

Abstract
The ubiquitin/proteasome pathway is a well characterized system for degrading intracellular proteins, although many aspects remain poorly understood. There is, for instance, a conspicuous lack of understanding of the site(s) where nuclear proteins are degraded because the subcellular distribution of peptidase activity has not been investigated systematically. Although nuclear proteins could be degraded by importing proteasomes into the nucleus, it is also evident that some nuclear proteins are degraded only after export to cytosolic proteasomes. Proteasomes and substrates are mobile, and consequently, the sites of degradation might not be static. We sought to identify the location of proteasomes to provide more conclusive evidence on the sites of protein degradation. We report that catalytically active proteasomes exist almost exclusively in the cytosol. The resulting lack of nuclear peptidase activity suggests that little, if any, degradation occurs in the nucleus. These and other studies suggest that the export of proteolytic substrates could define an important regulatory step in the degradation of nuclear proteins by cytosolic proteasomes.
Keywords: E3 ubiquitin ligase, nuclear envelope, nuclear pore, proteasome, protein degradation, protein export, proteolysis
Introduction
The ubiquitin/proteasome pathway is a major mechanism for eliminating regulatory and damaged proteins. The key enzymology is well understood, and many targeting factors that attach ubiquitin to proteolytic substrates have been identified and characterized (1). Despite these advances, there remain areas of ambiguity. A detailed understanding of the assembly of multiubiquitin chains, the transport of proteolytic substrates, and sites of intracellular protein turnover remain unclear.
Many proteins that are involved in cell cycle progression, DNA repair, and transcription are nuclear proteins that play a central role in cell growth and stress response. The stability of these proteins is controlled by the proteasome, and it is generally assumed that they are degraded inside the nucleus. This view is fostered by the detection of proteasome subunits in the nucleus (2, 3) and enrichment in the nuclear envelope (4). However, many of these studies examined the distribution of GFP-tagged proteins, which does not ensure that the tagged subunits are present in intact proteasomes. This is an important consideration because certain proteasome subunits and subcomplexes perform non-proteolytic roles in the nucleus (5, 6). We also note that certain GFP-tagged proteasome subunits are not efficiently assembled into intact complexes, and in some instances the fluorescence is reduced after assembly into the proteasome (7). Consequently, the signal observed could arise predominantly from the free form of proteasome subunits. Critically, there is no convincing evidence that peptidase activity is present in the nucleus.
We and others reported that the yeast DNA repair protein Rad4, DNA polymerase subunit Cdc17 (2), and HO endonuclease (8) are stabilized in nuclear export mutants, although proteasome assembly and catalytic activity are unaffected. The stabilizing effect of blocking export is not restricted to a few nuclear proteins because overall polyubiquitylated protein levels increased (2). Similarly, the degradation of mammalian nuclear proteins, including p53, β-catenin, TRIP-Br2, and hMSH5, requires nuclear export (9–12). Because import is unaffected in export mutants, proteasomes could have entered to degrade nuclear proteins. However, this was not observed, suggesting that nuclear proteins are exported and degraded by cytosolic proteasomes.
To test this hypothesis, we investigated the site of peptidase activity in both yeast and cultured human cells. We did not detect intact proteasomes in the nucleus, and virtually all peptidase activity was present in the cytosol. These findings diverge from the general opinion, which maintains that proteasomes degrade proteins inside the nucleus (4, 13–25). Our results predict an important regulatory role for export in the degradation of nuclear proteins.
Results
Purified Nuclei Lack Peptidase Activity
Actively growing yeast cells were lysed with zymolyase, and nuclei were separated by differential centrifugation. Prior to centrifugation, “total” cellular protein was prepared by lysing an aliquot of the unfractionated spheroplasts. We measured histone deacetylase (HDAC)3 activity to gauge the purity of the isolated nuclei (Fig. 1A). HDAC activity was detected only in purified nuclei (Nuc), whereas the hydrolysis of a fluorogenic proteasome substrate (LLVY-AMC) was found predominantly in the cytoplasm (Cyto). A low level of peptidase activity in the nuclear fraction is likely to originate from unlysed cells and endoplasmic reticulum that can co-sediment with nuclei. Fractionated yeast extracts were examined by immunoblotting to confirm that peptidase activity coincided with the location of proteasome subunits (Fig. 1B). High levels of proteasome subunits (Rpn11, Rpn10, and Rpt1) were detected in the cytosolic fraction, and significantly lower levels were present in purified nuclei. As expected, both histone (FLAG-H2B) and centrin (Cdc31 (26)) were present only in the nucleus. In contrast, Rad23, a shuttle factor that can translocate ubiquitylated substrates to the proteasome (27), was strongly enriched in the nucleus, and low levels were detected in the cytosol. We reported previously that the Ubc4 ubiquitin-conjugating enzyme can bind the proteasome (28) and is required for degrading damaged translational products (29). Ubc4 was detected in the cytosol, consistent with the cytosolic location of catalytically active proteasomes. To reinforce these findings, we examined the location of key GFP-tagged proteins in yeast cells (Fig. 1C). The nuclear localization of centrin/Cdc31, histone H2B, and Rad23 and the cytosolic localization of GFP-Ubc4 are consistent with the immunoblotting results (Fig. 1B). The nuclear localization of proteasome subunits (Pre6-GFP ; Rpn1-GFP) is consistent with previous studies but differs from the cytosolic localization observed in Fig. 1B. One interpretation of this finding is that proteasomes are present on the nuclear surface and are dissociated during fractionation.
FIGURE 1.
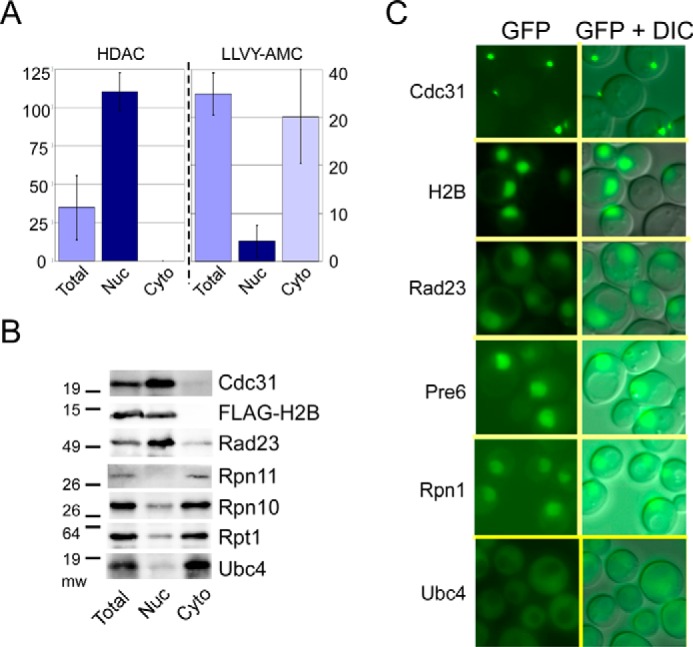
Intact and catalytically active proteasomes are located in the cytosol. A, yeast cells extracts were fractionated, and HDAC and proteasome peptidase activities were measured. HDAC activity was detected solely in the nuclear fraction, whereas LLVY-AMC hydrolysis was predominantly cytosolic. B, fractionated protein extracts were characterized by immunoblotting to determine the location of proteasome subunits. Antibody reactions against nuclear proteins FLAG-histone H2B and centrin/Cdc31 are shown. The filters were also treated with antibodies against proteasome subunits (Rpn11, Rpn10, and Rpt1), substrate shuttle factor (Rad23), and ubiquitin-conjugating enzyme Ubc4. C, GFP-tagged proteins were expressed in wild type yeast cells. Because a major fraction of Cdc31 is bound to the spindle pole-body, small foci of fluorescence are seen. GFP-Ubc4 shows cytosolic localization, consistent with the localization of native Ubc4 (B). DIC, differential interference contrast.
The time-consuming procedure required to isolate nuclei from yeast cells raised concern that proteasomes might have leaked out of the nucleus during purification. In addition, the small size of yeast cells presented difficulty in obtaining detailed microscopy images. To overcome these concerns, we characterized cultured human cells.
