

Abstract
Considerable evidence has demonstrated that metformin can activate 5’-AMP-activated protein kinase (AMPK) signaling pathway, which plays a critical role in protection of endothelial cell permeability. Hence, the present study evaluated the effects of metformin on blood brain barrier permeability and AQP4 expression in vitro, and assessed the effects of metformin treatment on tumor-induced brain edema in vivo. Hypoxia or VEGF exposure enhanced bEnd3 endothelial cell monolayer permeability and attenuated the expression of tight junction proteins including Occludin, Claudin-5, ZO-1, and ZO-2. However, 0.5 mM metformin treatment protected bEnd3 endothelial cell monolayer from hypoxia or VEGF-induced permeability, which was correlated with increased expression of tight junction proteins. Furthermore, metformin treatment attenuated AQP4 protein expression in cultured astrocytes. Such an effect involved the activation of AMPK and inhibition of NF-κB. Finally, metformin treatment dose-dependently reduced glioma induced vascular permeability and cerebral edema in vivo in rats. Thus, our results suggested that metformin may protect endothelial cell tight junction, prevent damage to the blood brain barrier induced by brain tumor growth, and alleviate the formation of cerebral edema. Furthermore, since the formation of cytotoxic edema and AQP4 expression was positively correlated, our results indicated that metformin may reduce the formation of cytotoxic edema. However, given that AQP4 plays a key role in the elimination of cerebral edema, attenuation of AQP4 expression by metformin may reduce the elimination of cerebral edema. Hence, future studies will be necessary to dissect the specific mechanisms of metformin underlying the dynamics of tumor-induced brain edema in vivo.
Keywords: Metformin, glioma, aquaporin 4, brain edema, blood brain barrier, vascular endothelial growth factor
Introduction
Brain edema is a result of an imbalance in the distribution of water in the brain, and is a common pathological phenomenon in many brain diseases. The causes of brain tumor-induced edema can be generally divided into vasogenic edema and cytotoxic edema [1,2]. These two forms of edema coexist and create a vicious cycle of the development of brain edema, leading to neurological deficits, which seriously affect the quality of patient life [1-3].
Under normal physiological conditions, the blood-brain barrier consists of endothelial cells, which form tight junctions [4,5]. Tight junction molecules, including Claudin 3, Claudin 5, and Claudin 12, and other transmembrane proteins such as occludin, play a crucial role in forming endothelial cell tight junctions [5]. The disturbance of tight junction molecules is the main cause of damaging effects on the blood-brain barrier by brain tumors [1-3]. Various studies have shown that malignant brain tumors can induce ischemic hypoxia due to the rapid growth of the tumor, and can increase the release of a large amount of vascular endothelial growth factor (VEGF), leading to the attenuation of the expression of tight junction molecules [1,2]. Furthermore, while there is no clear link between the expression of aquaporin-4 protein (AQP4) and the glioblastoma growth and proliferation, the formation of brain tumor-induced edema is highly correlated with the protein expression of AQP4 [6,7]. Under normal circumstances, AQP4 in astrocytes are expressed in the end foot area surrounding vessels. However, in human gliomas, the normal expression localization is disrupted. Instead, AQP4 expression is dispersed on the cell membrane of tumor cells [8]. In I-IV grade gliomas, the extent of AQP4 expression and edema formation is highly relevant [6]. Although the expression of AQP4 has no significant effect on tumor growth or metastasis, enhanced AQP4 expression can increase permeability and enhance tumor cell adhesion [9].
Metformin is a drug widely used to treat type II diabetes, and has been fully confirmed by the activation of 5’-AMP-activated protein kinase (AMPK). Clinically, the long-term metformin treatment can reduce the risk of stroke, and can reduce cardiovascular mortality by 24% [10,11]. In addition, in human umbilical vein endothelial cells, metformin may inhibit tumor necrosis factor-α (TNF-α) induced by IL-6, indicating that metformin has anti-inflammatory effect on the endothelial cells [12]. More importantly, recent studies have found that metformin can increase the expression of tight junction molecules on vascular endothelial cells [13]. Since the endothelial cell permeability plays a critical role in the formation of tumor-induced edema, we hypothesized that metformin may prevent the damage to the endothelial cell permeability and reduce the formation of tumor-induced brain edema.
Furthermore, it has been extensively shown that aquaporin 4 (AQP4) plays a critical role in the formation of tumor-associated brain edema. Recent studies have demonstrated that increase in permeability of blood brain barrier (BBB) is correlated with increased AQP4 expression, suggesting the potential involvement of AQP4 in the formation of tumor-associated brain edema [14]. Although few studies have investigated the effects of metformin on AQP4 expression, it has been shown that metformin treatment can reduce astrocyte edema in vitro [15]. Furthermore, 5-amino-imidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) can suppress AQP9 expression in HepG2 cells by activating AMPK [16]. Given that metformin can also activate AMPK signaling pathway, we hypothesized that metformin may attenuate AQP4 expression in astrocytes.
To test the above hypothesis, the present study evaluated the mechanisms underlying the effects of metformin on blood brain barrier permeability and AQP4 expression in vitro, and assessed the effects of metformin treatment on tumor-induced brain edema in vivo. To this end, we first assessed the effects of metformin treatment on bEnd3 endothelial cell monolayer permeability under hypoxia or VEGF exposure, and evaluated the expression of tight junction proteins including Occludin, Claudin-5, ZO-1, and ZO-2. Furthermore, we investigated the mechanisms underlying the effects of metformin treatment on AQP4 protein expression in cultured astrocytes. Finally, we evaluated the effects of metformin treatment on glioma induced cerebral edema in vivo in rats.
Materials and methods
Cell culture
bEnd3 cell lines were purchased from Shanghai Institute enzyme biotechnology company, and were grown in DMEM containing 4.3 g/l glucose, 3.5 g/l sodium bicarbonate, 4 mM glutamine, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. During the culture growth, bEnd3 cells were maintained in a humidified incubator at 37°C and 10% CO2. In all experiments, trypsin was used to digest bEnd3 cells, and cells were seeded at a density of 0.5-1.0 x 104 cells/cm2 onto 24-well plates or Transwell™ system to be allowed to reach confluence within 6-7 days. The medium was removed, the filter membrane was washed with PBS, and media was replaced with normal growth medium (10% FBS), followed by MTT assays, permeability assays, or protein analysis.
