

Abstract
Coenzyme engineering that changes NAD(P) selectivity of redox enzymes is an important tool in metabolic engineering, synthetic biology, and biocatalysis. Here we developed a high throughput screening method to identify mutants of 6-phosphogluconate dehydrogenase (6PGDH) from a thermophilic bacterium Moorella thermoacetica with reversed coenzyme selectivity from NADP+ to NAD+. Colonies of a 6PGDH mutant library growing on the agar plates were treated by heat to minimize the background noise, that is, the deactivation of intracellular dehydrogenases, degradation of inherent NAD(P)H, and disruption of cell membrane. The melted agarose solution containing a redox dye tetranitroblue tetrazolium (TNBT), phenazine methosulfate (PMS), NAD+, and 6-phosphogluconate was carefully poured on colonies, forming a second semi-solid layer. More active 6PGDH mutants were examined via an enzyme-linked TNBT-PMS colorimetric assay. Positive mutants were recovered by direct extraction of plasmid from dead cell colonies followed by plasmid transformation into E. coli TOP10. By utilizing this double-layer screening method, six positive mutants were obtained from two-round saturation mutagenesis. The best mutant 6PGDH A30D/R31I/T32I exhibited a 4,278-fold reversal of coenzyme selectivity from NADP+ to NAD+. This screening method could be widely used to detect numerous redox enzymes, particularly for thermophilic ones, which can generate NAD(P)H reacted with the redox dye TNBT.
Nicotinamide adenine dinucleotide (NAD, which includes NAD+ and NADH) and nicotinamide adenine dinucleotide phosphate (NADP, which includes NADP+ and NADPH) play distinctive roles in catabolism and anabolism, respectively. NAD and NADP differ in an additional phosphate group esterified at the 2′-hydroxyl group of adenosine monophosphate moiety of NADP (Fig. 1). Numerous redox enzymes use NAD(P) as a coenzyme, which is usually held within the Rossmann fold. Coenzyme engineering that changes coenzyme selectivity (i.e., NAD vs. NADP) of dehydrogenases and reductases is one of the important tools for metabolic engineering and synthetic biology. For example, to produce high-yield biofuels (e.g., butanol, fatty acid esters) under anaerobic conditions, it is essential to balance NADH generation and NAD(P)H consumption1,2,3. Besides the use of transhydrogenase to transfer the hydride from NADH to NADPH4,5, coenzyme engineering matching coenzyme selectivity of dehydrogenases and reductases is essential to achieve nearly theoretical product yields6,7,8. Coenzyme engineering is also essentially important in biocatalysis. Most times, changing the coenzyme selectivity of dehydrogenases from NADP to NAD is preferable due to (1) NAD is less costly than NADP9,10 and (2) NADH is more stable than NADPH11,12,13. Also, there are more NADH regeneration enzymes than NADPH regeneration enzymes14. Intensive studies have been conducted for changing coenzyme selectivity of dehydrogenases from NADP to NAD1,15,16 and from NAD to NADP17,18,19 as well as broadening coenzyme selectivity10. Recent coenzyme engineering studies have expanded the coenzyme selectivity of some redox enzymes to biomimetic coenzymes9,20,21,22.
Figure 1. Chemical structures of NADP+ and NAD+.

Structures of NADP+ and NAD+ were shown and the phosphate group on NADP+ was highlighted in gray.
Directed evolution is one of the powerful protein engineering tools that can change enzymes’ substrate selectivity. The most challenging task of directed evolution is the efficient identification of desired mutants from a large mutant library23. As for coenzyme engineering, the use of 96-well microplate screening based on the absorbency of NAD(P)H at 340 nm is a straightforward choice1. Also, the signal of NAD(P)H can be detected by colorimetric redox indicators. For example, the Arnold’s group utilized a redox dye nitroblue tetrazolium (NBT) plus catalyst phenazine methosulfate (PMS) to determine enhanced thermal stability of 6-phosphogluconate dehydrogenase (6PGDH) with the natural coenzyme (NADP+) in the cell lysate of E. coli24. Later, Zhao and his coworkers applied this method to find out dehydrogenase mutants with relaxed coenzyme selectivity10. However, the microplate-based screening is labor-intensive and time-consuming, involving colony picking, liquid cell culture, cell lysis, centrifugation, and enzyme activity assay. Due to high background noise of the intracellular reducing compounds and other redox enzymes in the cell lysate, Banta et al. utilized native gels to separate mutants of 2, 5-diketo-D-gluconic acid reductase from the cell lysate, followed by the measurement of UV absorbency changes25. However, this method required more steps and had lower capability of screening. Holbrook and his coworkers26 developed a method to duplicate colonies from Petri dishes to nitrocellulose paper followed by cell lysis by using lysozyme, detergent, and heat treatment. The targeted dehydrogenase activity was measured by the NBT-PMS assay. Later, Ellington’s group applied this method to identify lactate dehydrogenase mutants with their coenzyme preference change from NAD+ to NADP+ 27. Nevertheless, this screening method still requires a lot of steps and the throughput is modest due to smearing effect of colony duplication on nitrocellulose paper26. Therefore, it is urgently needed to develop a simple and effective high-throughput screening method to determine coenzyme selectivity change of dehydrogenases.
6-phosphogluconate dehydrogenase (6PGDH, EC 1.1.1.44), the third enzyme in the pentose phosphate pathway, converts the 6-phophogluconate and NADP+ to ribulose 5-phosphate, NADPH, and CO2. 6PGDH from a thermophilic bacterium Moorella thermoacetica was utilized to generate NADPH for the high-yield hydrogen production28 and generate NADH for electricity generation in biobattery29, but the catalytic efficiency (kcat/Km) for NADP+ was far higher than that for NAD+. Increasing this enzyme’s coenzyme selectivity for NAD+ could be important to decrease NADP+ use and increase lift-time of biobattery and other applications, such as low-cost biohydrogenation powered by sugars30.
In this study, we developed a simple Petri-dish-based double-layer screening for identifying mutants of thermophilic 6PGDH with enhanced catalytic efficiencies for NAD+, where the second agarose layer contained a redox dye tetranitroblue tetrazolium (TNBT), a catalyst PMS, 6-phophogluconate, and NAD+ and positive mutants were observed by darker color of heat treated colonies. Via this method, several 6GPDH mutants were identified with coenzyme selectivity reversed from NADP+ to NAD+.
