

Abstract
The unique cell wall of mycobacteria is essential to their viability and the target of many clinically used anti-tuberculosis drugs and inhibitors under development. Despite intensive efforts to identify the ligase(s) responsible for the covalent attachment of the two major heteropolysaccharides of the mycobacterial cell wall, arabinogalactan (AG) and peptidoglycan (PG), the enzyme or enzymes responsible have remained elusive. We here report on the identification of the two enzymes of Mycobacterium tuberculosis, CpsA1 (Rv3267) and CpsA2 (Rv3484), responsible for this function. CpsA1 and CpsA2 belong to the widespread LytR-Cps2A-Psr (LCP) family of enzymes that has been shown to catalyze a variety of glycopolymer transfer reactions in Gram-positive bacteria, including the attachment of wall teichoic acids to PG. Although individual cpsA1 and cpsA2 knock-outs of M. tuberculosis were readily obtained, the combined inactivation of both genes appears to be lethal. In the closely related microorganism Corynebacterium glutamicum, the ortholog of cpsA1 is the only gene involved in this function, and its conditional knockdown leads to dramatic changes in the cell wall composition and morphology of the bacteria due to extensive shedding of cell wall material in the culture medium as a result of defective attachment of AG to PG. This work marks an important step in our understanding of the biogenesis of the unique cell envelope of mycobacteria and opens new opportunities for drug development.
Keywords: cell wall, cryo-electron microscopy, Mycobacterium tuberculosis, outer membrane, peptidoglycan, polysaccharide, arabinogalactan, ligase
Introduction
The compositional and architectural complexity of the mycobacterial cell envelope distinguishes species of the Mycobacterium genus from other prokaryotes. It is the basis of many of the physiological and pathogenic features of mycobacteria and the site of susceptibility and resistance to many anti-tuberculosis drugs (1, 2). With this in mind, considerable effort has been placed on investigating the cell envelope structure and its biosynthesis to identify attractive drug targets. The mycobacterial cell envelope is made up of three major segments: the plasma membrane, the cell wall core and the outermost layer. The cell wall core, which is essential for viability, consists of peptidoglycan (PG)5 in covalent attachment via phosphoryl-N-acetylglucosaminosyl-rhamnosyl linkage units (P-GlcNAc-Rha)with the heteropolysaccharide arabinogalactan (AG); AG is in turn esterified at its non-reducing ends to long-chain (C70–C90) mycolic acids (Fig. 1). The latter form the bulk of the inner leaflet of the outer membrane (also referred to as mycomembrane), with the outer layer consisting of a variety of non-covalently attached (glyco)lipids, polysaccharides, lipoglycans, and proteins (3–7).
FIGURE 1.
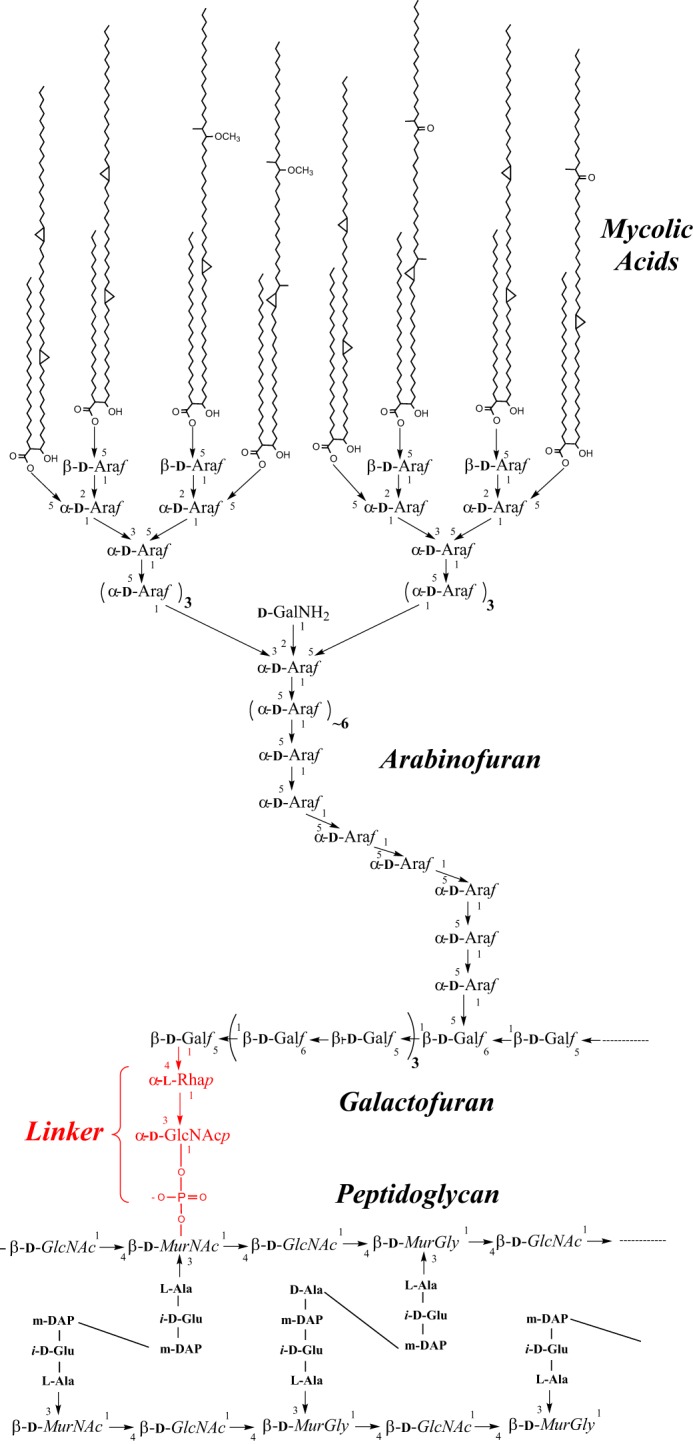
The M. tuberculosis cell wall core. PG from M. tuberculosis is composed of linear chains of N-acetyl-α-d-glucosamine and modified muramic acid substituted with peptide side chains that are heavily cross-linked (70–80%), providing added structural integrity to the bacterium. AG is attached to PG through a phosphodiester link to position 6 of some of the Mur residues. The specific linker unit ensuring its covalent attachment to PG is shown in red. One arabinan chain is shown here attached to the galactan domain. The characteristic Ara6 non-reducing termini of the arabinan domain of AG serve as the anchoring points for the mycolates. Only one arabinan chain is shown for clarity.
PG from Mycobacterium tuberculosis has been classified as A1γ as has that of Escherichia coli and Bacillus spp. (8), and its synthesis is for the most part similar to that in other bacteria (9). The synthesis of AG is initiated in the cytoplasm on a decaprenyl phosphate (Dec-P) carrier lipid with formation of the linker unit followed by the addition of Galf and Araf residues (10–12). Many of the enzymes involved in this process have been identified (7). A WecA-like transferase encoded by Rv1302 in the genome of M. tuberculosis H37Rv transfers GlcNAc 1-phosphate to Dec-P to form Dec-P-P-GlcNAc (GL-1). This step is followed by the attachment of a rhamnosyl residue from dTDP-Rha to the 3-position of GlcNAc in a reaction catalyzed by WbbL1 to form GL-2 (Dec-P-P-GlcNAc-Rha), the “linker unit” (Fig. 1). The galactan is biosynthesized in the cytoplasm by two bifunctional galactosyltransferases, with the first two Galf residues added to the linker unit by the galactosyltransferase GlfT1 (Rv3782) and the remaining alternating 5- and 6-linked Galf residues added by GlfT2 (Rv3808c). The arabinosylation of AG takes place on the periplasmic side of the plasma membrane catalyzed by membrane-associated decaprenyl phosphate arabinose-dependent glycosyltransferases (7). It is thought that the GlcNAc of the linker unit of the mature AG next forms a 1-O-phosphoryl linkage with the 6-position of a MurNAc residue of PG (12) and that this transfer reaction requires newly synthesized PG undergoing concomitant cross-linking (13). The fact that a Corynebacterium glutamicum mutant deficient in the arabinan domain of AG was found to assemble a simplified cell wall consisting of the galactan chain of AG attached to PG suggests, however, that neither the arabinosylation of AG nor its mycolylation are prerequisites for its attachment to PG (14, 15). Despite intensive efforts to identify the ligase(s) responsible for the covalent attachment of AG to PG, no enzymes had yet been categorically implicated in the ligation process.