We previously showed that treatment of yeast cells with high frequency sonication caused the release of proteasomes into the medium (30). Because nuclei remained intact, these findings suggested that proteasomes were located in the cytosol. In a similar strategy, we exposed HEK293T cells to digitonin to permeabilize the plasma membrane (31). Although the nuclear envelope is not disrupted under these mild conditions (25 μg/ml digitonin) (31), proteasome subunits and activity were rapidly released into the culture medium (Fig. 2A). A higher concentration of digitonin (50 μm), and longer incubation (20 and 30 min), did not increase this effect. Immunoblotting confirmed that proteasome subunits were present in the medium following digitonin treatment (Fig. 2B). The cytosolic localized tubulin was detected in the medium (Fig. 2B, lanes 5 and 7, M), whereas histone H3 was present in the cell pellet (lanes 4 and 6, P), which contained intact nuclei and lysed cells. In untreated cells, both histone H3 and proteasome subunits were present in the cell pellet (Fig. 2B, lanes 1 and 2), and peptidase activity was not detected in the culture supernatant (Fig. 2A). To verify these findings, we purified nuclei and cytosol from HEK293T and HCT116 cultured human cells (Fig. 2, C and D). Total protein extract was isolated by lysing unfractionated cells (Fig. 2, C and D, T). We used two methods to rapidly isolate nuclei, and both yielded similar results. Protein extracts were characterized by either using equal amount of protein (Fig. 2, C and D, Conc) or a proportional volume representative of the cytosol and nucleus (Fig. 2, C and D, Vol). Isolated nuclei (Fig. 2, C and D, N) showed very low levels of peptidase activity. In contrast, peptidase activity was detected predominantly in the cytosolic fraction (Fig. 2, C and D, C). To confirm that the peptidase activity detected in the cytosol corresponded to the location of the proteasome, we performed immunoblotting studies. Proteasome subunits were strongly enriched in the cytosolic fraction in the samples described above (Fig. 2D).
FIGURE 2.

Proteasome peptidase activity can be released into the medium. A, HEK293T cells were treated with varying concentrations of digitonin and pelleted. Following digitonin treatment, the peptidase activity that was released into the medium (M) was compared with that detected in the cell pellet (P). Prolonged exposure to digitonin (30 min) did not further increase peptidase activity in the medium. Total peptidase activity (T) was determined by lysing untreated cells and measuring LLVY-AMC hydrolysis. B, HEK293T cells were treated for 10 min with varying concentrations of digitonin (0, 25, and 50 μm), and immunoblotting was used to determine the proteasome subunit levels in the medium (M) and cell pellet (P). The detection of tubulin and histone H3 verified the location of a cytosolic and nuclear protein, respectively. Basal levels of Rpn2, Rpt6, and α-proteasome subunits were established using lysates prepared from untreated cells (lane 1). Histone H3 was recovered in the pellet in both treated and untreated cells (lanes 2, 4, and 6). Treatment with digitonin resulted in significant depletion of proteasome subunits from the pellet fraction (lanes 4 and 6) and their recovery in the medium (M, lanes 5 and 7). C, nuclear and cytosolic fractions were prepared from HEK293T and HCT116 cells. LLVY-AMC hydrolysis was measured in the fractionated lysates using either an equal amount of protein (Conc) or proportional volumes (Vol). Peptidase activity in both cell lines was predominantly cytosolic. The sum of cytosolic (C) activity was similar to total (T) activity, indicating that the proteasome activity is predominantly in the cytosol. D, protein extracts described in C were examined by immunoblotting. Rpn7 and multiple α-subunits (Alpha mix), as well as tubulin, were detected primarily in the cytosol (C) in both cell lines, whereas histone H3 was found in the nucleus (N). Analysis of equal protein (Conc) significantly over-represents the nuclear fraction, as evidenced by the high levels of histone H3 in the nuclear (N) fraction when an equal amount of protein (Conc) was examined. In contrast, proteasome subunits were not detected in the nuclear fraction when we examined proportional volumes of the fractionated lysates. Similar findings were observed in two independent cell lines (HEK293T and HCT116).
To demonstrate that peptidase activity was generated by intact proteasomes, we prepared total, cytosol, and nuclear fractions from HEK293T cells, as described above. We again measured peptidase activity using equal amount of protein (Fig. 3, conc.), and proportional volumes of lysate (vol.). Both methods confirmed that the nuclear fraction lacked peptidase activity (Fig. 3A). Significantly, the peptidase activity in the cytosol was comparable with that in total extract (when we used proportional volumes), indicating that almost all peptidase activity is cytosolic. Data representing four independent experiments were standardized to the peptidase activity in total extract. To determine whether the peptidase activity was generated by intact proteasomes, and not free catalytic (20S) particles, we separated lysates in native polyacrylamide gels (Fig. 3B). In-gel hydrolysis of LLVY-AMC revealed two bands corresponding to single capped (19S + 20S), and double-capped (19S + 20S + 19S) proteasomes in both total and cytosolic extracts. A significantly lower level of free 20S particle was also detected. Strikingly, none of these complexes was present in the nuclear fraction (Fig. 3B, lane 3), even when 5-fold excess nuclear extract was examined (lane 4).
FIGURE 3.

Proteasome localization in cultured human cells. A, mammalian HEK293T cells were fractionated, and proteasome peptidase activity was determined in the nuclear and cytosolic fractions. Error bars represent S.E. from four independent experiments. Peptidase activity was detected solely in the cytoplasmic lysate, using either equal amount of protein (conc.) or proportional volumes of lysates (vol.) following fractionation. B, total, cytoplasmic, and nuclear fractions were prepared from HEK293T cells and separated in a native polyacrylamide gel. LLVY-AMC hydrolysis was examined in situ. A 5-fold higher loading of the nuclear extract (5× Nuc) was also examined. The positions of the single-capped and double-capped proteasomes are shown. C, protein extracts from HEK293T cells were separated by SDS-PAGE and transferred to nitrocellulose, and the filters stained with Ponceau-S. Adjusting the lysates to equal protein concentrations resulted in significant over-representation of nuclear proteins (lanes 1–3), as noted above (Fig. 2). Antibody reactions against tubulin (cytosolic) and histone H3 (nuclear) showed that the fractionation yielded nuclei that were free of cytoplasmic proteins. An asterisk in lane 3 identifies an over-represented nuclear protein that cross-reacted with anti-Rpn12 antibody (see D, below). D, immunoblot in C was reacted with antibodies indicated on the right. Proteasome subunits (19S and 20S subunits) and tubulin were detected only in the cytosol, and nuclear proteins (XPC, p53, and H3) were detected only in the nuclear fraction.
Purified nuclear and cytosolic fractions were separated in SDS-polyacrylamide gels and probed with antibodies to confirm the location of proteasome subunits. An immunoblot was stained with Ponceau S (Fig. 3C) and then probed with antibodies against tubulin and histone H3 to gauge the purity of the cytosolic and nuclear preparations, respectively (bottom panel). Ponceau S staining showed much lower amounts of protein in the nuclear fraction when proportional volumes were examined (Fig. 3C, lane 5). In contrast, loading an equal amount of protein (Fig. 3C, conc.) significantly over-sampled nuclear proteins (lane 2), and an asterisk points to a particularly over-represented nuclear protein (Fig. 3C, compare lanes 1 and 2). Despite this over-sampling of the nuclear fraction, we detected virtually no peptidase activity in purified nuclei (Fig. 3A).
The filters were probed with antibodies against 19S (Rpt1, Rpn2, Rpn10, and Rpn12) and 20S subunits (α1, α2, α3, α5, α6, α7, β1, and β2) (Fig. 3D). High levels of proteasome subunits were detected in the cytosol (Fig. 3D, Cyto, lanes 3 and 6), consistent with the results shown in Fig. 1B. These subunits were not present in purified nuclei (Fig. 3D, Nuc, lanes 2 and 5). The levels of proteasome subunits in the cytosol and total fractions were equivalent (Fig. 3D, compare lanes 1 and 3, 4 and 6), demonstrating that proteasomes located in the cytosol represent the total cellular quantity. Similar findings were obtained whether we examined equal protein concentration (Fig. 3D, conc) or equal volume (vol).
The nuclear substrates XPC and p53 were strongly enriched in the nucleus. The retention of these proteins and histone H3 in isolated nuclei and the exclusion of cytosolic tubulin indicated that the nuclei were undamaged. Based on its known association with the nuclear envelope, it is likely that proteasomes are embedded within the nuclear pore complex (NPC) but remain on the cytoplasmic side (4).
