

Abstract
Ubiquitination of the TrkA neurotrophin receptor in response to NGF is critical in the regulation of TrkA activation and functions. TrkA is ubiquitinated, among other E3 ubiquitin ligases, by Nedd4-2. To understand mechanistically how TrkA ubiquitination is regulated, we performed a siRNA screening to identify deubiquitinating enzymes and found that USP36 acts as an important regulator of TrkA activation kinetics and ubiquitination. However, USP36 action on TrkA was indirect because it does not deubiquitinate TrkA. Instead, USP36 binds to Nedd4-2 and regulates the association of TrkA and Nedd4-2. In addition, depletion of USP36 increases TrkA·Nedd4-2 complex formation, whereas USP36 expression disrupts the complex, resulting in an enhancement or impairment of Nedd4-2-dependent TrkA ubiquitination, respectively. Moreover, USP36 depletion leads to enhanced total and surface TrkA expression that results in increased NGF-mediated TrkA activation and signaling that augments PC12 cell differentiation. USP36 actions extend beyond TrkA because the presence of USP36 interferes with Nedd4-2-dependent Kv7.2/3 channel regulation. Our results demonstrate that USP36 binds to and regulates the actions of Nedd4-2 over different substrates affecting their expression and functions.
Keywords: deubiquitylation (deubiquitination), neurite outgrowth, neurotrophic factor, neurotrophin, ubiquitylation (ubiquitination), Kv channels, Nedd4-2, TrkA, USP36
Introduction
Ubiquitination, a post-translational modification involving the covalent binding of the 76-amino acid protein ubiquitin to a substrate, plays a critical role in many different cellular functions including protein degradation. The attachment of ubiquitin requires three sequential enzymatic steps, the first being the binding and activation of ubiquitin by an ubiquitin-activating enzyme (E1), followed by the transfer of ubiquitin to an ubiquitin-conjugating enzyme (E2) forming a complex that interacts with an ubiquitin-ligase (E3), which finally transfers the ubiquitin to the substrate (1). Ubiquitination can be reversed through the action of deubiquitinating enzymes (DUBs)3 (2), and an intimate association of the different E3 ubiquitin ligases and DUBs has been described to fine-tune substrate ubiquitination (3). Therefore, DUBs provide an additional regulation to this post-translational modification.
The NGF receptor TrkA undergoes ubiquitination by Nedd4-2 in response to NGF binding (4, 5) and also by three different E3 ubiquitin ligases: TRAF6 (5, 6), c-Cbl (7), and Cbl-b (8). TrkA ubiquitination mediated by Nedd4-2 plays a seminal role in NGF-TrkA functions such as differentiation, survival, and nociception both in vitro and in vivo (4, 5, 9, 10). Regarding DUBs, in addition to the potential role of CYLD (cylindromatosis tumor suppressor) in TrkA ubiquitination in the brain (6, 11), where TrkA expression is restricted to a small number of neuronal populations (12), a recent report suggests that USP8 (the ubiquitin-specific peptidase 8) deubiquitinates TrkA to regulate receptor trafficking and, consequently, PC12 cell differentiation (13). Because very little is known about how ubiquitination of the TrkA receptor is regulated by DUBs, the identification of new DUBs modulating TrkA ubiquitination will be very relevant to understanding the molecular mechanisms underlying regulation of TrkA ubiquitination and NGF-mediated functions.
Nedd4-2 belongs to the Nedd4 family of HECT-type E3 ubiquitin ligases that include eight members in mice and nine members in humans. Nedd4 proteins have shown some redundancy but also exhibit specificity for different biological processes (14). Nedd4-2 is present in many different types of tissues but is most highly expressed in heart, kidney, brain, lung, and liver (15, 16). Several substrates have been described for Nedd4-2 including Kv7.1 (17), Kv7.2/3, and Kv7.3/5 heterotetramers (18, 19), epithelial sodium channel (20), serum and glucocorticoid kinase 1, and 14-3-3 (21), and TrkA (4, 5, 22), among others. Nedd4-2 activity and substrate binding is regulated by different mechanisms including intramolecular interactions (23), phosphorylation by serum and glucocorticoid kinase (24, 25) and by AMPK (26), and interactions with Ndfip (27, 28), 14-3-3 proteins (29), and USP2-45 (30, 31). However, the identification of new regulators is required to fully understand the mechanisms of action of Nedd4-2.
In this work, we have identified USP36 (ubiquitin-specific peptidase 36) as a key regulator of TrkA activation and ubiquitination in response to NGF. Interestingly, USP36 does not function as a direct deubiquitinating enzyme of TrkA but rather exerts its function through an interaction with Nedd4-2. The effect of USP36 over Nedd4-2 extends beyond TrkA because the Kv7.2/3 channels, substrates of Nedd4-2, are also regulated by USP36.
Results
A Functional Screen to Identify Deubiquitinases That Modulate TrkA Activation Kinetics
To identify DUBs involved in the regulation of TrkA, we first studied the activation kinetics of TrkA in response to NGF using different conditions in PC12-6/15, because they overexpress human TrkA and display more sensitivity to NGF (32). The cells were stimulated with a 10-min pulse of NGF (100 ng/ml), which was then removed, and samples were collected at different time points. Western blotting analyses showed a rapid increase in TrkA activation at 15 min that diminished over a time period of 60 min (Fig. 1A) and was a consequence of TrkA degradation (10). This variation allowed us to monitor changes in TrkA activation and degradation in response to the alteration of the levels of DUBs compared with that obtained in control conditions.
FIGURE 1.

Screening to identify DUBs regulating TrkA activation in response to NGF. A, kinetics of TrkA activation after a 10-min pulse of NGF. PC12-6/15 cells were serum-starved overnight, and the next morning NGF (100 ng/ml) was added for 10 min. Then fresh medium without NGF was added to the cells, and the samples were collected after 15, 30, and 60 min. Western blotting analyses were performed to detect active TrkA (pTrkA) and total TrkA. Tubulin was used as a loading control. Graph showing pTrkA activation in response to NGF (n = 3). B, Western blotting showing pTrkA activation in response to NGF in cells expressing USP36, USP25, and USP4 siRNAs. PC12-6/15 cells were transfected with individual pools of the DUB siRNA library. The cells were treated with NGF for 10 min, and samples were collected 15 and 60 min after NGF removal. TrkA levels are shown. Tubulin was used as a loading control. C, graph showing the relative decrease of TrkA activation after NGF treatment between 15 and 60 min in response to DUB depletion from two independent experiments. The horizontal and dotted lines indicate the averages and standard deviations, respectively, of the results obtained for all the genes assessed. The bars corresponding to TrkA activation average from USP36, USP25, and USP4 depleted cells are indicated. D, list of genes and quantification of the screening showing the means ± standard deviation of TrkA activation from all assessed DUBs.
To identify DUBs modulating TrkA activation kinetics, we performed a screen using a library of siRNAs targeting dub genes that has been previously used (33, 34). The DUB library consisted of pools of four nonoverlapping siRNAs, which targeted all known or putative DUBs (see “Experimental Procedures”). PC12-6/15 cells were transfected with the siRNA pools, and 48 h later the cells were serum-starved for an additional 24 h. The cells were then stimulated with NGF, and the samples were collected 15 and 60 min after NGF had been removed. TrkA activation was then assessed as already indicated above (Fig. 1A). The results indicated that depletion of several DUBs differently affected TrkA activation at early and late time points (Fig. 1B). The results of two independent experiments were quantified as the decrease of pTrkA at 60 min post-NGF stimulation (Fig. 1, C and D). The reliability of our screen is supported by the fact that other DUBs affecting TrkA activation kinetics, such as USP8 and CYLD, have previously shown to be involved in TrkA ubiquitination (11, 13). Therefore, the results of our screen using a siRNA library identified several potential DUBs that altered TrkA activation kinetics.
USP36 as a DUB Candidate That Regulates TrkA Activation Kinetics
First, we focused our attention on DUBs (USP11, USP36, and USP49), in which a larger reduction of pTrkA signal was detected (Fig. 1, C and D). We performed a thorough validation of the siRNAs for each of the above DUBs generating lentiviral vectors expressing individual shRNAs with the sequences used in the siRNA screen. USP11 was efficiently down-regulated by two independent shRNAs but did not alter TrkA activation levels in response to NGF (data not shown). In addition, overexpression assays of USP11 did not modify TrkA protein levels or TrkA ubiquitination (data not shown). Finally, USP49 expression was not affected by any of the shRNAs used (data not shown). Therefore, we discontinued the study of these DUBs and focused our attention on USP36 based on these results. Of the four shRNA vectors (vectors 1–4) used to target USP36 mRNA, we observed that shRNA-1 was able to efficiently down-regulate endogenous expression of the USP36 protein upon the infection of PC12-6/15 cells with the lentiviruses (Fig. 2A). To confirm the data obtained from the screen, we performed detailed analyses of pTrkA activation kinetics in response to NGF in PC12-6/15 cells infected with the lentivirus expressing USP36 shRNA-1. We observed an increase in TrkA activation at early time points (5–30 min) when USP36 was depleted as compared with that of control cells. In addition, an increase in Akt and MAPK activation was also present in USP36-depleted cells treated with NGF (Fig. 2B, top panel). The enhanced activation of TrkA depended on the observed increased levels of the receptor, because quantification of relative TrkA activation with respect to total TrkA levels showed no differences between control and USP36-depleted cells (Fig. 2B, bottom graphs). As an additional control, we performed similar experiments using lentivirus expressing USP36 shRNA-4, which did not affect USP36 protein levels. In this case, no differences in the levels, activation of TrkA, or downstream signaling pathways were observed (data not shown). Thus, these data suggested that USP36 modulates TrkA levels that affect TrkA activation kinetics in response to NGF.
