent molecules, are shown as dashed lines. C, stereo view of a section of the electron density map near the SR7-SR8 interface calculated using the SIRAS phases after density modification (contoured at 1σ). The refined structure is shown as sticks. D, feature-enhanced 2mFobs − DFmodel map calculated at the final stage of refinement (contoured at 1σ) of the region shown in C.
The orientation of SR8 is related to SR7 by a ∼46 Å translation and a ∼54° rotation around the longitudinal axis of the molecule. Consequently, helices SR7-A and SR8-C are on the same side of the molecule. The inter-repeat linker formed by the fusion of helices SR7-C and SR8-A participates in the hydrophobic cores of both repeats. Helices SR7-B and SR8-B extend toward the adjacent repeat, creating two short segments of four-helix bundles (Fig. 3B). The SR7-SR8 arrangement is further stabilized by the interaction between the AB loop of SR7 and the BC loop of SR8. These loops are connected by a network of polar contacts that include interactions of the side chains of Glu-1200 and Arg-1203 with backbone atoms of the AB loop of SR7, a salt bridge between Arg-1035 and Glu-1196, and several hydrogen bonds mediated by water molecules partially buried between both loops. Collectively, our data indicates that the SR7-SR8 region is a rigid segment. Owing to the lateral inter-repeat contacts at the linker region, we refer to SR7 and SR8 as overlapping repeats.
Crystal Structure of the SR7–SR9 Region of Plectin
Crystals of the SR7–SR9 region of plectin produced highly anisotropic diffraction. Anisotropy arises from the alignment of the elongated SR7–SR9 molecules in the crystal lattice (Fig. 4). The AU contains two SR7–SR9 molecules that are very similar (Fig. 5A). After superimposition of the individual repeats, the rmsd for the Cα atoms were 0.20, 0.33, and 0.54 Å for SR7, SR8, and SR9, respectively. The interdomain arrangement is also identical in both molecules; the complete monomers superpose with an rmsd for 341 Cα atoms of 0.63 Å. In addition, the structure of the isolated SR7-SR8 region closely resembles that of the corresponding region of the SR7–SR9; after superimposition the rmsd of the common Cα between SR7-SR8 and SR7–SR9 range between 0.77 and 0.86 Å.
FIGURE 4.

Anisotropic diffraction and lattice packing of the plectin SR7–SR9 crystals. A, representation of the reciprocal lattice points colored according to local mean value of I/σ(I) calculated with STARANISO for the zones (h0l), (hk0), and (0kl). A mean I/σ(I) ≥ 1.2 threshold was used to define the anisotropic cut-off surface. B, analysis of CC1/2 as a function of the resolution along the three principal directions of anisotropy, d1 (0.53 a* + 0.85 c*), d2 (b*), and d3 (−0.38 a* + 0.93 c*) (filled circles and solid lines), and the curves fitted to the experimental values (empty triangles and dashed lines). Analysis was done with the program AIMLESS (70). C, orthogonal views of the two molecules in the AU (blue and red) and symmetry-related neighboring molecules (yellow). The edges of the unit cell are shown in blue. The longitudinal axes of SR7–SR9 are aligned, favoring the crystal order in that direction, whereas weak lateral contacts result in poor diffraction in two other directions.
FIGURE 5.

Crystal structure of SR7–SR9 of plectin. A, orthogonal views of the ribbon representation of the SR7–SR9 structure. B, worm representation of the backbone of SR7 (green), SR8 (magenta), and SR9 (turquoise) superimposed. The arrowhead indicates a kink in helices SR8-B and SR9-B. The extra segments of helix B in SR8 and SR9, which protrude out of the helical bundle, are indicated. C, close-up view of the final part of helix SR9-B and its interaction with SR8 (dotted line square in A). The side chains of the main residues that participate in these inter-repeat contacts are shown as sticks. For clarity the backbone of helix SR9-B is represented as a worm. Electron density maps of this region are shown in Fig. 6. D, pseudo superhelical structure of the SR7–SR9 region. Uninterrupted helices are shown in a single color. Helices that belong to the same pseudo-thread are shown in similar colors. The four threads of the superhelix are highlighted by dashed lines, and their polarity is indicated by arrowheads at the C terminus.
The overall structure of SR9 is very similar to that of SR7 and SR8 (Fig. 5B). SR9 has a very long helix B (∼54 residues) that has two turns more at the C terminus than helix SR8-B. The final part of helix SR9-B contacts SR8 forming a four-helix bundle (Fig. 5C and Fig. 6). Residues Tyr-1309, Leu-1313, and Tyr-1316, which occupy positions d, a, and d of the heptad motif of helix SR9-B, face the groove formed by helices A and C and loop AB of SR8.
FIGURE 6.

Electron density maps of the SR7–SR9 crystal structure. A, stereo view of a 2mFobs − DFmodel simulated annealing omit map (contoured at 1σ) of the C-terminal part of helix SR9-B. The map was calculated using phases from a model from which the region 1298–1321 was excluded, and that was refined by simulated annealing (starting temperature, 3000 K). B, feature-enhanced 2mFobs − DFmodel map (contoured at 1σ) corresponding to the region shown in A.
The SR8 structure can be superimposed on that of SR9 by applying a ∼51 Å translation and a ∼69° rotation around the longitudinal axis of the molecule, which is similar to the relative arrangement of the SR7-SR8 pair (see above). Owing to the conserved inter-repeat rotation in the SR7–SR9 region, helices A–C and the unusually long B helices of SR8 and SR9 form a left-handed pseudo superhelix that resembles a twisted rope (Fig. 5D). Moreover, helix SR7-A is aligned with helix SR8-C/SR9-A forming a discontinuous thread of the superhelix that spans the full length of the molecule. Similarly, helix SR7-B is aligned with helix SR9-B, and helix SR7-C/SR8-A is aligned with helix SR9-C. Therefore, the superhelical architecture of the SR7–SR9 region consists of four antiparallel threads. In summary, the overlapping repeats of the SR7–SR9 region form a rigid structure stabilized by long helices and extensive inter-repeat contacts.
The SR7–SR9 Region Is Conserved in Other Plakins
We analyzed whether the SR7–SR9 region was also present in other plakins. Searching the human proteome with a profile hidden Markov model (HMM) built from the sequence of the SR7–SR9 region of plectin revealed the presence of homologous regions of similar extension in BPAG1, MACF1, and desmoplakin (Table 1). Because this initial search did not reveal similar regions in other plakins, we performed a new search using a profile HMM built from the SR7–SR9 sequences of plectin, BPAG1, MACF1 and desmoplakin, which unveiled the presence of an SR7–SR9 region in envoplakin and periplakin.
TABLE 1.
Identification of SR7-SR9 segments in plakins
UniProt accession codes are shown in parentheses. NA, not applicable. E-values are the expected number of hits to have a score equal or better by chance.
| Protein | Organism | E-value | Region | Repeats |
||
|---|---|---|---|---|---|---|
| SR7 | SR8 | SR9 | ||||
| Plectin (Q15149–2) | H. sapiens | NA | 1004–1372 | 1004–1116 | 1117–1234 | 1234–1372 |
| BPAG1 (Q03001–3) | H. sapiens | 8.1e-81 | 734–1103 | 734–846 | 847–964 | 965–1103 |
| MACF1 (Q9UPN3–2) | H. sapiens | 2.7e-71 | 1041–1414 | 1041–1157 | 1158–1275 | 1276–1414 |
| Desmoplakin (P15924) | H. sapiens | 1.4e-48 | 660–1025 | 660–768 | 769–886 | 887–1025 |
| Envoplakin (Q92817) | H. sapiens | 3.3e-31 | 520–889 | 519–632 | 633–750 | 751–889 |
| Periplakin (O60437) | H. sapiens | 7.2e-30 | 500–870 | 499–612 | 613–730 | 731–871 |
| VAB-10 (G5EDD3) | C. elegans | 1.8e-16 | 986–1345 | 981–1086 | 1087–1215 | 1216–1360 |
| Shot (A1Z9J3) | D. melanogaster | 1.7e-08 | 1039–1295 | 1039–1144 | 1145–1257 | 1258–1394 |
Next, we used a profile HMM built from the SR7–SR9 sequences of the six human plakins to analyze the presence of homologous regions in plakins of invertebrates. We identified a single SR7–SR9-like region in VAB-10 and Shot, the only plakins found in C. elegans and D. melanogaster, respectively. The region of Shot identified in the search (residues 1039–1295) included SR7-SR8 but only part of SR9, yet two predicted α-helices completed the SR9 region (residues 1258–1394), which is similar in length to other plakins. A multiple sequence alignment of the SR7–SR9 sequences of human plakins, VAB-10, and Shot is shown in Fig. 7. In all of the searches, a single SR7–SR9 region was identified in the C-terminal part of the plakin domain of each protein, and no homologous regions were detected in other proteins. Thus, our profile-based search was specific for the SR7–SR9 signature. In summary, the SR7–SR9 region is present in all plakins that contain a plakin domain, and this region is distinct from other tandem arrays of SRs.