Results
Dual promoter plasmid for screening and protein expression
For directed evolution, it is important to create the library with a large number of mutants and express enough recombinant proteins for characterization. In this study, the dual T7-tac promoter was constructed to control the expression of 6PGDH in both high transformation efficiency host E. coli TOP10 and high protein expression host E. coli BL21(DE3) (Fig. 2a). Plasmids and strains were listed in Table 1. Plasmid pET28a-Ptac-6pgdh consists of a strong inducible promoter T7, a modest inducible promoter tac, a lac operator, a ribosome binding site (RBS), and the 6pgdh gene. In E. coli TOP10, the modest expression of 6PGDH was accomplished by the tac promoter, while the T7 promoter was inactive due to a lack of T7 RNA polymerase. In E. coli BL21(DE3), high expression levels of 6PGDH was obtained under the control of both T7 and tac promoter. As SDS-PAGE analysis showed, although the 6PGDH expression was modest in E. coli TOP10, the 6PGDH expression level in E. coli BL21(DE3) was high and displayed 4.3-fold greater than that in E. coli TOP10 (Fig. 2b).
Figure 2. Validation of the dual T7-tac promoter for 6PGDH screening in E. coli TOP10 and protein expression in E. coli BL21(DE3).
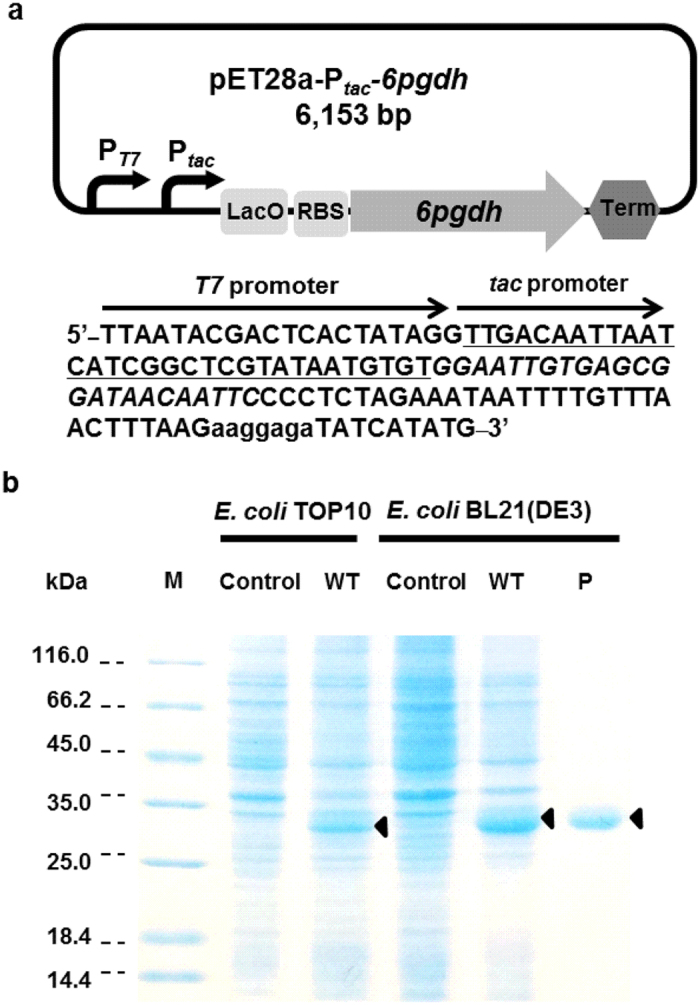
(a) The conceptual plasmid map of pET28a-Ptac-6pgdh. The DNA sequence of Ptac, lac operator, and RBS were shown as underlined, italic, and small, respectively. (b) SDS-PAGE analysis of 6PGDH expression in E. coli TOP10 and E. coli BL21(DE3). M, protein marker; Control, pET28a-Ptac; WT, pET28a-Ptac-6pgdh; P, purified 6PGDH. 6PGDH was indicated with an arrow.
Table 1. The strains, plasmids, and oligonucleotides used in this study.
| Description | Contents | Reference/sources |
|---|---|---|
| Strain | ||
| E. coli Bl21star(DE3) | B F− ompT gal dcm lon hsdSB(rB−mB−) rne131 (DE3) | Invitrogen |
| E. coli TOP10 | F– mcrA crmrr-hsdRMS-mcrBC) Φ80lacZac80 ΔlacX74 recA1 araD139 Δ(ara leu) 7697 galU galK rpsL (StrR) endA1 nupG | Invitrogen |
| Plasmid | ||
| pET28a | Invitrogen | |
| pET28a-6pgdh | Precusor of pET28a-Ptac-6pgdh | in this study |
| pET28a-Ptac-6pgdh | dual promoter (PT7and Ptac) and moth6pgdh | In this study |
| primers* | ||
| 28_back_F | 5′-CGACCAAGACCGACTAAGCCGATATGCATATGTATATCTCCTTCTTAAAG-3′ | pET28a |
| 28_back_R | 5′-GGCCACGACGTGGCCCGGAAACACCACCACCACCACCACTGAGAT-3′ | |
| 6PG_F | 5′-CTTTAAGAAGGAGATATACATATGCATATCGGCTTAGTCGGTCTTGGTCG-3′ | Moorella thermoacetica |
| 6PG_R | 5′-ATCTCAGTGGTGGTGGTGGTGGTGTTTCCGGGCCACGTCGTGGCC-3′ | |
| T7_Tac_F | 5′-Phos-GGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTC-3′ | pET28a-6pgdh |
| T7_Tac_R | 5′-Phos-GATGATTAATTGTCAACCTATAGTGAGTCGTATTAATTTCG-3′ | |
| 31_NNK_F | 5′-GAAGTGCGAGGATACGCCNNKACTAAGGCTACCGTGG-3′ | pET28a-Ptac-6pgdh |
| 31_NNK_R | 5′-CCACGGTAGCCTTAGTMNNGGCGTATCCTCGCACTTC-3′ | |
| 30_32_NNK_F | 5′-CATGGTCATGAAGTGCGAGGATACNNKATTNNKAAGGCTACCGTGGACAAAGC-3′ | pET28a-Ptac-6pgdh R31I |
| 30_32_NNK_R | 5′-GCTTTGTCCACGGTAGCCTTMNNAATMNNGTATCCTCGCACTTCATGACCATG-3′ | |
*Boldface nucleotide sequences indicate randomized positions.
