

Abstract
Acute myocardial infarction is one of the leading causes for death around the world. Although essential for successful interventional therapy, it is inevitably complicated by reperfusion injury. Thus effective approaches to reduce ischemia/reperfusion (I/R) injury are still critically needed. To test our hypothesis that intravenous administration of NAD+ can attenuate I/R injury by reducing apoptotic damage and enhancing antioxidant capacity, we used a rat mode of myocardial I/R. Our study found that administration of 10-20 mg/kg NAD+ can dose dependently reduce myocardial infarct induced by I/R, with an approximately 85% reduction of the infarct at the dosage of 20 mg/kg NAD+. We further found that the injection of NAD+ can significantly decrease I/R-induced apoptotic damage in the heart: NAD+ administration can both decrease the TUNEL signals, Bax, cleaved caspase-3 levels and increase the Bcl-XL levels in the rats that are subjected to myocardial I/R injury. NAD+ administration can also significantly attenuate I/R-induced decreases in SOD activity and SOD-2 protein levels in the hearts. NAD+ can profoundly decrease myocardial I/R injury at least partially by attenuating apoptotic damage and enhancing the antioxidant capacity, thus suggesting that NAD+ may become a promising therapeutic agent for myocardial I/R injury.
Keywords: Myocardial ischemia/reperfusion, infarction, NAD+, apoptosis, antioxidation
Introduction
Acute myocardial infarction is the leading cause of death with an estimated annual incidence of 0.735 million in the United States [1]. Although percutaneous coronary intervention (PCI) has been widely applied in clinical settings, myocardial ischemia/reperfusion (I/R) injury remains poorly solved, which somehow brings about acute adverse events such as ventricular fibrillation, acute heart failure and death [2]. Many approaches tried to reduce the infarct volume in acute myocardial infarction, but so far none has been approved by FDA [3]. Therefore, it is critical to establish novel therapeutic strategies for myocardial I/R injury.
It is generally accepted that the major pathological factors in myocardial I/R injury include energy metabolism dysfunction, oxidative stress, mitochondrial permeability transition pore (mPTP), inflammation and calculim overload [4-6]. All those factors jointly induced the apoptosis and necrosis of myocytes in I/R condition. Thus, finding an approach to decrease the death of myocyte may be the final goal in this direction.
Cumulative studies have uncovered that NAD+ mainly or partly involved in maintaining the balance of energy, calcium, redox, and organelles under stress [7-10]. Previous observations of our lab and other laboratories have suggested that NAD+ treatment can profoundly decrease brain I/R injury and the death of myocytes under insults, respectively [11,12]. However, to our best knowledge, there has been no in vivo studies described that directly intravenous supplementation of NAD+ can profoundly protected hearts from I/R damage to date. Therefore, we aim to test that exogenous NAD+ can profoundly protect hearts from I/R insult and delineate the possible underlying mechanisms in vivo. Our observations have suggested that exogenous NAD+ administration can profoundly decrease I/R hearts injury, at least partially by attenuating apoptotic cell death and enhancing antioxidant capacity.
Methods and materials
Materials
All of the chemicals were purchased from Sigma (St. Louis, Missouri, USA), except where specified.
Ethics statement
All aspects of animal protocols and experimentation were approved by Animal Study Committee of the School of Biomedical Engineering, Shanghai Jiao Tong University (Permit Number: 2014001). All surgical procedures were performed under sodium pentobarbital anesthesia (30 mg/kg i.p.), in which all efforts were made to minimize animal suffering.
Myocardial ischemia and reperfusion
A total of 102 adult male Wistar rats weighing 250-280 g were purchased from SLRC Laboratory (Shanghai, China). The trachea was cannulated with a PE-90 catheter, and then animal ventilator was used with FiO2 of 0.80, a frequency of 100 strokes/minutes, a tidal volume of 0.8-1.2 ml to maintain normal PO2, PCO2 and pH. Saline or NAD+ (5 mg/kg, 10 mg/kg, 20 mg/kg, dissovled in saline) were injected intravenously right before ischemia [11]. The left anterior descending (LAD) branch was occluded by ligation with a 5-0 silk suture after a left thoracotomy. After 60 minutes of ischemia, the ligation loosened. The sham control rats were subjected to all same surgical procedure except ligation. Rats were sacrificed 6 hours or 24 hours after reperfusion respectively as shown in Results except 7 rats died during the procedure.