Nuclear Proteins Are Retained in Purified Nuclei
We considered several approaches to address the concern that proteasomes could have been released into the cytosol if the nuclei were damaged during purification. We expressed bacterial derivatives of β-galactosidase in HEK293T cells; NLS-βGal-GFP contained the SV40 nuclear localization signal, whereas βGal-GFP lacked this targeting motif. NLS-βGal-GFP showed strong nuclear localization (Fig. 4A), as reported previously (31), and was retained in purified nuclei (lower panels). We note that the tetrameric form of GFP-βgal is ∼575 kDa, similar to the size of the 20S catalytic particle. βGal-GFP lacking the NLS was detected only in the cytosol and was not present in purified nuclei (Fig. 4B). Proteasome peptidase activity was measured in extracts prepared from cytosol and purified nuclei from cells expressing either NLS-βGal-GFP or βGal-GFP (Fig. 4C). Consistent with earlier results, LLVY-AMC hydrolysis was only seen in the cytosolic fraction, regardless of the location of the βGal derivatives.
FIGURE 4.

Purified nuclei are structurally intact. A, HEK293T cells were transfected with a vector expressing NLS-βGal-GFP, and fluorescence was detected predominantly in the nucleus (upper panels). Following fractionation, GFP fluorescence was retained in the purified nuclei (lower panels). B, βGal-GFP (lacking a nuclear localization signal) was similarly expressed in HEK293T cells, and cytosolic expression was observed (upper panels). Purified nuclei showed no evidence for GFP fluorescence (lower panels). C, we measured proteasome peptidase activity in cells described above. LLVY-AMC hydrolysis in the cytosol (C) and nucleus (N) was compared with activity present in total lysate (T). Error bars indicated S.E. D, nuclei were isolated from HEK293T cells and incubated with FITC-dextran in the sizes indicated. FITC 10-kDa dextran readily entered the nucleus, whereas FITC 2,000-kDa dextran was entirely excluded. Hoechst staining of nuclei and a merged differential interference contrast image are also shown.
To confirm that purified nuclei retained structural integrity after fractionation, we examined their permeability to FITC-dextran (Fig. 4D). FITC-conjugated to 10-kDa dextran entered the nucleus immediately (Fig. 4D, upper row). Much lower levels of 70-kDa dextran entered the nucleus (Fig. 4D, middle row), consistent with the ∼50-kDa diffusion limit for transit of a globular protein through the nuclear pore. In contrast 2,000-kDa FITC-dextran (Fig. 4D, lower row), which is almost as large as the 26S proteasome, was entirely excluded from the nucleus after 5 min of incubation. We also incubated purified nuclei with FITC-dextran for a significantly longer duration (3 h) in the presence and absence of 1% Triton X-100 (data not shown). After 3 h of incubation in PBS, 2,000-kDa FITC-dextran was not detected in the nucleus. However, treatment with Triton X-100 caused nuclear entry at 3 h. Collectively, these studies showed that NLS-βGal-GFP is retained in purified nuclei and 2,000-kDa FITC-dextran does not enter purified nuclei. We believe these compelling results support our hypothesis that nuclei are undamaged following purification. Consequently, the absence of catalytically active proteasomes in purified nuclei after fractionation suggests that proteasomes function in the cytosol.
The detection of GFP-tagged proteasome subunits in the nucleus has fostered the view that protein degradation occurs in the nucleus. We expressed proteasome subunit β7-GFP (7) in HEK293T cells and detected fluorescence in both the nucleus and cytosol (Fig. 5A). Fluorescence was also detected in purified nuclei (Fig. 5A, bottom row). However, purified nuclei did not contain peptidase activity (Fig. 5B) consistent with other studies described here. These results indicate that the nuclear localization of a GFP-tagged proteasome subunit does not conclusively establish the site of catalytically active proteasomes. Lysates were separated by SDS-PAGE (Fig. 5C), and immunoblotting showed that β7-GFP was present in purified nuclei (Fig. 5C, lanes 2 and 5), in agreement with the imaging results (Fig. 5A). However, native β7 was only detected in total lysate and in the cytosol (lanes 3 and 6) and not in the nuclear fraction (Fig. 5C). Thus, the location of native β7 differed from its GFP-tagged derivative. (As noted previously, the high level of β7-GFP and histone H3 in purified nuclei (Fig. 5C, lane 2) is due to oversampling of nuclear proteins when equal protein levels are examined.)
FIGURE 5.
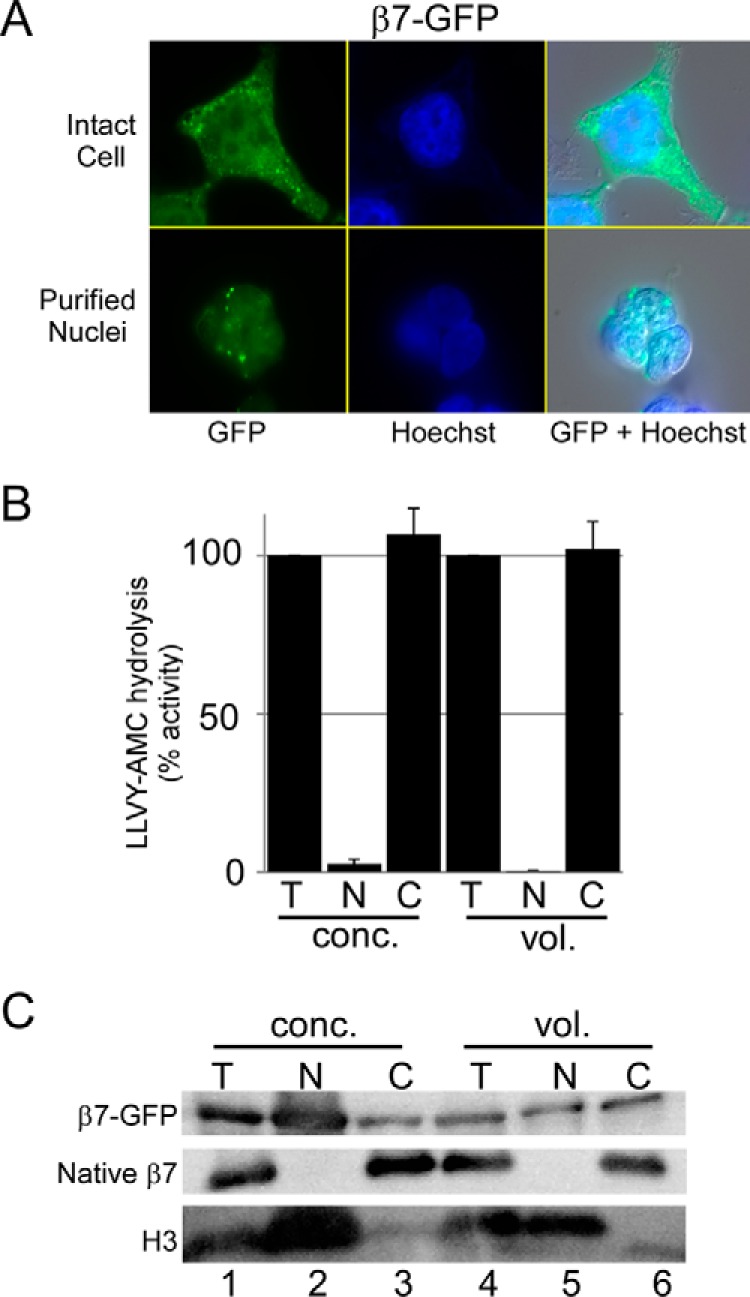
Proteasome subunit, but not peptidase activity, is detected in purified nuclei. A, 20S proteasome subunit β7-GFP was expressed in HEK293T cells and detected uniformly throughout the cells (upper panels). Purified nuclei contained β7-GFP (lower panels). B, peptidase activity was measured in total (T), nuclear (N), and cytosolic (C) fractions using either equal protein concentration (conc.) or equal lysate volume (vol.), and results from three independent studies are shown (error bars indicate S.E.). C, protein lysates were resolved by SDS-PAGE and immunoblotted. The filters were reacted to antibodies against GFP and native β7. Whereas β7-GFP is detected in purified nuclei, native β7 was only present in the cytosol. A reaction against histone H3 is also shown.
