

Abstract
Human nucleoside transporters (hNTs) mediate cellular influx of anticancer nucleoside drugs, including cytarabine, cladribine, and fludarabine. BCR-ABL tyrosine kinase inhibitors (TKIs) imatinib and dasatinib inhibit fludarabine and cytarabine uptake. We assessed interactions of bosutinib, dasatinib, imatinib, nilotinib, and ponatinib with recombinant hNTs (hENT1, 2; hCNT1, -2, and -3) produced individually in yeast Saccharomyces cerevisiae. Nilotinib inhibited hENT1-mediated uridine transport most potently (IC50 value, 0.7 μm) followed by ponatinib > bosutinib > dasatinib > imatinib. Imatinib inhibited hCNT2 with an IC50 value of 2.3 μm. Ponatinib inhibited all five hNTs with the greatest effect seen for hENT1 (IC50 value, 9 μm). TKIs inhibited [3H]uridine uptake in a competitive manner. Studies in yeast with mutants at two amino acid residues of hENT1 (L442I, L442T, M33A, M33A/L442I) previously shown to be involved in uridine and dipyridamole binding, suggested that BCR-ABL TKIs interacted with Met33 (TM1) and Leu442 (TM11) residues of hENT1. In cultured human CEM lymphoblastoid cells, which possess a single hNT type (hENT1), accumulation of [3H]cytarabine, [3H]cladribine, or [3H]fludarabine was reduced by each of the five TKIs, and also caused a reduction in cell surface expression of hENT1 protein. In conclusion, BCR-ABL TKIs variously inhibit five different hNTs, cause a decrease in cell surface hENT1 expression, and decrease uridine accumulation when presented together with uridine or when given before uridine. In experiments with mutant hENT1, we showed for the first time interaction of Met33 (involved in dipyridamole binding) with BCR-ABL inhibitors and reduced interaction with M33A mutant hENT1.
Keywords: drug resistance, drug transport, inhibition mechanism, membrane transport, nucleoside/nucleotide transport, receptor tyrosine kinase, Saccharomyces cerevisiae
Introduction
Several tyrosine kinase inhibitors (TKIs)2 have been in clinical use for treatment of chronic myelogenous leukemia, Philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia, and acute myelogenous leukemia (1). Philadelphia chromosome positive chronic myelogenous leukemia and acute lymphoblastic leukemia cells have a constitutively active BCR-ABL oncogene that is the therapeutic target of inhibition by BCR-ABL TKIs (2). Since the approval more than a decade ago of imatinib, a TKI that inhibits BCR-ABL, more potent BCR-ABL TKIs have been developed and approved because BCR-ABL-dependent and -independent resistance mechanisms occur in nearly 33% of patients treated with imatinib (3). Second generation BCR-ABL TKIs dasatinib and nilotinib target patients who are either imatinib resistant or imatinib intolerant. In vitro, nilotinib and dasatinib are 30 and 325 times more potent, respectively, than imatinib (4) and both are active in imatinib-resistant or imatinib-intolerant patients (1). Bosutinib and ponatinib are third generation potent BCR-ABL TKIs, with ponatinib being active against cells with BCR-ABL mutations including T315I (5).
One feature common to imatinib and more recently developed BCR-ABL TKIs is that they were designed to compete with ATP for the ATP binding pocket of a variety of tyrosine kinases that are expressed in different tumor types. Because of this feature several off-target effects have been discovered. Imatinib, nilotinib, and dasatinib were shown to interact with the ATP-binding cassette (ABC) transporter(s) ABCG2/BCRP protein at its substrate binding site (6–9). In other studies, interactions of imatinib, nilotinib, and dasatinib was also shown with ABCB1/P-glycoprotein/MDR1 thereby conferring resistance to these drugs (9–12). Organic cation transporter 1 and organic-anion transporting polypeptide 1A2 were shown to mediate imatinib uptake in chronic myelogenous leukemia cells (13–15). Nilotinib uptake was shown to be enhanced by organic cation transporter 1 but not by organic-anion transporting polypeptide 1A2 (16). The cellular uptake of dasatinib was mainly passive and not dependent on organic cation transporters (9). Ponatinib was a potent inhibitor of ABCG2 (17), whereas bosutinib was not a substrate of ABCB1 or ABCG2 (8).
Another group of potential off-target proteins are NTs. Inhibition of human equillibrative NT 1 (hENT1) mediated activity in K562 cells by p38 mitogen-activated protein kinase inhibitors (18) and murine equilibrative NT 1 (mENT1) (19) by imatinib were shown earlier. A more recent study showed inhibition of hENT1-mediated cytarabine uptake by imatinib and nilotinib in human leukemic cell lines (20).
Solute carrier transporters for physiologic nucleosides and nucleoside analogs include bidirectional human equilibrative, nucleoside transporters 1–4 (hENT1–4), which are members of the solute carrier 29 family of integral membrane proteins, and inwardly directive human concentrative nucleoside transporters (hCNT1–3), which are members of the solute carrier 28 family of integral membrane proteins. Detailed summaries of the roles of hNT in transport of nucleoside drugs can be found in several reviews (21, 22). hENT1 is predicted to have 11 transmembrane domains (TMs 1–11) wherein the N terminus is cytoplasmic and the C terminus is extracellular (23). Earlier studies (24) implicated a Met residue (residue 33 in TM1) predicted to lie at the extracellular end of TM1 and a Leu residue (residue 442) in TM11 in interacting with dipyridamole and nucleoside binding, respectively.
hENT1 transports both physiological purine and pyrimidine nucleosides as well as anticancer nucleoside drugs. Nitrobenzylmercaptopurine ribonucleoside, dipyridamole, and dilazep are structurally diverse inhibitors of hENT1. Pyrimidine and purine nucleoside analog drugs gemcitabine, cytarabine, cladribine, and fludarabine are used as anticancer chemotherapeutic drugs (25). Clinical efficacy of these nucleoside drugs, which act intracellularly, is dependent on activity of hNTs present in plasma membranes of tumor cells.
Although earlier literature reports showed interaction of imatinib with hENT1 (26), these studies lacked in depth analysis of interactions of the BCR-ABL class of TKIs with the five major human nucleoside transporters found in plasma membranes. In this study we assessed inhibitory properties of five BCR-ABL TKIs against each of five hNTs produced individually in a yeast model expression system to document their interactions. In addition, inhibition by BCR-ABL TKIs of mutant hENT1 proteins produced in yeast was examined to identify interactions with permeant and/or inhibitor binding sites. We also examined inhibition of hENT1-mediated transport of uridine in cultured human lymphoblastoid CEM cells and human lung cancer A549 cells by the BCR-ABL TKIs. Finally, we examined their effects on cytotoxic nucleoside drug accumulation as well as hENT1 protein levels in CEM cells to understand the lack of synergy seen in some clinical trials when the BCR-ABL TKIs are combined with nucleoside analog drugs.