FIGURE 7.

Multiple sequence alignment of the SR7–SR9 region of plakins. The sequences of human plectin (UniProt Q15149-2), BPAG1e (UniProt Q03001–3), MACF1 (UniProt Q9UPN3–2), desmoplakin (UniProt P15924), envoplakin (UniProt Q92817), and periplakin (UniProt O60437), as well as VAB-10 of C. elegans (UniProt G5EDD3) and Shot of D. melanogaster (UniProt A1Z9J3) were aligned with ClustalO (71). Secondary structure elements of the plectin structure and positions a and d of the helical heptad repeat are shown at the top of the alignment. Filled circles mark every tenth residue in plectin. Residues involved in inter-repeat contacts between loop AB of SR7 and loop BC of SR8 are highlighted by orange boxes. Residues involved in contacts between the final segment of helix SR9-B and SR8 are indicated by blue boxes.
Structure of the Plakin Domain of Plectin in Solution
To elucidate the structure of the plakin domain of plectin in solution we analyzed the SR3–SR9 segment and its two constituent moieties, SR3–SR6 and SR7–SR9, by SAXS (Fig. 8 and Table 2). The samples were monodisperse and monomeric, as suggested by the linearity of the Guinier plots of the scattering data (Fig. 8B) and the molecular masses estimated from the scattering data. The Guinier radii of gyration (Rg) of each protein remained constant within the experimental errors in the range of concentrations measured, indicating minimal contribution of interparticle effects. The pair-distance distribution functions, P(r), calculated from the scattering data had maxima at short interatomic distances and long tails extending to maximum distances (Dmax) of about 210, 170, and 350 Å for the SR3–SR6, SR7–SR9, and SR3–SR9 regions, respectively (Fig. 8C). This P(r) shape is characteristic of elongated rod-like particles. The Rg of the SR3–SR6, SR7–SR9, and SR3–SR9 regions determined from the P(r) were 53.9, 44.1, and 90.8 Å, respectively. These values were slightly greater than those obtained from the Guinier approximation (50.4, 42.3, and 85.4 Å, respectively) with the differences likely due to the inherently limited extension of the Guinier region caused by the rod-like shape of these fragments.
FIGURE 8.

SAXS analysis of the plakin domain of plectin in solution. A, SAXS profiles extrapolated to infinite dilution of the SR3–SR6 (blue), SR7–SR9 (violet), and SR3–SR9 (orange) regions. Curves are offset on the log scale; the same data color scheme is used in B–F. The scattering calculated for the atomic models in H are shown as dashed lines. B, Guinier plots of the scattering data shown in A. The Guinier Rg at several protein concentrations and the linear regression fit (dashed lines) are shown in the inset. C, P(r) functions determined from the scattering data (colored solid lines) and P(r) calculated for the atomic structures (dashed lines). D, cross-sectional Guinier plots of the scattering data. The linear fits to the intermediate-q data used to calculate Rc are shown as dashed lines. E, cross-sectional Pc(r) functions. For representational purposes, the P(r) and Pc(r) functions have been scaled. F, dimensionless Kratky plots. The crosshair indicates the expected position of the theoretical maxima of the plot for spherical compact particles (qRg ∼1.732 and (qRg)2I(q)/I(0) ∼1.104). G, Porod-Debye plots of the SAXS data. H, EOM analysis of the flexibility between the rigid segments SR3–SR6 and SR7–SR9. Frequency distributions of the Rg in a pool of models (black line) and in the selected ensemble that fits the SAXS data of the SR3–SR9 fragment (red line). I, ribbon representation of the atomic structures of SR3–SR6, SR7–SR9, and SR3–SR9 docked into their SAXS-derived molecular envelopes (semi-transparent surfaces). The low resolution structures are the average of 15 (SR3–SR6 or SR7–SR9) or six (SR3–SR9) independent bead models. Two orthogonal views are shown.
TABLE 2.
SAXS data collection and derived parameters
| Plectin SR3–SR6 | Plectin SR7–SR9 | Plectin SR3–SR9 | Desmoplakin SR7–SR9 | Desmoplakin SR3–SR9 | |
|---|---|---|---|---|---|
| Data collection | |||||
| Concentration range (mg ml−1) | 1.7–26.5 | 1.5–12.3 | 0.92–14.7 | 0.96–7.7 | 0.59–4.7 |
| Exposure time (s) | 20 × 0.05 | 20 × 0.05 | 20 × 0.05 | 30 × 0.05 | 30 × 0.05 |
| Structural parameters | |||||
| Guinier region qRg range | 0.21–1.20 | 0.22–1.00 | 0.46–1.07 | 0.39–1.30 | 0.45–1.30 |
| I(0)/c (10−2 cm2 mg−1) (from Guinier)a | 3.03 ± 0.01 | 2.53 ± 0.01 | 5.85 ± 0.04 | 2.91 ± 0.01 | 7.33 ± 0.01 |
| Rg (Å) (from Guinier) | 50.4 ± 0.6 | 42.3 ± 0.3 | 85.4 ± 2.0 | 43.7 ± 0.3 | 72.7 ± 1.0 |
| Rc (Å) (from modified Guinier) | 9.34 ± 0.01 | 9.23 ± 0.02 | 9.37 ± 0.01 | 9.33 ± 0.01 | 9.80 ± 0.02 |
| I(0) (10−2 cm2 mg−1)a (from P(r)) | 3.11 ± 0.01 | 2.55 ± 0.01 | 5.93 ± 0.02 | 2.96 ± 0.01 | 7.60 ± 0.01 |
| Rg (Å) (from P(r)) | 53.9 | 44.1 | 90.8 | 47.2 | 83.4 |
| Dmax (Å) (from P(r)) | 210 | 170 | 350 | 175 | 350 |
| Rc (Å) (from Pc(r)) | 9.4 | 10.0 | 10.3 | 10.5 | 11.4 |
| Dc (Å) (from Pc(r)) | 35 | 44 | 45 | 50 | 50 |
| Porod volume, Vp (Å3) | 90,624 | 57,400 | 135,300 | 69,400 | 136,000 |
| Excluded volume, Vex (Å3) | 98,350 ± 670 | 77,810 ± 460 | 187,500 ± 830 | 89,300 ± 470 | 234,100 ± 2,900 |
| Molecular mass (kDa) (from (Vp/1.6)) | 56.6 | 35.9 | 84.6 | 43.4 | 85.0 |
| Molecular mass (kDa) (from (Vp/2)) | 49.2 ± 0.3 | 38.9 ± 0.2 | 93.7 ± 0.4 | 44.7 ± 0.2 | 117.1 ± 1.5 |
| Monomeric mass from sequence (kDa) | 53.2 | 42.8 | 95.6 | 46.8 | 99.9 |
| SASBDB codeb | SASDBA4 | SASDBB4 | SASDBC4 | SASDBD4 | SASDBE4 |
a Absolute intensities were determined using water as a secondary standard.
b SASBDB, Small Angle Scattering Biological Data Bank.
We also analyzed the intermediate q-region of the scattering curves using a modified Guinier approximation (ln(I(q)q) versus q2) that revealed a linear correlation indicative of a rod-like shape (Fig. 8D). The radii of gyration of a cross-section (Rc), calculated from the modified Guinier plot, was very similar for the three constructs, ranging between 9.23 and 9.37 Å. In addition, the pair distribution functions, Pc(r), of the cross-sections of the three fragments were also very similar (Fig. 8E). In summary, the plectin fragments are rod-like particles of similar thickness.
The dimensionless Kratky plots of the scattering data have bell-shaped peaks indicating that the SR3–SR6, SR7–SR9, and SR3–SR9 regions are compact particles (Fig. 8F), yet the position and amplitude of the maxima deviate largely from the expected value for spherical particles due to the highly anisometric shape of these proteins. The Porod-Debye plots (I(q)q4 versus q4) (24) of the scattering data had clear plateaus (Fig. 8G) indicative of a sharp electron density contrast between the proteins and the solvent, further supporting the idea that the particles lack disordered segments.