Localization of Functional Immunoproteasome Resembles 26S Proteasome
A previous study (32) reported that chimeric proteasomes, comprising subunits from the 26S and immunoproteasome, can be detected in cultured cells. We therefore investigated immunoproteasome localization in HEK293T cells because the substrate selectivity of chimeric proteasomes is unclear. LMP2-GFP, encoding the β1i subunit of the immunoproteasome, was uniformly distributed in HEK293T cells (Fig. 6A) and was also recovered in purified nuclei (lower panels). Immunoblotting showed that both LMP2-GFP and native LMP2 were present in the nuclear fraction (Fig. 6B). In contrast, subunits of the 20S core particle, and LLVY-AMC hydrolysis, were detected primarily in the cytosolic fraction ((Fig. 6B, lanes 3 and 6). We measured immunoproteasome peptidase activity and detected Ac-PAL-AMC hydrolysis only in the cytosolic fraction (Fig. 6C), similar to the hydrolysis of LLVY-AMC (Fig. 6D). Thus, peptidase activity corresponding to both 26S proteasome and the immunoproteasome was only detected in the cytosol, irrespective of the subcellular distribution of individual GFP-tagged subunits.
FIGURE 6.

Catalytic activity of the immunoproteasome is restricted to the cytosol. A, LMP2-GFP was expressed in HEK293T cells, and distribution was observed throughout the cells (upper panels). Purified nuclei also showed LMP2-GFP fluorescence (lower panels). B, fractionated extracts were examined by SDS-PAGE and immunoblotting. LMP2-GFP and native LMP2 were both detected in nuclear (N) and cytosolic (C) fractions, whereas subunits of the 26S proteasome showed strong cytosolic localization (α-1, 2, 3, 5, 6, 7). Histone H3 was detected only in the nuclear fraction, and tubulin was found in the cytosol. Loading equal concentration (conc.) of protein over-samples nuclear proteins, as noted by the higher level of histone H3 in the nucleus (N, lane 2) compared with total lysate (T, lane 1). In contrast, when proportional volumes (vol.) are examined, nuclear (N) and total (T) levels of H3 are similar. C, hydrolysis of Ac-PAL-AMC, which measures immunoproteasome peptidase activity, was only detected in the cytosolic fraction. D, in agreement with earlier results, LLVY-AMC hydrolysis occurred exclusively in the cytosolic fraction. The level of LMP2-GFP fluorescence was determined in the same fractionated extracts, and localization of LMP2-GFP in the nuclear fraction was verified. (Quantitation reflects S.E. from three independent experiments.)
Cytosolic Proteasomes Are Quantitatively Intact
Cytosolic extracts were prepared from HEK293T cells and resolved in Sephacryl 300HR, and proteasome peptidase activity, subunit levels, and composition were examined. Peptidase activity was present in fractions representing high molecular weight species (Fig. 7A, fractions 13 and 14), consistent with the size of intact 26S proteasomes. Lower activity was present in subsequent fractions corresponding to free 20S particles (Fig. 7A, fractions 16 and 17). All proteasome activity that was applied to the column was recovered in these fractions, and none was detected in the trailing column fractions. Because proteasome subunits are present predominantly in intact complexes in vivo (18), the weak 20S, specific activity observed in fractions 16 and 17, could reflect post-lysis dissociation of the 26S proteasome. The fractionated proteins were examined by immunoblotting, and 20S subunits (α4, β7) were found in all fractions that contained peptidase activity (Fig. 7B). Subunits in the 19S regulatory particle (Rpn2, Rpt1, and Rpn7) were also detected primarily in fractions 13 and 14. To confirm that these subunits were present in intact 26S proteasomes, we separated proteins in a native polyacrylamide gel. In-gel hydrolysis of LLVY-AMC confirmed the presence of 26S proteasomes in fractions 13 and 14. Weak peptidase activity was detected in fractions 16 and 17, which contained 20S particles (Fig. 7C). We also separated 10-fold excess nuclear extract in the same column and detected no peptidase activity (data not shown), indicating that all proteasome activity is present in the cytosol.
FIGURE 7.

Cytosolic proteasomes are intact. A, cytoplasmic protein extracts prepared from fractionated HEK293T cells were resolved in Sephacryl 300HR. Two peaks of proteasome activity were detected. Fractions 13 and 14 were coincident with the exclusion volume. The data represent one of three independent fractionation experiments. B, immunoblotting showed the presence of both 19S (Rpn2, Rpt1, and Rpn7) and 20S subunits (α4 and β7) in fractions 13 and 14, which displayed peak LLVY-AMC peptidase activity. Very low levels of the 20S particle were detected in subsequent fractions. C, Sephacryl 300HR fractions 12–18 were resolved in a native polyacrylamide gel. An “in-gel” assay, performed by overlaying the gel with fluorogenic substrate, revealed intact proteasomes in fractions 13 and 14, consistent with A and B. A low level of the free 20S particle was detected in fractions 16 and 17. Both total (T) and nuclear (Nuc) extracts showed peptidase activity in the high molecular weight fractions. In contrast, 10-fold excess nuclear extract showed no peptidase activity.
The degradation of a number of nuclear proteins in yeast and human cells requires export. The proposed cytosolic localization of catalytically active proteasomes imposes an unexpected requirement for transporting substrates out of the nucleus. We examined the effect of nuclear export on p53 stability using both pharmacological and genetic approaches. Native p53 was not detected in untreated cells but was readily detected after 18 h of treatment with leptomycin B (LMB) (data not shown). Based on these findings, we compared the stability and localization of an export-deficient p53 mutant (GFP-p53(nes−)). The level and stability of native p53 were similar in HCT116 cells expressing either GFP-p53 (Fig. 8A, lanes 1–5), or GFP-p53(nes−) (lanes 7–11). Native p53 was undetected in the absence of LMB (Fig. 8A, lanes 6 and 12; UNT) but was readily detected after exposure to LMB (lanes 1 and 7; + LMB, 18 h). Both an export defect and treatment with LMB had a strong stabilizing effect on GFP-p53(nes−) levels (Fig. 8A, lanes 7 and 11). HCT116 cells were suspended in fresh medium either containing (+) or lacking (−) LMB, and protein levels were followed. In the presence of LMB, noticeably higher levels of native p53 remained after 24 h (Fig. 8A, lane 5). In contrast, the level of native p53 declined in the absence of LMB (Fig. 8A, lane 4). GFP-p53(nes−) levels did not decrease after the removal of LMB (Fig. 8A, lanes 8 and 10), as this mutant is already export-deficient. Protein levels were quantified by densitometry and adjusted to the abundance of the α7 proteasome subunit. Numerical values in parentheses indicate the fold change, compared with time 0 (Fig. 8A, lanes 1 and 7).
FIGURE 8.

Export defect stabilizes p53 and causes accumulation in the nucleus. A, we expressed GFP-p53 and GFP-p53(nes−), a mutant that is poorly exported, in HCT116 cells. GFP-p53 and native p53 are poorly detected in untreated cells (lane 7). Treatment of cells with leptomycin B (+LMB 18 h) resulted in elevated levels of native p53 and the GFP-tagged derivatives. The cells were placed in fresh medium either containing (+) or lacking (−) LMB, and extracts were prepared after 14 and 24 h. Native p53 levels decreased after 24 h in the absence of LBM but remained elevated in the presence of LMB (compare lanes 4 and 5). Similarly, we detected a modest decrease in the levels of GFP-p53 at 24 h (−LMB). In contrast, the levels of GFP-p53(nes−) were strongly increased following treatment with LMB, and removal of LMB did not result in any appreciable decrease in its levels, even after 24 h (lane 10). B and C, HCT116 cells were pretreated with LMB for 18 h, and the localization of GFP-p53 and GFP-p53(nes−) was examined. Both GFP-p53 and GFP-p53(nes−) were strongly enriched in the nucleus (upper panels). Cells were incubated in fresh medium either containing (+) or lacking (−) LMB, and the protein levels were examined after 24 h. GFP-p53 was entirely eliminated, whereas high levels of nuclear GFP-p53(nes−) remained. Unt, untreated.
We investigated where stabilized GFP-p53(nes−) accumulated in the cell. Fluorescence microscopy showed that both GFP-p53 and GFP-p53(nes−) were localized predominantly in the nucleus (Fig. 8, B and C, 18 h + LMB). To examine the effect of inhibiting export, we compared the levels of GFP-p53 and GFP-p53(nes−), and after removal of LMB from the growth medium GFP-p53 was entirely lost within 24 h (Fig. 8, B, 24 h − LMB). In contrast, if LMB was present in the medium, the nuclear levels of GFP-p53 persisted (Fig. 8, B, 24 h + LMB). In contrast, the nuclear levels of GFP-p53(nes−) remained high even after LMB was removed (Fig. 8, C, 24 h – LMB), consistent with the export deficiency of this mutant protein.