To obtain astrocytes in primary culture, cortical brain tissue was dissected out from new born Sprague-Dawley rat (Animal Center, School of Basic Medicine, Jilin University, China; license No. SCXK (Ji) 2003-001). The cells were isolated by incubation with 0.25% trypsin (Gibco, Invitrogen) for 15 min at 37°C, followed by filtration by stainless steel mesh. Cells were then isolated by centrifugation, re-suspended in DMEM/F12 (1:1; Gibco, Invitrogen) medium containing 10% FBS, and seeded in 75 cm2 flasks. Cells reached 100% confluence after 7-9 days. Flasks were then vigorously shaken manually for about 0.5 minutes in order to remove microglial cells and O-2A lineage cells from astrocyte monolayers. The medium was then replaced, and astrocytes were incubated overnight in a CO2 incubator at 37°C. This procedure was repeated three times in the next three days. Flasks were washed with EMEM, and cells were digested with trypsin-EDTA. Cell suspension was placed onto 10 cm dish, and incubated in a CO2 incubator at 37°C for 20 minutes. Almost all fibroblasts were attached within 20 min in a Petri dish. The cell cultures were then inoculated in 24-well plates for further experiments.
MTT analysis
Cell viability was assessed using the MTT assay. bEnd3 cells or astrocytes were seeded in 24-well plates, and incubated to 80% confluence. 48 h after seeding, cells were treated with different concentrations of metformin (repeated at least three times the concentration of each). After 24, 36, or 72 h, 20 μl MTT solution (5 mg/ml) in phosphate buffered saline (PBS) was added to each well, and plates were placed in incubator for 2 hours at 37°C. MTT containing medium was then removed, 100 μl buffer (2.5% acetic acid, 2.5% of 1N hydrochloric acid, 50% N,N-dimethylformamide, and 20% (w/v) sodium lauryl sulfate solution, pH = 4.7). Spectrophotometric absorbance of each sample was measured at 570 nmusing a microplate reader (Molecular Devices, USA).
Western blotting
TRIzol® reagent (Invitrogen, USA) was used to extract protein from bEnd3 cells or astrocytes. Then the lysate was centrifuged at 12000 rpm for 20 min at 4°C. Protein concentration was determined using the BCA protein assay kit (Pierce, USA) with bovine serum albumin as the standard. Final total protein in each sample was adjusted to a concentration of 5 μg/μl in 4× sample buffer, and then heated at 95°C for 10 min and was added to 4-20% SDS-PAGE gel (50 µg per lane) for electrophoresis (120 min, 100 V). Proteins were then transferred to a PVDF membrane (Millipore, USA) in transfer buffer (90 min, 250 mA). At room temperature, the PVDF membrane was placed in 5% skim milk for 2 h. Primary antibody (anti-Occludin, anti-ZO1, anti-ZO2, anti-Claudin 5, 1:250-1:1000 dilution, Zymed, USA; anti-GAPDH, 1:5000, Calbiochem, USA; anti-AQP4, anti-AMPK, anti-pAMPKα (Thr-172), anti-NF-κB, or anti-pNF-κB, 1:750-1:1000 dilution, Santa Cruz, USA) was incubated overnight with the PVDF membrane together in 0.5% BSA/PBS at 4°C. Membrane was then washed with PBS buffer, incubated with secondary antibody with horseradish peroxidase (HRP) (1:1000; Santa Cruz, USA) for 2 h, and finally washed with PBS buffer 3 times. Protein bands were visualized using ImageQuant LAS 500 (GE, China). Western blot image was analyzed using ImageQuant TL 8.1 software (GE, China). The experiment was repeated three times, protein expression was normalized to GAPDH or β-actin expression for statistical analysis.
Immunohistochemistry
Astrocytes culture was fixed in PBS containing 4% paraformaldehyde, followed by incubation with 4% goat serum containing 0.1% of Triton X-100 in phosphate buffer at room temperature for 1 h. Astrocytes were then plated onto agarose-coated glass coverslips with a 1:200 dilution of AQP4 primary antibody (Santa Cruz, USA) containing 1% goat serum and 0.05% Triton in PBS, and were incubated overnight at 4°C. All coverslips were then washed with PBS, and incubated with 1:200 dilution of goat anti-rabbit IgG-FITC secondary antibody (Invitrogen, USA) for 30 min at 37°C in the dark. The coverslips were washed three times with PBS, fixed on glass slides containing DAPI anti-fade solution. Astrocytes were visualized under confocal microscopy (LSM 510 META, Zeiss, USA).
Transendothelial resistance (TEER) and permeability test
TEER readings were measured using epithelial cell voltage resistance meter (WPI, China). Confluent bEnd3 monolayers were grown on a permeable support system. Electrical resistance values of bEnd3 cell monolayer were measured using chopsticks STX2 electrodes. The resistance (Ω·cm2) of the bEnd3 cell monolayer was determined using the following equation. TEER (bEnd3) = TEER (Total) - TEER (filter) × membrane area.
Prior to measuring the permeability, samples were exposed to 6 h of hypoxic conditions, by placing bEnd3 monolayers in an incubator containing 1% O2, or incubated with recombinant vascular endothelial growth factor (VEGF; 50 ng/ml) for 12 h. [14C] sucrose was used to determine the paracellular diffusion across bEnd3 monolayers, as described in a previous study [17]. Because [14C] sucrose with a molecular weight of 342 poorly permeates the blood-brain barrier, it does not diffuse across the tight junctions between endothelial cells. To determine apical-to-basolateral diffusion, the appearance rate of radioactive [14C] sucrose (pmol) appear in the receiving chamber in unit time (min) was measured. Permeability coefficient values were measure after 2 h of radioactive sucrose diffusion. The permeability coefficient was calculated using the following equation: Permeability coefficient (cm/min) = [(volume/(SA×CD)) × (CR/Time)]. The volume is measured as the volume of media in the receiving chamber, SA represents the activity of radioactive marker, CD represents the initial concentration of radioactive marker, and CR represents the concentration of the radioactive marker in the receiving chamber at a given time.
Animals
All animals were provided by the Animal Center, School of Basic Medicine, Jilin University, China (license No. SCXK (Ji) 2003-001). Male Sprague Dawley rats (n=80), weighing 200-250 g were housed in a temperature- and humidity-controlled vivarium under a 12 h light/dark cycle. All animals had free access to food daily with water ad libitum. Animal experiments were approved by the local Institutional Animal Care and Use Committee. The housing and treatment of the animals followed the Guidance Suggestions for the Care and Use of Laboratory Animals, formulated by the Ministry of Science and Technology of China.