Determination of myocardial infarct size
Hearts were harvested 24 hours after reperfusion and washed three times with saline. The tissue was sliced into 1-mm-thick transsection by a rodent model, and incubated in 1% 2,3,5-triphenyltetrazolium chloride (TTC) for 20 minutes at 37°C. Then all transsections were photographed. And the infarction was measured and analyzed using Image J (NIH, Bethesda, MD, USA).
Determination of serum cTni levels
Rats were sacrificed 6 hours after reperfusion, and the blood samples were obtained from the postcava vein with pro-coagulation tube (BD Biosciences, San Diego, CA, USA). Then the blood samples were centrifuged at 3000 RPM for 15 minutes after the blood condensed at room temperature. The concentration of cardiac troponin I (cTni) in serum was measured under the Access RImmunoassay System (Beckman Coulter Inc., Brea, CA) [13,14].
SOD activity assay
Tissue samples were obtained from the ischemia area for SOD activity using the SOD assay kit-WST (Dojindo Molecular Technology, Kumamoto, Japan) [15-17]. SOD activity was assayed by utilizing Dojindo’s highly water-soluble tetrazolium salt, WST-1 (2-(4-Iodophenyl)-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt) that produces a water-soluble formazan dye upon reduction with the superoxide anion. The rate of the reduction of WST-1 with O2 - is linearly related to the xanthine oxidase (XO) activity, and this reduction is inhibited by SOD. Therefore, the IC50 (50% inhibition activity of SOD or SOD-like materials) can be determined by the colorimetric method. We conducted the assay according to the manufacturer’s directions. The final SOD activity was normalized to tissue wet weight.
Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL)
The TUNEL assay (Millipore, Billerica, MA, USA) was used to assess apoptosis-like DNA fragmentation in situ. We conducted the assay according to the manual of the kit with minor modifications [18]. Briefly, frozen sections were fixed in 1% PFA for 15 minutes. The endogenous peroxidase activity of the sections was quenched by 3% hydrogen peroxide in PBS for 5 minutes at room temperature. The sections were applied with equilibration buffer and working-strength TdT enzyme, incubated in a humidified chamber at 37°C for one hour and then incubated with anti-digoxigenin peroxidase for 30 minutes at room temperature, which was detected by DAB. Sections were counterstained in 0.5% (w/v) methyl green for 10 seconds at room temperature. After three washes with distilled water, the sections were dipped into 100% N-butanol and dehydrated in xylene. After mounting, the sections were viewed under a microscope (Leica, Heidelberg, German). The slices of the brains obtained 24 hours after middle cerebral artery occlusion were used as positive controls. Negative control samples were prepared as other samples except that the TdT enzyme was omitted in the procedures.
Immunohistochemistry staining
Immunohistochemistry staining was conducted according to the protocol of Vectastain ABC kit (Vector Laboratories Inc., Burlingame, California, USA) with some modifications. The heart cryosections (20 μm) were fixed in 4% paraformaldehyde for 15 minutes. The endogenous peroxidase activity of the sections was quenched by 3% hydrogen peroxide in methyl alcohol for 10 minutes at room temperature, and nonspecific binding was blocked with 10% goat serum in PBS. Sections were then incubated overnight at 4°C with rabbit monoclonal anti-Bax antibody (1:50 dilution, Abcam, Cambridge, UK) and biotinylated secondary antibody (1:200 dilution, Vector Laboratories Inc.) in PBS containing 1% goat serum for 30 minutes at room temperature, followed by a further incubation with the Vectastain ABC Reagent (Vector Laboratories Inc.). Sections were counterstained in 0.5% (w/v) methyl green for 10 seconds at room temperature. After three washes with distilled water, the sections were dipped into 100% N-butanol and dehydrated in xylene. After mounting, the sections were viewed under a microscope (Leica).