Wall teichoic acids (WTA), in place of AG, are more classically found in covalent attachment to PG in the cell walls of many Gram-positive bacteria (16). Despite the fundamental structural differences that exist between these anionic glycopolymers and AG, the structure of the AG-PG linker shares similarity with that involved in the covalent attachment of WTA to PG (e.g. -N-acetylmannosaminyl-N-acetylglucosaminyl-phosphoryl- in Bacillus subtilis). Much like AG, the biosynthesis of WTA is initiated on a polyisoprenoid carrier (undecaprenyl phosphate) on the inner surface of the cytoplasmic membrane. The transfer of the two first sugar residues (N-acetylglucosaminyl and N-acetylmannosaminyl) from their nucleotide donors is followed by the assembly of the rest of the linker and the teichoic acid proper, still on the lipid carrier. The WTA precursors are then exported by an ABC (ATP-binding cassette) transporter to the cell surface, where the WTA are finally transferred from their lipid-linked precursor to the C-6 hydroxyl of MurNAc within PG (16). Recently, enzymes of the widespread LytR-Cps2A-Psr (LCP) family (17) have been proposed to carry out this function in B. subtilis and Staphylococcus aureus (18–21). LCP-like proteins have further been involved in the transfer of capsular polysaccharides to PG and the glycosylation of cell wall-associated proteins in a variety of other Gram-positive organisms (22–29). These reports led us to investigate the possible involvement and therapeutic potential of the three LCP-like proteins encoded by the M. tuberculosis genome in the ligation of AG to PG. Our results highlight the participation of two of these proteins in the phosphotransferase reaction leading to the ligation of AG and PG, a crucial step in the assembly of the entire complex cell wall of M. tuberculosis.
Results
Identification of LCP-like Proteins in the Genome of M. tuberculosis
LCP proteins share a conserved predicted secondary structure consisting of an N-terminal cytoplasmic tail, one to three transmembrane (TM) regions and an extracellular C-terminal region carrying the LytR-Cps2A-Psr domain (17). LCP-encoding genes further tend to cluster with the biosynthetic genes of the glycopolymers of which they catalyze the transfer (26). A search for proteins displaying the characteristic secondary structure and signature motif of LCP proteins in the translated genome of M. tuberculosis H37Rv yielded three candidates: Rv0822c, Rv3267 (herein renamed CpsA1), and Rv3484 (herein renamed CpsA2). The three proteins share between 21 and 29% identity (37–50% similarity) with LCP proteins from S. aureus (MsrR, SA0908, and SA2103), Streptococcus pneumoniae (CpsA2), and B. subtilis (TagT, TagU, and TagV) (Fig. 2A). Whereas both CpsA1 and CpsA2 are predicted to display a short (13–24-residue) N-terminal cytoplasmic domain followed by a single TM segment and a periplasmic catalytic domain (Fig. 2B), Rv0822c displays a significantly longer N-terminal domain (175 residues) and lacks a clear TM helix based on TMHMM server version 2.0 predictions. Importantly, cpsA1 maps to an AG biosynthetic gene cluster adjacent to wbbL1 (Rv3265c) and rmlD (Rv3266c) involved, respectively, in the formation of GL-2 and dTDP-Rha. This gene was reported to be induced by ethambutol (an inhibitor of AG synthesis) (30) and is also the most conserved candidate across the Mycobacterium genus and other AG-producing Actinobacteria, including C. glutamicum. CpsA2, in contrast, is apparently not well conserved in rapidly growing Mycobacterium species, and Rv0822c is a pseudogene in Mycobacterium leprae.
FIGURE 2.

Sequence and transmembrane topology of M. tuberculosis LCP protein candidates. A, alignment of the LytR-CpsA-Psr domains of LCP proteins and LCP protein candidates from M. tuberculosis, C. glutamicum, S. aureus, B. subtilis, and Streptococcus pneumoniae using Clustal Omega. Amino acids that are invariant in the alignment are colored white with a red background; homologous residues are in blue. Green diamonds, charged residues that contact the pyrophosphate headgroup of bound octaprenyl pyrophosphate in the crystal structure of CpsA2 from S. pneumoniae; orange diamonds, residues that coordinate a magnesium ion; crosses, conserved hydrophobic residues involved in the binding of the polyisoprenoid chain. B, transmembrane topology of Rv3267 (CpsA1), Rv3484 (CpsA2) and Rv0822c. The models were generated using TOPO2. C, topology of the C-terminal LCP domains of CpsA1, CpsA2, and Rv0822c in M. smegmatis. The LCP domains of the cpsA1, cpsA2, and Rv0822c genes were fused in frame with gfp in pJB(−) (red bars) and pJB(+) (blue bars). The positions of the GFP fusions in each protein are indicated by arrows and orange stars in the models shown in B. Fluorescence intensities were normalized to the A600 of the cultures, and the results shown represent the means and S.D. values of fluorescence intensities determined on at least 3–5 independent M. smegmatis transformants for each pJB(−) and pJB(+) plasmid. The addition of a single transmembrane domain from glycophorin A between the C-terminal fusion point of the protein of interest and the GFP in pJB(+) allows membrane-associated proteins with extracellular C-terminal fusions to be converted to proteins with intracellular C-terminal fusions. The native topology is reported with the fusion junction lacking the glycophorin A single transmembrane domain in the pJB(−) plasmid. Because GFP fluoresces in the cytoplasm but not in the periplasm, a high fluorescence signal in the pJB(+) version and background fluorescence in the pJB(−) version, as is the case with all three proteins here, are indicative of the C-terminal fusion of the protein being periplasmic. Error bars, S.D.
To experimentally validate the transmembrane topology of Rv0822c, CpsA1, and CpsA2, C-terminal GFP fusions were generated for each of the three genes in the pJB(−) and pJB(+) mycobacterial expression plasmids (31) and used to transform Mycobacterium smegmatis. In pJB(+), the addition of a single transmembrane domain from glycophorin A between the C-terminal fusion point of the protein of interest and the GFP converts membrane proteins with extracellular C-terminal fusions to proteins with intracellular C-terminal fusions (32). In pJB(−), the GFP fusion is direct and indicates the native topology. Because GFP fluoresces in the cytoplasm but not in the periplasm, a high fluorescence signal in the pJB(−) version and background fluorescence in the pJB(+) version are indicative of the C-terminal fusion of the protein being cytoplasmic. Opposite fluorescence intensities indicate, on the contrary, that the C-terminal fusion of the protein is localized in the periplasm. As shown in Fig. 2C, the LCP domains of all three fusion proteins clearly map to the periplasmic face of the plasma membrane of M. smegmatis. Ideally, topological determination of CpsA1, CpsA2, and Rv0822c would be improved by using more than one reporter to prepare fusion constructs. However, there was precedent in the GFP fusion approach that we followed, and our result was consistent with what has been reported in other studies involving proteins from Mycobacterium and Corynebacterium species (31).6
Purified Recombinant CpsA1 and CpsA2 and Rv0822c Display Pyrophosphatase Activity in Vitro
LCP proteins are thought to catalyze a phosphotransferase reaction in which polyprenyl phosphate-linked glycopolymer intermediates are transferred onto the C-6 hydroxyl of MurNAc residues within PG releasing polyprenyl phosphate. Accordingly, LCP proteins display pyrophosphatase activity (i.e. cleavage of the pyrophosphate group to release free phosphate) on geranyl pyrophosphate (GPP). This activity was shown to be magnesium-dependent, and conserved residues required for the binding of polyprenyl chains, pyrophosphate, and Mg2+ were identified within the primary sequence of these enzymes (19). These residues are well conserved in CpsA1, CpsA2, and Rv0822c (Fig. 2A). To determine whether the M. tuberculosis LCP candidates are endowed with pyrophosphatase activity, all three proteins (devoid of TM domain) were recombinantly expressed in E. coli, purified (Fig. 3A), and tested for activity on GPP by monitoring phosphate release. All three proteins displayed time-dependent and protein concentration-dependent pyrophosphatase activity on the generic substrate (Fig. 3, B and C). In all cases, activity was inhibited by EDTA, indicative of a requirement of the enzymes for magnesium ions (Fig. 3D).
FIGURE 3.

Pyrophosphatase activity of the three LCP proteins of M. tuberculosis recombinantly expressed and purified from E. coli. A, Coomassie Blue-stained SDS-polyacrylamide gel showing the purified CpsA1, CpsA2, and Rv0822c proteins devoid of transmembrane domain (CpsA1ΔTM, CpsA2ΔTM, and Rv0822cΔTM) upon affinity chromatography. The expected size of CpsA1ΔTM is 47.7 kDa, that of CpsA2ΔTM is 51.5 kDa, and that of Rv0822cΔTM is 52.4 kDa. Some degradation was consistently seen with CpsA1ΔTM and CpsA2ΔTM. The bottom bands have been confirmed to be proteolytic truncations of the target enzyme. B–D, pyrophosphatase assays. This assay monitors spectrophotometrically the hydrolysis of the pyrophosphate phosphoanhydride bond of GPP releasing Pi. B, the activity of the three purified LCP proteins is time-dependent. Reaction mixtures contained 5 μg (Rv0822cΔTM) or 10 μg (CpsA1ΔTM and CpsA2ΔTM) of recombinant proteins and were incubated for 15 min, 2 h, 5 h, and 16 h. C, protein concentration dependence. Reaction mixtures contained 0.5–16 μg of recombinant proteins and were incubated for 16 h. D, reaction mixtures contained 5 μg of recombinant protein and were incubated for 16 h in the absence (black bars) or presence (orange bars) of 10 mm EDTA. All assays were performed at least twice on independent recombinant protein preparations. Shown are the averages and S.D. values (error bars) of enzyme activities measured in duplicate in one representative experiment.
