

Abstract
This study was aimed to investigate the functional roles of cytokine receptor (CXCR) CXCR2 and CXCR7 in esophageal cancer (EC). Specific small interfering RNAs (siRNA) against CXCR2 and CXCR7 were transfected into EC cell lines TE-1, EC9706, and EC109 cells. Expression of CXCR2 and CXCR7 was validated, along with cell viability, chemotaxis, apoptosis rate, and ERK1/2 pathways associated protein after transfection. Moreover, EC9706 cells treated with or without CXCR2/7 siRNA were injected into athymic nude mice. Tumor volumes were measured. Besides, immunohistochemical (IHC) staining was performed to investigate the expression of CXCR2/7 in adjacent normal tissues and tumor tissues from esophageal squamous cell carcinoma (ESCC) patients. Also, the associations between CXCR2/7 expression and clinicopathological features and progression were explored. The mRNA levels of CXCR2 and CXCR7 were significantly reduced after transfection. Silencing of CXCR2 and CXCR7 statistically decreased cell viability and chemotaxis, and increased apoptotic rate. Cells invasion was significantly reduced by silencing of CXCR2, however, no significance was found in silencing of CXCR7. The protein levels of pERK1/2 were significantly decreased by silencing of CXCR2 and CXCR7. Besides, silencing of CXCR2 and CXCR7 significantly reduced tumor growth in vivo, and associated with clinicopathological features and progression. Silencing of CXCR2 and CXCR7 protects against EC by inhibiting cell growth and chemotaxis, and inducing apoptosis though ERK1/2 pathways. Silencing of CXCR2 and CXCR7 has potentially therapeutic target for EC.
Keywords: Esophageal cancer, CXCR2, CXCR7, SiRNA, clinicopathological features, progression
Introduction
Esophageal cancer (EC) is the eighth most common malignancy worldwide and the sixth leading cause of cancer mortality [1]. Whereas, EC has been ranked as the fourth most common cause of cancer death in China [2], and it accounts for approximately one-tenth of all malignancies mortality [2]. More than 90% EC patients fail to be diagnosed as a pre-malignant condition [3,4]. A randomized control trial indicated that endoscopic screening and surveillance for patients with esophageal neoplasia were unpractical due to the expensive cost and relatively low benefit for patients [5]. Most patients develop into advanced stage before this disease could be suspected or diagnosed. In spite of considerable progresses have been made in diagnosis and management, the overall prognosis of EC is still poor [6-9]. The 5-year survival rate of EC is about 15-25% [2]. Therefore, a better and a deeper understanding of the molecular mechanisms underlying EC is helpful to search novel and tailored therapeutic strategies.
It has been well demonstrated that both chemokines and their receptors play significant roles in tumor development [10-13]. Among the chemokines, chemokine ligand (CXCL) 12/stromal-derived factor (SDF)-1 and interleukin (IL) 8/CXCL-8 have been reported to be involved in tumor progression and metastasis of various entities [14,15]. Recently, the functional roles of CXCL12 and its receptor chemotaxis cytokine receptor (CXCR) 4 or CXCR7, and CXCL-8 and its receptor CXCR2 in EC have been well investigated [16,17]. Although several studies have investigated the function and clinical significance of CXCR2 and CXCR7 in EC, however, rare studies have been done concerning the impact of silencing of CXCR2 and CXCR7 on EC cells.
In the present study, we performed the small interfering RNAs (siRNA) to silence the expression of CXCR2 and CXCR7 into the three EC cell lines (TE-1, EC9706, and EC109 cells). The cell growth, chemotaxis, invasion, and apoptosis of these cells were explored, as well as the potential associated mechanism. Besides, animal experiment was used to analyze the effect of silencing of CXCR2 and CXCR7 on tumor growth in vivo. In addition to cell and animal researches, the associations between CXCR2/7 expression in human EC and clinicopathological features and progression were explored.
Materials and methods
Cell lines and cultures
The human EC cell lines TE-1, EC9706, and EC109 were provided by the American Type Culture Collection (ATCC, Manassas, VA, USA). These cells were cultured in Dualbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA), 100 μg/μL streptomycin (Gibco, Gaithersburg, MD, USA) and 100 μg/μL penicillin (Gibco, Gaithersburg, MD, USA) at 37°C in a humidified incubator with 5% CO2.
Transfection
After 24 h post-culturing in DMEM at an appropriate concentration (about 60-80%), the TE-1, EC9706, and EC109 cells were plated in 6-well plates, respectively. Then the specific siRNA against CXCR2, CXCR7, and nonspecific (NS)-siRNA (GenaPharma Shanghai, China) were respectively transfected into TE-1, EC9706, and EC109 cells at a final concentration of 60 nM using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. After 4 h of incubation, normal media was added. In addition, cells in control group received no special treatment. Then the cells were harvested after 48 h for further analysis.