Immunofluorescence assay
In brief, the frozen sections were fixed in 4% paraformaldehyde for 15 minutes, followed by three washes with PBS. The sections were incubated in 10% goat serum for 1 hour at room temperature and then incubated with rabbit monoclonal anti-cleaved caspase-3 antibody (1:300 dilution, Cell Signaling, Danvers, MA, USA) in PBS containing 1% goat serum overnight at 4°C and Alexa Fluor 488 goat anti-rabbit secondary antibody (1:500 dilution, Molecular Probes, Eugene, Oregon, USA) in PBS containing 1% goat serum for 1 hour at room temperature. Then the sections were counterstained with DAPI for 5 minutes at room temperature. After three washes with PBS, the fluorescence images of the slices were scanned under a Leica confocal fluorescence microscope.
Western blot
The heart tissues from the ischemia were collected 6 hours after reperfusion and lysed in Radio-Immunoprecipitation Assay (RIPA) buffer (Millipore) containing Complete Protease Inhibitor Cocktail (CWBIO, Shanghai, China) and 1 mM phenylmethanesulfonyl fluoride. Total protein (60 ug) were electrophoresed through a 10% SDS-PAGE and then transferred to a PVDF transfer membrane (0.22 μm, Millipore). Membranes were blocked at room temperature with 5% fat-free milk in TBST and then incubated overnight at 4°C with primary antibodies (rabbit monoclonal antirat Bax antibody, 1:1000 dilution, Abcam; rabbit monoclonal anti-rat Bcl-XL antibody, 1:1000 dilution, Abcam; rabbit polyclonal anti-rat SOD-2 antibody, 1:1000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit monoclonal anti-rat β-tubulin antibody, 1:1000 dilution, Abcam) in TBST containing 1% bovine serum albumin (BSA). The membranes were washed three times with TBST and incubated with goat anti-rabbit HRP-conjugated secondary antibody (1:2000 dilutions, HuaAn Biotechnology, Hangzhou, Zhejiang, China) in TBST containing 1% BSA at room temperature for 1 hour. Immunoreactive bands were visualized using an enhanced chemoluminescence (Thermo Scientific) and quantified by image analyzer (BioRad, Hercules, CA, USA). The intensity analysis was carried out using a Gel-Pro Analyzer (Media Cybernetics, Silver Spring, MD, USA).
Statistical analysis
All data are presented as means ± standard errors (SEM). Data were assessed by analysis of variance (ANOVA) followed by the Student-Newman-Keuls post hoc test. p values less than 0.05 were considered statistically significant.
Results
We studied the effects of three dosages of NAD+ (5 mg/kg, 10 mg/kg, 20 mg/kg) on the myocardial I/R injury of rats. The rats were injected with NAD+ intravenously. Subsequently, the rats were exposed to myocardial I/R injury. Twenty-four hours after reperfusion, the hearts were harvested for TTC staining which is widely used to identify infarct regions at 24 hours or later after ischemia [19]. We observed obvious protective effects of 10 mg/kg and 20 mg/kg NAD+ on the infarct formation (Figure 1A). Quantifications of the myocardial infarct volume indicate that NAD+ dose dependently decreased infarct formation (Figure 1B). We also found that the NAD+ administration markedly attenuated the I/R induced increase in cTni -a gold standard for myocardial injury in clinical settings (Figure 2).
Figure 1.
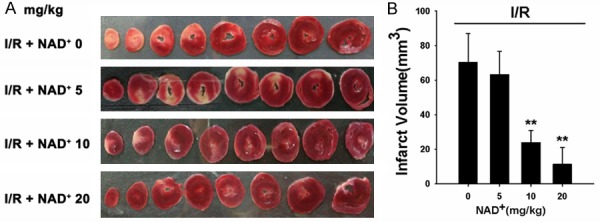
NAD+ administration dose-dependently attenuated myocardial I/R-induced heart injury. A. Representative TTC staining images of heart sections from the rats that were treated with either saline or three dosages of NAD+ (5 mg/kg, 10 mg/kg, 20 mg/kg) and subsequently underwent myocardial I/R. Twenty-four hours after the I/R, the heart sections were obtained for TTC staining. B. Quantifications of the myocardial infarct size. The NAD+ administration produced dose-dependent protection against I/R: Both 10 mg/kg and 20 mg/kg NAD+ significantly reduced the mycocardial infarct size. N = 5-9. **p < 0.01.