Generation and Characterization of C. glutamicum LCP Knock-out and Knockdown Mutants
The involvement of M. tuberculosis LCP proteins in the ligation of AG to PG was next assessed genetically by disrupting the genes of interest by allelic replacement and analyzing the consequences of these disruptions on growth and cell wall biosynthesis. As a first approach, the closely related Corynebacterianeae, C. glutamicum ATCC13032, was used as a model in these experiments in light of the known permissiveness of this bacterium to changes affecting its cell wall composition. Also, C. glutamicum displays two putative LCP proteins compared with three in M. tuberculosis. Cg0847 (cps2a) is orthologous to Rv3267 and, like its M. tuberculosis counterpart, maps to the same AG biosynthetic gene cluster in the C. glutamicum genome. Cg0847 and Rv3267 (M. tuberculosis CpsA1) share 50% identity and 63% similarity on a 452-amino acid residue overlap (Fig. 2A). The second putative LCP protein of C. glutamicum, Cg3210, has no clear ortholog in M. tuberculosis but most closely resembles Rv0822c (30% identity and 45% similarity on a 285-amino acid residue overlap). Both Cg0847 and Cg3210 display the conserved predicted secondary structure of LCP proteins consisting of an N-terminal cytoplasmic tail (28 and 119 amino acid residues long, respectively), one TM region, and an extracellular C-terminal region carrying the LytR-Cps2A-Psr domain (data not shown).
To investigate the putative involvement of Cg0847 and Cg3210 in the assembling of the cell wall core, their encoding genes were disrupted by allelic replacement in C. glutamicum. Whereas Cg3210 disruption was easily achieved (Fig. 4A), the screening of 210 Cg0847 candidate mutants by PCR failed to identify any null mutant, suggesting that Cg0847 may be essential for growth. Evidence for this assumption was obtained by constructing a conditional Cg0847 knockdown mutant, CGLcKD-0847, in which the endogenous Cg0847 gene was disrupted and replaced by a WT copy of this gene expressed from an IPTG-inducible promoter (Fig. 4B). As shown in Fig. 4C, whereas CGLcKD-0847 grew in medium containing IPTG, it totally failed to grow in the absence of inducer. Thus, Cg0847 is essential for C. glutamicum growth, and Cg3210 cannot compensate for a deficiency in Cg0847. The Cg3210 knock-out mutant (CGLΔ3210) grew similarly to the WT parent C. glutamicum strain (Fig. 4C).
FIGURE 4.

Inactivation of cg0847 and cg3210 in C. glutamicum. A, evidence for allelic replacement at the cg3210 locus of C. glutamicum. The deletion of the entire cg3210 ORF results in the replacement of the WT 2,157-bp amplification signal by a 1,033-bp fragment in the knock-out mutant. B, schematic representation of the chromosomal region of the cg0847 knockdown mutant (CGLcKD-0847) after integration of the pZEΔ847 plasmid into cg0847. Evidence for the correct insertion of the plasmid by PCR analysis using the two sets of primers, 847ver1/lacIver2 (resulting amplicon: 696 bp) and paraver1/847ver2 (resulting amplicon: 1,898 bp). C, growth characteristics of C. glutamicum WT, CGLΔ3210, and CGLcKD-0847. The strains were grown in LB broth at 30 °C with shaking. Inset, growth of CGLcKD-0847 in the presence of 0 (crosses), 12.5 μm (squares), 25 μm (triangles), or 1 mm (circles) IPTG; C. glutamicum WT (diamonds). The arrow indicates the time point at which samples were removed for the cell wall analyses described in this study. D, optical micrographs of C. glutamicum WT and CGLcKD-0847 cells cultivated overnight in BHI medium. One droplet of culture was absorbed onto a microscope slide coated with 1% agarose and visualized using a DMIRE2 optical microscope (Leica) equipped with a CCD camera (CoolSNAP HQ2, Roper Scientific). Scale bar, 5 μm.
Preliminary optical microscopic observations revealed that decreasing the concentration of IPTG in the culture medium led to important changes in the morphology of CGLcKD-0847, characterized by the appearance of more voluminous asymmetric cells (representing ∼90% of the cell population shown in Fig. 4D), most of which displayed several septa. To better visualize the effects of silencing Cg0847 on the cell envelope, CGLcKD-0847 cells were analyzed by cryo-TEM. Whereas WT cells were homogeneous and presented a typical C. glutamicum cell envelope with a continuous outer membrane (Fig. 5A), CGLcKD-0847 grown in the presence of a low concentration of IPTG (25 μm) revealed a heterogeneous population of cells, some of them clearly displaying a disrupted outer layer with large fragments detaching from the cell surface (Fig. 5B). Other cells in the CGLcKD-0847 culture presented phenotypes intermediate between that showed in Fig. 5B and that of WT C. glutamicum, possibly reflecting their different metabolic state and level of exposure to IPTG (data not shown).
FIGURE 5.
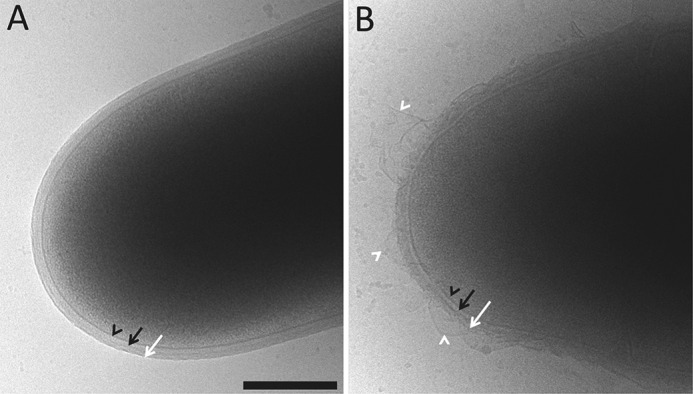
Cryo-TEM of WT C. glutamicum (A) and the conditional knockdown strain CGLcKD-0847 (B). CGLcKD-0847 was grown in the presence of 25 μm IPTG. Black arrowheads, black arrows, and white arrows, plasma membrane, cell wall, and outer membrane, respectively. White arrowheads, detached membrane fragments in the conditional knockdown. Scale bar, 300 nm.
The shedding of material by CGLcKD-0847 was evident from the recovery of a translucent pellet upon ultracentrifugation of culture supernatants whose abundance was inversely proportional to the concentration of IPTG added to the culture medium. Whereas a pellet was clearly visible in CGLcKD-0847 cultures grown in the presence of 0, 12.5, and 25 μm IPTG (Fig. 6A), no pellet was recovered from cultures containing 1 mm IPTG or from WT C. glutamicum cultures. Cryo-TEM analysis of the released material revealed the presence of fragments of different sizes and shapes in the conditional knockdown that were not seen in similarly processed WT C. glutamicum culture supernatants (Fig. 6, B and C). These fragments are reminiscent of those previously observed in a C. glutamicum aftB mutant deficient in the synthesis of the non-reducing arabinan termini of AG (33) and are suggestive of the release of cell wall and outer membrane materials by CGLcKD-0847. The finding of sugars, lipids, and proteins typifying the outer membrane and cell wall core of C. glutamicum in the ultracentrifugation pellet shown in Fig. 6A supports this assumption. Indeed, whereas WT C. glutamicum and cg0847 knockdown cells displayed very similar total lipid contents (supplemental Fig. S1), culture supernatants from the latter strain were clearly enriched in trehalose monocorynomycolates (TMCM) and trehalose dicorynomycolates (TDCM), showing only minimal amounts of inner membrane phospholipids (34) (Fig. 6D). Likewise, the protein profile of the released fragments resembled that obtained upon SDS treatment of the WT cells, a treatment known to extract cell wall but not inner membrane proteins (33, 35) (Fig. 6E). This profile is characterized, in particular, by the presence of the major outer membrane-associated mycolyltransferases, cMytA, cMytB, and cMytC (Fig. 6E and supplemental Fig. S2). Quantitative analysis of the alditol acetate derivatives of the pellet material further revealed significant amounts of rhamnose, arabinose, and galactose (typifying Corynebacterium AG (cAG)) as well as galactosamine, N-acetyl-glucosamine, and muramic acid (the latter two sugars typifying PG and, perhaps, other cell wall glycopolymers), whose relative abundance in the culture medium increased at lower IPTG concentrations (Fig. 6F). That the fragments released by CGLcKD-0847 did not originate from the inner membrane was further confirmed by the absence of detectable NADH dehydrogenase activity associated with this material (data not shown). Taken together, these results thus indicate that the cg0847 knockdown mutant sheds into the culture medium substantial amounts of outer membrane and cell wall materials.