Results
Effects of BCR-ABL TKIs on Uridine Uptake Mediated by Recombinant hNTs Produced in Saccharomyces cerevisiae
Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib (chemical structures shown in Fig. 1) were assessed for their relative abilities to inhibit [3H]uridine uptake by each of five hNTs in concentration-dependent inhibition experiments that yielded IC50 values (inhibitor concentration that produced 50% inhibition of transport). Representative concentration-effect curves for dasatinib, imatinib, and nilotinib inhibition of hENT1-mediated uridine transport in yeast are shown in Fig. 2A, and IC50 values obtained from such experiments with bosutinib, dasatinib, imatinib, nilotinib, and ponatinib in yeast producing each of the five recombinant NTs are presented in Table 1. For hENT1, bosutinib, dasatinib, imatinib, nilotinib, and ponatinib IC50 values (± S.E.) were 13 ± 1, 60 ± 3, 110 ± 26, 0.7 ± 0.1, and 9.0 ± 0.1 μm, respectively. Inhibition of hENT2, hCNT1, hCNT2, and hCNT3 was seen with IC50 values ranging from 24 to 260 μm with the exception of nilotinib (hCNT1, hCNT2, and hCNT3), bosutinib and dasatinib (hCNT2) for which IC50 values were >300 μm.
FIGURE 1.
Structures of BCR-ABL TKIs. Structures of bosutinib, dasatinib, imatinib, nilotinib, and ponatinib.
FIGURE 2.

Effects of BCR-ABL TKIs on [3H]uridine uptake in yeast and CEM cells. Yeast cells were incubated with 1 μm [3H]uridine for 10 min in the absence or presence of increasing concentrations of imatinib (●) (0–300 μm), dasatinib (○) (0–300 μm), or nilotinib (□) (0–30 μm) (panel A). Data presented are averages of three experiments each conducted with six replicates per concentration and are expressed as mean ± S.E. Uptake values represent % uridine uptake in the presence of BCR-ABL TKIs relative to that in its absence (control). Error bars are not shown where S.E. values are smaller than the size of the symbol. Panel B shows effects of increasing concentrations (0–30 μm) of imatinib (●), dasatinib (○), or nilotinib (□) on [3H]uridine uptake in CEM cells. Values with mean ± S.E. are shown in each panel. Each experiment was repeated three times with four replicates per condition.
TABLE 1.
Summary of IC50 values for inhibition of uridine transport in yeast and CEM cells
Inhibition of [3H]uridine uptake by BCR-ABL TKIs was assessed in yeast producing each of the five recombinant hNTs and in CEM cells, which have only hENT1 activity, in concentration-effect experiments as described under “Experimental Procedures.” IC50 values (mean ± S.E.) are listed below.
| Transporter yeast | IC50 |
||||
|---|---|---|---|---|---|
| Bosutinib | Dasatinib | Imatinib | Nilotinib | Ponatinib | |
| μm ± S.E. | |||||
| hENT1 | 13.0 ± 1.0 | 60.0 ± 3.0 | 110.0 ± 26.0 | 0.7 ± 0.1 | 9.0 ± 0.1 |
| hENT2 | 24.0 ± 2.0 | 240.0 ± 57.0 | 230.0 ± 11.0 | 150.0 ± 21.0 | 33.0 ± 1.0 |
| hCNT1 | 260.0 ± 6.0 | 84.0 ± 1.0 | 130.0 ± 5.3 | >300 | 22.0 ± 3.0 |
| hCNT2 | >300 | >300 | 2.3 ± 0.3 | >300 | 64.0 ± 6.0 |
| hCNT3 | 150.0 ± 3.0 | 170.0 ± 20.0 | 62.0 ± 2.0 | >300 | 70.0 ± 3.0 |
| CEM | 2.0 ± 0.4 | 8.0 ± 1.6 | 16.0 ± 2.0 | 0.7 ± 0.01 | 3.0 ± 0.2 |
Effects of BCR-ABL TKIs on Kinetics of Uridine Uptake in hENT1-producing Yeast
Kinetic studies of hENT1 with the BCR-ABL TKIs were undertaken in hENT1-producing yeast cells by studying effects of fixed bosutinib (Fig. 3, A and F), dasatinib (Fig. 3, B and G), imatinib (Fig. 3, C and H), nilotinib (Fig. 3, D and I), or ponatinib (Fig. 3, E and J) concentrations on varying concentrations of [3H]uridine uptake. Analysis of the results using Lineweaver-Burk plots (Fig. 3, A–E) and Dixon slope replots (Fig. 3, F–J) showed the competitive nature of uridine uptake inhibition by the BCR-ABL TKIs, suggesting binding of these compounds and uridine to the same or overlapping sites on hENT1.
FIGURE 3.