Glioma tumor animal model
Rat C6 glioma cells were cultured in DMEM medium containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin under 5% CO2 at 37°C. When reaching the log phase, cells were digested using 0.25% trypsin, and cell suspension was collected. The cell concentration was then adjusted to 2.5 × 106/ml, and cells were placed at 37°C for subsequent use. Rats were fasted 12 h before surgery, and were initially anesthetized with 3% isoflurane and placed onto the stereotactic frame with anesthesia be maintained using 1.5% isoflurane through a nose cone. A 1.5 × 1.5 cm area of hair on top of the rat head was removed, and the skin was disinfected. A 1 cm longitudinal scalp incision was made along the midline to expose the skull. C6 cells were injected into the right striatum. The coordinates used for the injection was 1.0 mm anterior to the bregma, 3 mm lateral to the midline, and 4 mm ventral to the surface of the skull. 25 μl of C6 cells suspension was slowly delivered into the caudate nucleus using a needle attach with a micro-syringe at a rate of 2.5 μl/min for a total 10 min. After injection, needle was removed and skin incision was sutured. Sham group (Sham) received the same surgery but was infused with an equal volume of saline.
Metformin therapy
After one week of C6 cell implantation, rats were randomly assigned to different group (n = 16/group) receiving either metformin (0.1, 0.3, and 1 mg/ml) or saline treatment through drinking water. Treatment lasted one week. Daily water intake (average 25.5 g; no significant differences between treatment groups) of each rat was calculated by weighing water bottle.
Evans blue staining
The permeability of the blood-brain barrier was evaluated using Evans blue dye (EB, Sigma Co. USA). In two weeks after tumor cell inoculation, animals were anesthetized using 3% isoflurane and injected with EB (2% EB, i.v., 2 ml/kg). After 30 min, animals were transcardially perfused with 0.9% saline until the perfusate from the right atrium came out clear. Animals were then quickly decapitated, and the brains were rapidly separated without olfactory bulb and cerebellum. Brains were divided into the left and right hemispheres and placed in two separate containers. Each sample was then weighed and manually homogenized in 3.75 ml PBS and 1.25 ml 100% trichloroacetic acid solution (TCA, Sigma Co. USA), followed by cooling overnight at 4°C. The next day, samples were centrifuged at 1000 g for 30 min. The supernatant of each sample was then measured at 620 nm using the Synergy™ Mx Multi-Mode Microplate analyzer (Bio-Tek, Inc. USA). The amount of EB in brain tissue (µg/g) was calculated based on standard curve of EB absorbance.
Brain water content
In two weeks after tumor cell inoculation, animals were decapitated after pentobarbital anesthesia (150-200 mg/kg, i.p.) Rat brains were quickly removed from the skull without the olfactory bulb and cerebellum. The brains were then divided into the left and right hemisphere. Each hemisphere was immediately weighed to obtain wet brain weight. Then the hemisphere was rapidly wrapped in aluminum foil, labeled, and placed in oven at 100°C for 72 h, followed by re-weighing to obtain the dry brain weight. Brain water content was calculated using the following equation. Brain water content = (wet weight - dry weight)/wet weight × 100%.
Data analysis
All values are presented as mean ± standard deviation (SD). Data were analyzed using one-way analyses of variance (ANOVA) or Multi-factor ANOVAs, where appropriate. Significant ANOVA main and interaction effects were further investigated using Tukey or Bonferonni post hoc tests, when appropriate. Alpha was set at 0.05.
Results
Effects of metformin treatments on bEnd3 cell and astrocytes viability
The effects of metformin on bEnd3 cell and astrocytes viability were assessed using MTT assay. After incubation with 0.1 mM or 0.5 mM metformin for 24 h, metformin did not alter bEnd3 cell viability as compared with control. However, 1 mM or 2 mM metformin decreased bEnd3 cell viability as compared with control (Figure 1A). Furthermore, 0.5 mM metformin did not alter bEnd3 cell viability after 24, 36 or 72 h, as compared with control (Figure 1B). Similarly, after incubation with 0.1 mM or 0.5 mM metformin for 24 h, metformin did not alter astrocytes viability as compared with control. However, 1 mM or 2 mM metformin decreased astrocytes viability as compared with control (Figure 1C). Furthermore, 0.5 mM metformin did not alter astrocytes viability after 24, 36 or 72 h, as compared with control (Figure 1D). Therefore, 0.5 mM metformin were selected in the rest of the present study.
Figure 1.
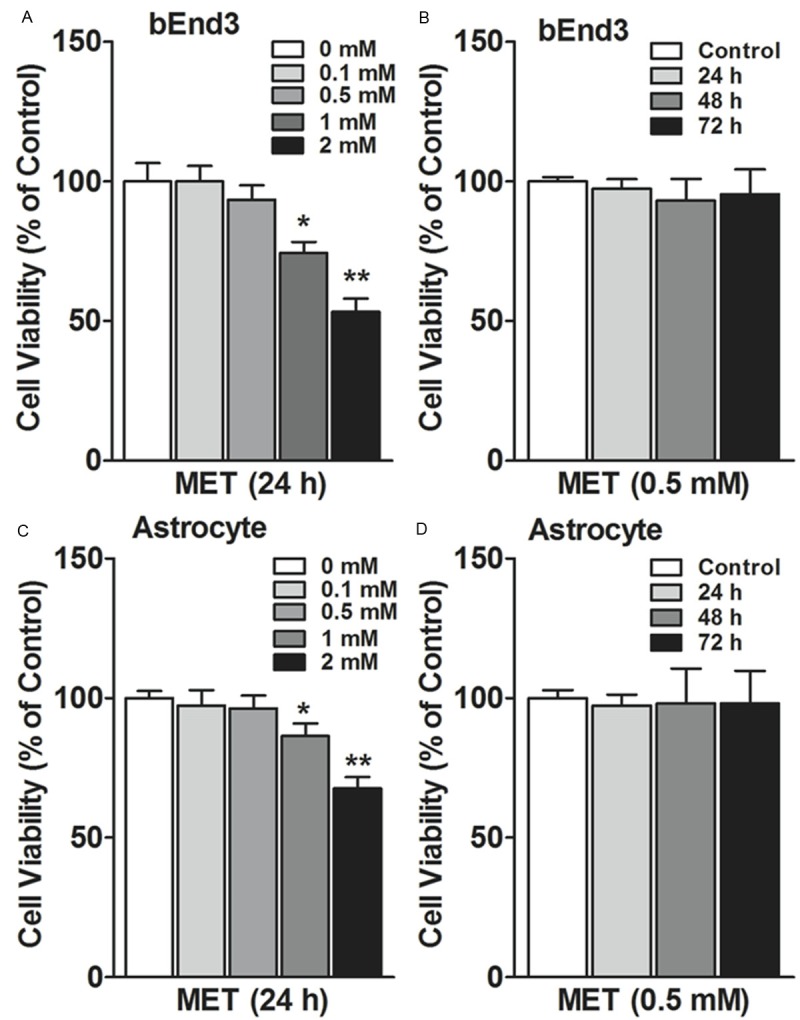
Effects of metformin on the viability of BEnd3 cells and astrocytes. A. bEnd3 cells (5 × 103 cells/well) were seeded in 24-well culture plates. After 48 hours, cells were incubated with metformin (MET: 0, 0.1, 0.5, 1, 2 mM), 24 hours later, cell viability was measured using the MTT assay. B. 24, 48 and 72 hours post-treatment of 0.5 mM metformin (MET), bEnd3 cell proliferation was measured using MTT assay. C. Astrocytes (5 × 103 cells/well) were seeded in 24-well culture plates. After 48 hours, cells were incubated with metformin (MET: 0, 0.1, 0.5, 1, 2 mM), 24 hours later, cell viability was measured using the MTT assay. D. 24, 48 and 72 hours post-treatment of 0.5 mM metformin (MET), astrocytes proliferation was measured using MTT assay. Results are expressed as a percentage of cell viability as compared with the control (0 mM). *p<0.05, **p<0.01. Each concentration takes an average of three independent experiments (Mean ± SEM).