Optimization of screening conditions
The mechanism of colorimetric assay in double-layer screening was shown in Fig. 3. The reduced NADH generated by 6PGDH reacts with TNBT in the presence of PMS, yielding a black TNBT-formazan. Heat-treatment was applied to reduce the background noise from host mesophilic enzymes and metabolites (e.g., NADPH and NADH)31 and disrupt cell membranes for NAD+ diffusion32,33. To find out the optimal heat-treatment temperature, two control colonies of E. coli TOP10 containing pET28a-Ptac-6pgdh (positive) and containing pET28a-Ptac (negative) were treated at 23, 60, 70 and 80 °C for 1 h. The colonies were then covered by the second layer containing the dye, PMS, NAD+, and substrate. As the result showed in Fig. 4a, the positive colonies and the negative colonies without heat treatment developed the same black color. When the heat-treatment temperature was greater than 70 °C, the colonies of the negative control did not develop the black color, indicating the decreased background noises. Positive control colonies expressing 6PGDH exhibited the darker color with haloes regardless of heat-treatment temperatures. Based on the result, the optimal heat-treatment temperature was 70 °C.
Figure 3. Scheme of the colorimetric assay for 6PGDH activity for NAD+.

6PGDH oxidizes 6-phosphogluconate into ribulose-5-phosphate and CO2, and reduces NAD+ to NADH. In the presence of phenazine methosulphate (PMS) and NADH, the colorless redox dye tetranitroblue tetrazolium (TNBT) was reduced to black TNBT-formazan.
Figure 4. Optimization of heat treatment temperature and color development time.

(a) Optimization of heat-treatment temperature for screening. E. coli TOP10 (pET28a-Ptac) was a negative control while E. coli TOP10 (pET28a-Ptac-6pgdh) was a positive control. Colonies were treated at 23, 60, 70 and 80 °C for 1 h, respectively. The color changes of the colonies overlaid by the second agarose layer were examined. (b) Optimization of color development time and NAD+ concentration. Heat-treated colonies of E. coli TOP10 (pET28a-Ptac-6pgdh) was overlaid by the second layer containing 0, 0.1, 0.3 or 1 mM NAD+. The color change profiles of colonies were photographed at 0, 0.5, 1 and 2 h.
The screening conditions were also influenced by NAD+ concentration and reaction time. As shown in Fig. 4b, the E. coli colonies expressing 6PGDH developed darker color and larger haloes with increasing NAD+ concentration and time interval. The colonies with the second layer containing 0 mM NAD+ started developing the dark color after 2 h, while E. coli TOP10 colonies (pET28a-Ptac) did not develop the color under the same condition (data not shown), implying that the heat-treatment was not enough to degrade E. coli NAD(P)+ completely34,35. To minimize the impact of E. coli inherent NAD(P)+, the screening time was recommended to be less than 2 h.
Screening 6PGDH mutants for increasing NAD+ activity
After optimization of heat-treatment temperature and color development time, the double-layer screening method was used to determine 6PGDH mutants’ coenzyme selectivity change. Figure 5 shows the image of a typical double-layer screening plate containing positive mutants, which were identified by darker color with haloes compared to wild-type and negative mutants. It was found that the color densities of colonies were related to mutant activities for NAD+ (data not shown).
Figure 5. Photo images of the double-layer screening of the mutant library containing two-site mutagenesis of A30/T32.

The second layer contained 0.1 mM NAD+. The color development time was 1 h. The positive mutants featuring darker colonies with haloes were identified by red arrows.
To make a reasonable size mutant library with 5-fold coverage, the 6PGDH mutant library was conducted through two-round saturation mutagenesis. In the first round, the site-directed mutagenesis of R31 was conducted and approximately 200 colonies were screened. Two positive mutants, R31T and R31I, were identified and characterized (Table 2). Starting from the best mutant R31I, the two-site-saturated mutagenesis library A30/T32 was constructed. After screening of 5,000 mutants, another four positive mutants, R31I/T32G, A30C/R31I/T32K, A30E/R31I/T32D and A30D/R31I/T32I were identified.
Table 2. Kinetics parameters of 6PGDH and mutants.
| Mutations | Km [μM] | kcat [s−1] | kcat/Km [mM−1 *s−1] | Ratio kcat/Km | |||
|---|---|---|---|---|---|---|---|
| NADP+ | NAD+ | NADP+ | NAD+ | NADP+ | NAD+ | NAD+/NADP+ | |
| WT | 13. 9 ± 1.1 | 1397 ± 111 | 8.73 ± 0.02 | 12.6 ± 0.4 | 628.8 | 9.0 | 0.014 |
| R31T | 35.5 ± 1.7 | 605 ± 47 | 10.83 ± 0.13 | 9.12 ± 0.23 | 305.6 | 15 | 0.049 |
| R31I | 26.5 ± 1.3 | 354 ± 12 | 11.53 ± 0.15 | 15.01 ± 0.17 | 435.0 | 42.4 | 0.097 |
| R31I/T32G | 104.4 ± 5.4 | 362 ± 23 | 11.9 ± 0.16 | 12.94 ± 0.27 | 114.0 | 35.8 | 0.31 |
| A30C/R31I/T32K | 698 ± 49 | 404 ± 33 | 6.23 ± 0.18 | 6.0 ± 0.2 | 8.9 | 14.8 | 1.66 |
| A30E/R31I/T32D | 660 ± 64 | 127 ± 7 | 3.1 ± 0.1 | 10.8 ± 0.2 | 4.7 | 85.1 | 18.1 |
| A30D/R31I/T32I | 228 ± 16 | 11.87 ± 0.55 | 1.81 ± 0.05 | 5.75 ± 0.05 | 7.9 | 484.2 | 61.1 |
Each value represents the average of three independent measurements.