FIGURE 6.

Analysis of the material released by CGLcKD-0847 in the culture medium. A, pellets obtained after ultracentrifugation of the culture supernatants from C. glutamicum WT or CGLcKD-0847 grown in the presence of 0, 0.025, or 1 mm IPTG. B, cryo-TEM of the material recovered upon ultracentrifugation of the culture supernatant of a C. glutamicum WT culture. Although there was no clearly visible pellet in this case, some material could be still recovered at the bottom of the tube. C, cryo-TEM of the material contained in the pellet shown in A for CGLcKD-0847. Vesicles seen in both WT and CGLcKD-0847 cultures are indicated by black arrows in B and C, and outer membrane fragments are indicated by white arrows in C. Scale bar, 100 nm. D, TLC analysis of total lipids extracted from whole C. glutamicum WT cells grown to stationary phase (lane 1) for comparison with lipids extracted from the pellet obtained in A (lane 2). TLC plates were developed in the solvent system CHCl3/CH3OH/H2O (65/25/4, by volume) and revealed by immersion in 10% H2SO4 in ethanol and heating. PG, phosphatidylglycerol; CL, cardiolipin; PIM, phosphatidylinositol mannosides; TMCM, trehalose monocorynomycolates; TDCM, trehalose dicorynomycolates. E, SDS-PAGE showing proteins extracted from the cell wall from WT C. glutamicum and CGLcKD-0847 grown in the presence of 1 mm IPTG or 25 μm IPTG and the protein composition of the pellet shown in A. MWM, molecular weight marker. F, sugar composition of the pellet obtained after ultracentrifugation of culture supernatants from CGLcKD-0847. Individual monosaccharides from the ultracentrifugation pellets of 300-ml CGLcKD-0847 cultures grown in the presence of 0 μm, 25 μm, and 1 mm IPTG were analyzed as their alditol acetate derivatives and their quantities (in μg) standardized to the A600 of the cultures. Although mannose was also detected, the precise quantification of this sugar was hampered by the large quantities of residual Glc, presumably coming from the culture medium.
To determine whether the failure of CGLcKD-0847 bacilli to retain their cell envelope resulted from a deficient AG/PG ligation activity, we finally sought to directly assess the abundance of linker units in WT C. glutamicum and cg0847 knockdown cells exposed to different concentrations of IPTG. To this end, the cells were submitted to two separate analyses for the presence or lack of attachment of cAG to PG. In the first experiment, the amount of cAG was compared with the amount of PG using primarily the ratio of rhamnose (Rha; a marker for cAG) to GlcNAc and MurNAc (markers for PG). As shown in Table 1, when cg0847 was not expressed (cultures devoid of IPTG), the ratio of cAG to PG changed dramatically with much larger amounts of both GlcNAc and MurNAc compared with Rha than present in the WT strain cultured under similar conditions. In an alternative procedure to measure attachment of cAG to PG, the ratio of MurNAc to N-acetylmuramic acid-6-phosphate (MurNAc-6P) was measured by LC/MS. The results fully corroborated the glycosyl composition results in that the ratio increased dramatically (i.e. less MurNAc-6P) when cg0847 was not expressed and returned toward and even beyond WT values in an IPTG concentration-dependent manner when cg0847 was expressed (see cultures grown in the presence of 0.025 and 1 mm IPTG) (Fig. 7A). In contrast, the cg3210 KO mutant displayed a Mur/MurNAc-6P ratio comparable with that of the WT parent strain (Fig. 7B), suggesting that Cg3210 does not contribute any significant PG-AG ligase activity under the growth conditions studied herein.
TABLE 1.
Sugar analysis of cell walls prepared from Cgl WT and the cg0847 conditional knockdown mutant
Cell walls were prepared and hydrolyzed with 2 m TFA as described (44), and alditol acetates derived thereof were subjected to GC/MS. The amounts of each monosaccharide in the samples are expressed relative to Rha set to be constant. Note that the 2 m TFA acid hydrolysis does not fully release the GlcNAc and Mur from the cell wall. An independent LC/MS-based method to measure cAG to PG ratio via the ratio MurNAc to MurNAc-6P (see Fig. 7) was also performed. The results are representative of two experiments performed on two independent culture batches.
| Cgl strain | Rha | Ara | Man | Gal | Glc | GalNAc | Mur | GlcNAc |
|---|---|---|---|---|---|---|---|---|
| WT (1 mm IPTG) | 3.1 | 78.4 | 1.6 | 23.9 | 3.7 | 22.6 | 15.2 | 41.2 |
| cKD-0847 (1 mm IPTG) | 3.1 | 99.5 | 4.1 | 34.6 | 10.4 | 9.3 | 13.0 | 38.1 |
| WT (0 mm IPTG) | 3.1 | 85.7 | 3.2 | 29.7 | 5.6 | 27.0 | 24.2 | 50.5 |
| cKD-0847 (0 mm IPTG) | 3.1 | 121.3 | 2.9 | 26.4 | 2.7 | 20.8 | 103.9 | 175.2 |
FIGURE 7.
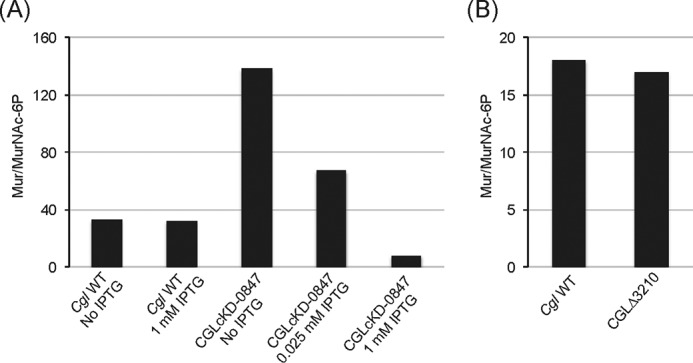
Analysis of the linker regions from C. glutamicum WT, the cg0847 knockdown strain, and the cg3210 knock-out mutant. Shown are the ratios of MurNAc to MurNAc-6P as determined by LC/MS in cell wall samples prepared from C. glutamicum WT and CGLcKD-0847 grown in LB medium containing 0–1 mm IPTG (see Fig. 4C, inset) (A) and C. glutamicum WT and CGLΔ3210 grown to mid-log phase (A600 = 1.6–1.7) in LB medium (B).
Generation and Characterization of M. tuberculosis LCP Knock-out Mutants
The question of function and redundancy of the three cell wall ligase candidates of M. tuberculosis was approached genetically (i.e. we used homologous recombination to knock out individually or in combination the cpsA1, cpsA2, and Rv0822c genes of M. tuberculosis H37Rv mc26206 (an avirulent auxotroph) and virulent M. tuberculosis CDC1551). Single mutants carrying transposon insertions in the Rv0822c or cpsA1 genes of M. tuberculosis CDC1551 were also obtained from BEI Resources. Whereas single mutants and a double Rv0822c/cpsA1 knock-out mutant were easily obtained in one or both M. tuberculosis isolates by allelic replacement (Fig. 8A) (or by transposition in the case of Rv0822c in M. tuberculosis CDC1551), the combined inactivation of cpsA1 and cpsA2 could not be achieved in either isolate, suggestive of synthetic lethality. Thus, CpsA1 and CpsA2 appeared to display partially overlapping essential activities in M. tuberculosis that could not be compensated by Rv0822c. These two M. tuberculosis enzymes were the objects of our further functional investigations.
FIGURE 8.

Growth characteristics and cording properties of cpsA1 and cpsA2 knock-out mutants of M. tuberculosis H37Rv mc26206. A, evidence for allelic replacement at the cpsA1 and cpsA2 loci of M. tuberculosis H37Rv mc26206. The WT 2,175-bp amplicon is replaced by a 4,241-bp PCR fragment in the cpsA1 mutant due to the replacement of 158 bp of the cpsA1 ORF flanked between two AfeI restriction sites with a 2-kb streptomycin resistance cassette. The replacement of the entire cpsA2 gene by a 1.2-kb kanamycin resistance cassette results in the replacement of the WT 3,734-bp amplification signal by a 3,420-bp fragment in the mutant. B, growth characteristics of the cpsA1 and cpsA2 mutants of M. tuberculosis H37Rv mc26206 WT, the WT parent strain, and the complemented cpsA1 mutant, M. tuberculosis H37RvΔcpsA1/pMVGH1-cpsA1. The strains were grown in 7H9-ADC-Tween 80 broth at 37 °C with shaking. C, cording properties of the cpsA1 and cpsA2 mutants of M. tuberculosis H37Rv mc26206. Shown are Ziehl-Neelsen smears of 37 °C 7H9-ADC-tyloxapol cultures. Scale bar, 50.2 μm.