We constructed atomic models of the SR3–SR6 region by combining the crystal structures of the overlapping segments SR3-SR4 (PDB code 3PDY), SR4-SR5-SH3 (PDB code 3PE0), and SR5-SR6-ΔSH3 (this work). Eight models were built to take into account the two and four interdomain conformations of the SR3-SR4 and SR5-SR6 pairs observed in the crystals. The calculated scattering profiles of the SR3–SR6 structures matched the experimental SAXS curve (Fig. 8A); χ2, a parameter that measures the quality of the fit, ranged between 1.2 and 1.3 for q ≤ 0.35 Å−1. Similarly, the scattering calculated for a model based on the crystal structure of SR7–SR9 reproduced the experimental SAXS data (χ2 =1.3). The P(r) value calculated from the atomic structures of SR3–SR6 and SR7–SR9 reproduced the corresponding P(r) estimated from the scattering data. Low resolution shapes of the SR3–SR6 and SR7–SR9 regions reconstructed from the SAXS data using ab initio methods could be superimposed closely on the respective atomic structures (Fig. 8I). In summary, the SR3–SR6 and SR7–SR9 segments are rigid rod-like structures in solution.
Next, we analyzed the possible segmental flexibility between the two rigid segments, SR3–SR6 and SR7–SR9, of the plakin domain by using an ensemble fitting method with the program EOM (optimization ensemble method). We generated a pool of 10,000 structures of the SR3–SR9 region that sample exhaustively the possible orientations of the two rigid segments, assuming a flexible linker between SR6 and SR7. The minimal ensemble that reproduces the SAXS data is mainly populated by similar conformations with a narrow range of Rg that is close to the largest Rg of the pool (Fig. 8H). In summary, the SR3–SR6 and SR7–SR9 regions adopt a linear arrangement with limited conformational variability.
The extended shape of the SR3–SR9 region precludes extensive contacts between the N- and C-terminal halves. Therefore, the apparent rigidity relies on local interactions at the SR6-SR7 interface, suggesting that these two repeats are connected by a well structured linker. As yet, the structure of the SR6-SR7 linker is unknown, and attempts to crystallize the SR6–SR8, SR6–SR9, and SR3–SR9 fragments were fruitless. Nonetheless, based on the similarity with other arrays of tandem SRs and the secondary structure prediction (see below), the linker was modeled as an α-helix. We generated models in which the SR3–SR6 and SR7–SR9 regions were positioned at different angles based on the four relative orientations of the SR5-SR6 region observed in the crystal structures. The scattering curves calculated for these models fitted the experimental SAXS data with χ2 values between 1.7 and 2.2 (q ≤ 0.35 Å−1). Next, we refined the structures by using an elastic network model-based normal modal analysis that explores large scale conformational changes without distorting local structures. Models were perturbed using the first nontrivial normal mode with the lowest frequency and were evaluated against the SAXS data. This led to a refined structure with an improved fit to the scattering profile (χ2 = 1.2), which superimposes closely with low resolution shapes reconstructed from the SAXS data using ab initio methods (Fig. 8I). In summary, the plakin domain of plectin resembles a slightly bent rod with a kink around SR6 and two rigid halves forming a ∼145° angle.
The bent shape of the plakin domain of plectin is not unusual in other SR arrays and bears a close resemblance to the curved structures of the SR14–SR16 region of β-spectrin (25–27) and the α-β-spectrin tetramerization domain (28) (Fig. 9).
FIGURE 9.
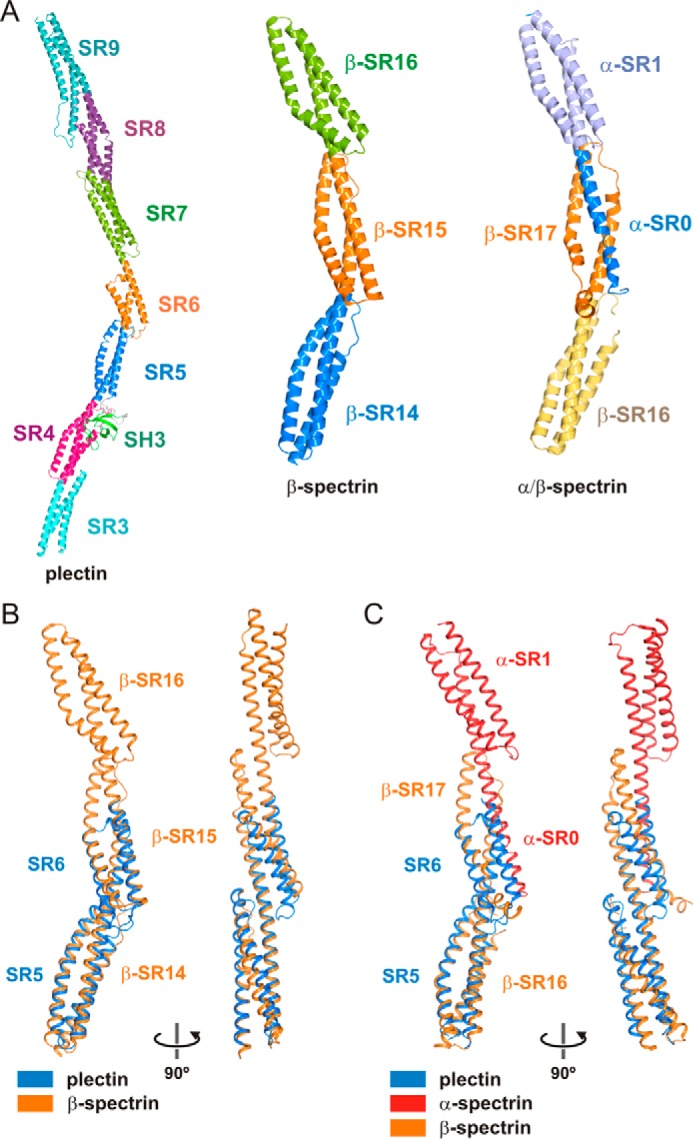
Comparison of the plakin domain of plectin with curved multi-repeat segments of spectrins. A, ribbon representation of the composite structure of the plakin domain of plectin, the crystal structure SR14–SR16 of human β2-spectrin (PDB code 3EDV), and the tetramerization domain complex of human erythroid spectrins formed by the partial SR0 and SR1 of α-spectrin and the SR6 and the partial SR17 of β-spectrin (PDB code 3LBX). B and C, superimposition of the structure of the SR5-SR6-ΔSH3-A of plectin onto repeats SR14-SR15 of β2-spectrin (B) or repeats β-SR16-β-SR17/α-SR0 of the tetramerization complex of α/β spectrins (C).
Structure of the Plakin Domain of Desmoplakin in Solution
It had been reported that the plakin domains of desmoplakin, envoplakin, and periplakin have articulated structures in which the regions upstream of SR7 and downstream of SR8 are highly flexible (21). In the light of the rigid structure of the SR7–SR9 region of plectin, which was unknown in previous studies, we analyzed the plakin domain of desmoplakin by SAXS under the same conditions used for plectin, which allowed a rigorous comparative characterization (Fig 10 and Table 2).
FIGURE 10.

SAXS analysis of the plakin domain of desmoplakin in solution. A, SAXS profiles extrapolated to infinite dilution of the SR7–SR9 (violet) and SR3–SR9 (orange). Curves are offset on the log scale; the same data color scheme is used in B–F. The scattering calculated for the homology atomic model of SR7–SR9 is shown as a dashed line. B, Guinier plots of the data in A. The Guinier Rg values at several protein concentrations and the linear regression fit (dashed lines) are shown in the inset. C, P(r) functions determined from the scattering data (colored solid lines). The P(r) value calculated from the SR7–SR9 atomic model is shown as a dashed line. D, cross-sectional Guinier plots of the scattering data. The linear fits to the intermediate-q data used to calculate Rc are shown as dashed lines. E, cross-sectional Pc(r) functions. For representational purposes, the P(r) and Pc(r) functions have been scaled. F, dimensionless Kratky plots. The crosshair indicates the expected position of the maxima for spherical compact particles. G, Porod-Debye plots of the SAXS data. H, ribbon representation of the atomic model of SR7–SR9 docked into the SAXS-derived molecular envelope (semi-transparent surfaces), which is the average of 15 independently reconstructed bead models. Two orthogonal views are shown. I, EOM analysis of the flexibility between the rigid segments SR3–SR6 and SR7–SR9. Frequency distributions of Rg in a pool of models (solid black line) and in the selected ensemble that fits the SAXS data of SR3–SR9 (solid red line). The distributions in the pool (dashed gray line) and the selected ensemble (dashed orange line) of the SR3–SR9 region of plectin are shown for comparison.