Characterization of 6PGDH mutants
The activity and kinetic constants for NAD(P)+ of wild-type 6PGDH and mutants were summarized in Table 2. Through the first round screening, the R31I had a double Km value (26.5 μM) for NADP+ and a one fourth Km value (354 μM) for NAD+ compared to those of wild-type. Similarly, the R31T exhibited a 3.5-fold reversal due to higher Km value for NADP+ and lower Km value for NAD+. Starting from R31I, the second round mutant R31I/T32G had higher Km of 104.4 μM for NADP+ than that of R31I but no significant change in Km for NAD+. The A30C/R31I/T32K had lower kcat of 6.23 s−1 but much higher Km of 698 μM on NADP+. Also, this mutant had a kcat value of 6.0 s−1 and Km value of 404 μM on NAD+. The A30E/R31I/T32D had a very low kcat value of 3.1 s−1 but a high Km value of 660 μM for NADP+, resulting in catalytic efficiency for NADP+ as low as 4.7 mM−1 s−1. However, the kcat and Km for NAD+ decreased to 10.8 s−1 and 127 μM, respectively, resulting in an increase in catalytic efficiency for NAD+ to 85.1 mM−1 s−1.
The best mutant was A30D/R31I/T32I in terms of kcat/Km for NAD+. Comparing with wild-type, the kcat value for NADP+ decreased to 1.81 s−1 but the Km value increased to 228 μM. On the other hand, the kcat value for NAD+ reduced to 5.75 s−1 and the Km value decreased to 11.87 μM, which was comparable to the Km value of wild-type for NADP+ (13.9 μM). The catalytic efficiency of A30D/R31I/T32I for NADP+ was decreased by 80-fold, while the catalytic efficiency for NAD+ was increased by 54-fold, from 9 to 484.2 mM−1 s−1, resulting in a 4,278-fold reversal of coenzyme selectivity from NADP+ to NAD+.
Discussion
Here we developed an easy high-throughput screening method based on double-layer Petri dishes for determining the coenzyme selectivity of thermophilic 6PGDH for NAD+. In this screening, the reduced NADH generated from 6-phosphogluconate catalyzed by 6PGDH mutants could react with TNBT, generating the black TNBT formazan. Although double-layer screening is a very classical enzyme- or microorganism-screening technique without costly instruments, it was surprising that there were few efforts in coenzyme engineering possibly due to multiple reasons. Compared to colony duplication developed by the Holbrook’s group26, our method avoided colony duplication and possible smear effects during colony duplication, resulting in less labor and higher throughput screening capacity (e.g., 800 colonies per dish). Furthermore, we applied heat-treatment to kill the E. coli cells, disrupt cell membrane33,36, degrade metabolites including NAD(P)H, and deactivate other E. coli enzymes that can work with NAD+, but retain intracellular thermophilic 6PGDH for a quick screening. This heat-treatment was efficient to decrease background interference and facilitate substrate mass transfer (Fig. 4) but it also killed the E. coli cells, resulting in a problem for recovering E. coli cells. To avoid living cell colony replication before heat-treatment as performed previously23, we developed an alternative technique to recover the plasmid from dead E. coli colonies – picking black dead-cell colonies for micro-plasmid purification followed by the transformation of E. coli TOP10. We had a high-throughput screening capacity without any colony replication associated with smear effects and possible cross contamination. As compared to 96- or 384-well plates with assistance of an automatic machine, this high-throughput screening method has several advantages: high screening capacity of approximately 800 colonies per Petri dish, less reagent consumption, less time used for cell cultures and enzyme activity assay, and no need for costly automatic machine that most labs cannot afford.
It was essentially important to find out a suitable redox dye that can react with NADH but have low interference by other compounds. We tested a few redox dyes, including methyl viologen37, benzyl viologen38, neutral red39, methylene blue40 and TNBT41,42. It was found that TNBT was the best because black formazan was very stable in the exposure of air and it had the strongest color change comparing with controls (data not shown). For example, oxidized methylene blue (blue color) is a pH-dependent redox dye that can react with NADH. But its reduced form (colorless) can react with oxygen in air, resulting in slow regeneration of blue color. As a result, this dye was not suitable for screening dehydrogenases whose specific activities were low on non-natural coenzymes.
The E. coli TOP10 is a good strain for mutant library construction because of the high transformation efficiencies (e.g., 108–9 cfu/μg plasmid DNA). However, its ability of recombinant protein expression is much lower than the E. coli BL21(DE3) utilizing the pET expression system, which suffers from low transformation efficiencies (e.g., 106 cfu/μg plasmid DNA) and possible undesired DNA recombination. A typical directed evolution protocol often involves screening in E. coli TOP10 followed by subcloning of mutant’s DNA sequences into pET plasmid and recombinant protein expression in E. coli BL21(DE3)43,44. To delete the subcloning step between screening and protein expression, we developed a dual promoter T7-tac (Fig. 2a). In E. coli TOP10 host growing on the LB medium, the tac promoter was responsible for modest expression of the target protein. In E. coli BL21(DE3) host plus IPTG, higher protein expression levels were achieved (Fig. 2b).
Six positive mutants were identified through two round mutant libraries. The arginine at position 31 of wild-type 6PGDH was critical to recognize 2′-phosphate of NADP+ and formed double hydrogen bonds with 2′-phosphate by the side chain, which was supported by previous study45. Similarly, T32 formed another hydrogen bond with 2′-phosphate through the side chain. Meanwhile, A30 was responsible for the formation of the NADP-binding pocket because of close proximity to 2′-phosphate in the structure model (Fig. 6a). After one-site mutation to isoleucine, the mutant R31I lost the ability of binding the 2′-phosphate of NADP+, resulting in a double increase in Km for NADP+ and a four-time decrease in NAD+ (Table 2). Similarly, after mutating to threonine, the R31T had a three-fold Km increase for NADP+ but a two-fold Km decrease for NAD+.
Figure 6.