Quantitative real-time polymerase chain reaction (qRT- PCR)
Total RNA was respectively extracted from TE-1, EC9706, and EC109 cells by TRIzol reagent (Invitrogen Corp., Carlsbad, CA) using a commercially available kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s recommendation. Complementary DNA (cDNA) was synthesized using the Reverse Transcription System (Promega, Madison, WI). The products of PCR were identified by an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, Calif). The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as a reference control. PCR amplification was carried out using LightCycler - FastStart DNA Master SYBR Green I (Roche Diagnostics, Tokyo, Japan). PCR conditions included 1 predenaturation cycle of 1 min at 95°C, 50 cycles of 95°C for 30 s, 58-62°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 10 min. The mRNA levels were expressed as threshold cycle (CT). The amount of target was measured using 2-ΔΔCT method. Reactions were carried out in triplicate.
The primers sequences used were as follows: CXCR2, 5’ primer-AGGCAC AGTGAAGACATCGG-3’ and reverse, 5’ primer-CAGCAGGCTCAGCAGGAATA-3’ [18]; CXCR7, 5’ primer-TGGGTGGTCAGTCTCGT-3’ and reverse, 5’ primer-CCGG CAGTAGGTCTCAT-3’ [19]; and GAPDH, 5’ primer-CGGAGTCAACGGATTG GTC-3’ and reverse, 5’ primer-AGCCTTCTCCAT GGTCGTGA-3’.
Cell proliferation, malignant and metastasis assays
For proliferation rate, cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) colorimetric assay. Briefly, TE-1, EC9706, and EC109 cells were respectively placed in 96-well plates at a final concentration of 5×104 per mL in 100 μL medium. Then the plates were maintained at 37°C in 5% CO2 incubator. After 24 h incubation, MTT (20 μL; 5 g/L) was added to each well and the plates were incubated at 37°C for another 4 h. The absorbance at 570 nm was determined with a microplate reader (Model Benchmark). Experiments were performed 3-5 times.
For cell invasion assays, Transwell chamber (Costar, Cambridge, MA) were used. Briefly, cells were harvested and then were suspended in serum-free dulbecco’s modified eagle medium (DMEM) media at a density of 1×105 cells/mL on 24-Transwell membranes (8 μm pores). The upper chamber of the transwell was coated with 70 μL Matrigel (1 mg/mL), and the lower chamber was filled with 10% FBS. Cell suspensions (100 μL) were added to the upper side, and then were disassembled after incubation at 37°C in 5% CO2 for 24 h. The membranes were fixed in 1% glutaraldehyde and then stained with 0.5% crystal violet reagent. The remaining cells were counted in eight random microscopic fields per membrane.
For chemotaxis assays, a modified Boyden chamber was performed. In brief, polycarbonate filter (8 μm pores) coated with collagen type IV was placed on a 48-well chamber. TE-1, EC9706, and EC109 cells (1×105 cells/well) were loaded into the upper chamber. The filter was disassembled after incubation at 37°C in 5% CO2 for 6 h. Then the cells were fixed and stained. The number of cells that moved through the chamber was counted by measuring absorbance at 595 nm.
Flow cytometry (FCM) detection
For the apoptosis rate, Annexin V-fluorescein-5-isothiocyanate (Annexin V-FITC) apoptosis detection kit (Biosea, China) was used in accordance with the manufacturer’s instruction. Briefly, cells (5×106) were harvested, mixed with 10 μL Annexin, and incubated at room temperature in the dark for 15 min. Then 5 μL 10 mg/L propidium iodide (PI) was added. Finally, the cells were read by FCM (Becton Dickinson, San Jose, CA, USA) with excitation wavelength at 488 nm and emission wavelength at 635 nm. CELLQuest 3.0 software (Becton Dickinson, San Jose, CA) was performed to analyze the numerical values.
Western blotting for signaling pathway analysis
For Western blotting analysis, TE-1 cells were harvested for protein extraction. Protein density was determined using a BCA assay kit (TaKaRa BIO INC, Japan). Proteins samples (20 μL) were resolved with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Bedford, MA, USA). The membranes were blocked with 5% defatted milk powder in phosphate buffer saline (PBS) for 2 h at room temperature and incubated overnight at 4°C with the following antibodies: anti-pERK1 (1:1,000), anti-pERK2 (1:1,000), anti-MMP-9 (1:1,000) and anti-β-actin (1:1,000). The membranes were then incubated with an appropriate secondary antibody for 2 h at room temperature. All the antibodies were purchased from Santa Cruz Biotechnology. Finally, enhanced chemiluminescence and densitometric analysis were performed according to the manufacturer’s instructions.