Effects of metformin treatments on bEnd3 cell monolayer permeability
Under 6 h hypoxic condition, the permeability of bEnd3 cell monolayer was increased, as compared with normoxic condition (p<0.01; Figure 2A). However, 6 h hypoxic condition did not alter bEnd3 monolayer permeability when cells were incubated with 0.5 mM metformin, as compared with normoxic condition (Figure 2A). In fact, 0.5 mM metformin treatment reduced bEnd3 monolayer permeability as compared with 0 mM control under 6 h hypoxic condition (p<0.01; Figure 2A). Furthermore, exposure to VEGF (50 ng/ml) for 12 h enhanced bEnd3 monolayer permeability, as compared with no VEGF control (p<0.01, Figure 2B). However, co-incubation of 0.5 mM metformin and VEGF (50 ng/ml) attenuated bEnd3 monolayer permeability, as compared with VEGF treatment alone (p<0.01, Figure 2B), to a similar level seen in no VEGF control treatment.
Figure 2.
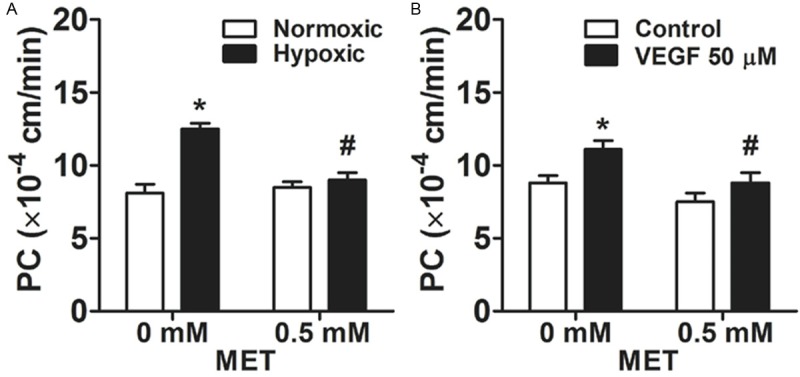
The protective effect of metformin on the permeability of bEnd3 cell monolayers. A. bEnd3 cell monolayers were exposed to 1% O2 hypoxic condition for six hours while incubated with metformin. B. bEnd3 cell monolayers were exposed to a recombinant vascular endothelial growth factor (VEGF; 50 ng/ml) while incubated with metformin for 12 h. *p<0.01 compared with the control group. #p<0.01 compared with 0 mM metformin group.
Effect of metformin treatment on expression of tight junction proteins
The expression of tight junction proteins including Occludin, Claudin-5, ZO-1, and ZO-2 was evaluated using Western blot after hypoxic or VEGF treatment. Specifically, after 6 h of hypoxic treatment, the expression of Occludin, Claudin-5, ZO-1, and ZO-2 proteins in bEnd3 monolayer was significantly reduced, as compared with normoxic control (p<0.01, Figure 3A). However, 0.5 mM metformin treatment increased the expression of Occludin, Claudin-5, ZO-1, and ZO-2 proteins after 6 h of hypoxia treatment in bEnd3 monolayer, as compared with 0 mM control under 6 h hypoxic condition (p<0.01; Figure 3A), to a similar level seen in normoxic control. Furthermore, exposure to VEGF (50 ng/ml) for 12 h decreased the expression of Occludin, Claudin-5, ZO-1, and ZO-2 proteins, as compared with no VEGF control (p<0.01, Figure 3B). However, co-incubation of 0.5 mM metformin and VEGF (50 ng/ml), enhanced the expression of Occludin, Claudin-5, ZO-1, and ZO-2 proteins as compared with VEGF treatment alone (p<0.01, Figure 3B), to a similar level seen in no VEGF control treatment.
Figure 3.
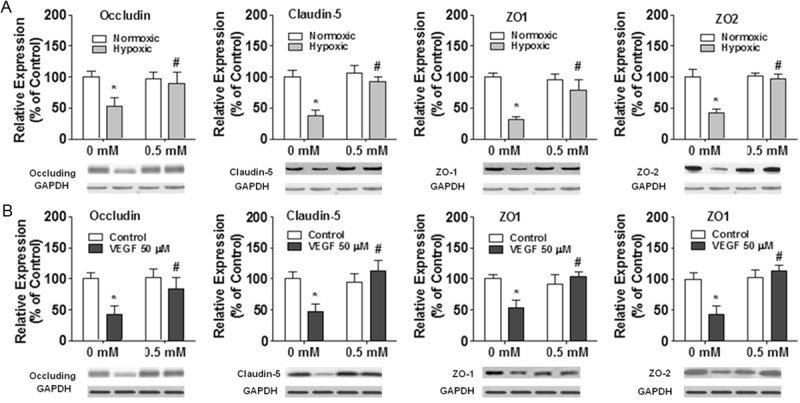
Effects of metformin on the expression of tight junction proteins in bEnd3 monolayers. A. bEnd3 cell monolayers were exposed to 1% O2 hypoxic condition for six hours while incubated with metformin. B. bEnd3 cell monolayers were exposed to a recombinant vascular endothelial growth factor (VEGF; 50 ng/ml) while incubated with metformin for 12 h. The results are displayed in percentage after protein expression level was normalized to GAPDH expression level *p<0.01 compared with the control group. #p<0.01 compared with 0 mM metformin group.