FIGURE 2.

USP36 as a regulator of TrkA levels, downstream signaling, and function. A, endogenous USP36 levels are reduced by shRNA-1. Western blotting showing USP36 levels in PC12-6/15 cells infected with lentivirus expressing control or USP36 shRNAs (shRNA-1–4) corresponding to the sequences of siRNAs present in the library used to perform the screening. B, USP36 protein regulates TrkA levels and the activation of the receptor and downstream signaling pathways in response to NGF. PC12-6/15 cells were infected with lentivirus expressing control shRNA or USP36 shRNA-1, and 5–7 days after infection, cells were treated as described in Fig. 1A. Active TrkA (pTrkA), Akt (pAkt), MAPK (pMAPK), and TrkA were detected using Western blotting. Tubulin was used as a loading control. A representative experiment is shown. Quantification of TrkA and pTrkA levels (bottom panels) is shown (means ± S.D.; n = 3; two-tailed unpaired Student's t test). C, a schematic diagram of the surface expression assay using the biotinylation procedure in response to NGF is shown (top panel). PC12 cells were infected with control and USP36 shRNA-1 lentivirus. The cells were treated with NGF as described in Fig. 1A and then biotinylated as described under “Experimental Procedures.” The cell lysates were prepared, surface proteins were subjected to precipitation with neutroavidin-agarose, and Western blotting analysis were performed with the corresponding antibodies (middle panel). Quantification of surface TrkA upon NGF treatment (means ± S.D.; n = 3, two-tailed unpaired Student's t test) is shown (bottom panel). D, PC12 cell differentiation in response to NGF is enhanced upon USP36 depletion. PC12 cells transfected with plasmids encoding for GFP and control shRNA, USP36 shRNA-1, or USP36 shRNA-6 were stimulated 2 days later with NGF (10 ng/ml) in serum-reduced medium to differentiate the cells. Western blotting analysis showing USP36 reduction in PC12 cells expressing USP36 shRNA-6 is shown (top panel). GFP-positive cells were scored for differentiation 72 h after the addition of NGF. The results are the means ± S.D. of five independent experiments counting at least 330 cells/condition (two-tailed unpaired Student's t test).
Because USP36 regulates total TrkA levels, we performed biotinylation experiments to address whether TrkA localization on the cell surface was also affected by USP36 at different time points upon NGF treatment (Fig. 2C, top panel). In the absence of NGF stimulation (zero time point), surface TrkA was increased in PC12-6/15 cells depleted of USP36 (Fig. 2C, middle and bottom panels). Furthermore, we observed a stronger reduction over time in the amount of surface TrkA in USP36-depleted cells, which at 60 min reached the same levels as biotinylated TrkA in control cells (Fig. 2C, middle and bottom panels). These data suggested that although there was more surface TrkA during basal conditions, when USP36 levels are reduced, TrkA seems to be degraded faster in response to NGF.
To address whether USP36 might have any physiological relevance on NGF-mediated function, we performed differentiation assays in PC12 cells transfected with plasmids expressing GFP and also control shRNA or USP36 shRNA-1. NGF stimulation (10 ng/ml) for 72 h evoked a significant increase of PC12 differentiation when USP36 was depleted using shRNA-1 (Fig. 2D). To have other independent shRNA against USP36 to validate the results obtained, we generated additional shRNAs and checked their effect on the levels of USP36. Among them, shRNA-6 was able to down-regulate USP36 levels (Fig. 2D, right panel). PC12 cells transfected with a plasmid encoding for GFP and shRNA-6 also enhanced cell differentiation in response to NGF compared with that of control shRNA (Fig. 2D). Therefore, USP36 control PC12 differentiation in response to NGF.
USP36 Interacts and Co-localizes with TrkA
Taking into account that previous reports using HeLa cells indicated that USP36 was localized to the nucleus (35), in mitochondria, and in the cytoplasm (36), we studied the localization of USP36 in PC12-6/15 cells. We obtained affinity-purified antibodies against USP36 (see “Experimental Procedures”) and performed immunofluorescence analyses. USP36 was present in nucleoli as previously described but also in other compartments outside of the nucleus (Fig. 3A, top panel). These results were further confirmed using another USP36 antibody (35) (Fig. 3A, bottom panel). In addition, fractionation experiments revealed that USP36 mainly accumulated in the non-nuclear compartments (Fig. 3B). Phospho-histone 3 was used as the control for nuclear fractions, whereas TrkA, tubulin, and GAPDH were utilized to monitor non-nuclear fractions (Fig. 3B). The lack of USP36 signal from the nuclear fractions using this approach was probably due to the low amount of USP36 coming from nucleoli. Therefore, USP36 is located mainly in non-nuclear fractions in PC12-6/15 cells.
FIGURE 3.
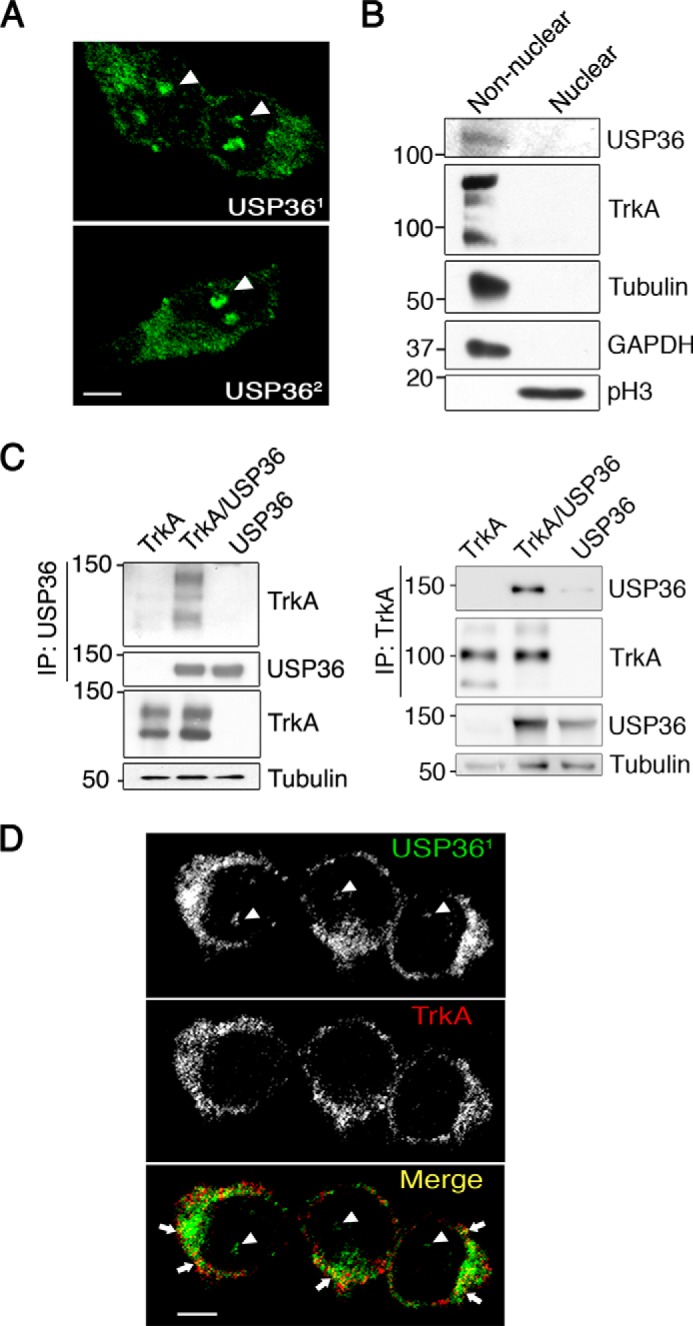
USP36 interacts and co-localizes with TrkA in PC12-6/15 cells. A, localization of USP36 in PC12-6/15 cells. USP36 immunofluorescence was detected using an antibody developed in our laboratory (top panel, USP361) and with an antibody obtained from Komada's laboratory (lower panel, USP362). Images were taken with a confocal microscope. Arrowheads indicate nucleoli staining. Scale bar, 5 μm. B, USP36 is mainly present in non-nuclear fractions in PC12-6/15 cells. Western blotting analyses were performed to detect USP36 in nuclear and non-nuclear fractions. TrkA, tubulin, and GAPDH were used as non-nuclear fractions, and phospho-Histone3 (pH3) was used as a nuclear marker. Note the presence of USP36 in non-nuclear fractions. C, USP36 interacts with TrkA. Lysates from HEK293 cells transfected with TrkA, FLAG-USP36, or TrkA and FLAG-USP36 were subjected to immunoprecipitation using FLAG antibodies (left panels) or TrkA antibodies (right panels). Western blotting was performed to assess co-immunoprecipitation of TrkA and USP36. The expression levels of TrkA, USP36, and tubulin as a loading control are shown. A representative experiment is shown (n = 3). D, co-localization of USP36 with TrkA. PC12-6/15 cells were seeded in coverslips, and immunofluorescence was performed using TrkA and USP36 antibodies. Images were taken with a confocal microscope. Arrows indicate co-localization of USP36 and TrkA, and arrowheads indicate nucleolar staining. Scale bar, 5 μm.