Mouse xenograft study
Male athymic nude mice (NCI-nu, 8-12 weeks of age) were purchased from the Institute of Experimental Animals, Chinese Academy of Medical Sciences. The mice were housed in a pathogen-free barrier facility. The use of animal was approved by the local Ethics Committee and was in line with the Guide for Care and Use of Laboratory Animals published by the China National Institutes of Health. Each mouse was subcutaneously injected in the right flank through a 22-gauge needle with EC9706 (1×106) cells treated with or without CXCR2 siRNA and CXCR7 siRNA. Mice injected with cell lines expressing green fluorescent protein (GFP) siRNA were regarded as controls. The mice (six in each group) were weighed, and tumor mass volumes were measured at 7, 14, 28, and 35 days throughout the study. The tumor mass volumes were recorded as follows: length × width2/2 with a vernier caliper. Five weeks after cell inoculation, the mice were killed by CO2 asphyxiation. The tumor xenografts were excised, weighed and recorded.
Human tissue and immunohistochemistry (IHC)
The use of human tumor specimen was approved by the local Ethics Committee and informed consents were obtained from all patients. Between April 2008 and December 2014, a total of 156 patients with esophageal squamous cell carcinoma (ESCC) who underwent surgery at our hospital were enrolled into the retrospective study. Samples including carcinoma tissue and paired adjacent normal tissues were selected from patients after obtaining the patients’ complete information on clinicopathologic characteristics. The ages of the patients ranged from 48 to 86 years (median, 67 years). Of the patients, 91 were men and 65 were women. Patients were diagnosed with ESCC according to the clinical pathological findings. None of the patients received preoperative special treatment (e.g. radiotherapy, chemotherapy, neoadjuvant or adjuvant treatment). According to Tumor Node Metastasis (TNM)/Union for International Cancer Control (UICC) classification 6th edition, the number of I:IIA:IIB:III was 32:20:48:56. The follow up for survival were recorded from all patients. By April 2015 (the time of data analysis), 66 patients had died and 90 patients were alive. The median survival time was 64 months.
The samples both from the tissue and surrounding normal tissue were embedded in paraffin. Tissue sections (4 μm) were prepared for IHC analyses of CXCR2 and CXCR7. In brief, the sections were de-paraffinized and, rehydrated, and were then subjected to antigen retrieval. Endogenous peroxidase was eliminated by the use of 3% H2O2 in PBS for 10 min. The slides were then incubated with normal horse or goat serum in PBS and anti-CXCR2 (1:100) or anti-CXCR7 (1:100) diluted in PBS overnight at 4°C. After being washed with PBS, the samples were incubated with a biotinylated second antibody (Horse anti-mouse IgG or Goat-anti rabbit IgG) (GBI) for 30 min at room temperature, developed with 3,3’-diaminobenzide tetrahydrochloride (DAB, Vector Laboratories Inc., Burlingame, CA, USA), counterstained with Mayer’s haematoxylin solution (Sigma) for 30 s, and scored under a microscope.
The staining intensity (0, 1+, 2+, 3+) and the positivity was quantified according to a previous study [20]. CXCR2/7 was recorded as negative when the spots without staining and spots with a staining intensity of 1+ accounted for ≤ 20% of tumor cells, and while was considered as positive when spots with a staining intensity of 1+ accounted for > 20% tumor cells, or spots with staining intensity ≥ 2.
Statistical analysis
The Kolmogorov-Smirnov (K-S) one-sample test was performed to test the normal distribution for all collected data. Chi-square test or rank-sum test was used to analyze enumeration data. Student t-test was used to evaluate the difference between two groups, while analysis of variance (ANOVA) was conducted to compare the difference for more than three groups. Post-hoc Tukey test was performed to examine significant pairwise comparisons. Univariate survival analysis was determined with Kaplan-Meier method, and log-rank test was performed to assess the difference between the survival curves. All the statistical analysis was determined by statistic package for social science (SPSS, version 17.0, Chicago, IL). A statistical significance was defined when P < 0.05.
Results
SiRNA reduced the expression of CXCR2 and CXCR7 in EC cell lines
To examine the impact of silencing of CXCR2 and CXCR7 on EC cells (TE-1, EC9706, and EC109 cells), CXCR2 siRNA and CXCR7 siRNA were used to down-regulate the expression of CXCR2 and CXCR7 in TE-1, EC9706 and EC109 cells. After 48 h transfection, expression mRNA levels of CXCR2 and CXCR7 was validated by qRT-PCR. The results showed that both the expression mRNA levels of CXCR2 and CXCR7 in TE-1, EC9706 and EC109 cells were significantly reduced after transfection of CXCR2 siRNA and CXCR7 siRNA, respectively (P < 0.05) compared with NS-siRNA and control group, respectively (P < 0.005). Besides, these cells transfected with NS siRNA had no evidence of knockdown of CXCR2 and CXCR7 (Figure 1A and 1B).
Figure 1.