Effects of metformin treatments on AQP4 expression in astrocytes in vitro
The expression of AQP4 in astrocytes in primary culture was visualized using immunohistochemistry. Representative photographs of AQP4 expression in astrocytes were shown in Figure 4A-C. 0.5 mM metformin (Figure 4C) but not 0 mM metformin (vehicle; Figure 4B) significantly attenuated AQP4 expression in astrocytes, as compared with no treatment control group (Figure 4A). This effect was further confirmed by Western blot results shown in Figure 4D.
Figure 4.
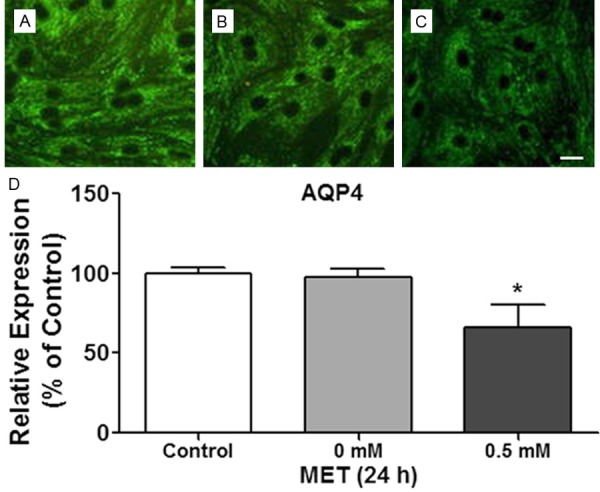
Representative photographs (×200) of AQP4 fluorescence immunolocalization of astrocytes after different treatment in (A) Control group, (B) Vehicle group (0 mM metformin), and (C) 0.5 mM metformin. Scale bar (shown in white line) is 50 μm. (D) Western blot results of AQP4 expression in astrocytes. *p<0.05 compared with the control group.
Role of AMPK and NF-κB in metformin-induced attenuation of AQP4 expression
To investigate the mechanisms underlying the effects of metformin on AQP4 expression in astrocytes, we treated astrocytes with 0.5 mM metformin for 24 h, and measured activity of AMPK and NF-κB. Specifically, 0.5 mM metformin treatment had no effect on total AMPK or NF-κB (Figure 5A), but significantly increased phosphorylation of AMPK (p<0.05; Figure 5A), and reduced phosphorylation of NF-κB (p<0.05; Figure 5A). The results suggested that metformin increased AMPK activation, and attenuated NF-κB activation in astrocytes. However, co-incubation 0.5 mM metformin with 0.1 mM dorsomorphin, an AMPK inhibitor, enhanced NF-κB activation (p<0.05; Figure 5B) and AQP4 protein expression (p<0.05; Figure 5C) in astrocytes respectively, as compared with 0.5 mM metformin treatment alone, to a similar level seen in no metformin control treatment.
Figure 5.
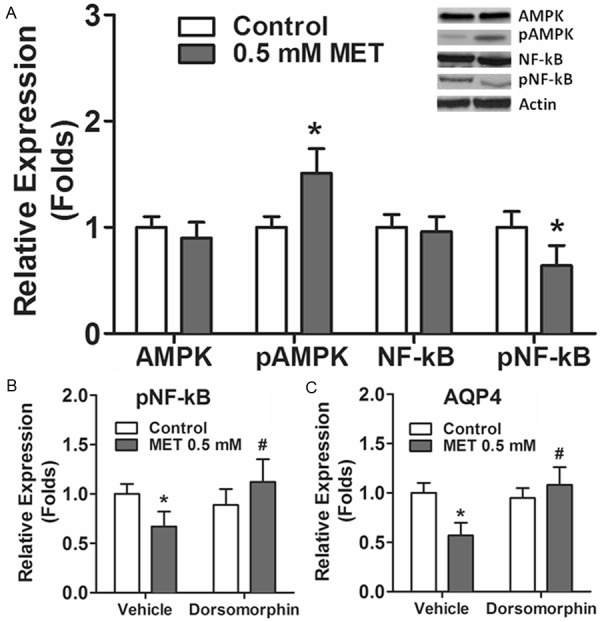
Role of AMPK-NF-κB activation in AQP4 expression in astrocytes. A. Effects of metformin on AMPK and NF-κB activation in astrocytes. B. Effects of dorsomorphin (0.1 mM) on NF-κB activation in astrocytes. C. Effects of dorsomorphin (0.1 mM) on metformin-induced attenuation of AQP4 expression in astrocytes. The results are displayed in folds after protein expression level was normalized to β-actin expression level. *p<0.05 compared with the control group. #p<0.05 compared with metformin group.
Effects of metformin treatments on glioma-induced brain edema in vivo
C6 glioma cells were injected into the right striatumon. Two weeks after inoculation, Evans blue content of right brain hemisphere in rats was evaluated. Specifically, glioma cells implantation increased Evans blue content in brain tissue in saline treated group, as compared with sham group, which received no glioma cells implantation (Figure 6A). However, one week of daily 0.3 or 1 mg/ml metformin treatment, but not 0.1 mg/ml metformin treatment, reduced Evans blue content in brain tissue, as compared with saline treated group (Figure 6A).
Figure 6.
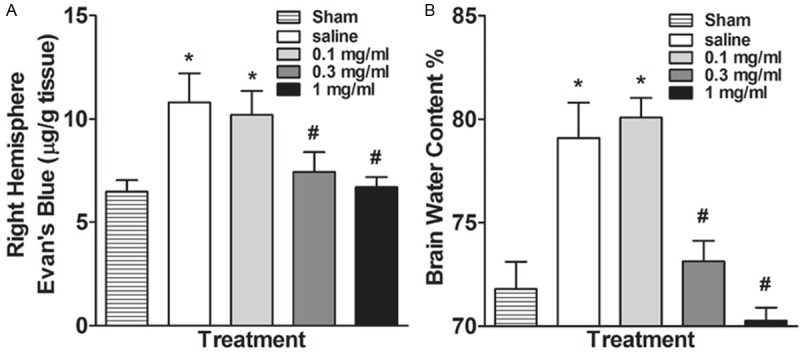
Effects of oral metformin treatment on (A) Evans blue content and (B) brain water content in the right brain hemisphere. *p<0.05 compared with the sham group (no C6 cell implantation). #p<0.05 compared with the saline group.