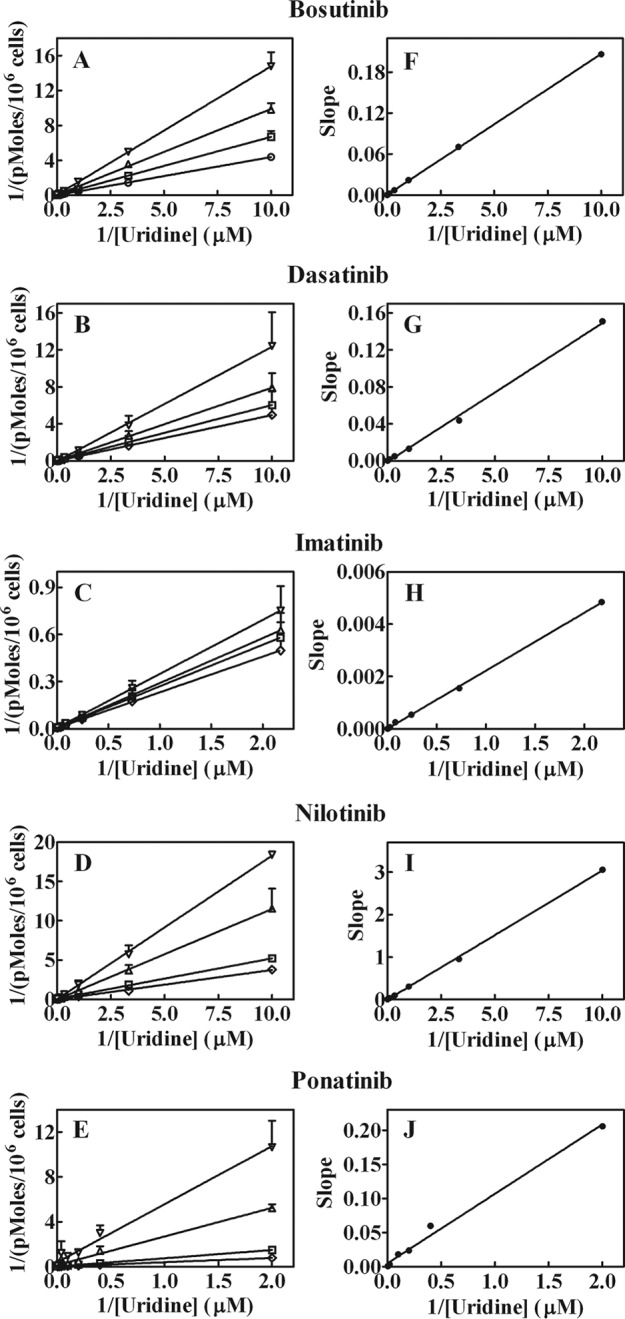
Inhibition of hENT1-mediated uridine uptake by BCR-ABL TKIs. Effects of fixed concentrations of bosutinib, dasatinib, imatinib, ponatinib (0 (○), 10 (▴), 25 (●), or 50 (□) μm) and nilotinib 0 (○), 1 (▴), 2.5 (●), or 5 (□) μm on varying concentrations of [3H]uridine uptake in hENT1-producing yeast cells. Panels A–E show Lineweaver-Burk transformations of the data and panels F–J show Dixon slope replots. Values with mean ± S.E. are shown in each panel. Each experiment was repeated three times with four replicates for each experiment.
Inhibition of Uridine Uptake in hENT1 Mutants by BCR-ABL TKIs
Previous studies with mutants of hENT1 have identified two amino acid residues involved in interaction of hENT1 with the well established NT inhibitor dipyridamole (24). To determine whether these amino acid residues (Leu442 and Met33) are also involved in interaction of hENT1 with BCR-ABL TKIs, previously characterized mutants L442I, L442T, M33A, and M33A/L442I were assessed for their sensitivity to the five BCR-ABL TKIs. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib were assessed for their relative abilities to inhibit [3H]uridine uptake by each of the four hENT1 mutants in concentration-dependent inhibition experiments (IC50 values are summarized in Table 2). For comparison IC50 values for inhibition of wild type hENT1 are also included. Major changes in IC50 values were noted for L442I and M33A mutants. L442I exhibited lower IC50 values, indicating increased sensitivities, and M33A exhibited increased IC50 values, indicating reduced sensitivities, with all five BCR-ABL kinase inhibitors.
TABLE 2.
Summary of IC50 values for inhibition of uridine transport in yeast containing recombinant hENT1 or one of four hENT1 mutants
Inhibition of [3H]uridine uptake by BCR-ABL TKIs was assessed in concentration-effect experiments as described under ”Experimental Procedures.“ IC50 values (mean ± S.E.) are listed below.
| Transporter yeast | IC50 |
||||
|---|---|---|---|---|---|
| Bosutinib | Dasatinib | Imatinib | Nilotinib | Ponatinib | |
| μm ± S.E. | |||||
| hENT1 | 13.0 ± 1.0 | 60.0 ± 3.0 | 110.0 ± 26 | 0.7 ± 0.1 | 9.0 ± 0.1 |
| L442I | 1.8 ± 0.2 | 11.0 ± 1.0 | 12.0 ± 0.3 | 0.2 ± 0.01 | 2.0 ± 0.1 |
| L442T | 8.0 ± 0.6 | 26.0 ± 1.0 | 26.0 ± 2 | 2.5 ± 0.3 | 7.0 ± 0.3 |
| M33A | 94.0 ± 8.0 | 170.0 ± 38 | >300 | 3.0 ± 0.4 | >100 |
| M33A/L442I | 9.0 ± 1.4 | 13.0 ± 1.4 | 21.0 ± 1.3 | 0.7 ± 0.08 | 5.0 ± 0.4 |
Inhibition of hENT1 Activity in CEM Cells by BCR-ABL TKIs
Because BCR-ABL TKIs are used routinely in treatment of several leukemias, their abilities to inhibit uridine transport activity in the human CEM (T-ALL) cell line, which possesses a single hNT activity (i.e. hENT1), were assessed. Inhibition of 1 μm [3H]uridine uptake by CEM cells by dasatinib, imatinib, or nilotinib is shown in Fig. 2B and the IC50 values for inhibition of [3H]uridine uptake mediated by hENT1 in CEM cells by all five TKIs are summarized in Table 1. hENT1 activity in CEM cells was inhibited by all five BCR-ABL TKIs with nilotinib being the most inhibitory TKI.
BCR-ABL TKIs Inhibit Chemotherapeutic Nucleoside Drug Uptake and Accumulation in CEM Cells
Imatinib and nilotinib were previously shown to inhibit uptake of cytarabine (20) and because failure of combination therapies with various TKIs and nucleoside analog drugs, effects of bosutinib, dasatinib, imatinib, nilotinib, or ponatinib on uptake of [3H]cytarabine, [3H]cladribine, and [3H]fludarabine were examined in CEM cells. Short-term (1 min, Fig. 4, A–C) and long-term (1 h, Fig. 4, D–F) uptake of 1 μm [3H]nucleoside drug, which assessed, respectively, intracellular uptake and accumulation of drugs plus their metabolites, were measured after incubation in the absence or presence of 10 μm bosutinib, dasatinib, imatinib, nilotinib, ponatinib, or dipyridamole, a potent hENT1 inhibitor (27) (Fig. 4, A–F). Nilotinib strongly inhibited uptake and accumulation of all three drugs, whereas other BCR-ABL TKIs inhibited uptake and accumulation to lesser and different extents.
FIGURE 4.

Effect of BCR-ABL TKIs on uptake and accumulation of cytotoxic nucleoside drugs. Uptake (1 min) and accumulation (60 min) of 1 μm [3H]cytarabine, [3H]cladribine, or [3H]fludarabine in CEM cells was examined in the absence or presence 10 μm bosutinib, dasatinib, imatinib, ponatinib, or dipyridamole or 2 μm nilotinib. Panels A–C show 1-min uptake and panels D–F show 60-min accumulation. The symbols (●, ▴, and ■) represent experiments done on different days. Values plotted are % control values obtained in the absence of BCL-ABL TKIs and values from two to three experiments each conducted with four replicates are shown in each panel as scatter plots.