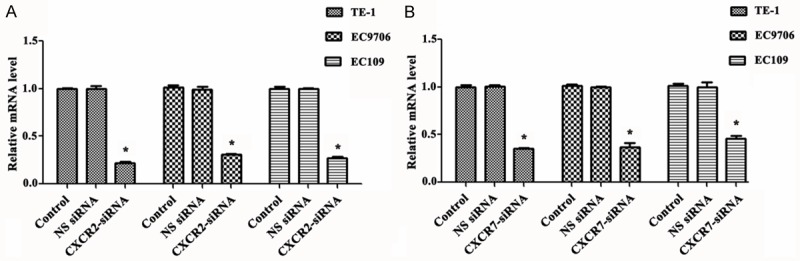
SiRNA reduces the expression of CXCR2 and CXCR7 in TE-1, EC9706 and EC109 cells. A. Relative expression of mRNA levels after transfection with CXCR2 siRNA; B. Relative expression of mRNA levels after transfection with CXCR7 siRNA. SiRNA, small interfering RNAs; CXCR, chemotaxis cytokine receptor. *P < 0.05 compared with control group and NS siRNA group.
Silencing of CXCR2 and CXCR7 decreased cell viability and chemotaxis, and increased apoptotic rate
To investigate the impact of silencing of CXCR2 and CXCR7 on EC cells, cell viability, chemotaxis, and apoptotic rate were evaluated in TE-1, EC9706 and EC109 cells. We found that both silencing of CXCR2 and CXCR7 could significantly decrease the cell viability (Figure 2A-C) and chemotaxis (Figure 2D) and increase the apoptotic rate (Figure 2E-G) compared with control group and NS siRNA group (P < 0.05).
Figure 2.

Silencing of CXCR2 and CXCR7 decreases cell viability and chemotaxis, and increases apoptotic rate. A-C. Silencing of CXCR2 and CXCR7 to TE-1, EC9706 and EC109 cells decreases cell viability; D. Silencing of CXCR2 and CXCR7 to TE-1, EC9706 and EC109 cells decreases cell chemotaxis; E-G, Silencing of CXCR2 and CXCR7 to TE-1, EC9706 and EC109 cells increases apoptotic rate. SiRNA, small interfering RNAs; CXCR, chemotaxis cytokine receptor; NS, nonspecific. *P < 0.05 compared with control group and NS siRNA group.
Silencing of CXCR2, but not CXCR7, induced TE-1 cells invasion
To explore the invasion ability of silencing of CXCR2 and CXCR7 on TE-1, EC9706 and EC109 cells, we used the matrigel invasion assay. We found that silencing of CXCR7 failed to induce these three kinds of cells invasion. There were no significant differences among the control group, NS siRNA group and CXCR7 siRNA group. However, silencing of CXCR2 induced significant invasion of these three kinds of cells compared with the control group and NS siRNA group (Figure 3A-C).
Figure 3.
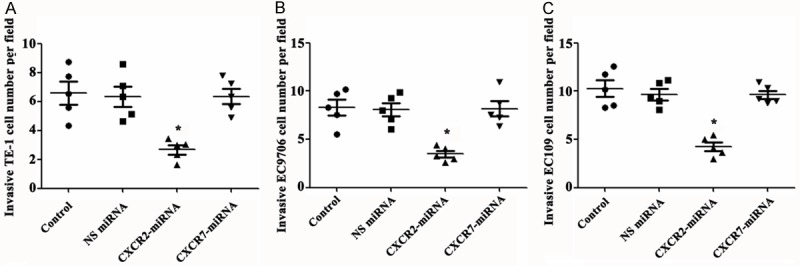
Silencing of CXCR2, but not CXCR7, induces cells invasion. A-C. Silencing of CXCR2, but not CXCR7, induces cells invasion in TE-1, EC9706 and EC109 cells, respectively. SiRNA, small interfering RNAs; CXCR, chemotaxis cytokine receptor; NS, nonspecific. *P < 0.05 compared with control group and NS siRNA group.
Silencing of CXCR2 and CXCR7 to TE-1 cells activated ERK1/2 pathways
To confirm whether silencing of CXCR2 and CXCR7 could induce ERK1/2 pathway activation in TE-1 cells, we measured associated protein expression of ERK1/2 pathway after transfection with siRNA. The results demonstrated that silencing of CXCR2 and CXCR7 both significantly decreased the expression protein levels of pERK1 and pERK2 compared with control group and NS siRNA group, indicating silencing of CXCR2 and CXCR7 was involved in activation of ERK1/2 pathways. In addition, the expression of MMP-9 was closely related with silencing of CXCR2, but was not related with silencing of CXCR7 (Figure 4A-D).
Figure 4.