The SR7–SR9 region of desmoplakin is monodisperse and monomeric in solution. The Rg value estimated by Guinier analysis (43.7 ± 0.3 Å) was similar to the Rg of the SR7–SR9 region of plectin. Similarly, the P(r) function, Dmax, Rc, and Pc(r) function of the SR7–SR9 of desmoplakin were also almost identical to those of the equivalent region of plectin (Fig 10, C–E). In addition, the dimensionless Kratky plot of the desmoplakin SR7–SR9 region has a maximum at the same position as the plectin fragment (Fig 10F). The scattering profile calculated for a homology model of desmoplakin, built based on the structure of plectin, reproduced the experimental SAXS curve (χ2 = 2.2) (Fig 10A), and there is a good correlation between the atomic homology model and the low resolution structure reconstructed from the SAXS curve (Fig 10H). Collectively, our sequence and SAXS analysis support the idea that the SR7–SR9 region of desmoplakin has a rigid rod-like structure similar to plectin.
Next, we analyzed the SR3–SR9 region of desmoplakin by SAXS, which was also monodisperse and monomeric in solution. The Rg values determined by Guinier analysis (72.7 ± 1.0 Å) or obtained from the P(r) (83.4 Å) were significantly shorter than the Rg of plectin. The P(r) of desmoplakin extends to a Dmax of 350 Å, similar to that of plectin; yet the P(r) has a wide plateau for distances, in the range of 20 to 75 Å (Fig 10C), suggesting that the SR3–SR9 region of desmoplakin is on average thicker than plectin. Similarly, the cross-sectional Rc value estimated from the modified Guinier (9.80 Å) and the Pc(r) (11.4 Å) was larger than for the SR7–SR9 region of plectin and desmoplakin or the SR3–SR9 region of plectin (Fig 10, D and E). The dimensionless Kratky plot has a bell shape that drops to zero at high qRg (Fig 10F), and the Porod-Debye plot shows a plateau (Fig 10G), indicating that the plakin domain of desmoplakin consists of compact domains and lacks large disordered regions. Nonetheless, the maximum in the dimensionless Kratky plot is located at lower values of qRg and (qRg)2I(q)/I(0) than the SR3–SR9 region of plectin, indicating that desmoplakin is on average less elongated than plectin. Finally, we analyzed the possible conformational heterogeneity of the desmoplakin SR3–SR9 region using EOM (Fig 10I). A pool of 10,000 structures was created by combining the crystal structure of the SR3–SR6 (PDB code 3R6N) (16) and the model of the SR7–SR9 region, which were treated as rigid bodies, whereas the 30-residue-long linker between SR6 and SR7 was allowed to be flexible. The ensemble that fits the experimental data contains conformations with a wide range of Rg values that spreads along all of the Rg values of the structures in the pool, although there is a higher abundance of compact (i.e. small Rg) conformations in the ensemble. A similar distribution is observed in the Dmax of the pool and the ensemble (data not shown). In summary, the plakin domain of desmoplakin is an articulated structure consisting of two rigid segments connected by a central flexible linker, which differs greatly from the extended structure of plectin.
Conservation of the SR6-SR7 Region in Other Plakins
The global differences between the extended plakin domain of plectin and the segmented domain of desmoplakin are due to local differences in the SR6-SR7 linker, or the SR5–SR7 linker in the case of periplakin, and envoplakin. Plectin, BPAG1e, MACF1, VAB-10, and Shot have short SR6-SR7 linkers of similar length, and a secondary structure prediction suggests that they have a helical structure (Fig 11). The SR6-SR7 linker of VAB-10 contains a cluster of three Pro residues that would interrupt the α-helix, yet the limited conformational freedom of Pro suggests that it would be a relatively rigid region. In summary, the plakin domains of BPAG1e, MACF1, Shot, and possibly VAB-10 are likely to adopt extended shapes similar to that observed in plectin.
FIGURE 11.

Analysis of the SR6-SR7 linker. Multiple sequence alignment of the region around the SR6-SR7 linker of plectin, BPAG1e, MACF1, VAB-10, and Shot. The extension of the α-helices into the crystal structures of plectin and positions a and d of the heptad repeats, which face the central hydrophobic core, are indicated at the top. The filled circles denote every tenth residue. The α-helical segments predicted from the sequences using the JPred4 server are indicated by rectangles colored according to their assignment to the SR6 domain (orange), the linker (gray), and the SR7 domain (green).
Pathogenic Missense Mutations in SR7–SR9 of Desmoplakin
Analysis of missense mutations linked to diseases provides insights into the functional and structural roles of specific residues. Although no such mutations have been described within the plakin domain of plectin to date, missense mutations in the plakin domain of desmoplakin cause a variety of cardiac and/or cutaneous diseases (29).
Most pathogenic mutations are located within the SR3–SR6 region of desmoplakin and target both buried (e.g. Ser-299, Asn-375, Ile-445, Ser-507, Ile-533, Leu-583, Ser-597, and Leu-622) and solvent-exposed (e.g. Arg-222, Asp-230, Asn-287, Glu-422, Asn-458, Lys-470, Thr-564, Ala-566, His-586, Gln-616, and His-618) residues. The structural effects of these mutations have been analyzed elsewhere (16, 30, 31).
In SR7–SR9 of desmoplakin, mutations N661I, Y787C, R808C, and R808H, have been linked to arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) (32–34), and the mutation I870M has been observed in a patient with dilated cardiomyopathy (35). Owing to the high similarity between the SR7–SR9 of plectin and desmoplakin, we analyzed the possible structural effects of these mutations in desmoplakin using the high resolution structure of plectin as a template (Fig 12).
FIGURE 12.

Analysis of missense mutation in SR7–SR9 of desmoplakin. A, ribbon representation of the SR7–SR9 region of plectin. The side chains of residues in positions equivalent to those of desmoplakin where missense mutations have been described are shown as sticks. Labels of residues in desmoplakin are shown in parentheses underneath the labels of the plectin residues. Blue boxes denote the positions of mutations not linked to diseases; orange boxes indicate residues mutated in arrhythmogenic right ventricular dysplasia/cardiomyopathy or dilated cardiomyopathy. B and C, close-up views of two areas of the SR8 region of plectin, which correspond to the areas of desmoplakin containing pathogenic missense mutations.
Asn-661 occupies a position equivalent to Glu-1005 of plectin, which lies upstream of helix SR7-A and makes no contact with other parts of the structure. Therefore, the mutation N661I is likely to affect the flexible SR6-SR7 linker. Tyr-787 is equivalent to His-1135 of plectin, which is in the helix SR8-A, buried in the hydrophobic core. Therefore, Y787C is likely to distort or destabilize SR8. Desmoplakin Arg-808 is equivalent to Lys-1156 in plectin, which is in the SR8-B helix on the surface of the domain; the aliphatic chain packs against Leu-1152 and Leu-1229 (Val-804 and Leu-881 in desmoplakin), and the amine group is flanked by the carboxylate groups of Glu-1153 and Glu-1230 (Glu-805 and Glu-882 in desmoplakin). The conserved local environment of plectin Lys-1156 and desmoplakin Arg-808 suggest that R808C might alter the helical bundle locally and expose a hydrophobic patch on the surface of the SR8. This is in agreement with the destabilizing effect of R808C when introduced in an SR7-SR8 construct (32). Finally, desmoplakin Ile-870 corresponds to Val-1218 of plectin, which is part of the hydrophobic core of the SR8; hence, the substitution I870M is likely to perturb the stability of this SR.
Other missense mutations within the SR7–SR9 region of desmoplakin have been observed in healthy individuals. These include T830I, R866C, E882K, R908H, and N956Y (36, 37), which are exposed to the solvent on the surface of the structure. In summary, disease-causing mutations are potentially linked to structural destabilization of the SR7–SR9 region, whereas changes on the surface apparently do not compromise its function.
Implications for the Role of the Plakin Domain in the Mechanical Properties of Plakins
Arrays of SRs form deformable structures that frequently participate in mechanically resilient cytoskeletal networks. This is depicted by the contribution of spectrins to the elasticity of red blood cells to withstand high shear stress during circulation. Plectin and other plakins are essential to maintain the integrity of tissues that suffer high mechanical stress. Junctional complexes that contain plakins have also been implicated in sensing biomechanical changes in the surrounding extracellular environment (38). Yet, it is unclear how the plakin domain might contribute to these functions.
Owing to the extended shape of the SR3–SR9 region of plectin and the juxtaposition of SR9 to the rod domain (∼190 nm long), the plakin domain acts as a spacer that further increases by at least ∼34 nm the distance between the IF-binding sites in the C-terminal region and the binding sites for junctional proteins, which are located mostly in the actin-binding domain. In addition, the plakin and rod domains form a continuous thread that transmits mechanical signals between distant regions of the molecule. Nonetheless, the elastic properties of these two domains are probably different. Coiled coils bend easily but are rather resistant to stretching (39). In contrast, arrays of SRs undergo moderate bending, but repeats unfold individually at low pulling forces (40). Hence, the plakin domain may work as a molecular shock absorber that dissipates elastic energy when cells are subjected to external forces.