BCR-ABL TKIs Reduce Cell Surface hENT1 Abundance in CEM Cells
To determine whether reduced NT activity was due to altered cell surface hENT1 protein levels, a non-permeable fluorescent hENT1 probe, 5′-S-[2-(6-aminohexanamido)]ethyl-6-N-(4-nitrobenzyl)-5′-thioadenosine-fluorescein-5-yl isothiocyanate (SAHENTA-FITC) was used to assess the relative abundance of hENT1 sites on cell surfaces as described earlier (28). CEM cells were treated overnight with non-toxic concentrations (1 μm) of bosutinib, dasatinib, imatinib, nilotinib, or ponatinib and were then stained with 100 nm SAHENTA-FITC. Fig. 5A shows unstained and stained cells that were either untreated or treated with ponatinib. The staining of bosutinib-treated cells decreased as seen by a shift to left in the histogram. Fig. 5B shows a transformation of the data for bosutinib, dasatinib, imatinib, nilotinib, and ponatinib expressed as percent of stained cells in treated versus untreated (control) cells. Ponatinib and bosutinib caused a greater decrease in cell surface hENT1, followed by dasatinib and nilotinib, whereas imatinib had no effect.
FIGURE 5.
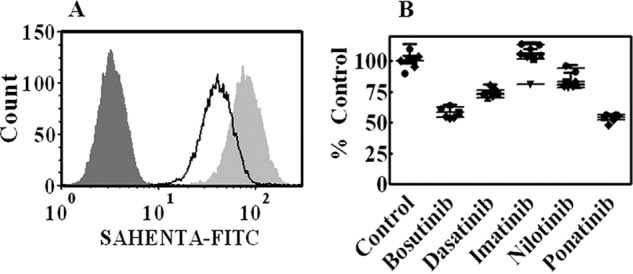
BCR-ABL TKIs decrease hENT1 surface expression. CEM cells were incubated overnight with 1 μm bosutinib, dasatinib, imatinib, nilotinib, or ponatinib after which cells were washed and incubated with 100 nm SAHENTA-FITC for 1 h. Fluorescence was then determined with a flow cytometer. Panel A shows histograms from ponatinib-treated cells from one of three independent experiments. Stained untreated cells are shown in light gray, stained ponatinib-treated cells in white, and unstained cells in dark gray histograms. Panel B shows the results plotted from such experiments with all five TKIs as % Control of untreated cells. The symbols (●, ▴, and ■) represent experiments done on different days. Values with mean ± S.D. are shown as scatter plot.
Effects of Sequencing of Administration of BCR-ABL TKIs and Uridine on Retention of [3H]Uridine in A549 Cells
The sequence of administration of BCR-ABL TKIs and uridine was examined in cultured human lung cancer A549 cells to see if changes in uridine uptake occurred when the two agents were added separately in sequence or simultaneously together. [3H]Uridine uptake was measured in A549 cells that were either treated without or with 10 μm of each BCR-ABL TKI for 15 min before, during, or after exposure to radiolabeled uridine for 15 min. Results of sequencing of administration on nucleoside accumulation are shown Fig. 6, A–E. The greatest inhibition of uridine accumulation occurred when the BCR-ABL TKIs were combined with uridine during simultaneous exposures and nilotinib was the most potent inhibitor.
FIGURE 6.

Effects of different modes of administration of [3H]uridine with BCR-ABL TKIs on accumulation of [3H]uridine in A549 cells. Panels A–E show [3H]uridine (1 μm) uptake in A549 that were either treated without or with 10 μm of each of the BCR-ABL TKIs (bosutinib, dasatinib, imatinib, or ponatinib) or 5 μm nilotinib for 15 min before, during, or after exposure to [3H]uridine for 15 min as described under ”Experimental Procedures.“ The symbols (●, ▴, and ■) represent experiments done on different days. Values plotted are % control values obtained in the absence of additives and values from three experiments each conducted with three replicates are shown in as scatter plots in each panel.
Discussion
In three of our recent studies (29–31) we presented results on interactions of epidermal growth factor receptor, vascular endothelial growth factor receptor, and multitargeted TKIs with human nucleoside and nucleobase transporters. We hypothesized that such interactions could be due to structural similarities of TKIs to potent and specific inhibitors of hENT1, such as nitrobenzylthioinosine and dipyridamole. We also showed competitive inhibition of uridine uptake by these TKIs in yeast producing recombinant hENT1. These studies suggested that TKIs, when combined with nucleoside or nucleobase chemotherapy, are likely to interfere with nucleoside chemotherapy. In this study we examined effects of BCR-ABL TKIs on five hNTs and also evaluated the role of two amino acid residues of hENT1 that were previously shown to be involved in uridine and dipyridamole binding on interaction with these TKIs.
Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib inhibited transport of uridine by recombinant hNTs produced individually in yeast to different extents. Nilotinib was the most potent inhibitor of hENT1, whereas its inhibition of hENT2 was poor and it had no effect on hCNTs. In contrast imatinib inhibited hCNT2 potently at low micromolar concentrations and also inhibited hCNT1/3 and hENT1, although to lesser extents. Bosutinib inhibited hENT1 and 2, dasatinib inhibited hENT1 and hCNT1, and ponatinib inhibited hENT1 and -2 and hCNT1, -2, and -3.
BCR-ABL TKIs inhibited uridine uptake competitively in hENT1-producing yeast cells thereby suggesting competition between uridine and bosutinib, dasatinib, imatinib, nilotinib, or ponatinib for interaction with hENT1. However, such inhibition could also be achieved by binding to a separate allosteric site that prevents binding of substrate (i.e. permeant) to its site (32).
Four mutants of hENT1 involving two amino acid residues (L442I, L442T, M33A, and M33A/L442I (24)) were studied in dose-response experiments to evaluate the importance of these residues for interaction with BCR-ABL TKIs. These mutations are in residues 33 of TM1 and residue 442 of TM11 and experimental evidence (23) suggests that TM1 and TM11 are in close proximity to each other. Conservative substitutions of Leu in hENT1 had minor effects on uridine transport activity, whereas substitution of Thr at residue 442 resulted in a 10-fold higher Km value for uridine. Earlier studies (24) implicated Met residue 33 in TM1 and Leu residue 442 in TM11 in interaction with dipyridamole and uridine, respectively. hENT1 binds dipyridamole with high affinity and competitively with uridine, suggesting that the dipyridamole (i.e. inhibitor) binding site overlaps with the uridine (i.e. permeant) binding site of hENT1 (33–35). Dipyridamole resistance was seen when Met33 was changed to Ala in TM1 of hENT1, thereby identifying it as a key residue for dipyridamole interaction with hENT1.
Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib inhibited transport of uridine by the conservative L442I mutant much more potently than by Leu442 containing hENT1, indicating that changing Leu to Ile improved the binding of the BCR-ABL TKIs to hENT1. Earlier studies (24) had shown that the L442I mutant exhibited minor changes in uridine transport activity but greatly reduced adenosine transport activity. The IC50 value for nilotinib with the L442T mutant was nearly four times higher compared with the IC50 value for hENT1, whereas for imatinib it was ¼ of the IC50 value for hENT1. All five BCR-ABL TKIs showed reduced interaction with the M33A mutant, which has 1000-fold greater resistance to dipyridamole with an unchanged uridine Km value. In the double mutant M33A/L442I the IC50 values for the BCR-ABL TKIs were similar or lower than that of hENT1. Taken together, these results suggest that BCR-ABL TKIs interact with the dipyridamole binding site of hENT1 and that Met33 of TM1 is a critical residue involved in dipyridamole interaction with hENT1. In addition, Leu442 in TM11 was also important for interaction with BCR-ABL TKIs. It appears that BCR-ABL TKIs are interacting with both dipyridamole and uridine binding sites in an overlapping manner.
Involvement of a critical residue, Met218 was shown in hydrogen bonding interactions of dasatinib, imatinib, nilotinib, and ponatinib with the Abl kinase domain in x-ray crystallographic studies (36–40). In addition, involvement of Ile and Leu residues was also shown in forming a hydrophobic pocket in the Abl kinase domain. Based on Abl kinase and our results we speculate that, BCR-ABL kinase inhibitors may be interacting with hENT1 through hydrogen bonding with Met33 and hydrophobic interactions with Leu442.
In uridine uptake experiments with CEM cells, which exhibit a single hNT activity (i.e. hENT1), nilotinib was the most potent inhibitor followed by bosutinib, ponatinib, dasatinib, and imatinib. The purine nucleoside analogs cladribine and fludarabine are used clinically for treatment of lymphoid malignancies and in combination with cytarabine are used routinely in relapsed or refractory acute myelogenous leukemia (41). Accumulation of [3H]cytarabine, [3H]cladribine, or [3H]fludarabine was inhibited in CEM cells by all five BCR-ABL TKIs but to different extents. Earlier results (26) showed inhibition of accumulation of fludarabine triphosphate in T-lymphocytes exposed to imatinib and fludarabine simultaneously and similar results were later shown by Naud et al. (20) with imatinib and nilotinib on cytarabine uptake in chronic myelogenous leukemia cell lines. Our results with first, second, and third generation BCR-ABL TKIs in a T-lymphocytic cell line are consistent, and illustrate potential difficulties in administering BCR-ABL TKIs with nucleoside drugs in combination therapies. Our sequencing studies, which were undertaken with uridine, a physiologic nucleoside permeant of the five hNTs used in this work, showed that the greatest inhibition of uptake occurred when the BCR-ABL TKI and uridine were administered together except with nilotinib and ponatinib for which the inhibitions were similar in conditions when the TKI was given before the nucleoside or together with the nucleoside. Attention should be paid to this during combination chemotherapy trials using ponatinib or nilotinib. In a recent clinical study, addition of cytarabine to high dose imatinib in treatment of chronic myelogenous leukemia showed no improvement in the major molecular response (42).
Results from cell surface staining of hENT1 by SAHENTA-FITC in flow cytometry experiments showed that the BCR-ABL TKIs caused a reduction in cell surface staining intensity of hENT1, thus suggesting that these TKIs reduced cell surface hENT1 abundance in addition to their direct inhibition of transport activity. Such loss of cell surface expression of hENT1 due to BCR-ABL TKI treatment would further enhance the inhibitory effects of BCR-ABL TKIs on hENT1 activity thereby decreasing the resulting cytotoxicity of nucleoside drugs. It is also possible that long-term treatment with BCR-ABL TKIs could lead to resistance to nucleoside drugs due to reduced cell surface expression of hENT1.
In summary, we have shown that all five BCR-ABL inhibitors inhibit hENT1, a ubiquitous hNT that is necessary for activity of many nucleoside chemotherapy drugs, and also other classes of nucleoside transporters to varying extents. Ponatinib inhibited five nucleoside transporters with different abilities. Nilotinib inhibited hENT1 the most. Incubation of CEM cells with these BCR-ABL inhibitors resulted in decreased accumulation of uridine, cytarabine, cladribine, and fludarabine. In addition, these inhibitors caused a decrease in cell surface expression of hENT1 in CEM cells. These results suggest that at concentrations achieved in plasma, ponatinib and bosutinib may exert their effects on hENT1 by down-regulating surface abundance of hENT1 over prolonged exposure times, whereas imatinib and dasatinib may have less effect on hENT1 activity and levels. Therefore imatinib may be safely combined with cytarabine or other nucleoside drugs, whereas other BCR-ABL kinase inhibitors (e.g. nilotinib, ponatinib and bosutinib) may not and should not be combined clinically. In experiments with mutant hENT1, we showed for the first time interaction of Met33 (a residue that is involved in dipyridamole binding) with BCR-ABL inhibitors because a mutation of this residue to Ala resulted in reduced interaction with the kinase inhibitors. Mutation of Leu442 of hENT1 to Ile resulted in enhanced interaction with the kinase inhibitors. Structure activity studies of this nature could help in the design of newer kinase inhibitors with reduced interaction with human nucleoside transporters.
Experimental Procedures
Yeast Strains and Media
Yeast strains were generated by transformation of the yeast/Escherichia coli shuttle vector pYPGE15 into KTK and fui:TRP1 and were separately transformed with plasmids (pYPhENT1, pYPhENT2, pYPhCNT1, pYPhCNT2, or pYPhCNT3) encoding hNTs (hENT1 or various hENT1 mutants, hENT2, hCNT1, hCNT2, or hCNT3, respectively), as described elsewhere (43, 44). These transfected yeast strains expressed each of the hNTs in isolation. The mutants of hENT1 (i.e. M33A, L442I, L442T, M33A/L442I) were produced and characterized earlier (24, 45). Yeast strains were maintained in complete minimal medium containing 0.67% yeast nitrogen base amino acids (as required to maintain auxotrophic selection) and 2% glucose (complete minimal medium/Glc).