Discussion
The presence of proteasome subunits and subcomplexes in the nucleus (2–4, 16, 17, 22, 24, 25) is consistent with the view that proteins can be degraded in the nucleus (14, 18–21, 23). However, the site of proteasome-specific peptidase activity has not been examined comprehensively. Although GFP-tagged proteasome subunits are detected in the nucleus (2, 3) and are enriched in the nuclear envelope (4, 33), we detected peptidase activity only in the cytosol. One interpretation of this result is that proteasomes are present on the nuclear surface and are dissociated during fractionation. This arrangement supports our hypothesis that the degradation of nuclear substrates requires an export mechanism (2).
The degradation of misfolded nuclear and cytosolic proteins has been investigated using CPY* and other engineered proteins that display a strong tendency to misfold (19–21). The degradation of some of these proteins, such as ΔssPrA, Δ2GFP (20), and CPY≠-GFP (34), required import into the nucleus. Although proteins can be conjugated to ubiquitin in the nucleus, there is insufficient evidence to conclude that they are degraded there, especially because export is required for the turnover of many nuclear proteins (2, 9–12, 35). Therefore, although misfolded cytosolic proteins may enter the nucleus, it less clear that they are degraded there. Intriguingly, the degradation of ΔssPrA, Δ2GFP, and CPY≠-GFP required the activity of nuclear ubiquitin E3 ligases (San1) and cytosolic E3 ligase (Ubr1) (14, 20, 34). Δ2GFP was strongly stabilized in san1 and accumulated in the nucleus. In contrast, Δ2GFP was only partially stabilized in ubr1Δ and was detected in both the nucleus and cytosol. Although San1 and Ubr1 may function independently, they could represent a two-step targeting paradigm in which San1 initiates ubiquitylation of a substrate in the nucleus that is followed by its cytosolic ubiquitylation by Ubr1. In agreement with this model, the attachment of an export signal rendered CPY≠-GFP vulnerable to ubiquitylation by cytosolic Ubr1 (34). Increased export could reduce nuclear retention and favor CPY≠-GFP ubiquitylation by cytosolic Ubr1. San1 might represent a rate-limiting or commitment step for the elimination of misfolded nuclear proteins. The distinct but coupled ubiquitylation of nuclear substrates by San1 and Ubr1 offers a coordinated mechanism for degrading misfolded nuclear proteins and mislocalized cytosolic proteins (34). It remains to be determined whether these E3 ligases function in concert to degrade physiological nuclear substrates.
The NPC plays a key role in DNA-related activities, including the degradation of transcription factors (36), DNA repair proteins (37), and in transcription silencing (38). Components in the NPC interact directly with chromatin (39) and gene promoters (16). Chromatin immunoprecipitation studies showed that proteasome subunits can be co-purified with the GAL10 gene and promoter (16), although the presence of proteasome peptidase activity was not verified. Whereas fluorescence imaging of nuclear pore subunits, such as Nup49-mCherry, reveals a ring circumscribing the nucleus, GFP-tagged proteasomes display a more dispersed signal across the nucleus, with intensification at the periphery. We note that certain individual proteasome subunits can perform non-proteolytic roles inside the nucleus, apart from intact proteasomes. In addition, proteasomes may partly enter into the volume of the nucleus without entering the nucleoplasm. These two observations could explain the diffuse nuclear localization of proteasome subunits using GFP-tagged subunits.
Niepel et al. (41) reported that a proteinaceous basket that is associated with the nuclear pore can extend ∼80 nm into the interior of the nucleus. Myosin-like proteins Mlp1 and Mlp2 are located at the base of this basket and provide a gateway between the nucleus and the cytosol (40). Mlp1 interacts with Esc1, a peripheral nuclear protein that binds proteasomes (41). This is significant because Mlp1 also binds promoters of many inducible genes, including GAL10 (40). The juxtaposition of active transcription units and proteasomes through their interactions with Mlp1 and Esc1 provides a path for degrading transcription factors (2, 36) and DNA repair proteins (2). In addition, both Mlp1 and Mlp2 bind Cdc31 (centrin) (41), which we reported can bind the proteasome and multiubiquitylated proteins (26). Cdc31 (centrin) is also linked to the nuclear export machinery (42). Collectively, these findings suggest that multiple NPC components could promote the degradation of nuclear proteins (see model, Fig. 9). Proteasomes are mobile and can be recruited from cytosolic storage granules to the nucleus within minutes (18, 43). Because proteasomes are assembled in the cytosol (18), their rapid mobilization to the nucleus might indicate localization within the NPC and not entry into the nucleoplasm. Based on this hypothesis, nuclear export could guide proteolytic substrates to proteasomes in the NPC-associated basket.
FIGURE 9.
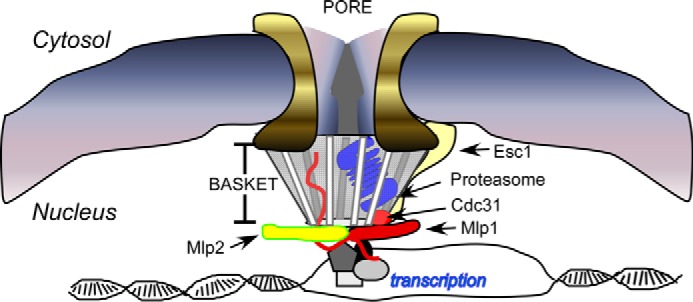
Model, a basket case. A number of independent observations have linked components in the nuclear pore complex to the proteasome. Specific components in the nuclear basket (including Mlp1 and Mlp2) can bind Esc1 and Cdc31/centrin (41). Esc1 can co-precipitate a large number of proteasome subunits, and we identified an interaction between Cdc31 (centrin) and the proteasome (26). The nuclear targeting of proteasomes is deficient in srp1-49 (48), an importin-α mutant that is import-proficient, suggesting that proteasomes are not imported. Because the degradation of many nuclear proteins requires export, these findings suggest that proteasomes reside near the nuclear surface, perhaps in the nuclear basket. This model reconciles many divergent findings on the mode of nuclear protein turnover, and specifically provides a mechanism whereby proteasomes may come in direct contact with chromatin.
Although we propose that proteasomes do not operate in the nucleus, we consider below alternative interpretations. We recognized that if nuclei were damaged during fractionation proteasomes could have leaked into the cytosol. To address this concern, we isolated nuclei using both isotonic and hypertonic lysis methods, and both approaches showed proteasome activity exclusively in the cytosol. We also used digitonin to selectively permeabilize the plasma membrane (31), and we determined that peptidase activity was released into the culture medium (Fig. 2, A and B). In contrast, no activity was detected in the pellet fraction of permeabilized cells that contained intact nuclei. We confirmed these findings in a different cell line (HTC116: Fig. 2, C and D). The integrity of purified nuclei was also confirmed by the retention of nuclear proteins (p53, XPC, and histone). Native p53 and GFP-p53 were stabilized when nuclear export was blocked in cultured human cells. The stabilization of GFP-p53 in the nucleus (Fig. 8D) is consistent with an export requirement for its degradation.
We examined the localization of nuclearly targeted bacterial β-galactosidase (31) to address the concern that proteins that were retained in the nucleus may have been pelleted in association with chromatin. βGal-GFP was attached to NLS, and NLS-βGal-GFP was quantitatively localized in the nucleus (Fig. 4A), whereas βGal-GFP was cytosolic (Fig. 4B). The retention of NLS-βGal-GFP, which is ∼575 kDa, in purified nuclei lends strong support to our view that if proteasomes (2,600 kDa) were present in the nucleus they would not have leaked out during fractionation. In addition, if proteasomes did function in the nucleus, peptidase activity should have been detected in purified nuclei. However, the absence of nuclear peptidase activity disputes this view. We propose that nuclear substrates are exported and degraded in the cytosol.
In a complementary approach, we incubated purified nuclei with FITC-dextran and found that 10-kDa dextran readily entered the nucleus, whereas the 2,000-kDa FITC-dextran was entirely excluded (Fig. 4D). However, high molecular mass dextran entered the nucleus after we permeabilized the nuclear envelope with detergent (data not shown). Using the yeast experimental model, we found HDAC activity only in purified nuclei, whereas peptidase activity was present in the cytosol (Fig. 1A), consistent with our hypothesis.