Figure 2.
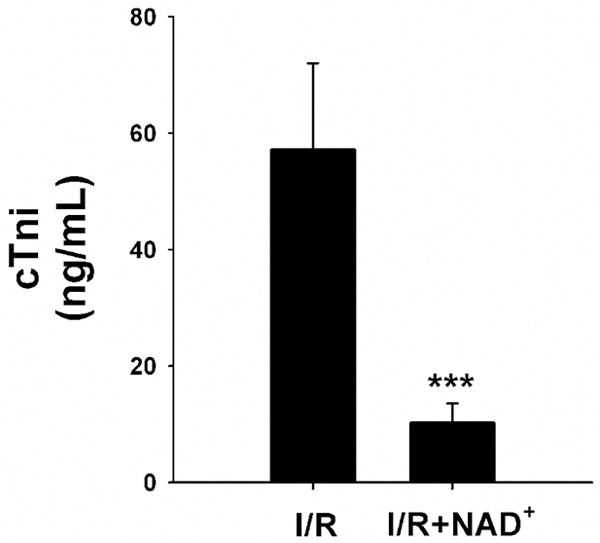
NAD+ administration attenuated the I/R-induced increase in the cTni levels in rat hearts. Rats were intravenously injected with saline or 20 mg/kg NAD+ and underwent I/R subsequently. Six hours after reperfusion, blood were collected for the cTni test. NAD+ administration significantly attenuated the I/R-induced increase in the cTni levels in rat hearts. N = 5-9. ***p < 0.001.
We conducted studies to test our hypothesis that NAD+ administration can attenuate myocardial I/R induced increases in apoptotic changes of the hearts. We found that the I/R induced a marked increase in TUNEL signals- an index of apoptosis-like changes, which was markedly attenuated by the NAD+ administration (Figure 3).
Figure 3.
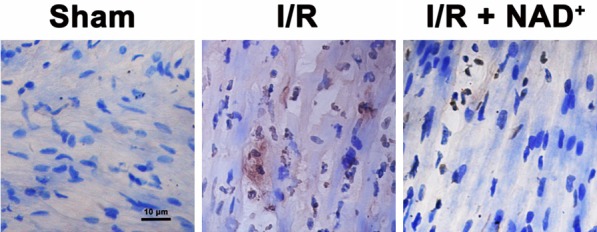
NAD+ administration attenuated myocardial I/R-induced increases in TUNEL signals in rat hearts. The rats were injected with saline or 20 mg/kg NAD+ intravenously. Subsequently, the rats were submitted to myocardial I/R surgery. Six hours after the reperfusion, the sections of rat hearts were stained for detecting TUNEL signals. NAD+ treatment markedly reduced the I/R-induced increases in TUNEL signals. N = 6.
Because cleaved caspase-3 is a key factor in apoptotic cascade, we conducted immunofluorescence staining assay to determine the effects of NAD+ administration on the levels of cleaved caspase-3 in the myocardial I/R injury. The I/R induced a notable increase in cleaved caspase-3 signals (green fluorescence), which was markedly blocked by the NAD+ administration (Figure 4). Our immunohistochemistry assay also showed that NAD+ administration decreased Bax signals in the rat hearts that underwent myocardial I/R (Figure 5A). Western blot assay for Bax in the rat hearts which were subjected to I/R also showed that NAD+ administration significantly attenuated the I/R induced increase in the Bax levels (Figure 5B). Bcl-XL is a key anti-apoptotic factor. We also found that NAD+ administration significantly attenuated the I/R induced decrease in the Bcl-XL levels in the rat hearts (Figure 6A and 6B).
Figure 4.