Intriguingly, whereas the inactivation of cpsA2 (and Rv0822c) had no effect on the axenic growth of M. tuberculosis H37Rv mc26206 and M. tuberculosis CDC1551, knocking out cpsA1 differently affected the growth of the two M. tuberculosis isolates, with H37RvΔcpsA1 but not CDC1551ΔcpsA1 displaying a significantly reduced growth rate compared with their WT parent (Fig. 8B and supplemental Fig. S3). This growth phenotype of H37RvΔcpsA1 was accompanied by a cording defect (Fig. 8C). Normal growth and cording were restored in the complemented H37RvΔcpsA1 mutant strain (Fig. 8, B and C). The reason for these phenotypic differences between M. tuberculosis CDC1551 and H37Rv mc26206 cpsA1 mutants is currently not known.
The glycosyl composition of the various M. tuberculosis H37Rv mc26206 and M. tuberculosis CDC1551 strains, where the amount of Rha (an AG marker) was compared with the amount of GlcNAc (a PG marker), was determined. The ratio of Mur to MurNAc-6P was also compared for the same cell walls. Although some fluctuation in the data were apparent (supplemental Table S1), there appeared, in general, to be a slight loss of AG attachment to PG when the data were examined as a whole but no major, consistent, differences (supplemental Table S1). Thus, CpsA1 appears to compensate for lack of CpsA2 and vice versa. These data further suggest that in the background of M. tuberculosis H37Rv mc26206, the cpsA1 knock-out mutant adjusts its growth rate to the total AG/PG ligase activity of the cells but maintains a constant cell wall core composition.
Consistently, metabolic labeling of H37Rv mc26206 WT, mutant, and complemented mutant cells with [14C]glucose followed by the analysis of total lipids, mycolic acids, and cell wall polysaccharides, as described previously (36), failed to reveal any significant changes in the mutants (data not shown). Finally, in contrast with the situation in C. glutamicum, neither the cpsA1 nor cpsA2 M. tuberculosis mutants released any detectable cell envelope material in the culture medium (data not shown). This absence of shedding probably reflects the ability of the mutants to negatively regulate the biosynthesis of cell wall constituents in response to a decrease in ligase activity, as reported earlier for B. subtilis LCP mutants (19).
Susceptibility of M. tuberculosis cpsA1 and cpsA2 Knock-out Mutants to Antibiotics
Despite the absence of any major change in the cell wall content of the cpsA1 and cpsA2 mutants of M. tuberculosis CDC1551 and H37Rv mc26206, the inactivation of cpsA1 in both backgrounds resulted in a significant decrease in the MIC of a number of antibiotics, including vancomycin, rifampicin, CPZEN-45, and β-lactams (used either alone or in combination with clavulanate) against M. tuberculosis (Table 2). Although changes in the permeability of the cell envelope of the cpsA1 mutants may account for the increased efficacy of some of these drugs (e.g. rifampicin), the inhibition by CPZEN-45, vancomycin, and β-lactams of biosynthetic pathways related to the ligation of AG to PG could explain the greater susceptibility of the cpsA1 knock-outs to these compounds. CPZEN-45 is indeed an inhibitor of WecA, an enzyme that transfers GlcNAc 1-phosphate to Dec-P to form Dec-P-P-GlcNAc and is thus directly involved in the formation of the linker unit (37). Likewise, the ligation reaction is thought to require the concomitant cross-linking of PG (13), a process inhibited by penicillins and vancomycin. It is possible that impeding the synthesis of the linker unit or the cross-linking of PG exacerbates the loss of fitness of cpsA1 mutants by further impairing PG-AG attachment, resulting in their hypersusceptibility to β-lactams, vancomycin, and CPZEN-45.
TABLE 2.
MIC values of various antibiotics against the cpsA1 and cpsA2 knock-out mutants of M. tuberculosis H37Rv mc26206 and M. tuberculosis CDC1551
MICs were determined in 7H9-ADC-tyloxapol (M. tuberculosis H37Rv mc26206) or 7H9-OADC-Tween 80 (M. tuberculosis CDC1551) broth. MIC values are in μg/ml. INH, isoniazid; EMB, ethambutol; CIP, ciprofloxacin; VAN, vancomycin; RIF, rifampicin; STR, streptomycin; AMP, ampicillin; PEN G, penicillin G; CLAV, clavulanate (used at 5 μg/ml). ND, not determined; NA, not applicable due to the fact that a streptomycin resistance cassette was used in the construction of the mutant strain. MIC determinations were performed at least twice on independent culture batches.
| Strains | MIC |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| INH | EMB | CIP | VAN | RIF | STR | CPZEN-45 | PEN G | PEN G/CLAV | AMP | AMP/CLAV | |
| μg/ml | |||||||||||
| M. tuberculosis H37Rv mc26206 | 0.02 | 4 | 0.8 | 25 | 0.1 | 8 | 0.2–0.4 | 600 | 37.5 | 150–300 | 37.5 |
| mc26206ΔRv3267 | 0.04 | 2 | 0.4 | 0.78 | 0.003 | NA | 0.025 | 300 | 9.4 | 75–150 | 9.4 |
| mc26206ΔRv3267-compl | 0.04 | 4 | 0.8 | 25 | 0.1 | NA | 0.4 | 600 | 37.5 | 300 | 37.5 |
| mc26206ΔRv3484 | 0.02 | 2 | 1.6 | 25 | 0.1 | 4 | 0.2 | 600 | 18.7 | 300 | 18.7 |
| mc26206ΔRv3484-compl | 0.02 | 4 | 0.8 | 25 | 0.1 | 8 | 0.2 | 600 | 37.5 | 300 | 9.4 |
| M. tuberculosis CDC1551 | ND | 2 | 0.4 | 3.12 | 0.012 | 4 | 0.4–0.8 | 600 | 9.4–18.8 | 150 | 18.8 |
| CDC1551ΔRv3267 | ND | 4 | 0.2 | 1.6 | 0.0063 | NA | 0.1 | 37.5 | 4.7 | 75 | 9.4 |
| CDC1551ΔRv3267-compl | ND | ND | ND | ND | 0.025 | NA | 0.4–0.8 | 600 | 37.5 | 150–300 | 37.5 |
| CDC1551(Rv3267::Tn) | ND | 1 | 0.2 | 1.6 | 0.0031 | 8 | 0.1 | ND | ND | ND | ND |
| CDC1551(Rv3267::Tn)-compl | ND | 1 | 0.4 | 6.25 | 0.05 | 4 | 0.4 | ND | ND | ND | ND |
| CDC1551ΔRv3484 | ND | 2 | 0.4 | 6.25 | 0.012 | 4 | 0.4–0.8 | ND | ND | ND | ND |
| CDC1551ΔRv3484-compl | ND | 2 | 0.4 | 6.25 | 0.012 | ND | 0.4 | ND | ND | ND | ND |
Discussion
LCP proteins are present, often as multiple paralogs, in almost all Gram-positive bacteria but are absent from the majority of Gram-negative organisms (17). A growing body of evidence indicates that these proteins play critical roles in the assembling of the cell wall of Gram-positive bacteria, catalyzing the transfer of a variety of glycopolymers from a lipid-linked polyprenyl pyrophosphate carrier to various cell envelope acceptors, including PG and surface proteins (19–29). Despite the growing interest in this family of proteins, technical challenges associated with the development of physiological assays for these enzymes have impeded thus far the reconstitution of the LCP-mediated attachment of capsules, protein glycosylation motifs, or WTA in vitro. Similarly, our efforts to directly measure the involvement of CpsA1 and CpsA2 in the attachment of AG to PG using different variants of the physiological ligase assay developed earlier by our laboratories (12) have so far failed to provide any conclusive results. Cell-free extracts prepared from our M. tuberculosis H37Rv mc26206 WT and mutant strains displayed comparable ligase activities in the assay described by Yagi et al. (12). Moreover, the recombinant CpsA1 and CpsA2 proteins, whether used as purified proteins devoid of transmembrane domain or recombinantly overexpressed under their full-size form in E. coli, failed to show any consistent hydrolytic activity on GL-1 and GL-2 whether or not soluble PG or PG precursors (lysozyme- or mutanolysin-digested PG, N-acetylmuramyl dipeptide, or MurNAc) were added as acceptors to the reaction mixture. Nonetheless, recombinant forms of CpsA1 and CpsA2 produced in E. coli demonstrated clear pyrophosphatase activity on the generic substrate, GPP. These results and the findings afforded by our other biochemical and genetic studies collectively indicate that these two LCP proteins are the long sought cell wall ligases of M. tuberculosis responsible for the covalent attachment of AG to PG, with CpsA1, whose gene maps to an AG-biosynthetic gene cluster, playing the predominant role. The participation of more than one LCP protein in the attachment of AG to PG is consistent with the involvement of multiple LCP proteins in the assembly of cell wall macromolecular complexes in Gram-positive bacteria and in line with our previous observation that a cpsA (orthologous to M. tuberculosis cpsA2) knock-out mutant of Mycobacterium marinum is only partially affected in AG attachment (38). In contrast, C. glutamicum exclusively relies on its CpsA1 ortholog (Cg0847) to carry out this function. Consistently, silencing cg0847 in C. glutamicum has profound effects on the ratio of cAG to PG and the morphology of the cells and leads to the shedding of abundant envelope material in the culture medium, as has been reported for LCP mutants of S. aureus and S. pneumoniae (20, 25).