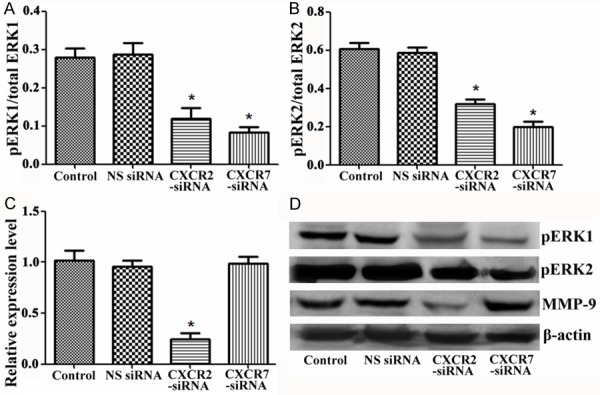
Silencing of CXCR2 and CXCR7 to TE-1cells activates ERK1/2 pathways. A. pERK1/total ERK1 protein level after transfection with siRNA; B. pERK2/total ERK2 protein level after transfection with siRNA; C. Relative expression level of MMP-9 after transfection with siRNA; D. The photo of Western blotting. SiRNA, small interfering RNAs; CXCR, chemotaxis cytokine receptor; NS, nonspecific; MMP, matrix metalloproteinases. *P < 0.05 compared with control group and NS siRNA group.
Silencing of CXCR2 and CXCR7 reduced in vivo tumor growth
To investigate whether silencing of CXCR2 and CXCR7 was associated with in vivo tumor growth in EC cells, we tested tumor sizes of mouse xenograft tumor growth after inoculation. As indicated in Figure 5, the subcutaneous tumor sizes in mice (Figure 5A and 5B) were both significantly declined in EC9706 cells treated with CXCR2 siRNA and CXCR7 siRNA compared with the controls (GFP siRNA).
Figure 5.
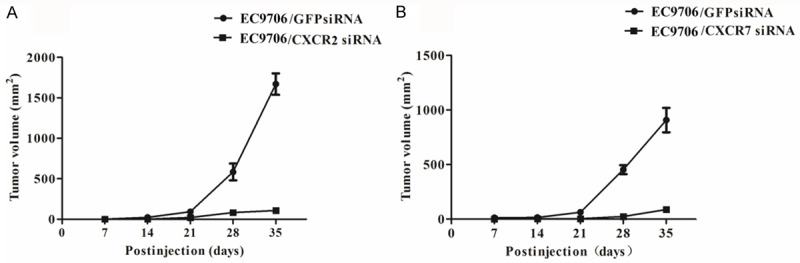
Silencing of CXCR2 and CXCR7 reduces in vivo tumor growth. SiRNA, small interfering RNAs; CXCR, chemotaxis cytokine receptor; GFP, green fluorescent protein.
Silencing of CXCR2 and CXCR7 associated with progression
To explore the role of CXCR2 and CXCR7 in EC, we collected 156 EC samples with paired adjacent normal tissues and performed IHC. The results showed that both CXCR2 and CXCR7 positive staining were observed in cytoplasm and membrane of EC cells (Figure 6A-D). Both the protein expression levels of CXCR2 (P = 0.006) and CXCR7 (P = 0.02) were significantly up-regulated in carcinoma tissues compared with adjacent normal tissues (Figure 6E). In addition, the results of follow-up data showed that the survival rate of patients with high expression of CXCR2 and CXCR7 was significantly lower than that with low expression of CXCR2 (P = 0.016) and CXCR7 (P = 0.023), respectively (Figure 6F and 6G).
Figure 6.
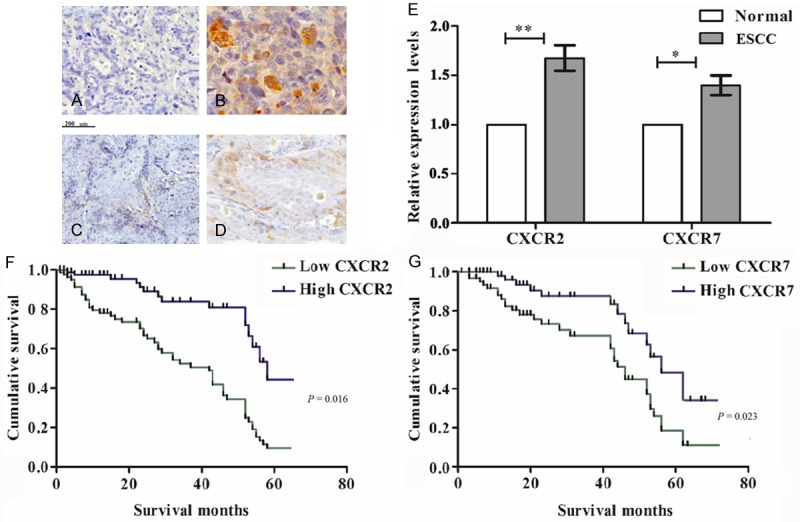
Silencing of CXCR2 and CXCR7 associates with progression. A and B. IHC of CXCR2 in normal tissue and cancer tissue, respectively; C and D. IHC of CXCR7 in normal tissue and cancer tissue, respectively; E. Quantitative analysis of IHC; F and G. Expression of CXCR2 and CXCR7 in EC tissues correlates with a lower cancer patient survival rate, respectively. IHC, immunohistochemistry; CXCR, chemotaxis cytokine receptor; ESCC, esophageal squamous cell carcinoma.