To address whether there was an interaction between TrkA and USP36, we performed co-immunoprecipitation assays in HEK293 cells transfected with plasmids expressing both proteins. TrkA and USP36 were pulled down after USP36 or TrkA immunoprecipitation, respectively (Fig. 3C). These data were further supported by the partial co-localization of TrkA and USP36 in PC12-6/15 cells (Fig. 3D, arrows). Altogether these results indicated that USP36 and TrkA interact and that USP36 is located mainly in non-nuclear compartments in PC12-6/15 cells, where a portion co-localizes with TrkA.
USP36 Modulates TrkA Ubiquitination Levels but Does Not Deubiquitinate TrkA
Considering that USP36 knockdown affects TrkA activation kinetics and USP36 interacts with TrkA, we tested the potential effect of USP36 on TrkA ubiquitination. Lysates from transfected HEK293 cells expressing TrkA with or without USP36, were subjected to immunoprecipitation with anti-TrkA antibodies to detect ubiquitinated TrkA by Western blotting analyses. The amount of ubiquitinated TrkA was significantly reduced in the presence of exogenous WT USP36 (Fig. 4A). To address whether the catalytic activity of USP36 was required, we generated a mutant C131S, because mutation of this residue has been reported to generate a catalytic inactive USP36 (35, 37). Interestingly, the expression of this mutant USP36 also rendered a similar significant reduction in TrkA ubiquitination compared with that of WT USP36 (Fig. 4A). To further address the role of USP36 on TrkA ubiquitination levels, we infected PC12-6/15 cells with lentivirus expressing control shRNA or USP36 shRNA-1. The cells with reduced levels of USP36 showed increased levels of TrkA ubiquitination in response to NGF compared with that of control cells (Fig. 4B). These data suggested that USP36 protein is instrumental in the regulation of TrkA ubiquitination levels upon activation of the receptor, although its activity seems not to be required.
FIGURE 4.

USP36 modulates TrkA ubiquitination in cells but not in vitro. A, TrkA ubiquitination is reduced in the presence of USP36 or USP36-C131S in HEK293 cells. Lysates from HEK293 cells transfected with different plasmids were subjected to immunoprecipitation (IP) using TrkA antibodies. Western blotting was performed to assess TrkA ubiquitination in the presence or absence of USP36. The expression levels of FLAG-USP36, TrkA, and tubulin, as the loading control, are shown. Note the reduced ubiquitination when TrkA is expressed together with WT USP36 or mutant USP36-C131S. A representative experiment is shown. Quantification of TrkA ubiquitination levels (bottom panel) is shown (means ± S.D.; n = 3; two-tailed unpaired Student's t test). B, USP36 depletion increases TrkA ubiquitination in response to NGF. PC12-6/15 cells infected with lentiviruses expressing control shRNA (C) or USP36 shRNA-1 (36) were serum-starved overnight and then stimulated with NGF (100 ng/ml) for the corresponding time. Lysates were subjected to immunoprecipitation using TrkA antibodies and subjected to Western blotting analysis to detect TrkA ubiquitination, TrkA activation, and TrkA. A representative experiment is shown. Quantification of TrkA ubiquitination levels in the above blot is shown. C, USP36 does not deubiquitinate TrkA in vitro. Lysates from HEK293 cells transfected with TrkA or with FLAG-USP36 were subjected to immunoprecipitation using antibodies TrkA or FLAG antibodies, respectively. FLAG-USP36 was eluted from the antibodies using FLAG peptide and added to one of the tubes containing TrkA. An in vitro deubiquitination reaction was performed as described under “Experimental Procedures” (left panels). As a positive control, in vitro deubiquitination assays were performed using FLAG-USP8 (right panels) using ubiquitinated TrkA from NGF-treated PC12-6/15 cells. A representative experiment for each DUB is shown (n = 3).
To address whether USP36 deubiquitinates TrkA, we carried out an in vitro deubiquitination assay. Ubiquitinated TrkA and USP36 protein were isolated by immunoprecipitation from transfected HEK293 cells, and both proteins were then co-incubated. TrkA ubiquitination levels were not altered by the presence of USP36 in the reaction (Fig. 4C, left panels), suggesting that USP36 does not deubiquitinate TrkA. To confirm that the deubiquitination reaction was done using the proper conditions, we performed additional experiments. Because USP8 has been previously reported to deubiquitinate TrkA (13), we repeated the assay using conditions similar to the ones used for USP36 in presence of purified USP8. TrkA ubiquitination was reduced in presence of USP8 (Fig. 4C, right panels). Therefore, altogether these data demonstrated that USP36 regulates TrkA ubiquitination but does not deubiquitinate TrkA.
USP36 Interacts with Nedd4-2 and Disrupts the TrkA·Nedd4-2 Complex
How can USP36 regulate TrkA ubiquitination without deubiquitinating TrkA? Several E3 ubiquitin ligases and DUBs act together to modulate substrate ubiquitination (3). To assess whether USP36 functions together with Nedd4-2, an E3 ubiquitin ligase that ubiquitinates TrkA (4, 5, 9, 10), we first performed interaction assays. Lysates from transfected HEK293 cells expressing FLAG-USP36, GFP-Nedd4-2, or both proteins together were subjected to immunoprecipitation. We observed that Nedd4-2 interacted with USP36 (Fig. 5A). To address whether there was an endogenous interaction between Nedd4-2 and USP36, co-immunoprecipitation experiments were performed in PC12-6/15 cells in the presence and absence of NGF. Nedd4-2 and USP36 interacted endogenously, and this association was independent of TrkA activation because NGF treatment did not alter it (Fig. 5B). To map the region of Nedd4-2 involved in the binding with USP36, we performed in vitro pulldown assays using lysates from HEK293 cells expressing FLAG-USP36 protein and different GST fusion proteins containing the WW domains of Nedd4-2 (GST-WW1, -WW2, -WW3, -WW4, and -WW3/4-HECT) (4), which are involved in protein interaction. The results showed that USP36 interacted with WW domains 1, 3, and 4 but not with other fusion proteins such as GST-Ral or GST-Raf (Fig. 5C). Altogether these results indicated that Nedd4-2 and USP36 interact independently of TrkA activation and that USP36 binds to the same WW domains that TrkA binds to (4). Subsequently, we asked whether there was competition between USP36 and TrkA for Nedd4-2 binding and performed pulldown assays that assessed the binding of TrkA to GST-WW3 in the absence or presence of USP36. The results obtained suggested that increasing amounts of USP36 disrupted the binding of TrkA to the GST-WW3 protein (Fig. 5D). Moreover, reduction of USP36 levels in PC12-6/15 cells enhanced the amount of TrkA bound to Nedd4-2 (Fig. 5E). Altogether these results suggested that USP36 interferes with TrkA·Nedd4-2 complex formation.
FIGURE 5.

USP36 interacts with Nedd4-2 through WW domains and competes TrkA binding to Nedd4-2. A, USP36 and Nedd4-2 interact. Lysates from HEK293 cells transfected with GFP-Nedd4-2, FLAG-USP36, or both plasmids together were subjected to immunoprecipitation (IP) using FLAG antibodies. Western blotting was performed to assess Nedd4-2 co-immunoprecipitation and USP36. The expression levels of Nedd4-2 and tubulin as a loading control are shown. A representative experiment is shown (n = 3). B, Nedd4-2 and USP36 interact independently of NGF. Cell lysates from PC12-6/15 cells stimulated or not with NGF for different time points were subjected to immunoprecipitation with Nedd4-2 antibodies and subjected to Western blotting analysis to detect the presence of USP36 in the immunoprecipitate. Active TrkA (pTrkA) and tubulin in cell lysates were probed to assess NGF treatment and loading, respectively. A representative experiment is shown (top panels). Quantification of USP36-Nedd4-2 interaction is shown (bottom panel) (means ± S.D.; n = 3). C, WW domains of Nedd4-2 mediate the interaction with USP36. Recombinant proteins containing the GST-WW domains (GST-WW1, GST-WW2, GST-WW3, and GST-WW4), WW3,4-HECT, and GST-Ral and GST-Raf binding domains were incubated with lysates containing USP36 and subjected to Western blotting analysis with FLAG antibodies (top panel). A Coomassie-stained gel is shown to detect GST fusion proteins (bottom panel). A representative experiment is shown (n = 2). Note the association of USP36 with WW1, 3, and 4 and WW3/4-HECT. D, USP36 competes with TrkA binding to Nedd4-2. A recombinant protein GST-WW3 domain was incubated with a fixed amount of TrkA and increasing amounts of USP36 and subjected to Western blotting analysis with different antibodies. A representative experiment is shown (n = 3). Note the displacement of TrkA binding to GST-WW3 and the increased binding of USP36 when increasing amounts of USP36 were added. E, TrkA and Nedd4-2 interaction is modulated by USP36 levels. Lysates from PC12-6/15 cells infected with lentivirus expressing control shRNA or USP36 shRNA-1 were subjected to immunoprecipitation using Nedd4-2 antibodies. Western blotting was performed to assess TrkA co-immunoprecipitation and Nedd4-2. The expression levels of TrkA, USP36, and tubulin as a control loading are shown. A representative experiment is shown (n = 3). Note the increased amount of TrkA co-immunoprecipitated with Nedd4-2 when USP36 levels are reduced.