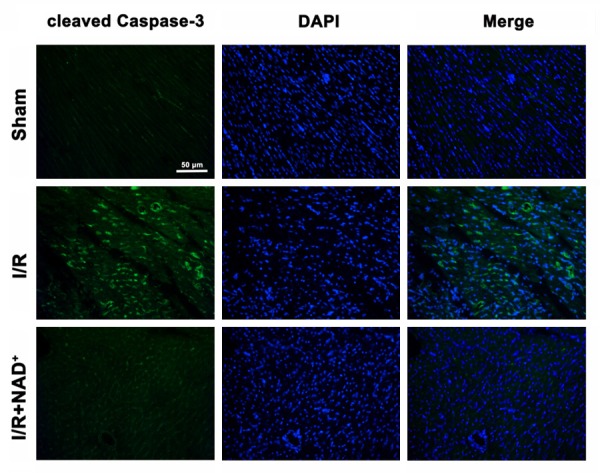
NAD+ administration attenuated myocardial I/R-induced increases in cleaved caspase-3 signals in rat hearts. The rats were administered with 20 mg/kg NAD+ intravenously. Subsequently, the rats were exposed to myocardial I/R surgery. Six hours after the reperfusion, the sections of rat hearts were stained for detecting cleaved caspase 3 (green fluorescence) positive signals. I/R induced a marked increase in cleaved caspase-3 signals, which was attenuated by the NAD+ treatment. N = 6.
Figure 5.
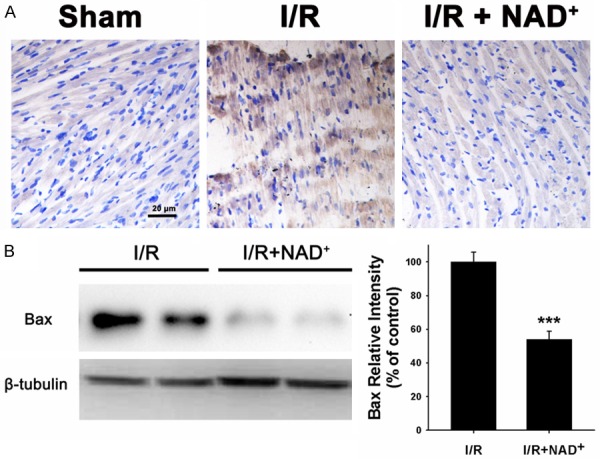
NAD+ administration profoundly decreased the Bax levels in the rat hearts that underwent myocardial I/R. A. Representative immunohistochemical images of Bax in rat hearts 6 hours after reperfusion. Rats were administered with saline or 20 mg/kg NAD+ intravenously. Subsequently, the rats were subjected to myocardial I/R surgery. Six hours after the reperfusion, the sections of rat hearts were stained for detecting Bax. N = 6. B. Western blot for Bax in rat hearts. Rats were treated with saline or 20 mg/kg NAD+ and underwent I/R subsequently. The heart tissues were harvested six hours after the reperfusion. NAD+ administration significantly decreased the protein level of Bax. N = 5. ***p < 0.001.
Figure 6.
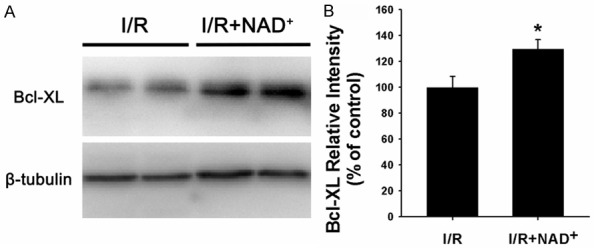
NAD+ administration markedly increased the Bcl-XL levels in rat hearts that were subjected to myocardial I/R. A. Western blot assay for Bcl-XL in rat hearts. Rats were administered with saline or 20 mg/kg NAD+ intravenously. Subsequently, the rats underwent myocardial I/R. Six hours after the reperfusion, the Bcl-XL levels in the homogenates of the rat hearts were determined by Western blot assay. B. Quantifications of the Bcl-XL protein level in the rat hearts. NAD+ administration significantly increased the protein level of Bcl-XL in the rat hearts. N = 5. *p < 0.05.
To determine the effects of NAD+ administration on the antioxidant capacity of rat hearts in myocardial I/R condition. We applied both SOD activity assay and Western blot assay to detect SOD-2 protein levels. We found that myocardial I/R induced a significant decrease in the SOD activity, which was significantly attenuated by the NAD+ administration (Figure 7A). Our Western blot assays also showed that NAD+ administration markedly increased SOD-2 levels in the rats that were subjected to myocardial I/R (Figure 7B and 7C).