Silencing of CXCR2 and CXCR7 associated with clinicopathological features of EC
To explore the relationship between CXCR2/CXCR7 protein expression and EC progression, we evaluated the correlation between CXCR2/CXCR7 expression and clinicopathological features of ESCC. For the clinical TNM/UICC stage, the strongly positive rate of CXCR2 protein was 10.6%, 30.6%, 46.9%, and 73.8% in stage I, IIA, IIB, and III, respectively. The strongly positive rate of CXCR7 protein was 8.4%, 25.9%, 44.7%, and 69.6% in stage I, IIA, IIB, and III, respectively. The strongly positive rate of CXCR2 and CXCR7 in stage IIA, IIB, and III were significantly higher than those in stage I (P = 0.000 and P = 0.021, respectively). These results suggested that the high expression of CXCR2 and CXCR7 were both closely associated with higher histological grade, advanced clinical stage in patients with ESCC.
Discussion
In the present study, we demonstrated that silencing of CXCR2 and CXCR7 could inhibit cell growth, reduce metastasis, induce apoptosis in EC cell lines, and reduce tumor growth in vivo. Besides, we found that high expression of CXCR2 and CXCR7 were closely associated with higher histological grade, advanced clinical stage, and lower survival in patients with ESCC. Our results suggest that silencing of CXCR2 and CXCR7 protects against EC. The effects may be considered to be associated with activation of ERK1/2 pathways.
Chemokines are low-molecular-weight cytokines and can be subdivided into CXC and CC chemokines. They could selectively modulate leukocyte extravasation to inflammation areas via chemoattraction [21]. In addition to their roles in inflammatory responses, a good deal of evidence has suggested that chemokines also play significant roles in tumor. Both CXC chemokines and their receptors CXCR regulate tumor development by regulating angiogenesis, activating a tumor-specific immune response and stimulating cell proliferation [22]. Recently, the functional roles of CXCR7 and CXCR-2 in tumorigenesis and tumor development have been widely studied. CXCR7, the second receptor of CXCL12 (SDF-1), is highly conserved in mammals. It is a membrane associated receptor protein located on chromosome 1 in mice and on chromosome 2 with CXCR4, CXCR2, and CXCR1. CXCR-2 is a G-protein-coupled receptor of CXCL-8 (formally known as IL-8) that functions as an autocrine growth factor in many various malignant melanoma cells [23]. It has been well demonstrated that CXCR7 and CXCR-2 express on many human tumor cell lines, such as breast cancer, prostate cancer, lung cancer, EC and so on [17,24-26]. Wang et al. [27] suggested that autocrine growth-related oncogene (GRO) α-CXCR2 and GROβ-CXCR2 signaling pathway may be involved in esophageal carcinogenesis. In addition, Meijer et al. [28] proposed that CXCR7 inhibited tumor tumorigenesis mainly in tumor tissue with high expression of CXCL12, in cells with CXCL12 autocrine signaling, or in tumors at specific stages. Hence, this autocrine signaling pathway contributes significantly to cancer cell proliferation and development.
Previous studies have indicated that CXCR2 is overexpressed in the cytoplasm and plasma membrane of ESCC, and is correlated with ESCC progression and metastasis [16,29], while CXCR7 is mainly expressed in ESCC and infrequently in adenocarcinoma [30]. Moreover, some researchers have found that the antagonists of CXCR2 (SCH-527123 and SCH-479833) protects against human colon cancer liver metastasis, indicating that inhibition of CXCR2 might be a potential therapeutic strategy for oncology [31,32]. Besides, targeting CXCR2 as a treatment of pancreatic cancer has been deeply explored [33,34]. To confirm the impact of suppression of CXCR2 and CXCR7 on EC, we depleted the expression of CXCR2 and CXCR7 though siRNA technology in TE-1, EC9706, and EC109 cells. Both the MTT and FCM results showed that silencing of CXCR2 and CXCR7 could statistically decrease the cell viability and chemotaxis, and increase the apoptotic rate of EC cell lines. Besides, our animal experimental research showed that silencing of CXCR2 and CXCR7 could significantly decrease the tumor growth at 7, 14, 28, and 35 days. In addition, we performed IHC on human tissues. Both CXCR2 and CXCR7 were expressed on cytoplasm and membrane of EC cells, which suggested that the source of ligands (both CXCR2 and CXCR7) were from the tumor cells. This autocrine signaling may be contributed to the tumor growth. Moreover, the higher expression of CXCR2 and CXCR7, the higher histological grade, advanced clinical stage, and lower survival were found in ESCC patients. These results were in line with previous studies, suggesting that CXCR2 and CXCR7 paly significant roles in EC and depletion of CXCR2 and CXCR7 may be potential treatments of EC. However, further studies should be performed to confirm the conclusion.