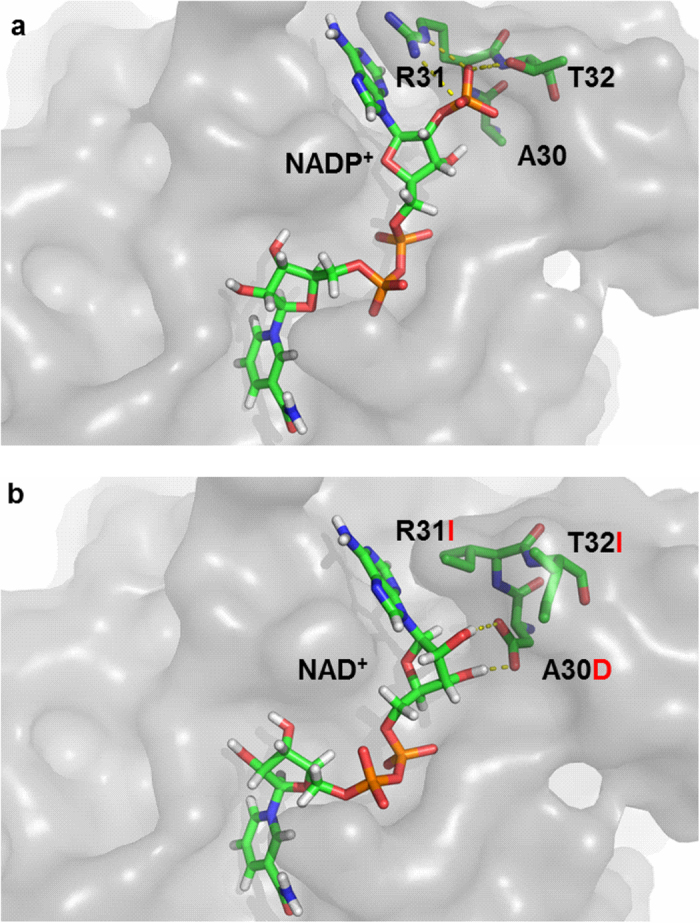
Surface view of wild-type 6PGDH with NADP+ (a) and mutant A30D/R31I/T32I with NAD+ (b). The amino acid residues A30, R31 and T32 of wild type 6PGDH and corresponding mutated residues of mutant A30D/R31I/T32I were depicted as sticks and the replacements were marked as red. Atoms were colored according to the types: N, blue; O, red; P, orange; C, green and H, white. Hydrogen bonding between residues and cofactor were shown as yellow line.
The A30D/R31I/T32I was the best mutant in terms of kcat/Km for NAD+. In addition to R31I, the extra mutation of alanine to aspartate at position 30 formed new hydrogen bonds with both 2′ and 3′-hydroxyl group of adenosine monophosphate moiety of NAD+ (Fig. 6b) and helped increasing the binding affinity for NAD+, further 30-fold decline in Km for NAD+ compared to R31I. The replacement to the other acidic amino acid glutamate at the same position was also found at A30E/R31I/T32D with 3-fold lower Km for NAD+ as compared to R31I. Recently, the mutant included replacement to aspartate at same position was reported for 6PGDH from G. stearothermophilus with slightly decreased Km46. The mutation threonine to isoleucine at position 32 broke the residual hydrogen bonds with 2′-phosphate of NADP+ and possibly decreased enzyme binding with NADP+, another 55-fold decrease in catalytic efficiency for NADP+. A decrease in binding affinity for NADP+ due to a mutation to a hydrophobic amino acid at the same threonine position was also reported for sheep liver 6PGDH mutant T34A47. Overall, a combination of the deletion of hydrogen bonds with 2′ phosphate of NADP+ at positions 31 and 32 and then addition of more hydrogen bonds with hydroxyl group of NAD+ at position 30 resulted in a more than 4,000-fold reversal of coenzyme selectivity from NADP+ to NAD+.
In conclusion, a high-throughput screening method was established for determining the NAD+ selectivity of thermophilic 6PGDH mutants. This double-layer method based on the colorimetric TNBT-PMS assay dramatically decreased dehydrogenase screening labor. The best 6PGDH mutant A30D/R31I/T32I showed a 4,278-fold reversal of coenzyme preference from NADP+ to NAD+.
Materials and Methods
Chemicals, plasmids and strains
All chemicals were reagent grade or higher, purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA), unless otherwise noted. The M. thermoacetica genomic DNA was purchased from the American Type Culture Collection (Manassas, VA). All enzymes for molecular biology experiments were purchased from New England Biolabs (NEB, Ipswich, MA). Strains, plasmids, and oligonucleotides used in this study are listed in Table 1.
Construction of pET28a-P tac -6pgdh
Plasmid pET28a-Ptac-6pgdh was constructed as follows. The inserted 6pgdh gene was amplified from M. thermoacetica genomic DNA by using a pair of primers 6PG_F/6PG_R and the linearized vector backbone was amplified from pET28a by using a pair of primers 28_back_F/28_back_R. The insertion and vector backbone were assembled into multimeric plasmids by prolonged overlap extension-PCR48. The PCR product was directly transformed into E. coli TOP10, yielding pET28a-6pgdh. To make the dual promoter plasmid pET28a-Ptac-6pgdh, the linear backbone of plasmid pET28a-Ptac-6pgdh was amplified based on pET28a-6pgdh by using a pair of 5′ phosphorylated primers T7_Tac_F/T7_Tac_R containing each half of the promoter Ptac and was self-ligated by NEB Quick Ligation™ Kit. After transformation into E. coli TOP10, plasmid pET28a-Ptac-6pgdh was obtained.
Construction of mutant libraries by saturation mutagenesis
The two-round DNA mutant libraries were constructed by the NEB Phusion site-directed mutagenesis kit. In the first round, the single-site saturation mutagenesis library R31 was amplified based on pET28a-Ptac-6pgdh by using a pair of degenerate primers 31_NNK_F/31_NNK_R. The two-site saturation mutagenesis library A30/T32 was amplified from plasmid of pET28a-Ptac-6pgdh (R31I) by using a pair of degenerate primers 30_32_NNK_F/30_32_NNK_R. PCR reaction solution (50 μL) containing 1 ng of plasmid template was conducted as follows: 98 °C denaturation for 1 min; 20 cycles of 98 °C denaturation for 30 s, 60 °C annealing for 30 s and 72 °C extension for 3 min; and 72 °C extension for 5 min. The PCR product was digested by DpnI followed by purification of gel electrophoresis and Zymoclean™ Gel DNA Recovery Kit (Zymo Research, Irvine, CA). The purified plasmid library was transformed into E. coli TOP10 for screening.