Our results have revealed that flexibility is not evenly distributed along the plakin domain. The unique structure of the SR7–SR9 region suggests that it is more resistant to mechanically induced deformation than other parts of the plakin domain. The prevailing structural role of the SR7–SR9 region is likely to be conserved in all plakins. For example, most of the disease-causing missense mutations in the SR7–SR9 region of desmoplakin are predicted to cause structural destabilization, and no specific protein-protein interactions have been mapped to this region. In contrast, inter-repeat bending has been observed only in the SR3-SR4 and SR5-SR6 pairs. Moreover, the plasticity of SR6 and its smaller size with respect to other SRs suggest that SR6 is mechanically weaker and might unfold at lower forces than other repeats.
The possibility of force-induced conformational changes in the plakin domain (e.g. bending/straightening at the inter-repeat level and unfolding of individual repeats) suggests that plectin not only transmits tension but might also act directly as a mechanosensor. The protein interaction sites in the plakin domain of plectin are located in the malleable N-terminal segment. This suggests that the affinity of plectin for some proteins might depend on the conformational state of the plakin domain, which in turn could be regulated mechanically. In this regard, the maintenance of hemidesmosomes and desmosomes apparently requires some tension through the attachment to a functional cytokeratin network (41).
Finally, our data suggest that there are two types of plakin domain architectures: (i) extended and (ii) those interrupted by central flexible hinges. These two types might sense and respond differently to mechanical cues. For example, the extended plakin domains of plectin and BPAG1e are likely to be under constitutive basal tension when they cross-link cytoskeletal structures, for example in hemidesmosomes. Thus, when cells are subjected to mechanical stress, forces would be readily transmitted to the IF cytoskeleton and would elicit additional changes in the plakin domain. On the other hand, the decoupling of the two halves of the plakin domain in desmosomal plakins suggest that an additional initial step, in which the central hinge is extended and strained, is required before the plakin domain can transmit tension or the arrays of SR undergo force-induced conformational changes. This is in agreement with the elongation of desmoplakin patches in desmosomes caused by external mechanical stretch (41).
Experimental Procedures
Protein Expression and Purification
The cDNA encoding the following fragments of human plectin were amplified by PCR and cloned in a modified pET15b vector using NdeI and BamHI restriction sites (42): residues 543–1006 (SR3–SR6), 1004–1233 (SR7-SR8), 1004–1372 (SR7–SR9), and 543–1372 (SR3–SR9). Two constructs of the SR5-SR6 region (residues 750–1006, Δ819–888) in which the SH3 was replaced by the short sequence GSGSG (SR5-SR6-ΔSH3-A) or GSG (SR5-SR6-ΔSH3-B) were created by a two-step PCR. The oligonucleotides used for cloning are described in supplemental Tables S3 and S4. Phe-752, which packs against SR4 in the SR4-SR5 structure (15), was replaced by Ala in the SR5-SR6-ΔSH3 constructs. The plectin fragments used herein are shared by all of the alternative splicing variants. We used the numbering of plectin 1c (UniProt Q15149-2) for consistency with our previous works. The cDNA fragments coding for regions 660–1025 (SR7–SR9) and 180–1025 (SR3–SR9) of human desmoplakin (UniProt P15924) were cloned in the pET15b derivative like the plectin constructs.
Proteins were expressed in Escherichia coli strain BL21(DE3), purified by immobilized metal ion affinity chromatography, and their N-terminal His tag was cleaved as described (43). Typical yields of the purified proteins are shown in supplemental Table S5.
Crystallization and Structure Determination of Fragment SR5-SR6-ΔSH3-A
Crystals of the protein SR5-SR6-ΔSH3-A (750–818-GSGSG-889–1006) were obtained by sitting drop vapor diffusion at 4 °C. A protein solution at 30 mg/ml in 10 mm Tris-HCl (pH 7.5), 50 mm NaCl, 1 mm DTT was mixed with an equal volume of the crystallization solution 0.2 m sodium potassium tartrate, 20% PEG 3350. Prior to data collection crystals were transferred to 0.2 m sodium potassium tartrate, 20% PEG 3350, 15% glycerol and flash-cooled in liquid nitrogen. A heavy atom derivative was obtained by soaking a native crystal in 50 mm Tris-HCl (pH 7.5), 0.2 m sodium potassium tartrate, 20% PEG 3350, 1 mm ethylmercurithiosalicylate (EMTS) for 6 h at 4 °C. The excess EMTS was removed during incubation in the same cryoprotectant solution as used for the native crystal. Data from a native crystal were collected on the X06DA beam line of the Swiss Light Source synchrotron (SLS, Villigen, Switzerland) at 105 K. Data from the EMTS-derivatized crystals were collected at 100 K using a Microstar-H rotating anode (Bruker AXS) and a mar345 detector (Marresearch GmbH). The diffraction data for these and all other crystals (see below) were processed with the XDS suite (44).
The crystals belong to space group H3 (R3:h) (Table 3) and contain two plectin molecules in the AU that correspond to a ∼63% solvent content. The structure was phased by single isomorphous replacement with anomalous scattering (SIRAS) using native and mercurial data sets. The heavy atom substructure, consisting of three mercury sites, was determined with ShelxC/D/E (45) and HKL2MAP (46). The phase probability distributions were further refined with autoSHARP (47). After phase improvement and extension with Solomon (48) an interpretable electron density map was obtained (Fig 13). Two copies of the SR5 domain from the structure of SR4-SR5 (PDB code 3PDY) were placed in the map using the program Molrep (49), and then the SR6 domains were built manually using the program Coot (50).
TABLE 3.
Crystallographic data collection and refinement statistics
| Protein |
SR5-SR6-ΔSH3-A |
SR5-SR6-ΔSH3-B |
SR7–SR8 |
SR7–SR9 |
||
|---|---|---|---|---|---|---|
| Data set | Native | EMTS | Native | Native | EMTS | Native |
| Data collection | ||||||
| Space group | H3 | H3 | C2 | P21 | P21 | C2 |
| Unit cell dimensions | ||||||
| a, b, c (Å) | 178.1, 178.1, 50.7 | 177.7, 177.7, 50.5 | 129.2, 47.8, 106.6 | 45.7, 115.9, 64.8 | 45.1, 115.9, 64.4 | 75.2, 90.7, 154.5 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 126.7, 90 | 90, 97.6, 90 | 90, 97.4, 90 | 90, 99.1, 90 |
| Wavelength (Å) | 1.0000 | 1.5418 | 1.5418 | 0.9792 | 1.5418 | 0.9330 |
| Resolution (Å) | 2.80 (2.87–2.80)a | 4.50 (4.62–4.50)a | 3.00 (3.08–3.00)a | 1.80 (1.85–1.80)a | 2.50 (2.60–2.50)a | 5.0/3.8/2.8b (2.87–2.80)a |
| Unique reflections | 14,753 | 7,046c | 10,510 | 61,449 | 42,726c | 14,129 |
| Average multiplicity | 10.2 (10.7)a | 4.5 (4.5)a | 3.9 (3.9) | 6.6 (6.5)a | 3.8 (4.0)a | 7.5 (6.2) |
| Completeness (%) | 100 (100)a | 99.8 (99.8)a | 97.7 (100) | 99.4 (98.3)a | 95.6 (94.1)a | 55.7 (11.4)a |
| Rmeasd | 0.122 (2.21)a | 0.127 (0.726)a | 0.093 (0.889) | 0.076 (1.65)a | 0.078 (0.457)a | 0.083 (1.38)a |
| CC1/2 | 0.999 (0.697) | 0.997 (0.898) | 0.998 (0.877) | 0.997 (0.888)a | 0.998 (0.941)a | 1.000 (0.555) |
| Mean I/σ(I) | 27.8 (2.0)a | 11.4 (2.7)a | 15.0 (2.0) | 12.0 (2.0)a | 14.4 (4.5)a | 17.2 (1.7)a |
| Risoe | 0.262 | 0.362 | ||||
| Refinement | ||||||
| Resolution range (Å) | 48–2.80 | 43–3.00 | 45–1.80 | 49–2.80 | ||
| Unique reflections, work/free | 13,976/714 | 9,989/510 | 58,161/3087 | 13,406/690 | ||
| Rwork/Rfree (%)f | 22.3/26.9 | 24.5/27.7 | 18.7/21.9 | 29.6/32.2 | ||
| Number | ||||||
| Residues | 183/173 | 188/188 | 228/229 | 346/345 | ||
| Waters | 2 | 349 | ||||
| PEGs | 3 | |||||
| Average B-value (Å2) | ||||||
| Wilson plot | 55.6 | 66.8 | 35.7 | 64.6 | ||
| Protein | 69.8/115.2 | 74.0/86.5 | 55.6/56.3 | 78.2/84.0 | ||
| Waters | 68.1 | 52.8 | ||||
| PEGs | 125.9 | |||||
| rmsd bond lengths (Å) | 0.002 | 0.002 | 0.008 | 0.005 | ||
| rmsd angles (°) | 0.45 | 0.56 | 0.94 | 0.76 | ||
| No. of residues in Ramachandran plotg | ||||||
| Favored regions | 342 (99.4%) | 370 (99.5%) | 506 (99.6%) | 672 (99.7%) | ||
| Additionally allowed | 2 (0.6%) | 2 (0.5%) | 2 (0.4%) | 2 (0.3%) | ||
| Outliers | 0 | 0 | 0 | 0 | ||
| PDB code | 5J1H | 5J1F | 5J1G | 5J1I | ||
a Numbers in parentheses correspond to the outer resolution shell.