It has been reported that expression of MMP-9 is involved in the differentiation, vessel permeation and lymph node metastasis of cancers because of its ability on degradation a basement membrane and extracellular matrix [35]. Moreover, some studies suggest the expression of MMP-9 play important roles in its malignant behavior, aggressiveness, and invasiveness on EC cells [36,37]. MMP-9 expression has been considered as a negative, independent prognostic factor in ESCC and may be a potential biomarker of ESCC diagnosis and treatment [38]. Furthermore, the release of MMP-9 could be significantly reduced by blocking the expression of CXCR2 [39]. In accordance with previous studies, we also found that expression of MMP-9 was significantly induced by silencing of CXCR2, but could not be induced by CXCR7 in our study. Additionally, many studies have suggested that CXCR2 signaling network is related to the activation of the mitogen-activated protein kinase (MAPK) MEK1/2, ERK1/2, and p38 [40,41]. Similarly, in our study ERK1/2 pathways were found to be activated by silencing of CXCR2. In addition to the results, we found that ERK1/2 pathways were also activated by silencing of CXCR7.
In conclusion, our results suggest that silencing of CXCR2 and CXCR7 inhibits cell growth, reduces metastasis, and induces apoptosis in EC cell lines, reduces tumor growth in vivo. Besides, both CXCR2 and CXCR7 are closely associated with the EC progression. These effects may be involved in activation of ERK1/2 pathways. Our results reflect a potential functional contribution of CXCR2 and CXCR7 in EC, and offer new insights into molecular basis for treatment of EC.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Ren Y, Zheng J, Wang L, Zhang W, Yin F, Zhou J, Ge X, Fan S, Tang R, Sun J. A study on EGFR gene amplification and protein expression in Chinese esophagus cancer patients and antitumor effect of an EGFR inhibitor in patient-derived esophagus cancer models. Cancer Res. 2014;74:1730–1730. [Google Scholar]
- 3.Dulai GS, Guha S, Kahn KL, Gornbein J, Weinstein WM. Preoperative prevalence of Barrett’s esophagus in esophageal adenocarcinoma: a systematic review. Gastroenterology. 2002;122:26–33. doi: 10.1053/gast.2002.30297. [DOI] [PubMed] [Google Scholar]
- 4.Hvid-Jensen F, Pedersen L, Drewes AM, Sørensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365:1375–1383. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 5.Spechler SJ, Sharma P, Souza RF, Inadomi JM, Shaheen NJ. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology. 2011;140:1084–1091. doi: 10.1053/j.gastro.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 6.Ando N, Kato H, Igaki H, Shinoda M, Ozawa S, Shimizu H, Nakamura T, Yabusaki H, Aoyama N, Kurita A, Ikeda K, Kanda T, Tsujinaka T, Nakamura K, Fukuda H. A randomized trial comparing postoperative adjuvant chemotherapy with cisplatin and 5-fluorouracil versus preoperative chemotherapy for localized advanced squamous cell carcinoma of the thoracic esophagus (JCOG9907) Ann Surg Oncol. 2012;19:68–74. doi: 10.1245/s10434-011-2049-9. [DOI] [PubMed] [Google Scholar]
- 7.Zhang JX, Tong ZT, Yang L, Wang F, Chai HP, Zhang F, Xie MR, Zhang AL, Wu LM, Hong H, Yin L, Wang H, Wang HY, Zhao Y. PITX2: a promising predictive biomarker of patients’ prognosis and chemoradioresistance in esophageal squamous cell carcinoma. Int J Cancer. 2013;132:2567–2577. doi: 10.1002/ijc.27930. [DOI] [PubMed] [Google Scholar]
- 8.Bonnetain F, Bouche O, Michel P, Mariette C, Conroy T, Pezet D, Roullet B, Seitz JF, Paillot B, Arveux P. A comparative longitudinal quality of life study using the Spitzer quality of life index in a randomized multicenter phase III trial (FFCD 9102): chemoradiation followed by surgery compared with chemoradiation alone in locally advanced squamous resectable thoracic esophageal cancer. Ann Oncol. 2006;17:827–834. doi: 10.1093/annonc/mdl033. [DOI] [PubMed] [Google Scholar]
- 9.Tahara M, Ohtsu A, Hironaka S, Boku N, Ishikura S, Miyata Y, Ogino T, Yoshida S. Clinical impact of criteria for complete response (CR) of primary site to treatment of esophageal cancer. Jpn J Clin Oncol. 2005;35:316–323. doi: 10.1093/jjco/hyi095. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Lee VC, Chevalier E, Hwang ST. Chemokine receptors as targets for cancer therapy. Curr Pharm Des. 2009;15:742–757. doi: 10.2174/138161209787582165. [DOI] [PubMed] [Google Scholar]