Furthermore, glioma cells implantation increased brain water content in right brain hemisphere in saline treated group, as compared with sham group, which received no glioma cells implantation (Figure 6B). However, one week of daily 0.3 or 1 mg/ml metformin treatment, but not 0.1 mg/ml metformin treatment, reduced brain water content in right brain hemisphere, as compared with saline treated group (Figure 6B).
Discussion
The present study evaluated the effects of metformin on blood brain barrier permeability and AQP4 expression in vitro, and assessed the effects of metformin treatment on glioma-induced brain edema in vivo. Specifically, we have found that hypoxia or VEGF exposure enhanced bEnd3 endothelial cell monolayer permeability and attenuated the expression of tight junction proteins including Occludin, Claudin-5, ZO-1, and ZO-2. However, 0.5 mM metformin treatment protected bEnd3 endothelial cell monolayer from hypoxia or VEGF-induced permeability, which was correlated with increased expression of tight junction proteins. Furthermore, metformin treatment attenuated AQP4 protein expression in cultured astrocytes. Such an effect involved the activation of AMPK and inhibition of NF-κB. Finally, metformin treatment dose-dependently reduced glioma induced vascular permeability and cerebral edema in vivo in rats. Thus, our results suggested that metformin may protect endothelial cell tight junction, prevent damage to the blood brain barrier induced by brain tumor growth, and alleviate the formation of cerebral edema. Furthermore, since the formation of cytotoxic edema and AQP4 expression was positively correlated, our results indicated that metformin may reduce the formation of cytotoxic edema. However, given that AQP4 plays a key role in the elimination of cerebral edema, attenuation of AQP4 expression by metformin may reduce the elimination of cerebral edema. Hence, future studies will be necessary to dissect the specific mechanisms of metformin underlying the dynamics of tumor-induced brain edema in vivo.
The experiments further confirmed the protective effect of metformin on blood vessels by up-regulating tight junction protein expression, thereby reducing hypoxia or VEGF induced endothelial cell permeability. In fact, our results were consistent with some of the previous findings. For example, in a rat model of ischemia induced by transient middle cerebral artery occlusion, metformin is shown to increase AMPK phosphorylation, inhibit NF-κB activation, and down-regulate cytokine (IL-1β, IL-6, TNF-α) and ICAM-1 expression [18]. In another study, there is evidence that metformin can activate AMPK in cultured microvascular endothelial cells, thus may increase blood-brain barrier function in rat brain [13]. Specifically, by measuring transendothelial resistivity (TEER) and fluorescein sodium permeability, previous studies have found that metformin significantly increases transendothelial resistivity and reduces sodium fluorescein penetration. Such a phenomenon is correlated with an increase in the expression of tight junction proteins including ZO-1 and occludin [13]. However, it should be noted that activation of AMPK signaling pathway can also enhance angiogenesis. For instance, long-term metformin therapy after stroke improves experimental angiogenesis and recovery [19]. Additionally, metformin can promote focal angiogenesis and neurogenesis in mice after cerebral artery occlusion [20]. In contrast, metformin seems to be able to decrease tumor angiogenesis, and plays a preventive role in the occurrence of breast cancer [21]. Furthermore, metformin can reduce the expression level of vascular endothelial growth factor receptor Flk-1 and inhibit angiogenesis after oxygen-induced retinopathy in mice [22]. Thus, cautions should be given to utilization of metformin for the treatment of tumor-induced brain edema. Therefore, cautions should be given to utilization of metformin for the treatment of brain edema. Future studies will be also necessary to evaluate the effects of metformin in brain tumor angiogenesis.
While dorsomorphin is considered as a selective AMPK inhibitor, and have been used in many studies to demonstrate AMPK-NF-κB signaling pathways, it is still not clear which AMPK isoforms are critical for regulating NF-κB activation and AQP4 expression in astrocytes. Using gene knockout techniques, previous studies have shown that AMPK α1 knockout eliminates anti-inflammatory effects of metformin [12]. Furthermore, while constitutively active AMPK α1 in macrophages can inhibit NF-κB signaling and reduce fatty acid-induced inflammation, dominant negative mutant AMPK α1 can reverse this phenomenon [23]. In addition, over-expression of AMPK α1 in aortic endothelial cells can reduce NF-κB activation [24]. Furthermore, NF-κB signaling is activated in aortic endothelial cells from AMPK α2 knockout mice, and AICAR treatment suppresses NF -κB activation in transgenic mice with constitutively active AMPK α2. Taken together, future studies may be important to investigate the role of specific AMPK catalytic α subunit in regulation of AQP4 expression in astrocytes.
Many studies have demonstrated the activation of AMPK pathway can reduce NF-κB activation [23,25-27]. Adding to this literature, the present study indicated that metformin can also attenuate NF-κB activation by activating AMPK. However, the exact mechanisms underlying this phenomenon remain unknown. Previous studies have shown that AMPK has a number of direct phosphorylation targets [28], and can inhibit NF-κB is through its downstream mediator, including SIRT1, FoxO family, and peroxisome proliferator-activated receptor γ [28]. In addition, a large number of studies have shown that activation of AMPK can inhibit NF-κB-mediated pro-inflammatory molecules, which in turn activate NF-κB, leading to the expression of anti-apoptotic proteins, such as Bcl-2 and survivin [29]. Furthermore, AMPK activation can inhibit TNF-α and fatty acid palmitate-induced NF-κB activation in endothelial cell [30]. Taken together, these studies suggest that metformin likely regulate the activity of NF-κB via activation of multiple AMPK-mediated downstream signaling pathways, which can ultimately regulate cell cycle and differentiation, metabolism, and inflammation. Thus, it will be necessary to dissect the AMPK-mediated signaling pathways that are critical for the effects of metformin on AQP4 expression in astrocytes in future studies.