Figure 7.
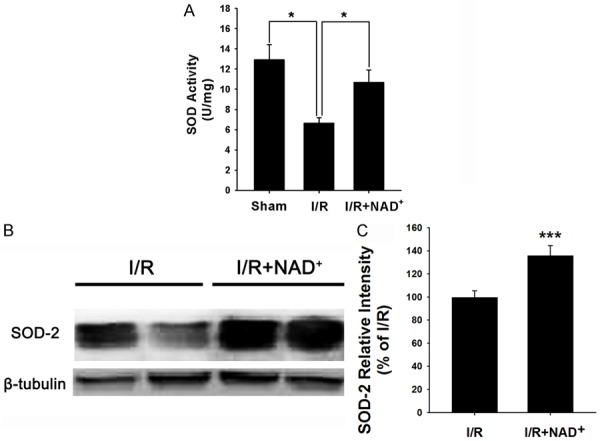
NAD+ administration attenuated myocardial I/R-induced decreases in SOD activity and SOD-2 protein levels in rat hearts. A. NAD+ administration significantly attenuated myocardial I/R-induced decrease in SOD activity in rat hearts. The rats were administered with saline or 20 mg/kg NAD+ intravenously. Subsequently, the rats were subjected to myocardial I/R injury. Six hours after the reperfusion, the SOD activity in the homogenates of the rat hearts were determined. NAD+ treatment significantly increased the SOD activity. B. NAD+ administration significantly increased SOD-2 levels in the rat hearts. The rats were administered with 20 mg/kg NAD+ intravenously. Subsequently, the rats underwent myocardial I/R. Six hours after the reperfusion, the SOD-2 protein levels in the homogenates of the rat hearts were determined. C. Quantifications of the SOD-2 protein levels in the rat hearts. NAD+ administration significantly increased the protein level of SOD-2. N = 5. *p < 0.05; ***p < 0.001.
Discussion
The major findings of this study include: First, NAD+ administration can profoundly attenuate the I/R induced myocardial infarct formation and the circulating cTni level; Second, NAD+ administration can markedly attenuate multiple I/R induced apoptotic changes in the heart tissue, including increased TUNEL signals, cleaved caspase-3 and Bax and decreased Bcl-XL levels; And third, NAD+ administration can prevent the I/R induced decreases in the SOD activity and SOD-2 protein level in the heart.
Acute myocardial infarction is always associated with myocardial ischemia and reperfusion injury. Although PCI can effectively provide blood reperfusion, there has been no effective therapy for myocardial I/R injury to date. Moreover, I/R injury can also frequently result from heart surgery [20] and cardiac arrest [21]. Therefore, it is of great clinical significance to find novel therapeutic strategies for this debilitating illness. Our study has provided the first in vivo evidence indicating that exogenous NAD+ administration can profoundly decrease myocardial I/R induced infarct formation. Compared with other drugs that have shown protective effects on myocardial I/R injury, the NAD+ produced 85% decrease in the infarct size has suggested that NAD+ is one of the drugs that have greatest capacity to decrease myocardial I/R damage [22-24].
Cumulating evidence has indicated that apoptosis-like changes play crucial roles in myocardial I/R injury [25,26]. There are multiple factors in the apoptotic machinery, including cleaved caspase-3, Bax and Bcl-XL. Our studies have provided several lines of evidence suggesting that NAD+ decreases myocardial I/R injury at least partially by decreasing apoptotic changes: 1) NAD+ treatment led to a marked decrease in TUNEL signals in the rat hearts that were subjected to myocardial I/R; 2) NAD+ administration exhibited a notable decrease in cleaved caspase-3 signals induced by myocardial I/R in the rat heart; 3) NAD+ profoundly attenuated the myocardial I/R induced increase in Bax levels in the rat hearts; and 4) NAD+ prevented the myocardial I/R induced decrease in the Bcl-XL levels.