USP36 and Nedd4-2 Regulate Each Other's Ubiquitination
Because USP36 regulates the ubiquitination of different substrates including SOD2, RNAP1, and H2B (36, 38, 39), we asked whether Nedd4-2 ubiquitination (23) could be modulated by USP36. Cell extracts from transfected HEK293 cells expressing Nedd4-2, USP36, or both together were subjected to immunoprecipitation with anti-Nedd4-2 antibodies to check the ubiquitination levels of Nedd4-2 by Western blotting analyses. Nedd4-2 protein seems to be less ubiquitinated in the presence of exogenous USP36 (Fig. 6A). To address whether Nedd4-2 expression levels were modulated by USP36, we carried out experiments in cells expressing Nedd4-2 and increasing amounts of USP36. Nedd4-2 protein levels were not affected by USP36 (Fig. 6B). Therefore, USP36 modulated Nedd4-2 ubiquitination status but not Nedd4-2 protein levels.
FIGURE 6.

USP36 and Nedd4-2 regulates each other ubiquitination. A, Nedd4-2 ubiquitination is reduced in presence of USP36. Lysates from HEK293 cells transfected with GFP-Nedd4-2, FLAG-USP36, or GFP-Nedd4-2 and FLAG-USP36 were subjected to immunoprecipitation (IP) using Nedd4-2 antibodies. Western blotting was performed to assess Nedd4-2 ubiquitination and Nedd4-2. The expression levels of USP36 and tubulin as a loading control are shown. A representative experiment is shown (n = 3). Note the reduced ubiquitination of Nedd4-2 when it is expressed together with USP36. B, USP36 expression does not affect Nedd4-2 levels. Lysates from HEK293 cells transiently transfected with Nedd4-2 and increasing concentrations of USP36 were subjected to Western blotting analysis to detect the amount of Nedd4-2. FLAG-USP36 protein levels from lysates are shown, and actin was used as a loading control. A representative experiment is shown (n = 3). C, USP36 polyubiquitination is modulated by Nedd4-2. Lysates from HEK293 cells transfected with GFP-Nedd4-2, USP36, or GFP-Nedd4-2 and USP36 were subjected to immunoprecipitation using FLAG antibodies. Western blotting was performed to assess USP36 ubiquitination and USP36. The expression levels of Nedd4-2 and actin as a loading control are shown. A representative experiment is shown (n = 2). Note the increased USP36 ubiquitination when Nedd4-2 is expressed together with USP36. D, Nedd4-2 expression down-regulates USP36 levels. Lysates from HEK293 cells transiently transfected with FLAG-USP36 and increasing concentrations of GFP-Nedd4-2 were subjected to Western blotting analysis to detect the amount of FLAG-USP36. GFP-Nedd4-2 protein levels from lysates are shown, and actin was used as a loading control. A representative experiment is shown (n = 3). Note the decrease in the levels of FLAG-USP36 with increasing amounts of GFP-Nedd4-2. E, Nedd4-2 ubiquitin ligase activity is required to down-regulate USP36 levels. Lysates from HEK293 cells transiently transfected with FLAG-USP36 and increasing concentrations GFP-Nedd4-2-C962S or YFP-Itch were subjected to Western blotting analysis to detect the amount of FLAG-USP36. GFP-Nedd4-2 protein levels and YFP-Itch are shown. Tubulin was used as a loading control. A representative experiment is shown (n = 3).
To address whether Nedd4-2 could ubiquitinate USP36, we performed additional experiments similar to the ones described above (Fig. 6A). In this case, USP36 was immunoprecipitated to detect its ubiquitination. An increase in USP36 polyubiquitination was detected in the presence of Nedd4-2 (Fig. 6C). To evaluate whether Nedd4-2 regulates USP36 protein levels, we carried out experiments in cells expressing USP36 and Nedd4-2. It was observed that increasing Nedd4-2 expression did significantly decrease USP36 levels (Fig. 6D). To test whether Nedd4-2 activity was required to regulate USP36 levels, the catalytically inactive Nedd4-2 mutant, C962S, which abolishes E3 ligase activity (4), was used. The expression of this mutant did not reduce USP36 levels (Fig. 6E). Moreover, the specificity of Nedd4-2 action over USP36 was assessed using Itch, another E3 ubiquitin ligase of the Nedd4 family, whose expression did not alter USP36 levels (Fig. 6E). Therefore, USP36 down-regulation mediated by Nedd4-2 requires the E3 ligase activity.
USP36 Regulates Nedd4-2-mediated Ubiquitination of TrkA
Considering that USP36 regulates TrkA ubiquitination (Fig. 4) and associates with Nedd4-2 modulating TrkA·Nedd4-2 binding (Fig. 5), we hypothesized that USP36 could regulate Nedd4-2-mediated ubiquitination of TrkA. To address this question, lysates from HEK293 cells expressing different combinations of the three proteins were subjected to immunoprecipitation with anti-TrkA antibodies. Whereas TrkA was efficiently ubiquitinated upon expression of Nedd4-2 (Fig. 7A, second lane), the co-expression of WT USP36 prevented TrkA ubiquitination mediated by Nedd4-2 (Fig. 7A, fourth and fifth lanes). The quantification of several experiments revealed a significant decrease in the TrkA ubiquitination induced by Nedd4-2 (Fig. 7A, right panel). These data suggested that USP36 competes with Nedd4-2-mediated ubiquitination of TrkA, most probably through an interaction with Nedd4-2 that prevents the access of Nedd4-2 to TrkA.
FIGURE 7.

USP36 competes with TrkA ubiquitination mediated by Nedd4-2. A, TrkA ubiquitination mediated by Nedd4-2 in HEK293 cells is impaired when USP36 is expressed. Lysates from HEK293 cells transfected with TrkA and/or GFP-Nedd4-2 and/or FLAG-USP36 were subjected to immunoprecipitation (IP) using TrkA antibodies. Western blotting was performed to assess TrkA ubiquitination and TrkA. The expression levels of Nedd4-2 and actin as a loading control are shown. A representative experiment is shown (n = 3). Quantification of TrkA ubiquitination levels in response to Nedd4-2 in the presence or absence of USP36 is shown (means ± S.D.; n = 3; one-tailed unpaired Student's t test). p values show the significance with respect to TrkA + Nedd4-2. B, USP36 controls TrkA levels competing the binding of Nedd4-2 and TrkA. Knockdown of USP36 increases TrkA protein levels leading to enhanced TrkA activation and signaling that results in augmented PC12 differentiation. USP36 ubiquitination and protein levels are regulated by Nedd4-2.
Expression of USP36 Competes with Kv7.2/3 Down-regulation and Restores Kv7.2/3 Inhibition Mediated by Nedd4-2
To evaluate whether USP36 could affect other Nedd4-2 substrates, we performed additional experiments. We used Kv7.2/3 channels, known substrates of Nedd4-2, which are tetramers conformed by Kv7.2 and Kv7.3 subunits that are the main constituent of the M current (14, 17, 40). The M current is a neuronal voltage-gated K+ conductance that controls resting membrane potential and excitability and is regulated by Nedd4-2 (18). First, we addressed whether Kv7.2/3 channels binding to Nedd4-2 were influenced by USP36. Pulldown assays performed as described in Fig. 5D, but in this case using Kv7.2/3 protein instead of TrkA, indicated that the binding of Kv7.2/3 channels to Nedd4-2 was diminished in the presence of USP36 (Fig. 8A). In addition, we assessed Kv7.2/3 expression levels upon co-transfection of the corresponding channels and Nedd4-2 or Nedd4-2, and USP36. Kv7.2/3 expression levels were decreased in the presence of Nedd4-2 (Fig. 8B, third lane). However, concomitant expression of USP36 restored channel expression levels to the ones observed in the absence of exogenous Nedd4-2 (Fig. 8B, fifth lane). Therefore, these data suggested that USP36 competes with Kv7.2/3 channel binding to Nedd4-2, preventing Nedd4-2-mediated down-regulation of the channels.
FIGURE 8.

USP36 regulates the protein levels and functions of Kv7.2/3 channels. A, USP36 competes with Kv7.2/3 binding to Nedd4-2. Recombinant protein GST-WW3 domain was incubated with a fixed amount of Kv7.2/3 channels and increasing amounts of USP36 and subjected to Western blotting analysis with different antibodies. A representative experiment is shown (n = 3). Note the displacement of Kv7.2/3 binding to GST-WW3 and the increased binding of USP36 when increasing amounts of USP36 were included. B, USP36 expression recues Kv7.2/3 down-regulation mediated by Nedd4-2. Lysates from HEK293 cells expressing Kv7.2/3 alone or in combination with GFP-Nedd4-2 and/or FLAG-USP36 were subjected to Western blotting analysis to detect the amount of Kv7.2/3 channels. GFP-Nedd4-2 and FLAG-USP36 protein levels from lysates are shown, and tubulin was used as a loading control. A representative experiment is shown (n = 3). Top and bottom asterisks indicate Kv7.2 and Kv7.3 subunits, respectively. Note that USP36 expression restores the levels of Kv7.2/3 channels in the presence of Nedd4-2. C, USP36 prevents Nedd4-2-mediated Kv7.2/3 current inhibition. Representative whole cell traces from HEK293T expressing Kv7.2/3, Nedd4-2, and USP36 proteins (top panels). D, normalized tail I-V relationships. The lines are fits of a Boltzmann equation to the data. E, graph showing the current densities of HEK293 cells expressing the corresponding constructs computed at −30 mV as the difference in quasi-instantaneous current after a prepulse to −110 mV (all channels closed) and +30 mV (all channels opened). The number of cells in each experiment is indicated in parentheses. The results are from two or more independent batches of cells and are shown as the means ± S.D. (unpaired Student's t test).