Nucleoside Transport in S. cerevisiae
Yeast cells containing pYPhENT1 or -2, pYPhCNT1, -2, or -3, or plasmids encoding various hENT1 mutant transporters were grown in complete minimal medium/Glc to an absorbance at 600 nm (A600) of 0.5 to 1.0/ml, washed twice, and resuspended to A600 of 4.0/ml.
Concentration-effect experiments were conducted at ambient temperature with graded concentrations (0–0.3 mm) of individual BCR-ABL TKIs in the presence of 1 μm [3H]uridine in yeast growth media at pH 7.4. Yeast were also incubated with 1 μm [3H]uridine in the absence of BCR-ABL TKIs as positive controls and values from these cultures were used to calculate the “% Control” observed in the presence of various BCR-ABL TKIs. Uridine self-inhibition was used as an internal control to define maximum inhibition of mediated transport. Transport reactions were initiated by rapid mixing of 50-μl yeast suspensions with or without graded concentrations of individual BCR-ABL TKIs with 50 μl of 2 × [3H]nucleoside in 96-well microtiter plates. Yeast cells were collected on filter mats using a Micro96 Cell Harvester and rapidly washed with deionized water. Individual filter circles, corresponding to wells of the microtiter plates, were removed from filter mats with forceps and transferred to vials for quantification of radioactivity by scintillation counting.
Data were subjected to nonlinear regression analysis using GraphPad Prism software (version 4.05; GraphPad Software Inc., San Diego, CA) to obtain the concentration of BCR-ABL TKI that inhibited growth of treated cells by 50% relative to that of untreated cells (IC50 values). IC50 values were determined from concentration-effect curves. Each experiment was conducted with nine concentrations and six replicates per concentration and was repeated three times.
Nucleoside Transport Inhibition Assays in CEM and A549 Cells
The human non-small cell lung cancer A549 cell line was obtained from American Type Culture Collection. The human CCRF-CEM leukemia, hereafter termed CEM/hENT1, cell line was obtained from William T. Beck (University of Illinois at Chicago, Chicago, IL), and cells were used between passages 15 and 30. Cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine. All cultures were kept at 37 °C in 5% CO2, 95% air and subcultured at 2–3-day intervals to maintain exponential growth. Transport and cytotoxicity experiments were conducted with cells in the exponential growth phase.
CEM cells were previously shown to possess a single nucleoside transport activity (i.e. hENT1) (46). Inhibition of 1 μm [3H]uridine uptake (30 s) was measured at ambient temperature in CEM cells in transport buffer (pH 7.4) containing 20 mm Tris, 3 mm K2HPO4, and 5 mm glucose with 144 mm NaCl in the absence or presence of graded concentrations (0–0.1 mm) of individual BCR-ABL TKIs. At the end of uptake intervals cells were spun down quickly through transport oil and permeant-containing solutions were removed by aspiration. Tubes were quickly rinsed twice with water after which the oil was aspirated and cell pellets were solubilized with 5% Triton X-100. Radioactivity in solubilized extracts was measured by liquid scintillation counting. Uptake values were expressed as pmol/106 cells and converted to % control activity and graphs were generated using GraphPad Prism. Each experiment was conducted three times with triplicate measurements for each condition.
Inhibition of uptake and accumulation of 1 μm [3H]cladribine, [3H]cytarabine, or [3H]fludarabine was assessed by exposing CEM cells to 10 μm bosutinib, dasatinib, imatinib, ponatinib, and dipyridamole or 2 μm nilotinib for 1 or 60 min after which uptake was terminated by spinning cells though transport oil. Cells were washed by centrifugation and cell-associated radioactivity was determined by scintillation counting. Each experiment was conducted with four replicates per condition and repeated at least three times.
For sequencing of exposure to nucleosides and TKIs, uptake experiments were conducted in A549 cells as follows. In the first set of experiments, A549 cells were incubated in buffer without drug for 15 min after which 10 μm [3H]uridine was added and uptake was measured for 15 min. In the second set of experiments, cells were incubated with 10 μm bosutinib, dasatinib, imatinib, and ponatinib or 5 μm nilotinib for 15 min after which the drug was removed and uptake of 10 μm [3H]uridine was measured for 15 min. In the third set of experiments, 10 μm [3H]uridine was added for 15 min after which media was removed and bosutinib, dasatinib, imatinib, ponatinib, or nilotinib was added. In the last set of experiments, cells were incubated in buffer without any drug for 15 min after which the buffer was removed and 10 μm [3H]uridine was added together with bosutinib, dasatinib, imatinib, ponatinib, or nilotinib and incubated for 15 min. At the end of these time points, media was removed and cells were processed for radioactivity as described above.
Staining and Flow Cytometric Analysis of hENT1 Abundance on CEM Cells with SAHENTA-FITC
Synthesis and use of a fluorescent probe for evaluation of the relative abundance of cell surface hENT1 sites was described earlier (28). CEM cells that were treated for 24 h with 0 or 1 μm bosutinib, dasatinib, imatinib, nilotinib, or ponatinib were washed free of growth medium, resuspended in PBS to a density of 2.5–105 cells/ml, and incubated at ambient temperature for 1 h with either no added compound (negative control) or 100 nm SAHENTA-FITC. Cells were then collected by centrifugation, washed twice with PBS, and analyzed for fluorescence by analytical flow cytometry.
Author Contributions
D. W. and M. K. conducted transport assays, cytotoxicity studies, and yeast inhibition experiments. V. L. D. designed and analyzed experiments and wrote the manuscript. M. B. S. and C. E. C. critically reviewed the manuscript. All authors approved the final version.
Acknowledgment
We acknowledge Delores Mowles for technical help.
This work was supported by grants from the Alberta Cancer Foundation and Alberta Innovates Health Solutions (to M. B. S.). V. L. D. and M. B. S. are listed on a patent “Combination treatments for cancer” held by Alberta Health Services.
- TKIs
- tyrosine kinase inhibitors
- BCR
- breakpoint cluster region
- ABL
- Abelson murine leukemia
- NTs
- nucleoside transporters
- h
- human
- ENT
- equilibrative nucleoside transporter
- CNT
- concentrative nucleoside transporter
- SAHENTA-FITC
- 5′-S-[2-(6-aminohexanamido)]ethyl-6-N-(4-nitrobenzyl)-5′-thioadenosine-fluorescein-5-yl isothiocyanate.