Despite the absence of any major changes in the cell envelope composition of the individual M. tuberculosis ligase mutants, the increased susceptibility of the M. tuberculosis H37Rv mc26206 and CDC1551 cpsA1 mutants to RIF, CPZEN-45, and penicillins and significant alterations in the growth rate and cording properties of the M. tuberculosis H37Rv mc26206 cpsA1 mutant are indicative of physiological and structural changes affecting the surface properties of the bacilli as well as their permeability and susceptibility to drugs. Of particular interest in terms of drug development is our observation that inhibitors of the PG-AG ligases may potentiate the effect of other drugs targeting the synthesis of the cell wall core, as illustrated by the dramatically increased susceptibility of M. tuberculosis cpsA1 mutants to penicillins, vancomycin, and CPZEN-45. Future inhibitors of cell wall ligation in M. tuberculosis, however, will need to target both CpsA1 and CpsA2 simultaneously to display anti-tuberculosis activity, a goal that may be achievable, considering the high degree of conservation of the active site of these enzymes.
Experimental Procedures
Bacterial Strains and Growth Conditions
M. tuberculosis H37Rv mc26206 (an avirulent ΔpanCDΔleuCD mutant of M. tuberculosis H37Rv; kind gift from Dr. W. R. Jacobs Jr., Albert Einstein College of Medicine, New York), M. tuberculosis CDC1551, and M. smegmatis mc2155 were grown in Middlebrook 7H9 broth with 10% OADC or ADC (BD Biosciences), 0.5% glycerol, and 0.05% Tween 80 or tyloxapol, glycerol-alanine-salts (GAS) medium or on Middlebrook 7H11 agar supplemented with 10% OADC (BD Biosciences) and 0.5% glycerol. All media used to grow M. tuberculosis H37Rv mc26206 were supplemented with 0.2% casaminoacids, 48 μg/ml pantothenate, and 50 μg/ml l-leucine. E. coli DH5α, the strain used for cloning, was grown in LB broth or agar (BD Biosciences). C. glutamicum strain RES167, a restrictionless derivative of C. glutamicum ATCC13032 (39), was cultured in brain-heart infusion (BHI) (3.7%, Difco) or LB medium at 30 °C. Kanamycin (Kan; 20–50 μg/ml), hygromycin (50–150 μg/ml), ampicillin (100 μg/ml), streptomycin (25 μg/ml), and 2–10% sucrose were added to the culture media as needed.
Construction of C. glutamicum Knock-out and Knockdown Strains
The strategy described by Schäfer et al. (40) was used to inactivate cg0847 and/or cg3210 in C. glutamicum RES167. Briefly, two DNA fragments overlapping the gene to be deleted at its 5′ and 3′ extremities were PCR-amplified from C. glutamicum RES167 genomic DNA and cloned in the non-replicative vector pK18mobSac. C. glutamicum RES167 was transformed with the resulting plasmids (pK18mobsacΔ847 and pK18mobsacΔ3210), and transformants having undergone single crossover events were selected on BHI-Kan agar. The second crossover events were selected by plating Kan-resistant clones on BHI plates containing 10% sucrose. Kan-susceptible/sucrose-resistant colonies were next screened by PCR and sequencing for allelic replacement at the target loci. For the construction of a cg0847 conditional knockdown, a suicide vector (pZEΔ847) capable of recombining with the first 400 bp of the cg0847 gene in C. glutamicum and containing the lacIq gene was constructed and used to electrotransform C. glutamicum RES167. Transformants were selected on BHI-Kan plates containing 1 mm IPTG. Plasmid integration by homologous recombination results in the replacement of cg0847 by an intact copy of this gene placed under control of the inducible Plac/ara-1 promoter. Details of primers and plasmid constructs are available upon request. cg0847 silencing in the conditional knockdown, CGLcKD-0847, was achieved as follows. One colony from a BHI plate supplemented with 1 mm IPTG was grown overnight in LB broth. Cells from this preculture were then washed twice with 2 volumes of LB and used to inoculate LB or BHI medium at an A600 of 0.2, in the absence or presence of different concentrations of IPTG.
Construction of M. tuberculosis Mutants and Complemented Mutant Strains
The construction of cpsA1 and cpsA2 knock-out mutants of M. tuberculosis H37Rv mc26206 involved replacing 158 bp of the coding sequence of Rv3267 flanked between two AfeI sites and the entire Rv3484 ORF by streptomycin and kanamycin resistance cassettes, respectively, following standard allelic replacement strategies with pPR27-xylE, a replicative plasmid harboring a temperature-sensitive origin of replication, the counterselectable marker sacB, and the colored marker xylE (41). Details of the plasmid constructs are available upon request. Complementation constructs for cpsA1, cpsA2, and Rv0822c consist of the full-size genes from M. tuberculosis H37Rv expressed under control of the phsp60 promoter from the replicative plasmid pMVGH1 (42).
Transmembrane Topology Mapping Using GFP Fusions
To establish the subcellular localization of the catalytic sites of CpsA1, CpsA2, and Rv0822c in mycobacteria, C-terminal GFP fusions were generated in the mycobacterial expression plasmids pJB(−) and pJB(+) (31) and used to transform M. smegmatis. Cultures of transformants grown to log phase resuspended in 100 μl of PBS were transferred to black 96-well plates with transparent bottoms (Corning), and their fluorescence was determined using a 2030 MultiLabel Reader Victor X5 plate reader (PerkinElmer Life Sciences) at excitation and emission wavelengths of 485 and 535 nm, respectively. The fluorescence value of each sample was normalized to the A600 of the culture.
Drug Susceptibility Testing
The MIC values of various antibiotics against the M. tuberculosis WT, mutant, and complemented mutant strains were determined in 7H9-OADC-Tween 80 and 7H9-ADC-tyloxapol broth at 37 °C in 96-well microtiter plates using the colorimetric resazurin microtiter assay (43) and visually scanning for growth.
Glycosyl Composition of Cell Walls
Cell walls were prepared as described (44) except that the cells were disrupted by probe sonication rather than by French press. The glycosyl composition was also determined as described (44). Mur and MurNAc-6P were released from cell walls by treatment with 6 m HCl at 95 °C for 6 h and then analyzed by LC/MS. LC/MS (ESI/APCI ionization) analysis was carried out on an Agilent 6220 TOF mass spectrometer equipped with a multimode source in negative mode and an Agilent 1200 binary pump HPLC. The liquid chromatography was performed using a Cogent Diamond Hydride column (150 × 2 mm) obtained from MicroSolv Technology Corp. (Eatontown, NJ). Solvent A was 10 mm ammonium acetate in water, and solvent B was 10 mm ammonium acetate in acetonitrile. The initial conditions were a flow rate of 0.4 ml/min and 95% solvent B, which was held for 3.1 min, followed by a gradient to 5% solvent B over the next 2.9 min at a flow rate of 0.7 ml/min, which was then held for 4 min, during which time the compounds of interest eluted. The LC column was then returned to 5% solvent B and washed for 5 min at a flow rate of 0.8 ml/min before returning to the original conditions for the next run. The mass spectrometer (negative mode) was set to monitor ions from m/z 100 to m/z 1,250 with a rate of 1.06 scans/s.
Other Analyses
Alditol Acetates and Culture Supernatant Materials
Total lipids were extracted with chloroform and methanol and analyzed by TLC. Alditol acetates were prepared and analyzed by GC/MS following procedures described earlier (44).
Purification of C. glutamicum Cell Envelope Fragments
Cell envelope fragments shed by the Cg0847 conditional knockdown in the culture medium were purified from 300-ml cultures containing either 0.025 or 1 mm IPTG. Cells were harvested by centrifugation at 5,000 × g, and the culture supernatants were centrifuged at least three additional times at 5,000 × g to eliminate intact cells. The resulting supernatants were then centrifuged at 100,000 × g for 1 h in a 45Ti rotor, and the centrifugation pellets were resuspended in 500 μl of 25 mm Tris, pH 8, for analysis of their protein, lipid, and sugar contents as described above.