To assess the functional consequences of USP36 on the effects of Nedd4-2 on channels, we performed additional experiments to study the impact of USP36 over Nedd4-2-mediated Kv7.2/3 inhibition co-expressing Kv7.2/3 channels with combinations of Nedd4-2 with or without USP36. First, we observed that the biophysical properties of the Kv7.2/3 channels suffered minor or no changes when co-expressed with USP36, Nedd4-2, or Nedd4-2 plus USP36 (Table 1). However, the current density decreased significantly when the Kv7.2/3 subunits were co-expressed with Nedd4-2 (Fig. 8C), concordant with previous reports (18). Interestingly, partial rescue of Kv7.2/3 current density inhibition by Nedd4-2 expression was observed when USP36 was also co-expressed (Fig. 8C). No changes in the current density of the Kv7.2/3 channels were observed by the co-expression of USP36 (data not shown). Therefore, USP36 competes with Nedd4-2 actions over Kv7.2/3 channels, suggesting that USP36 has a global effect on the actions of Nedd4-2 over different substrates.
TABLE 1.
Biophysical properties of heteromeric channels Kv7.2/3
| V½ | Slope (zF/RT) | |
|---|---|---|
| mV | ||
| Kv7.2/3 | −27.9 ± 0.9 | 5.7 ± 1.2 |
| Kv7.2/3 + USP36 | −24.5 ± 1.7 | 4.9 ± 1.3 |
| Kv7.2/3 + Nedd4 | −28.5 ± 0.6 | 9.4 ± 0.6 |
| Kv7.2/3 + Nedd4 + USP36 | −25.0 ± 0.9 | 8.9 ± 0.8 |
Discussion
In this report, we provide evidence that USP36 regulates TrkA levels, activation, and ubiquitination through a mechanism that involves Nedd4-2. We looked for DUBs that could alter TrkA activation kinetics in response to NGF performing a siRNA screen, an approach that has been successfully used by several groups trying to identify DUBs involved in the modulation of signaling cascades triggered by different receptors (33, 34). We identified USP36 as a DUB that affected not only TrkA signaling but also ubiquitination. Interestingly, the effect of USP36 over TrkA was indirect because USP36 did not deubiquitinate TrkA. Instead, USP36 interacted with Nedd4-2, competing with its association with TrkA and impairing TrkA ubiquitination mediated by Nedd4-2 (Fig. 7B). The effects of USP36 on Nedd4-2 extend beyond TrkA because the levels and function of Kv7.2/3 channels, known substrates of Nedd4-2, are also regulated by the expression of USP36.
To date, the knowledge regarding the ubiquitination/deubiquitination machinery on the physiology of TrkA receptor has been very limited. Through a functional screening, we identified many potential DUBs that alter the activation kinetics of TrkA receptor in response to NGF treatment. Further support for the overall results obtained in our screening comes from a recently published paper in which USP8, one of the DUBs that modified TrkA activation in our screening (Fig. 1D), has been implicated in TrkA deubiquitination, trafficking, and PC12 differentiation (13). In addition, CYLD, a DUB that interacts with TrkA in an NGF-dependent manner, has been implicated in the deubiquitination of TrkA (6, 11). CYLD was one of the DUBs whose depletion also affected TrkA activation kinetics (Fig. 1D). Therefore, it seems that the ubiquitination/deubiquitination machinery regulating TrkA may appear as complex as in the case of other receptor tyrosine kinases, such as EGFR and Met, for which many identified DUBs regulate in a direct or indirect way the response to their activation by their cognate ligands (34, 41). Additional studies need to be carried out to address the role of other DUBs identified in this work within the NGF-TrkA system.
Based on localization, expression pattern, and recent literature, we decided to first focus on USP11, USP36, and USP49. Because of the preliminary results obtained with USP11 and USP49, we decided to discontinue studying their relation to TrkA and to focus on USP36. USP36 homologues in Saccharomyces cerevisiae and Drosophila melanogaster are Ubp10 and dUSP36/Scny, respectively, and share different degrees of homology between them (38). Most of the functions already described for USP36 proteins have been localized to the nucleolus modulating different proteins such as nucleophosmin/B23 (35), RNA polymerase I (39), c-Myc (37), H2B (38), and proliferating cell nuclear antigen (42), although there is also regulation of SOD2 in the mitochondria (36). In our work we have identified for the first time that USP36 is able to function in other cellular compartments regulating TrkA and Kv7.2/3 channels. Our study indicates that USP36 levels have an impact on the levels of TrkA affecting the downstream signaling pathways in response to NGF, resulting in an increase in the PC12 cell differentiation when USP36 protein was depleted. We have observed that most of USP36 protein in PC12-6/15 cells is present in non-nuclear compartments, where a fraction co-localizes and interacts with TrkA, although USP36 does not directly deubiquitinate TrkA. These results are in agreement with the fact that USP36 activity was not required to alter TrkA ubiquitination because the expression of a catalytically inactive mutant had the same effect on TrkA ubiquitination as WT USP36. All these data suggested that an indirect effect was responsible for USP36 actions on TrkA.
TrkA ubiquitination is carried out by Nedd4-2 together with other E3 ubiquitin ligases (4, 5, 9, 10). E3 ubiquitin ligases can be regulated by different DUBs (43), and therefore, we sought to determine whether Nedd4-2 could be regulated by USP36. We have identified an interaction between USP36 and the WW domains of Nedd4-2 that displaces the binding of Nedd4-2 with TrkA, precluding its ubiquitination and leading to increased levels of total and surface TrkA in basal conditions. Upon NGF treatment, surface TrkA in the absence of USP36 is greatly reduced as compared with the control cells, suggesting that TrkA is rapidly degraded (Fig. 2C). This is further supported by the fact that depletion of endogenous USP36 results in enhanced ubiquitination of TrkA (Fig. 4B), probably mediated by Nedd4-2, because overexpression of USP36 competes with Nedd4-2-mediated ubiquitination of TrkA (Fig. 7A). Interestingly, Nedd4-2 and USP36 seem to regulate each other (Fig. 6); USP36 ubiquitination and levels depend directly on Nedd4-2, whereas USP36 affects Nedd4-2 ubiquitination without any significant effect on protein levels. Our data are in agreement with previous data reporting that USP2 binds to Kv7.1 channels counteracting Nedd4-2 actions on these channels (30) and that USP2 binds also to Nedd4-2 and epithelial sodium channels (31). The binding of USP2 requires the HECT domain of Nedd4-2, which interferes with Nedd4-2 activity (31). All these results together with the ones presented in this work suggest that Nedd4-2 is tightly regulated by different DUBs. The in vivo relevance of Nedd4-2 has been recently highlighted. Nedd4-2 knock-out mice die perinatally because of a failure to inflate the lungs (44, 45), Nedd4-2 shows a protective regulatory role against the development of cystic fibrosis in lung (45), and it has been directly implicated in the development of peripheral neuropathic pain (46). The role of TrkA on pain has been well established for a long time in mice (47) and humans (48). Moreover, our group has recently demonstrated the direct relationship between TrkA ubiquitination and pain sensation in mice (9). It is tempting to speculate that USP36 may play some role in the pathologies in which Nedd4-2 and TrkA have been implicated. Further studies in these pathological scenarios will be required to address the potential role of USP36.
Experimental Procedures
Materials
NGF was obtained from Alomone Labs (Israel). The following antibodies and the corresponding dilutions for Western blotting were used unless otherwise stated: anti-203 (1:2000), RTA (1:2000), and USP36 (1:500 and 1:100 for immunocytochemistry) (generous gifts from Dionisio Martín-Zanca, Louis Reichardt, and Masayuki Komada, respectively); Nedd4-2 (1:3000) was previously described (4); Trk (C-14) (1:400) and P4D1 (1:400) (Santa Cruz); TrkA (E7) (2 μg/ml) (Zymed Laboratories Inc.); anti-FLAG (3 μg/ml), GAPDH (1:10000), actin (1:5000), and βIII-tubulin (1:10000) (Sigma); phospho-Trk (Y490) (1:1000), phospho-Akt (1:1000), and phospho-MAPK (1:2000) (Cell Signaling); FK1 (1:500) (Enzo Life Sciences); and phospho-Histone 3 (1:500) (Abcam). Antibodies against USP36 (2 μg/ml for immunocytochemistry) and GST (0.5 μg/ml) were developed (see below). FLAG peptide was purchased from Sigma.