Different lines of evidence have pointed that I/R induced cell death can be mainly or at least partially explained by oxidative stress [27,28]. And our previous study has suggested that NAD+ administration can enhance the GSH levels in the liver of Dox-treated mice [18]. Therefore, in this study we determined if NAD+ can enhance the antioxidant capacity of hearts. We found that myocardial I/R induced significant decreases in both SOD activity and the protein levels of SOD-2, which were significantly attenuated by the NAD+ administration. Therefore, our study has suggested that NAD+ may also produce its protective effects by ameliorating oxidative stress.
Conclusion
Our study has suggested that NAD+ administration is a promising therapeutic strategy for myocardial ischemia. Our study has also suggested that NAD+ can profoundly decrease myocardial ischemic injury at least partially by attenuating apoptotic damage and enhancing the antioxidant capacity.
Acknowledgements
This study was supported by Shanghai Jiao Tong University Grants for Interdisciplinary Research on Medicine and Engineering (YG2014MS74 to X. Q and W. Y), Research Fund for the Scientific and Technical Project of Shanghai Chest Hospital (2014YZDH20300 to X. Q and W. Y), Natural Science Foundation of China (81370361 to X. Q), Natural Science Foundation of China (81171098 to W. Y.) and Natural Science Foundation of China (81271305 to W. Y.).
Disclosure of conflict of interest
None.
References
- 1.Writing Group Members; Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jiménez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB American Heart Association Statistics Committee; Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38–e360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Hausenloy DJ, Yellon DM. Targeting Myocardial Reperfusion Injury-The Search Continues. N Engl J Med. 2015;373:1073–1075. doi: 10.1056/NEJMe1509718. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Karcz MK, Nadtochiy SM, Brookes PS. High-throughput Screening Reveals the Mitochondrial Complex I Inhibitor Nornicotine is Cardioprotective in Ischemia-reperfusion Injury When Delivered at Reperfusion. Circ Res. 2015;117:A404–A404. [Google Scholar]
- 4.Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int J Cardiol. 2005;100:179–190. doi: 10.1016/j.ijcard.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+ and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J Biol Chem. 2000;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 6.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnone M, Bauer I, Poggi A, Mannino E, Sturla L, Brini M, Zocchi E, De Flora A, Nencioni A, Bruzzone S. NAD+ levels control Ca2+ store replenishment and mitogen-induced in crease of cytosolic Ca2+ by Cyclic ADP-ribose-dependent TRPM2 channel gating in human T lymphocytes. J Biol Chem. 2012;287:21067–21081. doi: 10.1074/jbc.M111.324269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J. Circadian Clock NAD+ Cycle Drives Mitochondrial Oxidative Metabolism in Mice. Science. 2013;342:1243417–1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Wang Y, Zhang J, Ma Y, Wang C, Zhou Y, Gu H, Ying W. NAD(+)-carrying mesoporous silica nanoparticles can prevent oxidative stress-induced energy failures of both rodent astrocytes and PC12 cells. PLoS One. 2013;8:e74100. doi: 10.1371/journal.pone.0074100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ying W, Wei G, Wang D, Wang Q, Tang X, Shi J, Zhang P, Lu H. Intranasal administration with NAD+ profoundly decreases brain injury in a rat model of transient focal ischemia. Front Biosci. 2007;12:2728–2734. doi: 10.2741/2267. [DOI] [PubMed] [Google Scholar]
- 12.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD Blocks Cardiac Hypertrophic Response via Activation of the SIRT3-LKB1-AMP-activated Kinase Pathway. J Biol Chem. 2009;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reagan WJ, York M, Berridge B, Schultze E, Walker D, Pettit S. Comparison of cardiac troponin I and T, including the evaluation of an ultrasensitive assay, as indicators of doxorubicin-induced cardiotoxicity. Toxicol Pathol. 2013;41:1146–1158. doi: 10.1177/0192623313482056. [DOI] [PubMed] [Google Scholar]