Optimization of heat treated temperature and time window of HTP screening
In order to find out the optimal heat treated temperature and time window for screening, the E. coli TOP10 carrying plasmid pET28a-Ptac(negative control) and E. coli TOP10 with pET28a-Ptac-6pgdh (positive control) were grown on the 1.5% agar LB medium with 50 μM kanamycin at 37 °C overnight and at room temperature for another day. Colonies of TOP10 (pET28a-Ptac) and TOP10 (pET28a-Ptac-6pgdh) were treated at 23, 60, 70 and 80 °C for 1 h, respectively. After cooling down to room temperature, 8 mL of 0.5% (wt/v) melted agarose solution (60 °C) containing 50 mM Tris-HCl (pH 7.5), 50 μM TNBT, 10 μM PMS, 2 mM 6-phosphogluconate, and 1 mM NAD+ was carefully poured on the heat-treated colonies. After incubation at room temperature for 1 h, the color change of colonies (to black) was examined on the white background. To find out the right time window for screening, colonies of E. coli TOP10 (pET28a-Ptac-6pgdh) were treated at 70 °C for 1 h. After cooling down to room temperature, the heat-treated colonies were overlaid by the melted 0.5% (wt/v) agarose solution containing 50 mM Tris-HCl (pH 7.5), 50 μM TNBT, 10 μM PMS, 2 mM 6-phosphogluconate, and a NAD+ concentration of 0, 0.1, 0.3 or 1 mM. The color change of different groups was photographed at room temperature at 0, 0.5, 1 and 2 h. Heat-treated colonies had three replicates.
High-throughput screening of 6PGDH mutants with enhanced NAD+ activities
The double-layer screening of 6PGDH mutants on NAD+ was performed as follows. After the transformation of the plasmid mutant library, the E. coli TOP10 cells were spread on the 1.5% agar LB medium containing 50 μM kanamycin with an expected colony number of 500–800 per Petri dish. The dishes were incubated at 37 °C overnight and at room temperature for another day to ensure enough recombinant 6PGDH expression49. The colonies on plates were treated at 70 °C for 1 h to kill cells, deactivate E. coli mesophilic enzymes, and degrade metabolites and intracellular coenzymes. Eight mL of 0.5% (wt/v) melted agarose solution (60 °C) containing 50 mM Tris-HCl (pH 7.5), 50 μM TNBT, 10 μM PMS, 2 mM 6-phosphogluconate, and 1 mM (for library R31) or 0.1 mM (for library A30/T32) NAD+ was carefully poured on the heat-treated colonies. After incubation at room temperature for 1 h, positive colonies were identified based on the formation of black haloes. The identified colonies embedded in the agarose gel were taken out by a sterile toothstick and then suspended in 200 μL of the P1 buffer of Zymo ZR Plasmid Miniprep™ kit followed by the Zymo ZR plasmid purification protocol. A tiny amount of plasmid extracted from the agarose gel was transformed into E. coli TOP10 for plasmid amplification, purification and DNA sequencing.
Overexpression and purification of wild-type 6PGDH and mutants
Plasmid pET28a-Ptac-6pgdh of wild-type or mutants was transformed to E. coli BL21(DE3) for protein overexpression in LB medium with 50 μM kanamycin at 37 °C. The protein expression was initiated by adding 0.1 mM IPTG until A600 reached ~0.6–0.8. The protein expression was induced at 37 °C for 6 h. Cell pellets were harvested by centrifugation and then were re-suspended in a 20 mM sodium phosphate and 0.3 M NaCl buffer (pH 7.5) containing 10 mM imidazole. After sonication and centrifugation, the supernatant was loaded onto the column packed with HisPur Ni-NTA Resin (Fisher Scientific, Pittsburgh, PA) and then eluted with 20 mM sodium phosphate buffer (pH 7.5) containing 300 mM NaCl buffer and 250 mM imidazole. Mass concentration of protein was determined by the Bradford assay using bovine serum albumin (BSA) as the standard and the 6PGDH expression levels and purified 6PGDH were checked by SDS-PAGE and analyzed by using densitometry analysis of the ImageJ software.
6PGDH activity assays
The activities of 6PGDH and mutants were measured in 100 mM HEPES buffer (pH 7.5) with final concentration of 2 mM 6-phosphogluconate, 2 mM NAD(P)+, 5 mM MgCl2 and 0.5 mM MnCl2 at 50 °C for 5 min, as described elsewhere29. The formation of NAD(P)H was measured at 340 nm by a UV/visible spectrophotometer (Beckman Coulter, Fullerton, CA). The enzyme unit was defined as one μmole of NAD(P)H produced per min. For determining enzyme kinetic parameters on coenzymes, the enzyme activity was measured in same buffer as described above except changing the concentration of NAD(P)+ from 5 to 5000 μM. The result was regressed by GraphPad Prism 5 (Graphpad Software Inc, La Jolla, CA) and apparent Km and kcat for NAD(P)+ of 6PGDH was given based on Michaelis-Menten nonlinear regression. All the reactions contained three independent replicates and fitted with linear range.
Structural analysis
The three-dimensional structure modeling of wild-type 6PGDH from M. thermoacetica and mutants were built by SWISS-MODEL based on the human 6PGDH (PDB: 2JKV) with 39.4% sequence identity. The structures of NADP+ and NAD+ were built by using Chemdraw (PerkinElmer, Waltham, MA). Starting from the initial protein and coenzyme structures, the conformation space accessible by NADP+ and NAD+ binding to the corresponding coenzyme binding area was analyzed by using the Autodock program (Scripps Research Institute, La Jolla, CA).
Additional Information
How to cite this article: Huang, R. et al. High-Throughput Screening of Coenzyme Preference Change of Thermophilic 6-Phosphogluconate Dehydrogenase from NADP+ to NAD+. Sci. Rep. 6, 32644; doi: 10.1038/srep32644 (2016).
Acknowledgments
This project cannot be carried out without the support of the Biological System Engineering Department, Virginia Polytechnic Institute and State University, Virginia, USA. This study is based upon work supported by the Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office under Award Number DE-EE0006968. Funding to YPZ for this work was partially supported by the Virginia Agricultural Experiment Station and the Hatch Program of the National Institute of Food and Agriculture, U.S. Department of Agriculture. Also, RH thanked Professor James Bowie for project discussion and thanked Professors Ryan Senger and Xueyang Feng for accessing some of their lab instruments.