Cell Cultures
HEK293 and HEK293FT cells were grown in DMEM supplemented with 10% bovine serum, 1% non-essential amino acids, 2 mm l-glutamine, and penicillin/streptomycin (100 units/ml). PC12 and PC12-6/15 cells, which stably overexpress human TrkA, (32) were grown in DMEM supplemented with 10% horse serum, 5% fetal bovine serum, 2 mm l-glutamine, and penicillin/streptomycin (100 units/ml).
DNA Transfections, Preparation of Cell Lysates, and Immunoblotting
Plasmid DNA was transiently transfected into HEK293 cells using the calcium phosphate method (49). Cells were lysed in lysis buffer (20 mm Tris, pH 8, 137 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 1 mm PMSF, 1 μg/ml aprotinin, 2 μg/ml leupeptin, 1 mm vanadate, 10 mm NaF, and 20 mm β-glycerophosphate) for 40 min at 4 °C with gentle shaking and centrifuged at 13,000 × g for 10 min to eliminate the debris. Lysates were resuspended in 2× Laemmli sample buffer and boiled for 7 min to denature the proteins. Proteins were resolved by SDS-PAGE, and Western blotting was performed with antibodies against different proteins. When co-immunoprecipitation assays were performed, cell lysates were incubated overnight with the corresponding antibody at 4 °C. Protein·antibody complexes were precipitated with protein A- or G-agarose, washed four times with lysis buffer, and detected by Western blotting assay. To avoid problems with the Ig chains in the immunoprecipitations, we used protein A- or G-conjugated HRP when the same species antibodies were used for both immunoprecipitation and Western blotting.
DUB siRNA Screen
Desiccated siRNA pools (0.25 nmol) targeting all known deubiquitinating enzymes were purchased from Dharmacon (SMARTpool siRNA catalog no. H-014700). The siRNA pools were diluted with OPTIMEM medium (25 μl/well), and Lipofectamine 2000 (2 μl/well) was added and incubated at 37 °C for 30 min. Lipofectamine·siRNA complexes were transferred to PC12-6/15 cells (5 × 105 cells/well in a 96-well plate) seeded the day before and then incubated for 48 h. The non-targeting siGlo RISC-Free siRNA was used as a negative control. Cell medium was replaced by serum-reduced medium (1/10 of normal amount), and cells were incubated for an additional 24 h. NGF (100 ng/ml) was added to the cells for 10 min, and then NGF-containing medium was removed and replaced by fresh serum-reduced medium. Cells were lysed in 2× Laemmli buffer 15 or 60 min after NGF was eliminated, and Western blotting analyses were performed to detect TrkA activation in response to DUB depletion.
Plasmids
The sequences of the mouse USP36 siRNAs 1–4 from the siRNA library were 5′-GAGCAAATATGTACTGTTG-3′, 5′-GAGAACGCCTATATGTGTG-3′, 5′-GAATGGCTATGCTAAGTTG-3′, and 5′-CGACAAGACTCTCTAATGA-3′. These sequences were used as templates to generate individual USP36 shRNAs against rat USP36, changing the non-conserved nucleotides using the pLVTHM lentiviral vector. In addition, the sequence 5′-GGAAGAAGAGGAAGAAGAAGA-3′ was also used to generate shRNA-6 against rat USP36. Control shRNA was generated using the sequence 5′-GCGCGCTTTGTAGGATTCG-3′ from Euglena gracillis chloroplast DNA between s16 S and 16 S rRNA genes.
The pcDNA3 expression vector containing the rat sequence of TrkA was used to express the TrkA protein (50). The pcDNA FLAG-HA-USP36-expressing plasmid was constructed after inserting FLAG-HA-USP36 DNA fragment from pDEST_LTR_N_FLAG_HA_USP36 (a gift from Wade Harper, Addgene plasmid no. 22579) in the pcDNA3 vector. The catalytically inactive USP36 mutant (USP36C131S) was generated by site-directed mutagenesis and confirmed by sequencing. The human isoform 3 Kv7.2 (Y15065) and Kv7.3 (NM004519) cDNA were provided by T. Jentsch (Leibniz-Institut für Molekulare Pharmakologie, Berlin, Germany). The subunits tagged at the N-terminal with mCFP or mYFP were cloned into pCDNA3.1. These N-terminal tags had been previously confirmed to have no impact on the electrophysiological properties of the channel (51).
Generation and Purification of anti-USP36 Antibodies
A GST fusion protein containing the first 120 amino acids of rat USP36 was produced in Escherichia coli and purified using glutathione-Sepharose affinity beads (ABT). Polyclonal antibodies against USP36 were generated after the immunization of two rabbits with a first injection of 500 μg of GST fusion protein with complete Freund adjuvant, followed by boosts of 250 μg with incomplete Freund adjuvant every 3 weeks. Bleedings were obtained 7–10 days after every boost, starting with the second boost. Antibodies generated against GST were depleted by incubation of sera with a GST affinity column. Antibodies against USP36 were purified using an affinity column coupled with the GST fusion protein used to inject the rabbits. Antibody elution was performed with 0.1 m glycine, pH 2.5, and the solution was neutralized and supplemented to a final concentration of 50 mm NaCl.
Lentivirus Generation and Infection
The lentiviruses used in this study were generated as previously described (52). Briefly, HEK293FT cells were transfected with 9 μg of pLVTHM control shRNA or pLVTHM USP36 shRNA-1, -2, -3, -4, or -6, together with 6 μg of psPAX2 and 5 μg of pMD.2G plasmids using the calcium phosphate method. The following day, the medium without antibiotics was changed, and the supernatant containing the lentiviruses was collected 48 h later, centrifuged at 5000 rpm (rotor ST16, Beckman) for 15 min, filtered (45 μm), and stored in aliquots at −80 °C. The viral medium was used to infect cells, which were monitored by the expression of GFP. In some preparations, lentiviruses were concentrated through ultracentrifugation or using LENTI-X solution (Clontech) as indicated by the manufacturer. Following infection, USP36 levels decreased by at least 70% using USP36 shRNA-1 or shRNA-6, but not with shRNA-2, -3, or -4, within 5–7 days.
Preparation of GST Fusion Proteins and in Vitro Binding Assays
The following GST-Nedd4-2 recombinant proteins were generated as previously described (4): GST-WW1 (amino acids 231–266), GST-WW2 (amino acids 406–438), GST-WW3 (amino acids 518–550), GST-WW4 (amino acids 569–601), and GST-WW3/4-HECT (amino acids 518–995). In addition, GST-RalBD and GST-RafBD were generated in a similar way. GST fusion proteins (20 μg) immobilized on glutathione-agarose beads were incubated with 800 μg of the corresponding FLAG-HA-USP36-transfected HEK293 lysates. Western blotting was performed with antibodies to detect USP36, and Coomassie Blue staining or Western blotting analyses were used to verify the equivalent loading of GST fusion proteins.
Cellular Fractionation
For nuclear and non-nuclear fractionation, PC12-6/15 cells were resuspended using cytosolic buffer (10 mm HEPES, pH 7, 1.5 mm MgCl2, 10 mm KCl, 1 mm EDTA, and 0.1% Nonidet P-40) with protease and phosphatase inhibitors and homogenized mechanically as described (53). Lysates were centrifuged at 500 × g for 10 min at 4 °C. The supernatant contains the non-nuclear fraction, and the pellet containing the nuclear fraction was resuspended in nuclear buffer (50 mm HEPES, pH 7, 1.5 mm MgCl2, 10 mm KCl, 10 mm NaCl, 1 mm EDTA, and 1% Nonidet P-40), incubated for 1 h on ice, frozen overnight at −80 °C, and then boiled for 5 min. Proteins were quantified, and protein loading buffer was added. p-Histone 3 was used as a nuclear marker.
Immunofluorescence
The PC12-6/15 cells used to detect USP36 localization were cultured on coverslips coated with 1 mg/ml poly-d-lysine. The cells were fixed with 4% paraformaldehyde in the presence of 20% sucrose in PBS for 5 min and washed with PBS for 5 min. Coverslips were incubated with 50 mm NH4Cl in PBS for 10 min to quench paraformaldehyde autofluorescence, and the cells were blocked in blocking solution (PBS with 10% FBS, 2% BSA, 0.1% Tween 20) with 0.1% Triton X-100 for 1 h at room temperature. Primary antibodies were added in the blocking solution plus 0.1% Triton X-100 and incubated at 4 °C overnight. Coverslips were washed with PBS, incubated with the fluorescent secondary antibody for 40 min in blocking buffer, and washed with PBS. Coverslips were mounted in Prolong Gold medium (Life Technologies). Images were collected with a Leica confocal microscope.
In Vitro Ubiquitination Assay
The experiments to perform in vitro ubiquitination assays were done as described (4), with slight modifications. Purified TrkA and FLA-HA-USP36 were immunoprecipitated from HEK293 cell lysates expressing TrkA or FLAG-HA-USP36. USP36 was eluted using FLAG peptide. Immunoprecipitated TrkA was incubated with 1 μg of GST or GST-WW3–4HECT with or without FLAG-HA-USP36 and in the presence of rabbit E1 (150 ng) and UbcH5b (300 ng) in 25 mm Tris-HCl, pH 7.5, 120 mm NaCl, 2 mm MgCl2, 2 mm ATP, 500 μm dithiothreitol, and 500 ng/μl bovine ubiquitin (Sigma) for 2 h at 30 °C. The reactions (70 μl) were stopped by the addition of 2× SDS buffer, resolved by SDS-PAGE, and visualized by Western blotting.