The lack of nuclear peptidase activity could be the result of selective proteasome inactivation in the nucleus. Regulatory factors are known to modulate proteasome function, and therefore, it was possible that nuclear proteasomes are only conditionally activated. To investigate this possibility, we separated 10-fold excess nuclear extract by gel exclusion chromatography to detect intact proteasomes. We found that none of the fractions corresponding to high molecular weight complexes contained proteasome subunits. We note that cytosolic proteasome activity is essentially identical to total activity. We conclude that cytosolic proteasomes contribute the major fraction of peptidase activity.
An important distinction between our findings and recent reports (19–21) is that we examined proteasome localization and not substrate turnover. Although native and misfolded proteins could be degraded by mechanistically distinct paths, it is germane that catalytically active proteasomes were not detected in the nucleus, and thus export must contribute significantly to the turnover of many types of nuclear proteins. Because multiple pathways facilitate nuclear export, the degradation of specific classes of nuclear substrates might involve distinct mechanisms. Consequently, inhibiting a single export pathway might not unequivocally demonstrate whether a nuclear substrate is degraded in the cytosol. For instance, we determined that the yeast XpoI export factor is required for the degradation of Rad4 but not Matα2 (data not shown). The degradation of another nuclear protein, Ho endonuclease, specifically requires the Msn5 export factor (8).
GFP tags are valuable for identifying the subcellular localization of proteins. However, it is less clear whether the location of a GFP-tagged proteasome subunit provides unambiguous evidence for the site of peptidase activity, because imaging studies cannot distinguish between intact proteasomes and free subunits. This concern is relevant because proteasome subunits, such as Rpn4, can perform non-proteolytic roles in transcription (6); other components (Rpn10) are expressed at super-stoichiometric levels (44, 45) and are not present exclusively in the proteasome (46). Consequently, proteasome subunits that are detected in the nucleus may be engaged in non-proteolytic functions. It has also been reported that certain GFP-tagged subunits (α7, α5, and β7) are unable to assemble efficiently into proteasomes (47), and in some instances, the fluorescence generated is reduced following assembly (7). These observations raise concern that GFP fluorescence arises disproportionately from free subunits. Pack et al. (18) expressed fusions of regulatory particle (RP) and catalytic particle (CP) proteasome subunits and found that the chimeras could replace the wild type proteins. The co-localization of these chimeras with the nucleus suggested that intact proteasomes operated in the nucleus. However, different proteasome subunits were tagged to GFP to define proteasome localization, and it is unclear whether they were entirely localized with intact proteasomes. These and other studies did not establish whether proteasomes entered the nucleus or remained at the nuclear surface. We report here that peptidase activity is detected in the cytosol, suggesting that proteasomes may be bound to the nuclear surface.
The presence of NLS motifs in certain proteasome subunits suggested that they facilitate entry into the nucleus (25). However, we reported that the targeting of proteasomes to the nucleus is entirely blocked in sts1-2, although Sts1 plays no role in nuclear import. The targeting of proteasomes to the nucleus also requires Srp1, a yeast importin-α protein. However, in contrast to other reports we determined that srp1-49 can successfully import NLS-bearing proteins but is specifically defective in targeting proteasomes to the nucleus (48). We speculate that Srp1 guides proteasomes to the nuclear pore complex but does not facilitate entry into the nucleus.
Our studies agree with the results of Kulichkova et al. (7), who detected human GFP-β7 in both the nucleus and cytosol. They also reported that intact proteasomes were present exclusively in the cytosol in fractionated HeLa cells (7). In agreement, we detected GFP-β7 in the nucleus and cytosol, but found peptidase activity only in the cytosol, in both HEK293T and HCT116 cells (Fig. 2, C and D). These findings suggest that β7 in the nucleus is not present in intact proteasomes. It is also significant that multiple proteasome subunits were detected only in the cytosol in primary mouse cardiac myocytes (49), demonstrating that our findings in yeast and cultured cells are conserved in other animal models.
Proteasome trafficking is dynamically regulated in response to growth conditions (43). Similarly, the degradation of certain substrates requires movement into and out of the nucleus. As many nuclear functions occur in the vicinity of the nuclear pore, a model envisioning proteolysis in the NPC would not be unorthodox. Stationing proteasomes in the nuclear pore complex would allow degradation of nuclear proteins without requiring entry into the nucleus. To reconcile the divergent views on nuclear protein turnover, we propose that export pathways traffic substrates to the NPC where proteasomes are located (Fig. 9). Further study will be required to establish the veracity of this paradigm.
Experimental Procedures
Yeast Strains and Methods
Exponential phase wild type cells expressing FLAG-H2B were pelleted, suspended at 1 ml/g in pretreatment buffer (PB: 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1 m sorbitol, and 60 mm 2-β mercaptoethanol), and incubated at room temperature for 15 min with gentle mixing. Cells were collected by centrifugation for 5 min at 1,500 × g, suspended in 3 ml/g digestion buffer (DB: PB + 5 mm 2-β mercaptoethanol, 2 mg/ml zymolyase 20T), and incubated at 37 °C for 60 min. Spheroplasts were collected by centrifugation at 4,000 × g for 10 min and then washed three times with cold DB. Spheroplasts were suspended in 0.5 ml/g DB and lysed by dropwise addition to 20 volumes of cold lysis buffer (LB: 18% Ficoll, 10 mm Tris-HCl, pH 7.5, 20 mm KCl, 5 mm MgCl2, 5 mm 2-β mercaptoethanol, 1 mm EDTA and protease inhibitors). Unlysed cells and debris were removed by centrifugation for 30 min at 3,000 × g. An aliquot of the suspension (3 ml) was removed and lysed by sonication to provide a sample representing total (Nuc + Cyto) cellular protein. The remaining supernatant was centrifuged for 20 min at 20,000 × g to pellet the nuclei. The nuclear pellet was suspended in LB at ∼1/7th of the cytoplasmic volume, as the yeast nucleus represents ∼15% of total cell volume (50). Nuclear proteins were recovered following disruption by sonication (Nuc). An equal volume (20 μl) of total, Nuc, and Cyto was used to measure HDAC (Sigma catalog no. CS1010) and proteasome activities (see below and Ref. 51). One hundred microliters of each fraction was resolved in a 12% Tricine-SDS gel and examined by immunoblotting.
Human Cell Lines and Plasmids
HEK293T cells were provided by B. Firestein (Rutgers University). HCT116 cells were provided by W. Hu (Cancer Institute of New Jersey). A plasmid encoding human pQCXIP-β7-EGFP-HTBH was a gift from A. Tsimokha (Russian Academy of Sciences). PSMB4-GFP, encoding β7-GFP, was provided by E. A. J. Reits (Academic Medical Center, Netherlands). Plasmids encoding β-galactosidase derivatives were obtained from U. Stochaj (McGill University), and LMP2-GFP was provided by J. Neefjes (Netherlands Cancer Institute). Plasmids expressing GFP-p53 and derivatives were purchased from Addgene (52).
Cell Culture and Transfection
HEK293T and HCT116 cells were cultured at 37 °C in Dulbecco's modified Eagle's medium containing 25 mm d-glucose and 4 mm l-glutamine (DMEM; Life Technologies, Inc., catalog no. 11965-092) and supplemented with 10% (v/v) fetal calf serum, 100 units/ml penicillin G, and 100 μg/ml streptomycin. HEK293T cells were transfected using Lipofectamine 3000 and seeded in a 10-cm plate with fibronectin-coated cover slides.
Digitonin Lysis
HEK293T cells were trypsinized, centrifuged at 500 × g for 5 min, and resuspended in 1 ml of culture medium or PBS (containing protease inhibitors) at 4 °C. Digitonin solution was added to 100 μl of cell suspension to achieve a final concentration of 0, 25, and 50 μg/ml. Samples were incubated at room temperature for 10, 20, and 30 min and centrifuged at 500 × g for 5 min at 4 °C, following which the supernatant was removed to assay proteasome peptidase activity.
Nuclei Purification
HEK293T and HCT116 cells were grown to ∼70–80% confluence. Media were removed, and cells were washed twice with PBS. EZ-lysis buffer (500 μl; Sigma catalog no. NUC-101), containing 5 mm ATP, 1 mm DTT, and protease inhibitors, was added to each flask. Lysed cells were dislodged and transferred to a single microcentrifuge tube (yielding ∼1.7 ml of cell suspension). Lysis was completed on ice for 5 min, following which 500 μl was removed and sonicated, serving as a total protein sample. The remainder was centrifuged at 500 × g for 5 min at 4 °C. The supernatant, containing cytoplasmic proteins, was withdrawn, and the pellet was washed in lysis buffer to remove residual cytoplasm. The pellet containing purified nuclei was suspended in 120 μl of lysis buffer and sonicated. Total, cytosolic, and nuclear fractions were examined biochemically and by gel filtration and electrophoresis.