b Data were processed using anisotropic resolution limits.
c Calculated treating Bijvoet pairs as independent reflections.
d Rmeas is the multiplicity-independent R-factor (72).
e Riso = Σ‖Fder|−|Fnat‖/Σ|Fnat|, where Fder is the heavy atom derivative structure factor and Fnat is the protein structure factor.
f Calculated using 5% of the reflections that were not included in the refinement.
g As defined in the program MolProbity (73).
FIGURE 13.

Electron density maps of the SR5-SR6-ΔSH3-A crystal structure. A, stereo view of a representative section of a map calculated with the phases obtained by SIRAS with autoSHARP after improvement by density modification with Solomon. The density is contoured a 1σ. The Cα trace of this section of the structure is shown. B, stick representation of the refined structure of the region shown in A, superimposed onto a feature-enhanced 2mFobs − DFmodel map calculated at the final stage of refinement (contoured at 1σ).
The structure was refined against native data extending to 2.8 Å resolution with phenix.refine (51) alternating with model building with Coot. Refinement included overall anisotropic and bulk solvent corrections, positional refinement, individual B-factor restrained refinement, and refinement of the translation/libration/screw-rotation (TLS) parameters of three groups in each molecule. Torsion angle non-crystallographic symmetry (NCS) restraints (i.e. local NCS restraints) were included. The final model includes residues 750–818 and 889–1000 in one molecule and residues 750–818, 889–970, and 974–996 in the second molecule; 99.4% of the main-chain torsion angles are in the favored regions of the Ramachandran plot, and the other 0.6% are in additionally allowed regions.
Crystallization and Structure Determination of the Fragment SR5-SR6-ΔSH3-B
Crystals of the SR5-SR6-ΔSH3-B (750–818-GSG-889–1006) region were obtained by hanging drop vapor diffusion at 4 °C by mixing equal volumes of a protein solution at 30 mg/ml in 10 mm Tris-HCl (pH 7.5), 50 mm NaCl, 1 mm DTT and crystallization solution consisting of 0.1 m bis-Tris propane (pH 6.0), 18% PEG 3350, 0.2 m sodium malonate. For data collection, crystals were transferred to the crystallization solution supplemented with 15% glycerol and flash-cooled in liquid nitrogen. Diffraction data were measured at 100 K using a rotating anode generator.
Crystals belong to the C2 space group (Table 3) and contain two molecules in the AU (∼58% solvent content). The structure was phased by molecular replacement with the program Phaser (52) using the structure of the SR5-SR6-ΔSH3-A region as a search model. The structure was refined with phenix.refine against data to 3.0 Å in a similar manner as used for the SR5-SR6-ΔSH3-A structure. Five TLS groups, three in one molecule and two in the other, were refined. The final model includes residues 750–818, the GSG linker, and residues 889–1000 in one molecule and residues 750–818, the GSG linker, and residues 889–1001 in the second molecule of the AU. The main-chain torsion angles of 370 residues (99.5%) occupy the favored regions of the Ramachandran plot, and the two remaining residues are in additionally allowed regions.
Crystallization and Structure Determination of the Fragment SR7-SR8
Crystals of the SR7-SR8 (1004–1233) region of plectin were obtained by mixing a protein solution at 27 mg/ml in 10 mm Tris-HCl (pH 7.5), 50 mm NaCl with an equal volume of crystallization solution (50 mm Tris-HCl (pH 7.7), 0.2 m NaF, 18% PEG 3350). The drop was vapor-equilibrated at 25 °C against 1 ml of the crystallization solution. A mercurial derivative was obtained by soaking a crystal for 15 h in crystallization solution supplemented with 2 mm EMTS. Prior to data collection, the crystals were transferred to the cryosolution (50 mm Tris-HCl (pH 7.7), 0.2 m NaF, 19% PEG 3350, 20% glycerol) and flash-cooled in liquid nitrogen. Data from a native crystal were collected at 100 K on the Xaloc beam line of the ALBA synchrotron (Barcelona, Spain) (53). Data from the EMTS-derivatized crystal were collected at 100 K using a rotating anode generator.
Crystals belong to space group P21 (Table 3) and contain two SR7-SR8 molecules in the AU (∼64% solvent content). The structure was phased by SIRAS using the native and EMTS data sets, similarly as for the SR5-SR6-ΔSH3-A structure. The mercury substructure (12 sites) was solved with ShelxC/D/E (45) and HKL2MAP (46). The phases were further refined with autoSHARP (47) and improved and extended to 1.8 Å with Solomon (48). A high quality map was calculated using the SIRAS phases (Fig. 3, C and D), which allowed for the automatic building of 450 (96%) residues using ARP/wARP (54). The model was refined against the native data to 1.8 Å using phenix.refine combined with manual model building using Coot. Five TLS groups in each molecule were refined. The final model includes residues 1004–1231 of protein molecule A and 1004–1233 of molecule B, 349 water molecules, and three PEG fragments. The model has excellent geometry; all main-chain torsion angles are in the favored regions of the Ramachandran plot, with the exception of D1039 that is located in an additionally allowed region of the plot.
Crystallization and Structure Determination of the Fragment SR7–SR9
Crystals of the SR7–SR9 (residues 1004–1372) region of plectin were obtained by vapor diffusion using 0.1 m bis-Tris propane (pH 8.0), 16% PEG 3350, and 0.3 m sodium potassium tartrate as the crystallization solution. Drops containing 3 μl of the SR7–SR9 protein at 12 mg/ml in 10 mm Tris-HCl (pH 7.5), 50 mm NaCl, 1 mm DTT, and 2 μl of crystallization solution were equilibrated at room temperature against 1 ml of the latter solution. Crystals were transferred to crystallization solution supplemented with 20% glycerol and flash-cooled in liquid nitrogen. Data were collected at 100 K on the ID 14.2 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France).
Crystals belong to space group C2 (Table 3) and contain two SR7–SR9 monomers in the AU (∼60% solvent content). Diffraction data were highly anisotropic with approximate diffraction limits of ∼2.8Å along the best direction but only ∼3.8 Å and ∼5.0 Å in the weakly diffracting directions (Fig. 4, A and B). Therefore, data were processed using the STARANISO server (Global Phasing Ltd.), which applies non-elliptical anisotropic limits based on a locally averaged mean I/σ(I) cut-off, performs a Bayesian estimation of structure amplitudes, and applies an anisotropic correction to the data. Detailed crystallographic statistics are shown in supplemental Table S6.
The structure was phased by molecular replacement using Phaser. First, two monomers of the SR7-SR8 region were located. Next, we located two copies of a partial model of the SR9 region based on the structure of SR1 of erythroid α-spectrin (PDB code 3LBX) (28). The structure was refined with phenix.refine against the anisotropically scaled data, similar to that described above. Briefly, torsion non-crystallographic symmetry restraints were used. In addition, dihedral angles of the SR7-SR8 domains were restrained to those derived from the high resolution structure of this region. Two B-factors were refined for each residue, one for the main-chain atoms and the other for the side-chain atoms. Two TLS groups, one for each protein chain, were refined. Owing to the limited resolution, the side chains of 39% of the residues of the SR9 were truncated after the Cβ atom. The final model includes residues 1007–1368 and 1009–1371 of molecules A and B present in the AU, respectively. Loops BC of the SR7 and loops AB and BC of SR9 in both monomers and part of loop AB of SR8 in monomer B were not modeled due to weak electron density in these regions. The model has very good geometry; 99.7% of the residues fall in the most favored regions of the Ramachandran plot and the remaining in additionally allowed regions.