References
- 1. Ohanian M., Cortes J., Kantarjian H., and Jabbour E. (2012) Tyrosine kinase inhibitors in acute and chronic leukemias. Expert. Opin. Pharmacother. 13, 927–938 [DOI] [PubMed] [Google Scholar]
- 2. Druker B. J., Sawyers C. L., Kantarjian H., Resta D. J., Reese S. F., Ford J. M., Capdeville R., and Talpaz M. (2001) Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N. Engl. J. Med. 344, 1038–1042 [DOI] [PubMed] [Google Scholar]
- 3. Jabbour E., Parikh S. A., Kantarjian H., and Cortes J. (2011) Chronic myeloid leukemia: mechanisms of resistance and treatment. Hematol. Oncol. Clin. North Am. 25, 981–995, v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ravandi F. (2011) Managing Philadelphia chromosome-positive acute lymphoblastic leukemia: role of tyrosine kinase inhibitors. Clin. Lymphoma Myeloma Leuk. 11, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang K., and Fu L. W. (2015) Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: a review. Crit. Rev. Oncol. Hematol. 93, 277–292 [DOI] [PubMed] [Google Scholar]
- 6. Brendel C., Scharenberg C., Dohse M., Robey R. W., Bates S. E., Shukla S., Ambudkar S. V., Wang Y., Wennemuth G., Burchert A., Boudriot U., and Neubauer A. (2007) Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 21, 1267–1275 [DOI] [PubMed] [Google Scholar]
- 7. Burger H., van Tol H., Boersma A. W., Brok M., Wiemer E. A., Stoter G., and Nooter K. (2004) Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 104, 2940–2942 [DOI] [PubMed] [Google Scholar]
- 8. Hegedus C., Ozvegy-Laczka C., Apáti A., Magócsi M., Német K., Orfi L., Kéri G., Katona M., Takáts Z., Váradi A., Szakács G., and Sarkadi B. (2009) Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 158, 1153–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hiwase D. K., Saunders V., Hewett D., Frede A., Zrim S., Dang P., Eadie L., To L. B., Melo J., Kumar S., Hughes T. P., and White D. L. (2008) Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin. Cancer Res. 14, 3881–3888 [DOI] [PubMed] [Google Scholar]
- 10. Illmer T., Schaich M., Platzbecker U., Freiberg-Richter J., Oelschlägel U., von Bonin M., Pursche S., Bergemann T., Ehninger G., and Schleyer E. (2004) P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 18, 401–408 [DOI] [PubMed] [Google Scholar]
- 11. Mahon F. X., Belloc F., Lagarde V., Chollet C., Moreau-Gaudry F., Reiffers J., Goldman J. M., and Melo J. V. (2003) MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood 101, 2368–2373 [DOI] [PubMed] [Google Scholar]
- 12. Mahon F. X., Hayette S., Lagarde V., Belloc F., Turcq B., Nicolini F., Belanger C., Manley P. W., Leroy C., Etienne G., Roche S., and Pasquet J. M. (2008) Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 68, 9809–9816 [DOI] [PubMed] [Google Scholar]
- 13. Thomas J., Wang L., Clark R. E., and Pirmohamed M. (2004) Active transport of imatinib into and out of cells: implications for drug resistance. Blood 104, 3739–3745 [DOI] [PubMed] [Google Scholar]
- 14. White D. L., Saunders V. A., Dang P., Engler J., Zannettino A. C., Cambareri A. C., Quinn S. R., Manley P. W., and Hughes T. P. (2006) OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 108, 697–704 [DOI] [PubMed] [Google Scholar]
- 15. Yamakawa Y., Hamada A., Shuto T., Yuki M., Uchida T., Kai H., Kawaguchi T., and Saito H. (2011) Pharmacokinetic impact of SLCO1A2 polymorphisms on imatinib disposition in patients with chronic myeloid leukemia. Clin. Pharmacol. Ther. 90, 157–163 [DOI] [PubMed] [Google Scholar]
- 16. Yamakawa Y., Hamada A., Uchida T., Sato D., Yuki M., Hayashi M., Kawaguchi T., and Saito H. (2014) Distinct interaction of nilotinib and imatinib with P-glycoprotein in intracellular accumulation and cytotoxicity in CML cell line K562 cells. Biol. Pharm. Bull. 37, 1330–1335 [DOI] [PubMed] [Google Scholar]
- 17. Sen R., Natarajan K., Bhullar J., Shukla S., Fang H. B., Cai L., Chen Z. S., Ambudkar S. V., and Baer M. R. (2012) The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol. Cancer Ther. 11, 2033–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang M., Wang Y., Collins M., Gu J. J., Mitchell B. S., and Graves L. M. (2002) Inhibition of nucleoside transport by p38 MAPK inhibitors. J. Biol. Chem. 277, 28364–28367 [DOI] [PubMed] [Google Scholar]
- 19. Leisewitz A. V., Zimmerman E. I., Jones S. Z., Yang J., and Graves L. M. (2008) Imatinib-resistant CML cells have low ENT activity but maintain sensitivity to gemcitabine. Nucleosides Nucleotides Nucleic Acids 27, 779–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naud J. S., Ghani K., de Campos-Lima P. O., and Caruso M. (2012) Nilotinib and imatinib inhibit cytarabine cellular uptake: implications for combination therapy. Leuk. Res. 36, 1311–1314 [DOI] [PubMed] [Google Scholar]
- 21. Damaraju V. L., Sawyer M. B., Mackey J. R., Young J. D., and Cass C. E. (2009) Human nucleoside transporters: biomarkers for response to nucleoside drugs. Nucleosides Nucleotides Nucleic Acids 28, 450–463 [DOI] [PubMed] [Google Scholar]
- 22. Zhang J., Visser F., King K. M., Baldwin S. A., Young J. D., and Cass C. E. (2007) The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 26, 85–110 [DOI] [PubMed] [Google Scholar]
- 23. Sundaram M., Yao S. Y., Ingram J. C., Berry Z. A., Abidi F., Cass C. E., Baldwin S. A., and Young J. D. (2001) Topology of a human equilibrative, nitrobenzylthioinosine (NBMPR)-sensitive nucleoside transporter (hENT1) implicated in the cellular uptake of adenosine and anti-cancer drugs. J. Biol. Chem. 276, 45270–45275 [DOI] [PubMed] [Google Scholar]