In Vitro Deubiquitination Assay
In vitro deubiquitination assays were carried out using a similar protocol as described (54), with slight modifications. Ubiquitinated TrkA was isolated from HEK293 cells transfected with an expression vector for TrkA and His6-Myc-ubiquitin. After incubation with proteasome inhibitor MG132 (20 μm) and chloroquine (50 μm) for 5–6 h, ubiquitinated TrkA was purified from cells extracts with anti-TrkA antibody (anti-203 antibody) covalently coupled to protein A-agarose in a lysis buffer containing 20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 0.2% sarkosyl, 10% glycerol, and protease inhibitors, except PMSF. FLAG-HA-USP36 was purified from HEK293 transfected cells using anti-FLAG M2-agarose and eluted with FLAG peptide. For in vitro deubiquitination assay, ubiquitinated TrkA protein was incubated with purified USP36 protein in the deubiquitination buffer (40 mm Tris-HCl, pH 8, 2 mm DTT, 5 mm MgCl2) for 4 h at 37 °C.
Surface Assays
PC12-6/15 cells were infected with control and USP36 shRNA lentivirus for 6 days. The cells were washed with PBS, chilled on ice, and biotinylated using 0.5 μg/ml Sulfo-NHS-SS-biotin for 20 min at 4 °C. The cells were sequentially washed with cold and room temperature PBS and incubated in prewarmed medium at 37 °C with or without NGF (50 ng/ml) for 10 min. Then NGF was removed and collected at different times (15 and 60 min) to allow the biotinylated receptors to become internalized and degraded. Subsequently, the cells were washed and lysed using lysis buffer, and the biotinylated proteins were precipitated with NeutroAvidin-beads, washed, subjected to SDS-PAGE, and immunoblotted using different antibodies. With this method it was possible to detect surface-labeled proteins that had not been degraded at different time points, regardless of whether the proteins had been internalized or whether they had returned to the plasma membrane.
PC12 Differentiation Assays
PC12 cells were transfected with lentiviral plasmids expressing GFP and control shRNA or USP36 shRNA-1 or -6. The medium was changed to serum-reduced medium 48 h late and stimulated with NGF (10 ng/ml) for an additional 72 h. Neurite-bearing cells expressing GFP and with the longest neurite, at least twice the size of the cell soma, were scored as positive.
Electrophysiological Measurements
Transient transfection for electrophysiology was performed using 25-kDa polyethylenimine (PolySciences reference no. 23966-2 2g, Eppelheim, Germany). All experiments were performed 48 h after transfection. Whole cell patch recordings of HEK293T cells were obtained at room temperature (21–25 °C) using a VE-2 amplifier (Alembic Instruments) equipped with an Rs Compensator. The cells were bathed in the following solution: 140 mm NaCl, 4 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm Na-HEPES, and 5 mm d-glucose, adjusted to pH 7.4 with NaOH. The osmolarity was adjusted with mannitol to ∼315 mOsm. Pipettes were pulled from borosilicate glass capillaries (Sutter Instruments) using a Narishige micropipette puller (PC-10; Narishige Instrument Company). Pipettes were filled with an internal solution containing 125 mm KCl, 5 mm MgCl2, 5 mm EGTA, 5 mm Na2ATP, and 10 mm K-HEPES, adjusted to pH 7.2 with KOH, and the osmolarity was adjusted to ∼300 mOsm with mannitol. The amplitude of the Kv7 current was defined as the peak difference in current relaxation measured at −30 mV after 500–1500-ms pulses to −110 mV (all channels closed) and to +30 mV (all channels opened). The data were acquired and analyzed using pCLAMP software (version 8.2).
Author Contributions
B. A., C.M.-R., A. V., and J. C. A. designed work; B. A., C. M.-R., C. G.-P., L. C., S. L.-B., A. A. C.-G., C. V.-G., and J. C. A. performed research; B. A., C.M.-R., C. G.-P., A. V., and J. C. A. analyzed the data; J. C. A. wrote the manuscript; and all authors commented on the manuscript.
Acknowledgments
We thank Dionisio Martín-Zanca, Louis Reichardt, Masayuki Komada, and Thomas Jentsch for the 203, RTA, and USP36 antibodies and the Kv7.2 and Kv7.3 plasmids, respectively.
This work was supported by Spanish Ministry of Science and Innovation Grant BFU2008-00162, by Spanish Ministry of Economy and Competitivity Grants BFU2011-22898 and BFU2014-51846-R, and in part by FP7-PAINCAGE Integrative Project Grant Agreement 603191 (to J. C. A.). The authors declare that they have no conflicts of interest with the contents of this article.
- DUB
- deubiquitinating enzyme.
References
- 1. Hershko A., Ciechanover A., and Varshavsky A. (2000) Basic medical research award: the ubiquitin system. Nat. Med. 6, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 2. Clague M. J., Coulson J. M., and Urbé S. (2012) Cellular functions of the DUBs. J. Cell Sci. 125, 277–286 [DOI] [PubMed] [Google Scholar]
- 3. Komander D. (2009) The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 37, 937–953 [DOI] [PubMed] [Google Scholar]
- 4. Arévalo J. C., Waite J., Rajagopal R., Beyna M., Chen Z. Y., Lee F. S., and Chao M. V. (2006) Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50, 549–559 [DOI] [PubMed] [Google Scholar]
- 5. Georgieva M. V., de Pablo Y., Sanchis D., Comella J. X., and Llovera M. (2011) Ubiquitination of TrkA by Nedd4-2 regulates receptor lysosomal targeting and mediates receptor signaling. J. Neurochem. 117, 479–493 [DOI] [PubMed] [Google Scholar]
- 6. Geetha T., Kenchappa R. S., Wooten M. W., and Carter B. D. (2005) TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 24, 3859–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takahashi Y., Shimokawa N., Esmaeili-Mahani S., Morita A., Masuda H., Iwasaki T., Tamura J., Haglund K., and Koibuchi N. (2011) Ligand-induced downregulation of TrkA is partly regulated through ubiquitination by Cbl. FEBS Lett. 585, 1741–1747 [DOI] [PubMed] [Google Scholar]
- 8. Emdal K. B., Pedersen A. K., Bekker-Jensen D. B., Tsafou K. P., Horn H., Lindner S., Schulte J. H., Eggert A., Jensen L. J., Francavilla C., and Olsen J. V. (2015) Temporal proteomics of NGF-TrkA signaling identifies an inhibitory role for the E3 ligase Cbl-b in neuroblastoma cell differentiation. Sci. Signal. 8, ra40. [DOI] [PubMed] [Google Scholar]
- 9. Yu T., Calvo L., Anta B., Lopez-Benito S., Lopez-Bellido R., Vicente-Garcia C., Tessarollo L., Rodriguez R. E., and Arevalo J. C. (2014) In vivo regulation of NGF-mediated functions by Nedd4-2 ubiquitination of TrkA. J. Neurosci. 34, 6098–6106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu T., Calvo L., Anta B., López-Benito S., Southon E., Chao M. V., Tessarollo L., and Arévalo J. C. (2011) Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic 12, 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wooten M. W., Geetha T., Babu J. R., Seibenhener M. L., Peng J., Cox N., Diaz-Meco M. T., and Moscat J. (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 283, 6783–6789 [DOI] [PubMed] [Google Scholar]
- 12. Sobreviela T., Clary D. O., Reichardt L. F., Brandabur M. M., Kordower J. H., and Mufson E. J. (1994) TrkA-immunoreactive profiles in the central nervous system: colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J. Comp. Neurol. 350, 587–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ceriani M., Amigoni L., D'Aloia A., Berruti G., and Martegani E. (2015) The deubiquitinating enzyme UBPy/USP8 interacts with TrkA and inhibits neuronal differentiation in PC12 cells. Exp. Cell Res. 333, 49–59 [DOI] [PubMed] [Google Scholar]
- 14. Yang B., and Kumar S. (2010) Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 17, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Araki N., Umemura M., Miyagi Y., Yabana M., Miki Y., Tamura K., Uchino K., Aoki R., Goshima Y., Umemura S., and Ishigami T. (2008) Expression, transcription, and possible antagonistic interaction of the human Nedd4L gene variant: implications for essential hypertension. Hypertension 51, 773–777 [DOI] [PubMed] [Google Scholar]
- 16. Kumar S., Harvey K. F., Kinoshita M., Copeland N. G., Noda M., and Jenkins N. A. (1997) cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics 40, 435–443 [DOI] [PubMed] [Google Scholar]
- 17. Jespersen T., Membrez M., Nicolas C. S., Pitard B., Staub O., Olesen S. P., Baró I., and Abriel H. (2007) The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 74, 64–74 [DOI] [PubMed] [Google Scholar]
- 18. Ekberg J., Schuetz F., Boase N. A., Conroy S. J., Manning J., Kumar S., Poronnik P., and Adams D. J. (2007) Regulation of the voltage-gated K+ channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J. Biol. Chem. 282, 12135–12142 [DOI] [PubMed] [Google Scholar]