Production and Purification of CpsA1, CpsA2, and Rv0822c in E. coli
Recombinant forms of CpsA1, CpsA2, and Rv0822c devoid of the N-terminal domain harboring the TM segment, hereafter referred to as CpsA1ΔTM (corresponding to residues 38–498 of the full-size protein), CpsA2ΔTM (corresponding to residues 49–512 of the full-size protein), and Rv0822cΔTM (corresponding to residues 195–684 of the full-size protein), were produced in E. coli BL21(DE3) using the pET32b and pET42b expression systems (EMD Biosciences). All recombinant proteins harbor an N-terminal hexahistidine tag. Following an overnight induction with 1 mm IPTG at 16 °C in LB broth (plus kanamycin or ampicillin), E. coli BL21(DE3) cells expressing each of the three candidate ligases were harvested, washed, and resuspended in lysis buffer consisting of 50 mm Tris-HCl, pH 8.0, 500 mm NaCl, and 10% glycerol for CpsA1ΔTM and CpsA2ΔTM or 20 mm Tris, pH 8.0, 250 mm NaCl, 5% glycerol, 5 mm imidazole, and 5 mm β-mercaptoethanol for Rv0822cΔTM. DNase I (Roche Applied Science) at a concentration of 0.1 mm was also added to the resuspended cells. Cells were disrupted by sonication, and the lysates were further centrifuged at 18,514 × g at 4 °C for 45 min. The recombinant proteins were then purified by applying the resulting cell lysates to a HisTrap TALON® chelating column (GE Healthcare) equilibrated with lysis buffer. Columns were washed with lysis buffer with a final wash in lysis buffer containing 0.15 mm dodecyl maltopyranoside until no absorbance at 280 nm was detected. Elution was performed with buffers consisting of 50 mm Tris, pH 8.0, 5% glycerol, and 150 mm imidazole (CpsA1ΔTM and CpsA2ΔTM) or 20 mm Tris, pH 8.0, 250 mm NaCl, 5% glycerol, 5 mm β-mercaptoethanol, and 150 mm imidazole (Rv0822cΔTM). Affinity tags were cleaved by enzymatic digestion with the human Rhinovirus 3C protease during a 16-h dialysis against lysis buffer. Protein samples were again applied to a HisTrap TALON® chelating column equilibrated with lysis buffer to remove the cleaved affinity tag and the protease. The final protein preparations were dialyzed against 20 mm Tris, pH 8.0, 150 mm NaCl, and 10% glycerol (CpsA1ΔTM and CpsA2ΔTM) or 20 mm Tris, pH 8.0, 5% glycerol, 5 mm imidazole, and 5 mm β-mercaptoethanol (Rv0822cΔTM). Enzymes were stored at −80 °C until used in enzyme assays.
Enzyme Assays
The pyrophosphatase activity of the M. tuberculosis candidate ligases was tested using GPP (Sigma) as the substrate. The standard reaction mixture contained 50 mm Tris-HCl (pH 8.0), 20 mm MgCl2, 270 μm GPP (Sigma), and 5–10 μg of recombinant LCP proteins (devoid of the TM domain) purified from E. coli. The reaction was incubated at 37 °C for up to 16 h. Inorganic phosphate concentrations were determined using the PiPerTM phosphate assay kit (Molecular Probes) with absorbance measured at 560 nm in a microplate reader.
Cryo-TEM
4-μl aliquots of samples (suspension of bacteria or membrane fragments) were adsorbed onto glow-discharged holey carbon-coated grid (Quantifoil Micro Tools GmbH, Grosslöbichau, Germany), blotted with Whatman filter paper, and plunge-frozen into liquid ethane at −180 °C using a Vitrobot (FEI Co., Eindhoven, Netherlands). Frozen grids were transferred onto a Philips CM200-FEG electron microscope using a Gatan 626 cryo-holder. Electron micrographs were recorded at an accelerating voltage of 200 kV using a low dose system (20 e−/Å2) and keeping the samples at −173 °C. Defocus values were around −2.5 μm. The micrographs were recorded with a 4,000 × 4,000-pixel CMOS camera (Tietz Video and Image Processing Systems GmbH, Gauting, Germany).
Author Contributions
M. J., M. R. M., P. J. B., K. M., and C. H. conceived the study. M. J., Q. G., M. R. M., D. R. R., and C. H. coordinated the study. M. J., M. R. M., D. R. R., A. E. G., C. S. A., and C. H. wrote the paper. A. E. G. and E. H.-C. constructed and analyzed recombinant mycobacterial strains. C. S. A. generated and analyzed recombinant corynebacterial strains. V. J., J. Z., and S. K. A. performed MIC determinations and analyzed the cell wall composition of M. tuberculosis and C. glutamicum strains. A. E. G., Q. W., and Y. I. performed enzymatic assays with the purified ligases and recombinant strains. A. E. G., Y. I., and C. P. produced and purified CpsA1, CpsA2, and Rv0822c from E. coli. J. M. B. participated in the topology studies. M. C. conducted the cryo-TEM experiments. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Dr. W. R. Jacobs Jr. (Albert Einstein College of Medicine, New York) for the kind gift of M. tuberculosis H37Rv mc26206. The following reagents were obtained through BEI Resources, NIAID, National Institutes of Health: M. tuberculosis CDC1551 transposon mutants 1718 (NR-18207) and 1859 (NR-18801). We thank Prof. Henning Stahlberg (Director of C-Cina, Biozentrum, University of Basel) for continuous support. We thank Magali Prigent (I2BC) for help with optical microscopy analyses.
This work was supported by NIAID, National Institutes of Health, Grant AI119670 and the College of Veterinary Medicine and Biomedical Sciences Research Council (Colorado State University). The electron microscopy work at C-CINA (Biozentrum) was supported in part by the Swiss National Science Foundation (SystemsX.ch RTD CINA and NCCR TransCure). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1 and Figs. S1–S3.
J. M. Belardinelli and M. Jackson, manuscript in preparation.
- PG
- peptidoglycan
- AG
- arabinogalactan
- cAG
- Corynebacterium AG
- Dec-P
- decaprenyl phosphate
- Mur
- muramic acid
- MurNAc
- N-acetylmuramic acid
- MurNAc-6P
- MurNAc 6-phosphate
- WTA
- wall teichoic acids
- LCP
- LytR-Cps2A-Psr
- TM
- transmembrane
- GPP
- geranyl pyrophosphate
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- TEM
- transmission EM
- MIC
- minimum inhibitory concentration
- OADC
- oleic albumin dextrose catalase
- ADC
- albumin dextrose catalase
- BHI
- brain-heart infusion
- Kan
- kanamycin.