Nuclear Permeabilization Studies
HEK293T cells were grown to ∼70% confluence in a 150-cm2 flask. Cells were collected and fractionated to separate nuclei from cytosol, as described above. Purified nuclei were suspended in 100 μl of PBS containing a protease inhibitor mixture. FITC-dextran (5 μl at 1 mg/ml) and Hoechst-33342 (1 μl; NucBlue Live Stain from Life Technologies, Inc., catalog no. R37605) were added to 5% of the purified nuclei (5 μl) and incubated on ice for 5 min. Microscopy images were captured using appropriate optical filters (excitation 430 nm; emission 535 nm). Derivatives of FITC-dextran (representing 10, 70, and 2,000 kDa of dextran) were purchased from Sigma.
Gel Exclusion Chromatography
Purified cytosolic proteins (∼2 mg) were clarified by brief centrifugation and resolved in Sephacryl S-300 HR at 4 °C. The column was equilibrated in 50 mm potassium phosphate, 5 mm EDTA, 100 mm NaCl, 10 mm MgCl2, 2 mm ATP, 1 mm DTT, and 5% glycerol. The sample represents ∼1% of column volume, and 1-ml fractions were collected at a rate of ∼0.5 column volume/h. Proteasome peptidase activity was measured in the fractions, and those containing peak activity were further characterized in native in-gel assays.
Western Blotting Analysis and Immunoprecipitation
Protein samples were resolved in a 12% SDS-Tricine/polyacrylamide gel, transferred to 0.45-μm nitrocellulose, and examined by immunoblotting. Chemiluminescence was captured using Kodak GelLogic Imaging software. Antibodies against histone H3 and p53 were purchased from BioLegend and Origene, respectively. Tubulin antibody was purchased from Life Technologies, Inc.; XPC antibody was purchased from Novus; proteasome and immunoproteasome antibodies were purchased from Enzo Biochem.
Native Gel Analysis
Proteasome-specific peptidase activity in total, nuclear, and cytosolic fractions was measured as described previously (51). Native polyacrylamide gels were incubated with buffer containing Suc-LLVY-AMC and 0.05% SDS, and the fluorescence signal was detected with Kodak GelLogic Imager.
Microscopy
Transfected cells were fixed and permeabilized with 4% paraformaldehyde in culture medium and 0.15% Triton X-100 in PBS, respectively. Hoechst was added directly to each slide prior to microscopy. Images were captured using Zeiss Imager.M1 microscope, an AxioCam MRm camera, Plan-Apochromatic 100×/1.4 objective lens, and Zeiss AxioVision software.
Biochemical Assays
For the proteasome activity measurement, 5 μl of epoxomicin (prepared in 50% DMSO at 50 ng/μl) was preloaded in duplicate in a 96-well plate. Similarly, 5 μl of vehicle (50% DMSO) was loaded in duplicate. Protein fractions (25 μl) were added to the aforementioned four wells. The proteasome assay buffer (200 μl: 25 mm Tris, pH 7.5, 1 mm EDTA) containing 40 μm Suc-LLVY-AMC was added to each well. A TECAN fluorometer was used to detect fluorescence generated by the hydrolysis of Suc-LLVY-AMC at 37 °C, using excitation wavelength 360 nm and emission wavelength 465 nm. Results were captured at 5-min intervals for at least 30 min. (The background value was determined by measuring epoxomicin-insensitive activity, which was typically <5% of total fluorescence.) GFP fluorescence was measured using a TECAN fluorometer using excitation wavelength 430 nm and emission wavelength 535 nm.
Immunoproteasome Activity Measurement
Total, cytosolic, and nuclear protein extracts were prepared and examined as described above. Immunoproteasome Assay Buffer contained 14.38 μm Ac-PAL-AMC, and fluorescence was detected using the TECAN fluorometer. Data were captured at 5-min intervals (excitation wavelength 360 nm/emission wavelength 465 nm).
p53 Chase with Leptomycin B
HCT116 cells were grown to ∼70% confluence. The growth medium was removed and replaced with fresh media containing 10 nm LMB. Following incubation for 18 h the medium was replaced and half the dishes were supplemented with LMB. Cells were harvested, pelleted, and frozen at the times indicated. GFP derivatives of wild type p53 and an export mutant (p53nes−) were transfected into HCT116 cells 48 h prior to treatment with LMB.
Author Contributions
K. M. and L. C. designed and coordinated the study. The manuscript was prepared by K. M. Figures were prepared by F. D. Data shown in Fig. 1, were generated by L. C. All other data were generated by F. D. Protocols for in-gel analysis were refined by L. C. K. M. guided studies involving gel filtration. All authors proofread and approved the final version of this manuscript. We thank members of the laboratory for critical review of this manuscript.
Acknowledgments
We thank N. Dantuma, B. Firestein, S. Kohn, W. Hu, J. Neefjes, U. Stochaj, and A. Tsimokha for generously providing plasmids and cell lines.
This work was supported by National Institutes of Health Grant GM104968 and Grant CA083875 from National Cancer Institute (to K. M.). K. M. is a founder of CellXplore LLC whose objectives are unrelated to the content of this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HDAC
- histone deacetylase
- Nuc
- nucleus
- Cyto
- cytoplasm
- LMB
- leptomycin B
- NPC
- nuclear pore complex
- NLS
- nuclear localization signal
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Suc
- N-succinyl
- AMC
- 7-amino 4-methylcoumarin.
References
- 1. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen L., and Madura K. (2014) Degradation of specific nuclear proteins occurs in the cytoplasm in Saccharomyces cerevisiae. Genetics 197, 193–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Enenkel C., Lehmann A., and Kloetzel P. M. (1999) GFP-labelling of 26S proteasomes in living yeast: insight into proteasomal functions at the nuclear envelope/rough ER. Mol. Biol. Rep. 26, 131–135 [DOI] [PubMed] [Google Scholar]
- 4. Enenkel C., Lehmann A., and Kloetzel P. M. (1998) Subcellular distribution of proteasomes implicates a major location of protein degradation in the nuclear envelope-ER network in yeast. EMBO J. 17, 6144–6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Russell S. J., Reed S. H., Huang W., Friedberg E. C., and Johnston S. A. (1999) The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol. Cell 3, 687–695 [DOI] [PubMed] [Google Scholar]
- 6. Xie Y., and Varshavsky A. (2001) RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc. Natl. Acad. Sci. U.S.A. 98, 3056–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kulichkova V. A., Artamonova T. O., Zaykova J. J., Ermolaeva J. B., Khodorkovskii M. A., Barlev N. A., Tomilin A. N., and Tsimokha A. S. (2015) Simultaneous EGFP and tag labeling of the β7 subunit for live imaging and affinity purification of functional human proteasomes. Mol. Biotechnol. 57, 36–44 [DOI] [PubMed] [Google Scholar]
- 8. Bakhrat A., Baranes-Bachar K., Reshef D., Voloshin O., Krichevsky O., and Raveh D. (2008) Nuclear export of Ho endonuclease of yeast via Msn5. Curr. Genet. 54, 271–281 [DOI] [PubMed] [Google Scholar]
- 9. Freedman D. A., and Levine A. J. (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 18, 7288–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiechens N., and Fagotto F. (2001) CRM1- and Ran-independent nuclear export of β-catenin. Curr. Biol. 11, 18–27 [DOI] [PubMed] [Google Scholar]
- 11. Zang Z. J., Gunaratnam L., Cheong J. K., Lai L. Y., Hsiao L. L., O'Leary E., Sun X., Salto-Tellez M., Bonventre J. V., and Hsu S. I. (2009) Identification of PP2A as a novel interactor and regulator of TRIP-Br1. Cell. Signal. 21, 34–42 [DOI] [PubMed] [Google Scholar]
- 12. Lahaye F., Lespinasse F., Staccini P., Palin L., Paquis-Flucklinger V., and Santucci-Darmanin S. (2010) hMSH5 is a nucleocytoplasmic shuttling protein whose stability depends on its subcellular localization. Nucleic Acids Res. 38, 3655–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blondel M., Galan J. M., Chi Y., Lafourcade C., Longaretti C., Deshaies R. J., and Peter M. (2000) Nuclear-specific degradation of Far1 is controlled by the localization of the F-box protein Cdc4. EMBO J. 19, 6085–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardner R. G., Nelson Z. W., and Gottschling D. E. (2005) Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815 [DOI] [PubMed] [Google Scholar]