Analysis of Atomic Structures
Superimposition of atomic structures was done with the program LSQKAB (55) or Theseus (56). The relative orientation of adjacent SRs in terms of three simultaneous rotations around the Cartesian axes was calculated as described elsewhere (22, 57). The rotation angles around the x, y, and z axes are termed tilt, roll, and twist, respectively. Principal component analysis of the structures of the SR5-SR6 region was done with the Bio3D package for the R statistical software (58). Molecular figures 1–6, 8–10, 12, and 13 were created with PyMOL, version 1.6.0 (Schrödinger).
SAXS Measurements and Analysis
Proteins for SAXS analysis were equilibrated in 20 mm sodium phosphate (pH 7.5), 150 mm NaCl, 5% glycerol, and 3 mm DTT by size exclusion chromatography using HiPrep Sephacryl S300 26/60 or Superdex 200 10/300 GL columns (GE Healthcare). Then, samples were concentrated by ultrafiltration using Amicon stirred cells (Merck Millipore) and centrifuged at 100,000 × g 30 min at 4 °C to remove possible aggregates. SAXS data were measured at the EMBL beamline P12 in the Deutches Elektronen Synchrotron (DESY, Hamburg) using radiation of wavelength (λ) 1.24 Å and a Pilatus 2 m detector (Dectris).
Protein samples and their corresponding buffers were measured consecutively at 10 °C. For each protein, the scattering data were measured at several sample concentrations obtained by 2-fold serial dilution in the ranges indicated in Table 2. SAXS data were collected over a scattering vector (q = (4πsinθ)/λ, where 2θ is the scattering angle) range from 0.005 to 0.45 Å−1, except for plectin SR3–SR9, for which data were measured in the range of 0.005 < q < 0.35 Å−1. Data were processed and analyzed with the ATSAS package (59). Unless otherwise indicated, analysis was done on the scattering extrapolated to an infinite dilution of the sample. Guinier and modified Guinier analysis were done with the program Primus QT (59). Pair-distance distribution functions, P(r), and cross-sectional distance distribution functions, Pc(r), were calculated with GNOM (60). Ab initio shape reconstructions were calculated with the program DAMMIF (61); multiple reconstructions were superimposed, averaged, and filtered with DAMAVER (62). Interdomain flexibility was analyzed using the program EOM (63, 64). For flexible fitting of the high-resolution structures of the SR3–SR9 region of plectin to the SAXS data, multiple conformations that represent possible large scale movements were calculated by normal modal analysis employing an elastic network model using the ElNémo server (65). Scattering profiles and distribution of intramolecular distances of atomic structures were calculated with CRYSOL (66) and HYDROPRO(67), respectively.
Sequence Analysis
Profile HMMs were used to search for regions similar to SR7–SR9 in other plakins with the suite HMMER (v3.1b2) (68). Briefly, the UniProt proteomes of Homo sapiens, C. elegans, and D. melanogaster, were searched with profile HMMs built from a single sequence or from multiple sequence alignments using the programs phmmer and hmmsearch, respectively. Secondary structure prediction was done with the JPred4 server (69).
Author Contributions
E. O. did the crystallographic analysis of the plectin fragments. E. O. and R. M. B. did the initial SAXS analysis of plectin. J. A. M. performed the SAXS analysis. A. M. C. and A. C. produced the desmoplakin fragments and did their SAXS analysis. A. S. assisted in the writing of the paper. J. M. dP conceived the study, assisted in the data analysis, and wrote the paper, which was read and approved by all authors.
Supplementary Material
Acknowledgments
We thank the ESRF, the Swiss Light Source synchrotron, ALBA-CELLS, and the EMBL for providing synchrotron radiation facilities.
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO) and the European Regional Development Fund, Grants BFU2012-32847 and BFU2015-69499-P (to J. M. dP), as well as by the Netherlands Science Foundation and the Dutch Cancer Society (to A. S.). This work also was supported by the European Community's Seventh Framework Programme (FP7/2007–2013) under BioStruct-X Grant Agreement 283570. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S6.
The atomic coordinates and structure factors (codes 5J1H, 5J1F, 5J1G, and 5J1I) have been deposited in the Protein Data Bank (http://wwpdb.org/).
SAXS data and derived models have been deposited in the Small Angle Scattering Biological Data Bank (SASBDB) under codes SASDBA4, SASDBB4, SASDBC4, SASDBD4, and SASDBE4.
- IF
- intermediate filament
- AU
- asymmetric unit
- BPAG
- bullous pemphigoid antigen
- EMTS
- ethylmercurithiosalicylate
- EOM
- optimization ensemble method
- HMM
- hidden Markov model
- MACF1
- microtubule and actin cross-linking factor 1
- PC
- principal component
- PDB
- Protein Data Bank
- SAXS
- small angle X-ray scattering
- SH3
- Src homology 3
- rmsd
- root mean square deviation
- SIRAS
- single isomorphous replacement with anomalous scattering
- SR
- spectrin repeat
- TLS
- translation/libration/screw-rotation
- bis-Tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane.
References
- 1. Bouameur J. E., Favre B., and Borradori L. (2014) Plakins, a versatile family of cytolinkers: roles in skin integrity and in human diseases. J. Invest. Dermatol. 134, 885–894 [DOI] [PubMed] [Google Scholar]
- 2. Sonnenberg A., and Liem R. K. (2007) Plakins in development and disease. Exp. Cell Res. 313, 2189–2203 [DOI] [PubMed] [Google Scholar]
- 3. Wiche G., Osmanagic-Myers S., and Castañón M. J. (2015) Networking and anchoring through plectin: a key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 32, 21–29 [DOI] [PubMed] [Google Scholar]
- 4. Natsuga K. (2015) Plectin-related skin diseases. J. Dermatol. Sci. 77, 139–145 [DOI] [PubMed] [Google Scholar]
- 5. de Pereda J. M., Lillo M. P., and Sonnenberg A. (2009) Structural basis of the interaction between integrin α6β4 and plectin at the hemidesmosomes. EMBO J. 28, 1180–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geerts D., Fontao L., Nievers M. G., Schaapveld R. Q., Purkis P. E., Wheeler G. N., Lane E. B., Leigh I. M., and Sonnenberg A. (1999) Binding of integrin α6β4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J. Cell Biol. 147, 417–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilhelmsen K., Litjens S. H., Kuikman I., Tshimbalanga N., Janssen H., van den Bout I., Raymond K., and Sonnenberg A. (2005) Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J. Cell Biol. 171, 799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fontao L., Geerts D., Kuikman I., Koster J., Kramer D., and Sonnenberg A. (2001) The interaction of plectin with actin: evidence for cross-linking of actin filaments by dimerization of the actin-binding domain of plectin. J. Cell Sci. 114, 2065–2076 [DOI] [PubMed] [Google Scholar]
- 9. García-Alvarez B., Bobkov A., Sonnenberg A., and de Pereda J. M. (2003) Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and to integrin a6b4. Structure 11, 615–625 [DOI] [PubMed] [Google Scholar]
- 10. Rezniczek G. A., Konieczny P., Nikolic B., Reipert S., Schneller D., Abrahamsberg C., Davies K. E., Winder S. J., and Wiche G. (2007) Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with β-dystroglycan. J. Cell Biol. 176, 965–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ketema M., Secades P., Kreft M., Nahidiazar L., Janssen H., Jalink K., de Pereda J. M., and Sonnenberg A. (2015) The rod domain is not essential for the function of plectin in maintaining tissue integrity. Mol. Biol. Cell 26, 2402–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nikolic B., Mac Nulty E., Mir B., and Wiche G. (1996) Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J. Cell Biol. 134, 1455–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonnenberg A., Rojas A. M., and de Pereda J. M. (2007) The structure of a tandem pair of spectrin repeats of plectin reveals a modular organization of the plakin domain. J. Mol. Biol. 368, 1379–1391 [DOI] [PubMed] [Google Scholar]
- 14. Jefferson J. J., Ciatto C., Shapiro L., and Liem R. K. (2007) Structural analysis of the plakin domain of bullous pemphigoid antigen1 (BPAG1) suggests that plakins are members of the spectrin superfamily. J. Mol. Biol. 366, 244–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ortega E., Buey R. M., Sonnenberg A., and de Pereda J. M. (2011) The structure of the plakin domain of plectin reveals a non-canonical SH3 domain interacting with its fourth spectrin repeat. J. Biol. Chem. 286, 12429–12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi H. J., and Weis W. I. (2011) Crystal structure of a rigid four-spectrin-repeat fragment of the human desmoplakin plakin domain. J. Mol. Biol. 409, 800–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koster J., van Wilpe S., Kuikman I., Litjens S. H., and Sonnenberg A. (2004) Role of binding of plectin to the integrin β4 subunit in the assembly of hemidesmosomes. Mol. Biol. Cell 15, 1211–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koster J., Geerts D., Favre B., Borradori L., and Sonnenberg A. (2003) Analysis of the interactions between BP180, BP230, plectin, and the integrin α6β4 important for hemidesmosome assembly. J. Cell Sci. 116, 387–399 [DOI] [PubMed] [Google Scholar]