Footnotes
Author Contributions Y.-H.P.Z. and R.H. wrote the main manuscript text, table and figures. R.H. conducted major experiments. H.C. conducted experiments of structure modeling of 6PGDH in Figure 6. C.Z. and J.E.K. were involved project discussion. All authors reviewed the manuscript.
References
- Brinkmann-Chen S. et al. General approach to reversing ketol-acid reductoisomerase cofactor dependence from NADPH to NADH. Proc. Natl. Acad. Sci. USA 110, 10946–10951 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.-D. & Zhang Y.-H. P. Analysis of biofuels production from sugar based on three criteria: Thermodynamics, bioenergetics, and product separation. Energy. Environ. Sci. 4, 784–792 (2011). [Google Scholar]
- Bastian S. et al. Engineered ketol-acid reductoisomerase and alcohol dehydrogenase enable anaerobic 2-methylpropan-1-ol production at theoretical yield in Escherichia coli. Metab. Eng. 13, 345–352 (2011). [DOI] [PubMed] [Google Scholar]
- Hou J., Lages N. F., Oldiges M. & Vemuri G. N. Metabolic impact of redox cofactor perturbations in Saccharomyces cerevisiae. Metab. Eng. 11, 253–261 (2009). [DOI] [PubMed] [Google Scholar]
- Gameiro P. A., Laviolette L. A., Kelleher J. K., Iliopoulos O. & Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 288, 12967–12977 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- King Z. A. & Feist A. M. Optimal cofactor swapping can increase the theoretical yield for chemical production in Escherichia coli and Saccharomyces cerevisiae. Metab. Eng. 24, 117–128 (2014). [DOI] [PubMed] [Google Scholar]
- Bommareddy R. R., Chen Z., Rappert S. & Zeng A. P. A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab. Eng. 25, 30–37 (2014). [DOI] [PubMed] [Google Scholar]
- Ehsani M., Fernandez M. R., Biosca J. A. & Dequin S. Reversal of coenzyme specificity of 2,3-butanediol dehydrogenase from Saccharomyces cerevisae and in vivo functional analysis. Biotechnol. Bioeng. 104, 381–389 (2009). [DOI] [PubMed] [Google Scholar]
- Rollin J. A., Tam T. K. & Zhang Y.-H. P. New biotechnology paradigm: cell-free biosystems for biomanufacturing. Green. Chem. 15, 1708–1719 (2013). [Google Scholar]
- Woodyer R., van der Donk W. A. & Zhao H. Relaxing the nicotinamide cofactor specificity of phosphite dehydrogenase by rational design. Biochemistry 42, 11604–11614 (2003). [DOI] [PubMed] [Google Scholar]
- Wu J. T., Wu L. H. & Knight J. A. Stability of NADPH: effect of various factors on the kinetics of degradation. Clin. Chem. 32, 314–319 (1986). [PubMed] [Google Scholar]
- Banta S., Swanson B. A., Wu S., Jarnagin A. & Anderson S. Alteration of the specificity of the cofactor-binding pocket of Corynebacterium 2,5-diketo-D-gluconic acid reductase A. Protein. Eng. 15, 131–140 (2002). [DOI] [PubMed] [Google Scholar]
- Wong C.-H. & Whitesides G. M. Enzyme-catalyzed organic synthesis: NAD (P) H cofactor regeneration by using glucose-6-phosphate and the glucose-6-phosphate dehydrogenase from Leuconostoc mesenteroides. J. Am. Chem. Soc. 103, 4890–4899 (1981). [Google Scholar]
- van der Donk W. A. & Zhao H. Recent developments in pyridine nucleotide regeneration. Curr. Opin. Biotechnol. 14, 421–426 (2003). [DOI] [PubMed] [Google Scholar]
- Lerchner A., Jarasch A., Meining W., Schiefner A. & Skerra A. Crystallographic analysis and structure-guided engineering of NADPH-dependent Ralstonia sp. alcohol dehydrogenase toward NADH cosubstrate specificity. Biotechnol. Bioeng. 110, 2803–2814 (2013). [DOI] [PubMed] [Google Scholar]
- Scrutton N. S., Berry A. & Perham R. N. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343, 38–43 (1990). [DOI] [PubMed] [Google Scholar]
- Hoelsch K., Suhrer I., Heusel M. & Weuster-Botz D. Engineering of formate dehydrogenase: synergistic effect of mutations affecting cofactor specificity and chemical stability. Appl. Microbiol. Biotechnol. 97, 2473–2481 (2013). [DOI] [PubMed] [Google Scholar]
- Zheng H., Bertwistle D., Sanders D. A. & Palmer D. R. Converting NAD-specific inositol dehydrogenase to an efficient NADP-selective catalyst, with a surprising twist. Biochemistry 52, 5876–5883 (2013). [DOI] [PubMed] [Google Scholar]
- Johannes T. W., Woodyer R. D. & Zhao H. Efficient regeneration of NADPH using an engineered phosphite dehydrogenase. Biotechnol. Bioeng. 96, 18–26 (2007). [DOI] [PubMed] [Google Scholar]
- Ji D. et al. Creation of bioorthogonal redox systems depending on nicotinamide flucytosine dinucleotide. J. Am. Chem. Soc. 133, 20857–20862 (2011). [DOI] [PubMed] [Google Scholar]
- Paul C. E., Arends I. W. & Hollmann F. Is simpler better? Synthetic nicotinamide cofactor analogues for redox chemistry. ACS Catalysis 4, 788–797 (2014). [Google Scholar]
- Zhang L., Yuan J., Xu Y., Zhang Y. H. & Qian X. New artificial fluoro-cofactor of hydride transfer with novel fluorescence assay for redox biocatalysis. Chem. Commun. 52, 6471–6474 (2016). [DOI] [PubMed] [Google Scholar]
- Liu W., Hong J., Bevan D. R. & Zhang Y. H. Fast identification of thermostable beta-glucosidase mutants on cellobiose by a novel combinatorial selection/screening approach. Biotechnol. Bioeng. 103, 1087–1094 (2009). [DOI] [PubMed] [Google Scholar]
- Mayer K. M. & Arnold F. H. A colorimetric assay to quantify dehydrogenase activity in crude cell lysates. J. Biomol. Screen. 7, 135–140 (2002). [DOI] [PubMed] [Google Scholar]