Molecular mechanisms underlying AQP4 expression have been extensively reported in various studies. Activation of protein kinase C can reduce the expression of AQP4 in cultured rat astrocytes [31]. On the contrary, activation of protein kinase A is not able to alter the expression of AQP4 in astrocytes [31]. Furthermore, it has been reported that IL-1β can induce AQP4 expression in astrocytes via NF-kB activation rather than MEK1/2 activation [32], even though activation of IL-1β can activate protein kinase C in astrocyte, which in turn can activate MEK1/2-mediated ERK1/2 signaling pathway [33]. In addition, high osmotic pressure can stimulate the expression of aquaporin AQP4 via p38 MAPK-dependent signaling pathways in rat astrocytes [34]. Adding to this literature, the present study has demonstrated that metformin decrease AQP4 expression in rat astrocyte likely by activating AMPK, thereby inhibiting NF-κB activation. While AMPK-NF-κB signaling pathway is likely to be an important mechanism underlying the regulation of AQP4 expression, there have been studies showing metformin can inhibit IL-1β-mediated signaling pathway [35,36], suggesting the effects of metformin on AQP4 expression may rely on multiple signaling pathways.
In summary, the present study has demonstrated the effects of metformin on glioma-induced brain edema may rely on it protective effects on endothelial cell permeability and inhibitory effects on AQP4 expression. However, metformin may also reduce the formation of tumor-induced brain edema by suppressing tumor cell malignancy via attenuation of the expression of AQPs, which are critical for tumor growth, angiogenesis, and metastatic process [37,38]. For instance, higher expression of AQP4 in glioma tumor cells has been reported [7,39,40], and previous studies have demonstrated AQP4 is critical in the control of glioblastoma cell migration and invasion [41,42]. Thus, metformin may also suppress glioma malignancy by reducing AQP4 expression. Furthermore, while the present study focused on AQP4, it is likely that metformin can alter the expression of other aquaporins. For example, AICAR can inhibit AQP9 expression by activating AMPK [16], a similar effect of metformin. While it is not clear whether metformin can alter the expression of AQP1, studies have demonstrated AQP1 expression is increased in glioma cells in response to higher glucose [43], which might be indirectly reduced by metformin [44]. In general, the present study has demonstrated that metformin can reduce the formation of glioma-induced cerebral edema in vivo. This phenomenon may rely on the multiple pharmacological effects of metformin. In support of this, the present study has shown that metformin treatment can protect endothelial cell tight junction and prevent damage to the blood brain barrier induced by hypoxia or VEGF exposure, as well as reduce AQP4 expression in vitro. However, given the complexity of the dynamics of tumor-induced brain edema in vivo, future studies will be necessary to explore other putative mechanisms of metformin in regulation of tumor-induced brain edema. Such a line of research would be necessary for the development of effective pharmacotherapy for tumor-induced brain edema.
Acknowledgements
This study was supported by the Jilin Provincial Program for Development of Science and Technology (Grant No. 200505119).
Disclosure of conflict of interest
None.
References
- 1.Papadopoulos MC, Saadoun S, Davies DC, Bell BA. Emerging molecular mechanisms of brain tumour oedema. Br J Neurosurg. 2001;15:101–108. doi: 10.1080/02688690120036775. [DOI] [PubMed] [Google Scholar]
- 2.Stummer W. Mechanisms of tumor-related brain edema. Neurosurg Focus. 2007;22:E8. doi: 10.3171/foc.2007.22.5.9. [DOI] [PubMed] [Google Scholar]
- 3.Kaal EC, Vecht CJ. The management of brain edema in brain tumors. Curr Opin Oncol. 2004;16:593–600. doi: 10.1097/01.cco.0000142076.52721.b3. [DOI] [PubMed] [Google Scholar]
- 4.Saunders NR, Dreifuss JJ, Dziegielewska KM, Johansson PA, Habgood MD, Mollgard K, Bauer HC. The rights and wrongs of blood-brain barrier permeability studies: a walk through 100 years of history. Front Neurosci. 2014;8:404. doi: 10.3389/fnins.2014.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haseloff RF, Dithmer S, Winkler L, Wolburg H, Blasig IE. Transmembrane proteins of the tight junctions at the blood-brain barrier: structural and functional aspects. Semin Cell Dev Biol. 2015;38:16–25. doi: 10.1016/j.semcdb.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Lin ZX. Glioma-related edema: new insight into molecular mechanisms and their clinical implications. Chin J Cancer. 2013;32:49–52. doi: 10.5732/cjc.012.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mou K, Chen M, Mao Q, Wang P, Ni R, Xia X, Liu Y. AQP-4 in peritumoral edematous tissue is correlated with the degree of glioma and with expression of VEGF and HIF-alpha. J Neurooncol. 2010;100:375–383. doi: 10.1007/s11060-010-0205-x. [DOI] [PubMed] [Google Scholar]
- 8.Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat. 2002;200:639–646. doi: 10.1046/j.1469-7580.2002.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCoy E, Sontheimer H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia. 2007;55:1034–1043. doi: 10.1002/glia.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roussel R, Travert F, Pasquet B, Wilson PW, Smith SC Jr, Goto S, Ravaud P, Marre M, Porath A, Bhatt DL, Steg PG. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch Intern Med. 2010;170:1892–1899. doi: 10.1001/archinternmed.2010.409. [DOI] [PubMed] [Google Scholar]
- 11.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 12.Huang NL, Chiang SH, Hsueh CH, Liang YJ, Chen YJ, Lai LP. Metformin inhibits TNF-alpha-induced IkappaB kinase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol. 2009;134:169–175. doi: 10.1016/j.ijcard.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 13.Takata F, Dohgu S, Matsumoto J, Machida T, Kaneshima S, Matsuo M, Sakaguchi S, Takeshige Y, Yamauchi A, Kataoka Y. Metformin induces up-regulation of blood-brain barrier functions by activating AMP-activated protein kinase in rat brain microvascular endothelial cells. Biochem Biophys Res Commun. 2013;433:586–590. doi: 10.1016/j.bbrc.2013.03.036. [DOI] [PubMed] [Google Scholar]