- 24. Visser F., Baldwin S. A., Isaac R. E., Young J. D., and Cass C. E. (2005) Identification and mutational analysis of amino acid residues involved in dipyridamole interactions with human and Caenorhabditis elegans equilibrative nucleoside transporters. J. Biol. Chem. 280, 11025–11034 [DOI] [PubMed] [Google Scholar]
- 25. Galmarini C. M., Mackey J. R., and Dumontet C. (2001) Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 15, 875–890 [DOI] [PubMed] [Google Scholar]
- 26. Woodahl E. L., Wang J., Heimfeld S., Ren A. G., and McCune J. S. (2008) Imatinib inhibition of fludarabine uptake in T-lymphocytes. Cancer Chemother. Pharmacol. 62, 735–739 [DOI] [PubMed] [Google Scholar]
- 27. Visser F., Vickers M. F., Ng A. M., Baldwin S. A., Young J. D., and Cass C. E. (2002) Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J. Biol. Chem. 277, 395–401 [DOI] [PubMed] [Google Scholar]
- 28. Robins M. J., Peng Y., Damaraju V. L., Mowles D., Barron G., Tackaberry T., Young J. D., and Cass C. E. (2010) Improved syntheses of 5′-S-(2-aminoethyl)-6-N-(4-nitrobenzyl)-5′-thioadenosine (SAENTA), analogues, and fluorescent probe conjugates: analysis of cell-surface human equilibrative nucleoside transporter 1 (hENT1) levels for prediction of the antitumor efficacy of gemcitabine. J. Med. Chem. 53, 6040–6053 [DOI] [PubMed] [Google Scholar]
- 29. Damaraju V. L., Kuzma M., Cass C. E., and Sawyer M. B. (2015) Inhibition of sodium-independent and sodium-dependent nucleobase transport activities by tyrosine kinase inhibitors. Cancer Chemother. Pharmacol. 76, 1093–1098 [DOI] [PubMed] [Google Scholar]
- 30. Damaraju V. L., Kuzma M., Mowles D., Cass C. E., and Sawyer M. B. (2015) Interactions of multitargeted kinase inhibitors and nucleoside drugs: achilles heel of combination therapy? Mol. Cancer Ther. 14, 236–245 [DOI] [PubMed] [Google Scholar]
- 31. Damaraju V. L., Scriver T., Mowles D., Kuzma M., Ryan A. J., Cass C. E., and Sawyer M. B. (2014) Erlotinib, gefitinib, and vandetanib inhibit human nucleoside transporters and protect cancer cells from gemcitabine cytotoxicity. Clin. Cancer Res. 20, 176–186 [DOI] [PubMed] [Google Scholar]
- 32. Segel I. H. (1975) Enzyme Kinetics, Wiley-Interscience, New York [Google Scholar]
- 33. Jarvis S. M. (1986) Nitrobenzylthioinosine-sensitive nucleoside transport system: mechanism of inhibition by dipyridamole. Mol. Pharmacol. 30, 659–665 [PubMed] [Google Scholar]
- 34. Jarvis S. M., McBride D., and Young J. D. (1982) Erythrocyte nucleoside transport: asymmetrical binding of nitrobenzylthioinosine to nucleoside permeation sites. J. Physiol. 324, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paterson A. R., Lau E. Y., Dahlig E., and Cass C. E. (1980) A common basis for inhibition of nucleoside transport by dipyridamole and nitrobenzylthioinosine? Mol. Pharmacol. 18, 40–44 [PubMed] [Google Scholar]
- 36. Asaki T., Sugiyama Y., Hamamoto T., Higashioka M., Umehara M., Naito H., and Niwa T. (2006) Design and synthesis of 3-substituted benzamide derivatives as Bcr-Abl kinase inhibitors. Bioorg. Med. Chem. Lett. 16, 1421–1425 [DOI] [PubMed] [Google Scholar]
- 37. Breccia M., and Alimena G. (2010) Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia. Leuk. Res. 34, 129–134 [DOI] [PubMed] [Google Scholar]
- 38. Eck M. J., and Manley P. W. (2009) The interplay of structural information and functional studies in kinase drug design: insights from BCR-Abl. Curr. Opin. Cell Biol. 21, 288–295 [DOI] [PubMed] [Google Scholar]
- 39. Manley P. W., Cowan-Jacob S. W., and Mestan J. (2005) Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia. Biochim. Biophys. Acta 1754, 3–13 [DOI] [PubMed] [Google Scholar]
- 40. Manley P. W., Stiefl N., Cowan-Jacob S. W., Kaufman S., Mestan J., Wartmann M., Wiesmann M., Woodman R., and Gallagher N. (2010) Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg. Med. Chem. 18, 6977–6986 [DOI] [PubMed] [Google Scholar]
- 41. Robak P., and Robak T. (2013) Older and new purine nucleoside analogs for patients with acute leukemias. Cancer Treat. Rev. 39, 851–861 [DOI] [PubMed] [Google Scholar]
- 42. Thielen N., van der Holt B., Verhoef G. E., Ammerlaan R. A., Sonneveld P., Janssen J. J., Deenik W., Falkenburg J. H., Kersten M. J., Sinnige H. A., Schipperus M., Schattenberg A., van Marwijk Kooy R., Smit W. M., et al. (2013) High-dose imatinib versus high-dose imatinib in combination with intermediate-dose cytarabine in patients with first chronic phase myeloid leukemia: a randomized phase III trial of the Dutch-Belgian HOVON study group. Ann. Hematol. 92, 1049–1056 [DOI] [PubMed] [Google Scholar]
- 43. Vickers M. F., Kumar R., Visser F., Zhang J., Charania J., Raborn R. T., Baldwin S. A., Young J. D., and Cass C. E. (2002) Comparison of the interaction of uridine, cytidine, and other pyrimidine nucleoside analogues with recombinant human equilibrative nucleoside transporter 2 (hENT2) produced in Saccharomyces cerevisiae. Biochem. Cell Biol. 80, 639–644 [DOI] [PubMed] [Google Scholar]
- 44. Zhang J., Visser F., Vickers M. F., Lang T., Robins M. J., Nielsen L. P., Nowak I., Baldwin S. A., Young J. D., and Cass C. E. (2003) Uridine binding motifs of human concentrative nucleoside transporters 1 and 3 produced in Saccharomyces cerevisiae. Mol. Pharmacol. 64, 1512–1520 [DOI] [PubMed] [Google Scholar]
- 45. Paproski R. J., Visser F., Zhang J., Tackaberry T., Damaraju V., Baldwin S. A., Young J. D., and Cass C. E. (2008) Mutation of Trp29 of human equilibrative nucleoside transporter 1 alters affinity for coronary vasodilator drugs and nucleoside selectivity. Biochem. J. 414, 291–300 [DOI] [PubMed] [Google Scholar]
- 46. Crawford C. R., Ng C. Y., Ullman B., and Belt J. A. (1990) Identification and reconstitution of the nucleoside transporter of CEM human leukemia cells. Biochim. Biophys. Acta 1024, 289–297 [DOI] [PubMed] [Google Scholar]