- 19. Schuetz F., Kumar S., Poronnik P., and Adams D. J. (2008) Regulation of the voltage-gated K+ channels KCNQ2/3 and KCNQ3/5 by serum- and glucocorticoid-regulated kinase-1. Am. J. Physiol. Cell Physiol. 295, C73–C80 [DOI] [PubMed] [Google Scholar]
- 20. Staub O., Gautschi I., Ishikawa T., Breitschopf K., Ciechanover A., Schild L., and Rotin D. (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 16, 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhalla V., Daidié D., Li H., Pao A. C., LaGrange L. P., Wang J., Vandewalle A., Stockand J. D., Staub O., and Pearce D. (2005) Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4–2 by inducing interaction with 14-3-3. Mol. Endocrinol. 19, 3073–3084 [DOI] [PubMed] [Google Scholar]
- 22. Persaud A., Alberts P., Amsen E. M., Xiong X., Wasmuth J., Saadon Z., Fladd C., Parkinson J., and Rotin D. (2009) Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol. Systems Biol. 5, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bruce M. C., Kanelis V., Fouladkou F., Debonneville A., Staub O., and Rotin D. (2008) Regulation of Nedd4-2 self-ubiquitination and stability by a PY motif located within its HECT-domain. Biochem. J. 415, 155–163 [DOI] [PubMed] [Google Scholar]
- 24. Boehmer C., Wilhelm V., Palmada M., Wallisch S., Henke G., Brinkmeier H., Cohen P., Pieske B., and Lang F. (2003) Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 57, 1079–1084 [DOI] [PubMed] [Google Scholar]
- 25. Snyder P. M., Olson D. R., and Thomas B. C. (2002) Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 277, 5–8 [DOI] [PubMed] [Google Scholar]
- 26. Bhalla V., Oyster N. M., Fitch A. C., Wijngaarden M. A., Neumann D., Schlattner U., Pearce D., and Hallows K. R. (2006) AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 281, 26159–26169 [DOI] [PubMed] [Google Scholar]
- 27. Harvey K. F., Shearwin-Whyatt L. M., Fotia A., Parton R. G., and Kumar S. (2002) N4WBP5, a potential target for ubiquitination by the Nedd4 family of proteins, is a novel Golgi-associated protein. J. Biol. Chem. 277, 9307–9317 [DOI] [PubMed] [Google Scholar]
- 28. Konstas A. A., Shearwin-Whyatt L. M., Fotia A. B., Degger B., Riccardi D., Cook D. I., Korbmacher C., and Kumar S. (2002) Regulation of the epithelial sodium channel by N4WBP5A, a novel Nedd4/Nedd4-2-interacting protein. J. Biol. Chem. 277, 29406–29416 [DOI] [PubMed] [Google Scholar]
- 29. Ichimura T., Yamamura H., Sasamoto K., Tominaga Y., Taoka M., Kakiuchi K., Shinkawa T., Takahashi N., Shimada S., and Isobe T. (2005) 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280, 13187–13194 [DOI] [PubMed] [Google Scholar]
- 30. Krzystanek K., Rasmussen H. B., Grunnet M., Staub O., Olesen S. P., Abriel H., and Jespersen T. (2012) Deubiquitylating enzyme USP2 counteracts Nedd4-2-mediated downregulation of KCNQ1 potassium channels. Heart Rhythm 9, 440–448 [DOI] [PubMed] [Google Scholar]
- 31. Oberfeld B., Ruffieux-Daidie D., Vitagliano J. J., Pos K. M., Verrey F., and Staub O. (2011) Ubiquitin-specific protease 2–45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am. J. Physiol. Renal Physiol. 301, F189–F196 [DOI] [PubMed] [Google Scholar]
- 32. Hempstead B. L., Rabin S. J., Kaplan L., Reid S., Parada L. F., and Kaplan D. R. (1992) Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 9, 883–896 [DOI] [PubMed] [Google Scholar]
- 33. Eichhorn P. J., Rodón L., Gonzàlez-Juncà A., Dirac A., Gili M., Martínez-Sáez E., Aura C., Barba I., Peg V., Prat A., Cuartas I., Jimenez J., García-Dorado D., Sahuquillo J., Bernards R., et al. (2012) USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat. Med. 18, 429–435 [DOI] [PubMed] [Google Scholar]
- 34. Savio M. G., Wollscheid N., Cavallaro E., Algisi V., Di Fiore P. P., Sigismund S., Maspero E., and Polo S. (2016) USP9X controls EGFR fate by deubiquitinating the endocytic adaptor Eps15. Curr. Biol. 26, 173–183 [DOI] [PubMed] [Google Scholar]
- 35. Endo A., Matsumoto M., Inada T., Yamamoto A., Nakayama K. I., Kitamura N., and Komada M. (2009) Nucleolar structure and function are regulated by the deubiquitylating enzyme USP36. J. Cell Sci. 122, 678–686 [DOI] [PubMed] [Google Scholar]
- 36. Kim M. S., Ramakrishna S., Lim K. H., Kim J. H., and Baek K. H. (2011) Protein stability of mitochondrial superoxide dismutase SOD2 is regulated by USP36. J. Cell. Biochem. 112, 498–508 [DOI] [PubMed] [Google Scholar]
- 37. Sun X. X., He X., Yin L., Komada M., Sears R. C., and Dai M. S. (2015) The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc. Natl. Acad. Sci. U.S.A. 112, 3734–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buszczak M., Paterno S., and Spradling A. C. (2009) Drosophila stem cells share a common requirement for the histone H2B ubiquitin protease scrawny. Science 323, 248–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Richardson L. A., Reed B. J., Charette J. M., Freed E. F., Fredrickson E. K., Locke M. N., Baserga S. J., and Gardner R. G. (2012) A conserved deubiquitinating enzyme controls cell growth by regulating RNA polymerase I stability. Cell Reports 2, 372–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Donovan P., and Poronnik P. (2013) Nedd4 and Nedd4-2: ubiquitin ligases at work in the neuron. Int. J. Biochem. Cell Biol. 45, 706–710 [DOI] [PubMed] [Google Scholar]
- 41. Buus R., Faronato M., Hammond D. E., Urbé S., and Clague M. J. (2009) Deubiquitinase activities required for hepatocyte growth factor-induced scattering of epithelial cells. Curr. Biol. 19, 1463–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gallego-Sánchez A., Andrés S., Conde F., San-Segundo P. A., and Bueno A. (2012) Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 8, e1002826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Komander D., Clague M. J., and Urbé S. (2009) Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 [DOI] [PubMed] [Google Scholar]
- 44. Boase N. A., Rychkov G. Y., Townley S. L., Dinudom A., Candi E., Voss A. K., Tsoutsman T., Semsarian C., Melino G., Koentgen F., Cook D. I., and Kumar S. (2011) Respiratory distress and perinatal lethality in Nedd4-2-deficient mice. Nat. Commun. 2, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kimura T., Kawabe H., Jiang C., Zhang W., Xiang Y. Y., Lu C., Salter M. W., Brose N., Lu W. Y., and Rotin D. (2011) Deletion of the ubiquitin ligase Nedd4L in lung epithelia causes cystic fibrosis-like disease. Proc. Natl. Acad. Sci. U.S.A. 108, 3216–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laedermann C. J., Cachemaille M., Kirschmann G., Pertin M., Gosselin R. D., Chang I., Albesa M., Towne C., Schneider B. L., Kellenberger S., Abriel H., and Decosterd I. (2013) Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4–2 in neuropathic pain. J. Clin. Invest. 123, 3002–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smeyne R. J., Klein R., Schnapp A., Long L. K., Bryant S., Lewin A., Lira S. A., and Barbacid M. (1994) Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368, 246–249 [DOI] [PubMed] [Google Scholar]
- 48. Indo Y., Tsuruta M., Hayashida Y., Karim M. A., Ohta K., Kawano T., Mitsubuchi H., Tonoki H., Awaya Y., and Matsuda I. (1996) Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat. Genet. 13, 485–488 [DOI] [PubMed] [Google Scholar]
- 49. Sambrok J. R., and Russell D. W. (2001) Molecular Cloning: A Laboratory Manual, pp. 16.14–16.19, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 50. Arevalo J. C., Conde B., Hempstead B. L., Chao M. V., Martin-Zanca D., and Perez P. (2000) TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol. Cell. Biol. 20, 5908–5916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gomis-Perez C., Alaimo A., Fernandez-Orth J., Alberdi A., Aivar-Mateo P., Bernardo-Seisdedos G., Malo C., Areso P., Felipe A., and Villarroel A. (2015) An unconventional calmodulin-anchoring site within the AB module of Kv7.2 channels. J. Cell Sci. 128, 3155–3163 [DOI] [PubMed] [Google Scholar]
- 52. Arévalo J. C., Wu S. H., Takahashi T., Zhang H., Yu T., Yano H., Milner T. A., Tessarollo L., Ninan I., Arancio O., and Chao M. V. (2010) The ARMS/Kidins220 scaffold protein modulates synaptic transmission. Mol. Cell. Neurosci. 45, 92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Calvo L., Anta B., López-Benito S., Martín-Rodriguez C., Lee F. S., Pérez P., Martín-Zanca D., and Arévalo J. C. (2015) Bex3 dimerization regulates NGF-dependent neuronal survival and differentiation by enhancing trkA gene transcription. J. Neurosci. 35, 7190–7202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu J., Xia H., Kim M., Xu L., Li Y., Zhang L., Cai Y., Norberg H. V., Zhang T., Furuya T., Jin M., Zhu Z., Wang H., Yu J., Li Y., et al. (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]