- 14.Tomas-Camardiel M, Venero JL, Herrera AJ, De Pablos RM, Pintor-Toro JA, Machado A, Cano J. Blood-brain barrier disruption highly induces aquaporin-4 mRNA and protein in perivascular and parenchymal astrocytes: protective effect by estradiol treatment in ovariectomized animals. J Neurosci Res. 2005;80:235–246. doi: 10.1002/jnr.20443. [DOI] [PubMed] [Google Scholar]
- 15.Prakash R, Li WG, Qu Z, Johnson MA, Fagan SC, Ergul A. Vascularization Pattern After Ischemic Stroke is Different in Control Versus Diabetic Rats Relevance to Stroke Recovery. Stroke. 2013;44:2875–2882. doi: 10.1161/STROKEAHA.113.001660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoyama Y, Iguchi K, Usui S, Hirano K. AMP-activated protein kinase modulates the gene expression of aquaporin 9 via forkhead box a2. Arch Biochem Biophys. 2011;515:80–88. doi: 10.1016/j.abb.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Brown RC, Morris AP, O’Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007;1130:17–30. doi: 10.1016/j.brainres.2006.10.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Tang G, Li Y, Wang Y, Chen X, Gu X, Zhang Z, Yang GY. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J Neuroinflammation. 2014;11:177. doi: 10.1186/s12974-014-0177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. Eur J Neurosci. 2014;39:2129–2138. doi: 10.1111/ejn.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Tang G, Zhang Z, Wang Y, Yang GY. Metformin promotes focal angiogenesis and neurogenesis in mice following middle cerebral artery occlusion. Neurosci Lett. 2014;579:46–51. doi: 10.1016/j.neulet.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Gao S, Jiang J, Li P, Song H, Wang W, Li C, Kong D. Attenuating tumour angiogenesis: a preventive role of metformin against breast cancer. Biomed Research International. 2015;2015:592523. doi: 10.1155/2015/592523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joe SG, Yoon YH, Choi JA, Koh JY. Anti-Angiogenic Effect of Metformin in Mouse Oxygen-Induced Retinopathy Is Mediated by Reducing Levels of the Vascular Endothelial Growth Factor Receptor Flk-1. PLoS One. 2015;10:e0119708. doi: 10.1371/journal.pone.0119708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang ZG, Kahn BB, Shi H, Xue BZ. Macrophage alpha 1 AMP-activated Protein Kinase (alpha 1AMPK) Antagonizes Fatty Acid-induced Inflammation through SIRT1. J Biol Chem. 2010;285:19051–19059. doi: 10.1074/jbc.M110.123620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katerelos M, Mudge SJ, Stapleton D, Auwardt RB, Fraser SA, Chen CG, Kemp BE, Power DA. 5-aminoimidazole-4-carboxamide ribonucleoside and AMP-activated protein kinase inhibit signalling through NF-kappaB. Immunol Cell Biol. 2010;88:754–760. doi: 10.1038/icb.2010.44. [DOI] [PubMed] [Google Scholar]
- 25.Bai A, Ma AG, Yong M, Weiss CR, Ma Y, Guan Q, Bernstein CN, Peng Z. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol. 2010;80:1708–1717. doi: 10.1016/j.bcp.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- 28.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. doi: 10.1007/s00018-010-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem. 2010;285:15346–15355. doi: 10.1074/jbc.M110.102491. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Cacicedo JM, Yagihashi N, Keaney JF Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004;324:1204–1209. doi: 10.1016/j.bbrc.2004.09.177. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto N, Sobue K, Miyachi T, Inagaki M, Miura Y, Katsuya H, Asai K. Differential regulation of aquaporin expression in astro cytes by protein kinase C. Brain Res Mol Brain Res. 2001;95:110–116. doi: 10.1016/s0169-328x(01)00254-6. [DOI] [PubMed] [Google Scholar]
- 32.Ito H, Yamamoto N, Arima H, Hirate H, Morishima T, Umenishi F, Tada T, Asai K, Katsuya H, Sobue K. Interleukin-1beta induces the expression of aquaporin-4 through a nuclear factor-kappaB pathway in rat astrocytes. J Neurochem. 2006;99:107–118. doi: 10.1111/j.1471-4159.2006.04036.x. [DOI] [PubMed] [Google Scholar]
- 33.Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131:152–159. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arima H, Yamamoto N, Sobue K, Umenishi F, Tada T, Katsuya H, Asai K. Hyperosmolar mannitol stimulates expression of aquaporins 4 and 9 through a p38 mitogen-activated protein kinase-dependent pathway in rat astrocytes. J Biol Chem. 2003;278:44525–44534. doi: 10.1074/jbc.M304368200. [DOI] [PubMed] [Google Scholar]
- 35.Zhao Z, Cheng X, Wang Y, Han R, Li L, Xiang T, He L, Long H, Zhu B, He Y. Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS One. 2014;9:e95884. doi: 10.1371/journal.pone.0095884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schonbeck U, Libby P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26:611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- 37.Ribatti D, Ranieri G, Annese T, Nico B. Aquaporins in cancer. Biochim Biophys Acta. 2014;1840:1550–1553. doi: 10.1016/j.bbagen.2013.09.025. [DOI] [PubMed] [Google Scholar]
- 38.Nico B, Ribatti D. Role of aquaporins in cell migration and edema formation in human brain tumors. Exp Cell Res. 2011;317:2391–2396. doi: 10.1016/j.yexcr.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Zhao WJ, Zhang W, Li GL, Cui Y, Shi ZF, Yuan F. Differential expression of MMP-9 and AQP4 in human glioma samples. Folia Neuropathol. 2012;50:176–186. [PubMed] [Google Scholar]
- 40.Noell S, Ritz R, Wolburg-Buchholz K, Wolburg H, Fallier-Becker P. An allograft glioma model reveals the dependence of aquaporin-4 expression on the brain microenvironment. PLoS One. 2012;7:e36555. doi: 10.1371/journal.pone.0036555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding T, Ma Y, Li W, Liu X, Ying G, Fu L, Gu F. Role of aquaporin-4 in the regulation of migration and invasion of human glioma cells. Int J Oncol. 2011;38:1521–1531. doi: 10.3892/ijo.2011.983. [DOI] [PubMed] [Google Scholar]
- 42.Ding T, Gu F, Fu L, Ma YJ. Aquaporin-4 in glioma invasion and an analysis of molecular mechanisms. J Clin Neurosci. 2010;17:1359–1361. doi: 10.1016/j.jocn.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi Y, Edwards NA, Proescholdt MA, Oldfield EH, Merrill MJ. Regulation and function of aquaporin-1 in glioma cells. Neoplasia. 2007;9:777–787. doi: 10.1593/neo.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aldea M, Tomuleasa C, Petrushev B, Susman S, Kacso GI, Irimie A, Florian IS, Soritau O. Antidiabetic pharmacology: a link between metabolic syndrome and neuro-oncology? J BUON. 2011;16:409–413. [PubMed] [Google Scholar]