- 14.Apple FS, Murakami MM, Ler R, Walker D, York M. Analytical characteristics of commercial cardiac troponin I and T immunoassays in serum from rats, dogs, and monkeys with induced acute myocardial injury. Clin Chem. 2008;54:1982–1989. doi: 10.1373/clinchem.2007.097568. [DOI] [PubMed] [Google Scholar]
- 15.Snow-Lisy DC, Sabanegh ES Jr, Samplaski MK, Morris VB, Labhasetwar V. Superoxide dismutase-loaded biodegradable nanoparticles targeted with a follicle-stimulating hormone peptide protect Sertoli cells from oxidative stress. Fertil Steril. 2014;101:560–567. doi: 10.1016/j.fertnstert.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 16.Savalia K, Manickam DS, Rosenbaugh EG, Tian J, Ahmad IM, Kabanov AV, Zimmerman MC. Neuronal uptake of nanoformulated superoxide dismutase and attenuation of angiotensin II-dependent hypertension after central administration. Free Radic Biol Med. 2014;73:299–307. doi: 10.1016/j.freeradbiomed.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, Villegas LR, Stenmark KR, Harrison DG, Majka SM, Irwin D, Farrow KN. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;307:L868–876. doi: 10.1152/ajplung.00096.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang B, Ma Y, Kong X, Ding X, Gu H, Chu T, Ying W. NAD(+) administration decreases doxorubicin-induced liver damage of mice by enhancing antioxidation capacity and decreasing DNA damage. Chem Biol Interact. 2014;212:65–71. doi: 10.1016/j.cbi.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Tseliou E, Kanazawa H, Dawkins J, Gallet R, Kreke M, Smith R, Middleton R, Valle J, Marban L, Kar S, Makkar R, Marban E. Widespread Myocardial Delivery of Heart-Derived Stem Cells by Nonocclusive Triple-Vessel Intracoronary Infusion in Porcine Ischemic Cardiomyopathy: Superior Attenuation of Adverse Remodeling Documented by Magnetic Resonance Imaging and Histology. PLoS One. 2016;11:e0144523. doi: 10.1371/journal.pone.0144523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hausenloy DJ, Candilio L, Evans R, Ariti C, Jenkins DP, Kolvekar S, Knight R, Kunst G, Laing C, Nicholas J, Pepper J, Robertson S, Xenou M, Clayton T, Yellon DM. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N Engl J Med. 2015;373:1408–1417. doi: 10.1056/NEJMoa1413534. [DOI] [PubMed] [Google Scholar]
- 21.Tomaselli GF, Barth AS. Sudden cardio arrest: oxidative stress irritates the heart. Nat Med. 2010;16:648–649. doi: 10.1038/nm0610-648. [DOI] [PubMed] [Google Scholar]
- 22.Qiao Z, Ma J, Liu H. Evaluation of the antioxidant potential of Salvia miltiorrhiza ethanol extract in a rat model of ischemia-reperfusion injury. Molecules. 2011;16:10002–10012. doi: 10.3390/molecules161210002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu L, Li F, Zhao G, Yang Y, Jin Z, Zhai M, Yu W, Zhao L, Chen W, Duan W, Yu S. Protective effect of berberine against myocardial ischemia reperfusion injury: role of Notch1/Hes1-PTEN/Akt signaling. Apoptosis. 2015;20:796–810. doi: 10.1007/s10495-015-1122-4. [DOI] [PubMed] [Google Scholar]
- 24.De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, Dietrich B, Rottensteiner H, Scheiflinger F, Wagner DD. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120:5217–5223. doi: 10.1182/blood-2012-06-439935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J. Silent Information Regulator 1 Protects the Heart From Ischemia/Reperfusion. Circulation. 2010;122:2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pu J, Yuan A, Shan P, Gao E, Wang X, Wang Y, Lau WB, Koch W, Ma XL, He B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J. 2013;34:1834–1845. doi: 10.1093/eurheartj/ehs011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Lau W, Du Y, Gao E, Koch W, Ma XX. AdipoRon, the First Orally Active Adiponectin Receptor Activator, Attenuates Post-ischemic Myocardial Apoptosis via AMPK Activation and Oxidative Stress Reduction. Circ Res. 2015;117:A114–A114. [Google Scholar]
- 28.Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, Liu Y, Zheng W, Shang H, Zhang J, Zhang M, Wu H, Guo J, Zhang X, Hu X, Cao CM, Xiao RP. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]