- 19. Hijikata T., Nakamura A., Isokawa K., Imamura M., Yuasa K., Ishikawa R., Kohama K., Takeda S., and Yorifuji H. (2008) Plectin 1 links intermediate filaments to costameric sarcolemma through β-synemin, α-dystrobrevin, and actin. J. Cell Sci. 121, 2062–2074 [DOI] [PubMed] [Google Scholar]
- 20. Lunter P. C., and Wiche G. (2002) Direct binding of plectin to Fer kinase and negative regulation of its catalytic activity. Biochem. Biophys. Res. Commun. 296, 904–910 [DOI] [PubMed] [Google Scholar]
- 21. Al-Jassar C., Bernad P., Chidgey M., and Overduin M. (2013) Hinged plakin domains provide specialized degrees of articulation in envoplakin, periplakin and desmoplakin. PloS One 8, e69767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grum V. L., Li D., MacDonald R. I., and Mondragón A. (1999) Structures of two repeats of spectrin suggest models of flexibility. Cell 98, 523–535 [DOI] [PubMed] [Google Scholar]
- 23. Kusunoki H., Minasov G., Macdonald R. I., and Mondragón A. (2004) Independent movement, dimerization and stability of tandem repeats of chicken brain alpha-spectrin. J. Mol. Biol. 344, 495–511 [DOI] [PubMed] [Google Scholar]
- 24. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ipsaro J. J., Huang L., and Mondragón A. (2009) Structures of the spectrin-ankyrin interaction binding domains. Blood 113, 5385–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stabach P. R., Simonović I., Ranieri M. A., Aboodi M. S., Steitz T. A., Simonović M., and Morrow J. S. (2009) The structure of the ankyrin-binding site of β-spectrin reveals how tandem spectrin-repeats generate unique ligand-binding properties. Blood 113, 5377–5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davis L., Abdi K., Machius M., Brautigam C., Tomchick D. R., Bennett V., and Michaely P. (2009) Localization and structure of the ankyrin-binding site on β2-spectrin. J. Biol. Chem. 284, 6982–6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ipsaro J. J., Harper S. L., Messick T. E., Marmorstein R., Mondragón A., and Speicher D. W. (2010) Crystal structure and functional interpretation of the erythrocyte spectrin tetramerization domain complex. Blood 115, 4843–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Samuelov L., and Sprecher E. (2015) Inherited desmosomal disorders. Cell Tissue Res. 360, 457–475 [DOI] [PubMed] [Google Scholar]
- 30. Al-Jassar C., Bikker H., Overduin M., and Chidgey M. (2013) Mechanistic basis of desmosome-targeted diseases. J. Mol. Biol. 425, 4006–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boyden L. M., Kam C. Y., Hernández-Martín A., Zhou J., Craiglow B. G., Sidbury R., Mathes E. F., Maguiness S. M., Crumrine D. A., Williams M. L., Hu R., Lifton R. P., Elias P. M., Green K. J., and Choate K. A. (2016) Dominant de novo DSP mutations cause erythrokeratodermia-cardiomyopathy syndrome. Hum. Mol. Genet. 25, 348–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Al-Jassar C., Knowles T., Jeeves M., Kami K., Behr E., Bikker H., Overduin M., and Chidgey M. (2011) The nonlinear structure of the desmoplakin plakin domain and the effects of cardiomyopathy-linked mutations. J. Mol. Biol. 411, 1049–1061 [DOI] [PubMed] [Google Scholar]
- 33. Ohno S., Nagaoka I., Fukuyama M., Kimura H., Itoh H., Makiyama T., Shimizu A., and Horie M. (2013) Age-dependent clinical and genetic characteristics in Japanese patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. J. 77, 1534–1542 [DOI] [PubMed] [Google Scholar]
- 34. Cox M. G., van der Zwaag P. A., van der Werf C., van der Smagt J. J., Noorman M., Bhuiyan Z. A., Wiesfeld A. C., Volders P. G., van Langen I. M., Atsma D. E., Dooijes D., van den Wijngaard A., Houweling A. C., Jongbloed J. D., Jordaens L., et al. (2011) Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 123, 2690–2700 [DOI] [PubMed] [Google Scholar]
- 35. Elliott P., O'Mahony C., Syrris P., Evans A., Rivera Sorensen C., Sheppard M. N., Carr-White G., Pantazis A., and McKenna W. J. (2010) Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 3, 314–322 [DOI] [PubMed] [Google Scholar]
- 36. Kapplinger J. D., Landstrom A. P., Salisbury B. A., Callis T. E., Pollevick G. D., Tester D. J., Cox M. G., Bhuiyan Z., Bikker H., Wiesfeld A. C., Hauer R. N., van Tintelen J. P., Jongbloed J. D., Calkins H., Judge D. P., et al. (2011) Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J. Am. Coll. Cardiol. 57, 2317–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quarta G., Muir A., Pantazis A., Syrris P., Gehmlich K., Garcia-Pavia P., Ward D., Sen-Chowdhry S., Elliott P. M., and McKenna W. J. (2011) Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation 123, 2701–2709 [DOI] [PubMed] [Google Scholar]
- 38. Osmani N., and Labouesse M. (2015) Remodeling of keratin-coupled cell adhesion complexes. Curr. Opin. Cell Biol. 32, 30–38 [DOI] [PubMed] [Google Scholar]
- 39. Adamovic I., Mijailovich S. M., and Karplus M. (2008) The elastic properties of the structurally characterized myosin II S2 subdomain: a molecular dynamics and normal mode analysis. Biophys. J. 94, 3779–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rief M., Pascual J., Saraste M., and Gaub H. E. (1999) Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol. 286, 553–561 [DOI] [PubMed] [Google Scholar]
- 41. Russell D., Andrews P. D., James J., and Lane E. B. (2004) Mechanical stress induces profound remodelling of keratin filaments and cell junctions in epidermolysis bullosa simplex keratinocytes. J. Cell Sci. 117, 5233–5243 [DOI] [PubMed] [Google Scholar]
- 42. Alonso-García N., Inglés-Prieto A., Sonnenberg A., and de Pereda J. M. (2009) Structure of the Calx-β domain of the integrin β4 subunit: insights into function and cation-independent stability. Acta Crystallogr. D Biol. Crystallogr. 65, 858–871 [DOI] [PubMed] [Google Scholar]
- 43. Manso J. A., García Rubio I., Gómez-Hernández M., Ortega E., Buey R. M., Carballido A. M., Carabias A., Alonso-García N., and de Pereda J. M. (2016) Purification and structural analysis of plectin and BPAG1e. Methods Enzymol. 569, 177–196 [DOI] [PubMed] [Google Scholar]
- 44. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pape T., and Schneider T. R. (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 47. Vonrhein C., Blanc E., Roversi P., and Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 48. Abrahams J. P., and Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 49. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 50. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Juanhuix J., Gil-Ortiz F., Cuní G., Colldelram C., Nicolás J., Lidón J., Boter E., Ruget C., Ferrer S., and Benach J. (2014) Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 21, 679–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kabsch W. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A 32, 922–923 [Google Scholar]
- 56. Theobald D. L., and Steindel P. A. (2012) Optimal simultaneous superpositioning of multiple structures with missing data. Bioinformatics 28, 1972–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Babcock M. S., Pednault E. P., and Olson W. K. (1994) Nucleic acid structure analysis. Mathematics for local Cartesian and helical structure parameters that are truly comparable between structures. J. Mol. Biol. 237, 125–156 [DOI] [PubMed] [Google Scholar]
- 58. Skjærven L., Yao X. Q., Scarabelli G., and Grant B. J. (2014) Integrating protein structural dynamics and evolutionary analysis with Bio3D. BMC Bioinformatics 15, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 61. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., and Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 64. Tria G., Mertens H. D., Kachala M., and Svergun D. I. (2015) Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Suhre K., and Sanejouand Y. H. (2004) ElNemo: a normal mode Web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL: a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 67. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eddy S. R. (1998) Profile hidden Markov models. Bioinformatics 14, 755–763 [DOI] [PubMed] [Google Scholar]
- 69. Drozdetskiy A., Cole C., Procter J., and Barton G. J. (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 43, W389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Diederichs K., and Karplus P. A. (1997) Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269–275 [DOI] [PubMed] [Google Scholar]
- 73. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.