- Banta S. & Anderson S. Verification of a novel NADH-binding motif: combinatorial mutagenesis of three amino acids in the cofactor-binding pocket of Corynebacterium 2,5-diketo-D-gluconic acid reductase. J. Mol. Evol. 55, 623–631 (2002). [DOI] [PubMed] [Google Scholar]
- El Hawrani A. S., Sessions R. B., Moreton K. M. & Holbrook J. J. Guided evolution of enzymes with new substrate specificities. J. Mol. Biol. 264, 97–110 (1996). [DOI] [PubMed] [Google Scholar]
- Flores H. & Ellington A. D. A modified consensus approach to mutagenesis inverts the cofactor specificity of Bacillus stearothermophilus lactate dehydrogenase. Protein. Eng. Des. Sel. 18, 369–377 (2005). [DOI] [PubMed] [Google Scholar]
- Rollin J. A. et al. High-yield hydrogen production from biomass by in vitro metabolic engineering: Mixed sugars coutilization and kinetic modeling. Proc. Natl. Acad. Sci. USA 112, 4964–4969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Kin Tam T., Sun F., You C. & Zhang Y.-H. P. A high-energy-density sugar biobattery based on a synthetic enzymatic pathway. Nat. Commun. 5, 3026 (2014). [DOI] [PubMed] [Google Scholar]
- Wang Y., Huang W., Sathitsuksanoh N., Zhu Z. & Zhang Y. H. Biohydrogenation from biomass sugar mediated by in vitro synthetic enzymatic pathways. Chem. Biol. 18, 372–380 (2011). [DOI] [PubMed] [Google Scholar]
- Berridge M. V., Herst P. M. & Tan A. S. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol. Annu. Rev. 11, 127–152 (2005). [DOI] [PubMed] [Google Scholar]
- Zhou Y. et al. Determining the Extremes of the Cellular NAD(H) Level by Using an Escherichia coli NAD+ -Auxotrophic Mutant. Appl. Environ. Microbiol. 77, 6133–6140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninh P. H., Honda K., Sakai T., Okano K. & Ohtake H. Assembly and multiple gene expression of thermophilic enzymes in Escherichia coli for in vitro metabolic engineering. Biotechnol. Bioeng. 112, 189–196 (2015). [DOI] [PubMed] [Google Scholar]
- Honda K. et al. In vitro metabolic engineering for the salvage synthesis of NAD +. Metab. Eng. 35, 114–120 (2016). [DOI] [PubMed] [Google Scholar]
- Hofmann D., Wirtz A., Santiago-Schübel B., Disko U. & Pohl M. Structure elucidation of the thermal degradation products of the nucleotide cofactors NADH and NADPH by nano-ESI-FTICR-MS and HPLC-MS. Anal. Bioanal. Chem. 398, 2803–2811 (2010). [DOI] [PubMed] [Google Scholar]
- Ninh P. H. et al. Development of a continuous bioconversion system using a thermophilic whole-cell biocatalyst. Appl. Environ. Microbiol. 79, 1996–2001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do P. M. et al. Engineering Escherichia coli for fermentative dihydrogen production: potential role of NADH-ferredoxin oxidoreductase from the hydrogenosome of anaerobic protozoa. Appl. Biochem. Biotechnol. 153, 21–33 (2009). [DOI] [PubMed] [Google Scholar]
- Mihara H. et al. The iscS gene is essential for the biosynthesis of 2-selenouridine in tRNA and the selenocysteine-containing formate dehydrogenase H. Proc. Natl. Acad. Sci. USA 99, 6679–6683 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D. H. & Zeikus J. G. Electricity generation in microbial fuel cells using neutral red as an electronophore. Appl. Environ. Microbiol. 66, 1292–1297 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilner O. I. et al. Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nano. 4, 249–254 (2009). [DOI] [PubMed] [Google Scholar]
- Kugler P. A gel-sandwich technique for the qualitative and quantitative determination of dehydrogenases in the enzyme histochemistry. I. Development of the new methods on the example of LDH (E.C. 1.1.1.27). Histochemistry 60, 265–293 (1979). [DOI] [PubMed] [Google Scholar]
- Ishizuka H., Tokuoka K., Sasaki T. & Taniguchi H. Purification and some properties of an erythrose reductase from an Aureobasidium sp. mutant. Biosci. Biotechnol. Biochem. 56, 941–945 (1992). [DOI] [PubMed] [Google Scholar]
- Weiß M. S., Pavlidis I. V., Vickers C., Höhne M. & Bornscheuer U. T. Glycine oxidase based high-throughput solid-phase assay for substrate profiling and directed evolution of (R)-and (S)-selective amine transaminases. Anal. Chem. 86, 11847–11853 (2014). [DOI] [PubMed] [Google Scholar]
- Shin H. et al. Exploring the functional residues in a flavin-binding fluorescent protein using deep mutational scanning. PloS one 9, e97817 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetaud E. et al. 6-Phosphogluconate dehydrogenase from Lactococcus lactis: a role for arginine residues in binding substrate and coenzyme. Biochem. J. 338(Pt 1), 55–60 (1999). [PMC free article] [PubMed] [Google Scholar]
- Opgenorth P. H., Korman T. P. & Bowie J. U. A synthetic biochemistry module for production of bio-based chemicals from glucose. Nat. Chem. Biol. 12, 393–395 (2016). [DOI] [PubMed] [Google Scholar]
- Li L. & Cook P. F. The 2′-phosphate of NADP is responsible for proper orientation of the nicotinamide ring in the oxidative decarboxylation reaction catalyzed by sheep liver 6-phosphogluconate dehydrogenase. J. Biol. Chem. 281, 36803–36810 (2006). [DOI] [PubMed] [Google Scholar]
- You C., Zhang X. Z. & Zhang Y.-H. P. Simple cloning via direct transformation of PCR product (DNA Multimer) to Escherichia coli and Bacillus subtilis. Appl. Environ. Microbiol. 78, 1593–1595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Banerjee A., Pan S. H. & Li Z. J. Galactose can be an inducer for production of therapeutic proteins by auto-induction using E. coli BL21 strains. Protein Expr. Purif. 83, 30–36 (2012). [DOI] [PubMed] [Google Scholar]