References
- 1. Barry C. E., Crick D. C., and McNeil M. R. (2007) Targeting the formation of the cell wall core of Mycobacterium tuberculosis. Infect. Disord. Drug Targets 7, 182–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackson M., McNeil M. R., and Brennan P. J. (2013) Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Fut. Microbiol. 8, 855–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Daffé M., and Draper P. (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39, 131–203 [DOI] [PubMed] [Google Scholar]
- 4. Hoffmann C., Leis A., Niederweis M., Plitzko J. M., and Engelhardt H. (2008) Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U.S.A. 105, 3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zuber B., Chami M., Houssin C., Dubochet J., Griffiths G., and Daffé M. (2008) Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190, 5672–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sani M., Houben E. N. G., Geurtsen J., Pierson J., de Punder K., van Zon M., Wever B., Piersma S. R., Jiménez C. R., Daffé M., Appelmelk B. J., Bitter W., van der Wel N., and Peters P. J. (2010) Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog. 6, e1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angala S. K., Belardinelli J. M., Huc-Claustre E., Wheat W. H., and Jackson M. (2014) The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit. Rev. Biochem. Mol. Biol. 49, 361–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schleifer K. H., and Kandler O. (1972) Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36, 407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pavelka M. S. Jr., Mahapatra S., and Crick D. C. (2014) Genetics of peptidoglycan biosynthesis. Microbiol. Spectr. 2, MGM2-0034-2013 [DOI] [PubMed] [Google Scholar]
- 10. Mikusová K., Mikus M., Besra G. S., Hancock I., and Brennan P. J. (1996) Biosynthesis of the linkage region of the mycobacterial cell wall. J. Biol. Chem. 271, 7820–7828 [DOI] [PubMed] [Google Scholar]
- 11. Mikusová K., Yagi T., Stern R., McNeil M. R., Besra G. S., Crick D. C., and Brennan P. J. (2000) Biosynthesis of the galactan component of the mycobacterial cell wall. J. Biol. Chem. 275, 33890–33897 [DOI] [PubMed] [Google Scholar]
- 12. Yagi T., Mahapatra S., Mikusova K., Crick D. C., and Brennan P. J. (2003) Polymerization of mycobacterial arabinogalactan and ligation to peptidoglycan. J. Biol. Chem. 278, 26497–26504 [DOI] [PubMed] [Google Scholar]
- 13. Hancock I. C., Carman S., Besra G. S., Brennan P. J., and Waite E. (2002) Ligation of arabinogalactan to peptidoglycan in the cell wall of Mycobacterium smegmatis requires concomitant synthesis of the two wall polymers. Microbiology 148, 3059–3067 [DOI] [PubMed] [Google Scholar]
- 14. Alderwick L. J., Radmacher E., Seidel M., Gande R., Hitchen P. G., Morris H. R., Dell A., Sahm H., Eggeling L., and Besra G. S. (2005) Deletion of Cg-emb in Corynebacterianeae leads to a novel truncated cell wall arabinogalactan, whereas inactivation of Cg-ubiA results in an arabinan-deficient mutant with a cell wall galactan core. J. Biol. Chem. 280, 32362–32371 [DOI] [PubMed] [Google Scholar]
- 15. Alderwick L. J., Dover L. G., Seidel M., Gande R., Sahm H., Eggeling L., and Besra G. S. (2006) Arabinan-deficient mutants of Corynebacterium glutamicum and the consequent flux in decaprenylmonophosphoryl-d-arabinose metabolism. Glycobiology 16, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 16. Swoboda J. G., Campbell J., Meredith T. C., and Walker S. (2010) Wall teichoic acid function, biosynthesis, and inhibition. Chembiochem 11, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hübscher J., Lüthy L., Berger-Bachi B., and Stutzmann Meier P. (2008) Phylogenetic distribution and membrane topology of the LytR-CpsA-Psr protein family. BMC Genomics 9, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hübscher J., McCallum N., Sifri C. D., Majcherczyk P. A., Entenza J. M., Heusser R., Berger-Bächi B., and Stutzmann Meier P. (2009) MsrR contributes to cell surface characteristics and virulence in Staphylococcus aureus. FEMS Microbiol. Lett. 295, 251–260 [DOI] [PubMed] [Google Scholar]
- 19. Kawai Y., Marles-Wright J., Cleverley R. M., Emmins R., Ishikawa S., Kuwano M., Heinz N., Bui N. K., Hoyland C. N., Ogasawara N., Lewis R. J., Vollmer W., Daniel R. A., and Errington J. (2011) A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dengler V., Meier P. S., Heusser R., Kupferschmied P., Fazekas J., Friebe S., Staufer S. B., Majcherczyk P. A., Moreillon P., Berger-Bächi B., and McCallum N. (2012) Deletion of hypothetical wall teichoic acid ligases in Staphylococcus aureus activates the cell wall stress response. FEMS Microbiol. Lett. 333, 109–120 [DOI] [PubMed] [Google Scholar]
- 21. Chan Y. G., Frankel M. B., Dengler V., Schneewind O., and Missiakas D. (2013) Staphylococcus aureus mutants lacking the LytR-CpsA-Psr family of enzymes release cell wall teichoic acids into the extracellular medium. J. Bacteriol. 195, 4650–4659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cieslewicz M. J., Kasper D. L., Wang Y., and Wessels M. R. (2001) Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J. Biol. Chem. 276, 139–146 [DOI] [PubMed] [Google Scholar]
- 23. Bender M. H., Cartee R. T., and Yother J. (2003) Positive correlation between tyrosine phosphorylation of CpsD and capsular polysaccharide production in Streptococcus pneumoniae. J. Bacteriol. 185, 6057–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hanson B. R., Runft D. L., Streeter C., Kumar A., Carion T. W., and Neely M. N. (2012) Functional analysis of the CpsA protein of Streptococcus agalactiae. J. Bacteriol. 194, 1668–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eberhardt A., Hoyland C. N., Vollmer D., Bisle S., Cleverley R. M., Johnsborg O., Håvarstein L. S., Lewis R. J., and Vollmer W. (2012) Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb. Drug Resist. 18, 240–255 [DOI] [PubMed] [Google Scholar]
- 26. Chan Y. G., Kim H. K., Schneewind O., and Missiakas D. (2014) The capsular polysaccharide of Staphylococcus aureus is attached to peptidoglycan by the LytR-CpsA-Psr (LCP) family of enzymes. J. Biol. Chem. 289, 15680–15690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu C., Huang I. H., Chang C., Reardon-Robinson M. E., Das A., and Ton-That H. (2014) Lethality of sortase depletion in Actinomyces oris caused by excessive membrane accumulation of a surface glycoprotein. Mol. Microbiol. 94, 1227–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liszewski Zilla M., Chan Y. G., Lunderberg J. M., Schneewind O., and Missiakas D. (2015) LytR-CpsA-Psr enzymes as determinants of Bacillus anthracis secondary cell wall polysaccharide assembly. J. Bacteriol. 197, 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liszewski Zilla M., Lunderberg J. M., Schneewind O., and Missiakas D. (2015) Bacillus anthracis lcp genes support vegetative growth, envelope assembly, and spore formation. J. Bacteriol. 197, 3731–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boshoff H. I., Myers T. G., Copp B. R., McNeil M. R., Wilson M. A., and Barry C. E. 3rd (2004) The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279, 40174–40184 [DOI] [PubMed] [Google Scholar]
- 31. Belardinelli J. M., Larrouy-Maumus G., Jones V., Sorio de Carvalho L. P., McNeil M. R., and Jackson M. (2014) Biosynthesis and translocation of unsulfated acyltrehaloses in Mycobacterium tuberculosis. J. Biol. Chem. 289, 27952–27965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hsieh J. M., Besserer G. M., Madej M. G., Bui H. Q., Kwon S., and Abramson J. (2010) Bridging the gap: a GFP-based strategy for overexpression and purification of membrane proteins with intra and extracellular C-termini. Protein Sci. 19, 868–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bou Raad R., Méniche X., de Sousa-d'Auria C., Chami M., Salmeron C., Tropis M., Labarre C., Daffé M., Houssin C., and Bayan N. (2010) A deficiency in arabinogalactan biosynthesis affects Corynebacterium glutamicum mycolate outer membrane stability. J. Bacteriol. 192, 2691–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bansal-Mutalik R., and Nikaido H. (2011) Quantitative lipid composition of cell envelopes of Corynebacterium glutamicum elucidated through reverse micelle extraction. Proc. Natl. Acad. Sci. U.S.A. 108, 15360–15365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peyret J. L., Bayan N., Joliff G., Gulik-Krzywicki T., Mathieu L., Schechter E., and Leblon G. (1993) Characterization of the cspB gene encoding PS2, an ordered surface-layer protein in Corynebacterium glutamicum. Mol. Microbiol. 9, 97–109 [DOI] [PubMed] [Google Scholar]
- 36. Mikusová K., Slayden R. A., Besra G. S., and Brennan P. J. (1995) Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 39, 2484–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ishizaki Y., Hayashi C., Inoue K., Igarashi M., Takahashi Y., Pujari V., Crick D. C., Brennan P. J., and Nomoto A. (2013) Inhibition of the first step in synthesis of the mycobacterial cell wall core, catalyzed by the GlcNAc-1-phosphate transferase WecA, by the novel caprazamycin derivative CPZEN-45. J. Biol. Chem. 288, 30309–30319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang Q., Zhu L., Jones V., Wang C., Hua Y., Shi X., Feng X., Jackson M., Niu C., and Gao Q. (2015) CpsA, a LytR-CpsA-Psr family protein in Mycobacterium marinum, is required for cell wall integrity and virulence. Infect. Immun. 83, 2844–2854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dusch N., Pühler A., and Kalinowski J. (1999) Expression of the Corynebacterium glutamicum panD gene encoding l-aspartate-α-decarboxylase leads to pantothenate overproduction in Escherichia coli. Appl. Environ. Microbiol. 65, 1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schäfer A., Tauch A., Jäger W., Kalinowski J., Thierbach G., and Pühler A. (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 [DOI] [PubMed] [Google Scholar]
- 41. Jackson M., Camacho L. R., Gicquel B., and Guilhot C. (2001) Gene replacement and transposon delivery using the negative selection marker sacB. in Mycobacterium tuberculosis Protocols (Parish T., and Stocker N. G., eds) pp. 59–75, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 42. Grzegorzewicz A. E., Pham H., Gundi V. A. K. B., Scherman M. S., North E. J., Hess T., Jones V., Gruppo V., Born S. E. M., Korduláková J., Chavadi S. S., Morisseau C., Lenaerts A. J., Lee R. E., McNeil M. R., and Jackson M. (2012) Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8, 334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin A., Camacho M., Portaels F., and Palomino J.-C. (2003) Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 47, 3616–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bhamidi S., Shi L., Chatterjee D., Belisle J. T., Crick D. C., and McNeil M. R. (2012) A bioanalytical method to determine the cell wall composition of Mycobacterium tuberculosis grown in vivo. Anal. Biochem. 421, 240–249 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.