- 15. Garrenton L. S., Braunwarth A., Irniger S., Hurt E., Künzler M., and Thorner J. (2009) Nucleus-specific and cell cycle-regulated degradation of mitogen-activated protein kinase scaffold protein Ste5 contributes to the control of signaling competence. Mol. Cell. Biol. 29, 582–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Geng F., and Tansey W. P. (2012) Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc. Natl. Acad. Sci. U.S.A. 109, 6060–6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lehmann A., Janek K., Braun B., Kloetzel P. M., and Enenkel C. (2002) 20 S proteasomes are imported as precursor complexes into the nucleus of yeast. J. Mol. Biol. 317, 401–413 [DOI] [PubMed] [Google Scholar]
- 18. Pack C. G., Yukii H., Toh-e A., Kudo T., Tsuchiya H., Kaiho A., Sakata E., Murata S., Yokosawa H., Sako Y., Baumeister W., Tanaka K., and Saeki Y. (2014) Quantitative live-cell imaging reveals spatio-temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat. Commun. 5, 3396. [DOI] [PubMed] [Google Scholar]
- 19. Park S. H., Kukushkin Y., Gupta R., Chen T., Konagai A., Hipp M. S., Hayer-Hartl M., and Hartl F. U. (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154, 134–145 [DOI] [PubMed] [Google Scholar]
- 20. Prasad R., Kawaguchi S., and Ng D. T. (2010) A nucleus-based quality control mechanism for cytosolic proteins. Mol. Biol. Cell 21, 2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rosenbaum J. C., Fredrickson E. K., Oeser M. L., Garrett-Engele C. M., Locke M. N., Richardson L. A., Nelson Z. W., Hetrick E. D., Milac T. I., Gottschling D. E., and Gardner R. G. (2011) Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 41, 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Russell S. J., Steger K. A., and Johnston S. A. (1999) Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem. 274, 21943–21952 [DOI] [PubMed] [Google Scholar]
- 23. Tsuchiya H., Arai N., Tanaka K., and Saeki Y. (2013) Cytoplasmic proteasomes are not indispensable for cell growth in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 436, 372–376 [DOI] [PubMed] [Google Scholar]
- 24. Weberruss M. H., Savulescu A. F., Jando J., Bissinger T., Harel A., Glickman M. H., and Enenkel C. (2013) Blm10 facilitates nuclear import of proteasome core particles. EMBO J. 32, 2697–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wendler P., Lehmann A., Janek K., Baumgart S., and Enenkel C. (2004) The bipartite nuclear localization sequence of Rpn2 is required for nuclear import of proteasomal base complexes via karyopherin αβ and proteasome functions. J. Biol. Chem. 279, 37751–37762 [DOI] [PubMed] [Google Scholar]
- 26. Chen L., and Madura K. (2008) Centrin/Cdc31 is a novel regulator of protein degradation. Mol. Cell. Biol. 28, 1829–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen L., and Madura K. (2002) Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 22, 4902–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tongaonkar P., Chen L., Lambertson D., Ko B., and Madura K. (2000) Evidence for an interaction between ubiquitin-conjugating enzymes and the 26S proteasome. Mol. Cell. Biol. 20, 4691–4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chuang S. M., Chen L., Lambertson D., Anand M., Kinzy T. G., and Madura K. (2005) Proteasome-mediated degradation of cotranslationally damaged proteins involves translation elongation factor 1A. Mol. Cell. Biol. 25, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen L., Romero L., Chuang S. M., Tournier V., Joshi K. K., Lee J. A., Kovvali G., and Madura K. (2011) Sts1 plays a key role in targeting proteasomes to the nucleus. J. Biol. Chem. 286, 3104–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sánchez L., Kodiha M., and Stochaj U. (2005) Monitoring the disruption of nuclear envelopes in interphase cells with GFP-β-galactosidase. J. Biomol. Tech. 16, 235–238 [PMC free article] [PubMed] [Google Scholar]
- 32. Shibatani T., Carlson E. J., Larabee F., McCormack A. L., Früh K., and Skach W. R. (2006) Global organization and function of mammalian cytosolic proteasome pools: Implications for PA28 and 19S regulatory complexes. Mol. Biol. Cell 17, 4962–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wilkinson C. R., Wallace M., Morphew M., Perry P., Allshire R., Javerzat J. P., McIntosh J. R., and Gordon C. (1998) Localization of the 26S proteasome during mitosis and meiosis in fission yeast. EMBO J. 17, 6465–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heck J. W., Cheung S. K., and Hampton R. Y. (2010) Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kaplun L., Tzirkin R., Bakhrat A., Shabek N., Ivantsiv Y., and Raveh D. (2005) The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol. Cell. Biol. 25, 5355–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muratani M., and Tansey W. P. (2003) How the ubiquitin-proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 4, 192–201 [DOI] [PubMed] [Google Scholar]
- 37. Gillette T. G., Huang W., Russell S. J., Reed S. H., Johnston S. A., and Friedberg E. C. (2001) The 19S complex of the proteasome regulates nucleotide excision repair in yeast. Genes Dev. 15, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andrulis E. D., Neiman A. M., Zappulla D. C., and Sternglanz R. (1998) Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 394, 592–595 [DOI] [PubMed] [Google Scholar]
- 39. Capelson M., Liang Y., Schulte R., Mair W., Wagner U., and Hetzer M. W. (2010) Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dieppois G., and Stutz F. (2010) Connecting the transcription site to the nuclear pore: a multi-tether process that regulates gene expression. J. Cell Sci. 123, 1989–1999 [DOI] [PubMed] [Google Scholar]
- 41. Niepel M., Molloy K. R., Williams R., Farr J. C., Meinema A. C., Vecchietti N., Cristea I. M., Chait B. T., Rout M. P., and Strambio-De-Castillia C. (2013) The nuclear basket proteins Mlp1p and Mlp2p are part of a dynamic interactome including Esc1p and the proteasome. Mol. Biol. Cell 24, 3920–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fischer T., Rodríguez-Navarro S., Pereira G., Rácz A., Schiebel E., and Hurt E. (2004) Yeast centrin Cdc31 is linked to the nuclear mRNA export machinery. Nat. Cell Biol. 6, 840–848 [DOI] [PubMed] [Google Scholar]
- 43. Laporte D., Salin B., Daignan-Fornier B., and Sagot I. (2008) Reversible cytoplasmic localization of the proteasome in quiescent yeast cells. J. Cell Biol. 181, 737–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Nocker S., Sadis S., Rubin D. M., Glickman M., Fu H., Coux O., Wefes I., Finley D., and Vierstra R. D. (1996) The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol. 16, 6020–6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elsasser S., Chandler-Militello D., Müller B., Hanna J., and Finley D. (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822 [DOI] [PubMed] [Google Scholar]
- 46. Apcher G. S., Maitland J., Dawson S., Sheppard P., and Mayer R. J. (2004) The α4 and α7 subunits and assembly of the 20S proteasome. FEBS Lett. 569, 211–216 [DOI] [PubMed] [Google Scholar]
- 47. Livinskaya V. A., Barlev N. A., and Nikiforov A. A. (2014) Immunoaffinity purification of the functional 20S proteasome from human cells via transient overexpression of specific proteasome subunits. Protein Expr. Purif. 97, 37–43 [DOI] [PubMed] [Google Scholar]
- 48. Chen L., and Madura K. (2014) Yeast importin-alpha (Srp1) performs distinct roles in the import of nuclear proteins and in targeting proteasomes to the nucleus. J. Biol. Chem. 289, 32339–32352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hedhli N., Wang L., Wang Q., Rashed E., Tian Y., Sui X., Madura K., and Depre C. (2008) Proteasome activation during cardiac hypertrophy by the chaperone H11 kinase/Hsp22. Cardiovasc. Res. 77, 497–505 [DOI] [PubMed] [Google Scholar]
- 50. Jorgensen P., Edgington N. P., Schneider B. L., Rupes I., Tyers M., and Futcher B. (2007) The size of the nucleus increases as yeast cells grow. Mol. Biol. Cell 18, 3523–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chandra A., Chen L., Liang H., and Madura K. (2010) Proteasome assembly influences interaction with ubiquitinated proteins and shuttle factors. J. Biol. Chem. 285, 8330–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boyd S. D., Tsai K. Y., and Jacks T. (2000) An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol. 2, 563–568 [DOI] [PubMed] [Google Scholar]