- 11.Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226:148–157. doi: 10.1002/path.3029. [DOI] [PubMed] [Google Scholar]
- 12.Rodero MP, Combadière C, Boissonnas A. Cancer Immunology. Springer; 2015. Role of Chemokines and Chemokine Receptors in Cancer; pp. 121–142. [Google Scholar]
- 13.Khurram SA, Bingle L, McCabe BM, Farthing PM, Whawell SA. The chemokine receptors CXCR1 and CXCR2 regulate oral cancer cell behaviour. J Oral Pathol Med. 2014;43:667–674. doi: 10.1111/jop.12191. [DOI] [PubMed] [Google Scholar]
- 14.Maksym RB, Tarnowski M, Grymula K, Tarnowska J, Wysoczynski M, Liu R, Czerny B, Ratajczak J, Kucia M, Ratajczak MZ. The role of stromal-derived factor-1-CXCR7 axis in development and cancer. Eur J Pharmacol. 2009;625:31–40. doi: 10.1016/j.ejphar.2009.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ, Wang J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–722. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogura M, Takeuchi H, Kawakubo H, Nishi T, Fukuda K, Nakamura R, Takahashi T, Wada N, Saikawa Y, Omori T, Miyasho T, Yamada S, Kitagawa Y. Clinical significance of CXCL-8/CXCR-2 network in esophageal squamous cell carcinoma. Surgery. 2013;154:512–520. doi: 10.1016/j.surg.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Tachezy M, Zander H, Gebauer F, von Loga K, Pantel K, Izbicki JR, Bockhorn M. CXCR7 expression in esophageal cancer. J Transl Med. 2013;11:238. doi: 10.1186/1479-5876-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Yang L, Xu J, Zhang X, Wang B. Enhanced expression and clinical significance of chemokine receptor CXCR2 in hepatocellular carcinoma. J Surg Res. 2011;166:241–246. doi: 10.1016/j.jss.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 19.Zheng K, Li HY, Su XL, Wang XY, Tian T, Li F, Ren GS. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2010;29:31. doi: 10.1186/1756-9966-29-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon R, Mirlacher M, Sauter G. Immunohistochemical analysis of tissue microarrays. Methods Mol Biol. 2010;664:113–126. doi: 10.1007/978-1-60761-806-5_12. [DOI] [PubMed] [Google Scholar]
- 21.Ebnet K, Vestweber D. Molecular mechanisms that control leukocyte extravasation: the selectins and the chemokines. Histochem Cell Biol. 1999;112:1–23. doi: 10.1007/s004180050387. [DOI] [PubMed] [Google Scholar]
- 22.Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256:137–165. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schadendorf D, Moller A, Algermissen B, Worm M, Sticherling M, Czarnetzki BM. IL-8 produced by human malignant melanoma cells in vitro is an essential autocrine growth factor. J Immunol. 1993;151:2667–2675. [PubMed] [Google Scholar]
- 24.Wang J, Shiozawa Y, Wang J, Wang Y, Jung Y, Pienta KJ, Mehra R, Loberg R, Taichman RS. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J Biol Chem. 2008;283:4283–4294. doi: 10.1074/jbc.M707465200. [DOI] [PubMed] [Google Scholar]
- 25.Iwakiri S, Mino N, Takahashi T, Sonobe M, Nagai S, Okubo K, Wada H, Miyahara R. Higher expression of chemokine receptor CXCR7 is linked to early and metastatic recurrence in pathological stage I nonsmall cell lung cancer. Cancer. 2009;115:2580–2593. doi: 10.1002/cncr.24281. [DOI] [PubMed] [Google Scholar]
- 26.Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172:2853–2860. doi: 10.4049/jimmunol.172.5.2853. [DOI] [PubMed] [Google Scholar]
- 27.Wang B, Hendricks DT, Wamunyokoli F, Parker MI. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006;66:3071–3077. doi: 10.1158/0008-5472.CAN-05-2871. [DOI] [PubMed] [Google Scholar]
- 28.Meijer J, Ogink J, Roos E. Effect of the chemokine receptor CXCR7 on proliferation of carcinoma cells in vitro and in vivo. Br J Cancer. 2008;99:1493–1501. doi: 10.1038/sj.bjc.6604727. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Wang B, Hendricks DT, Wamunyokoli F, Parker MI. A growth-related oncogene/CXC chemokine receptor 2 autocrine loop contributes to cellular proliferation in esophageal cancer. Cancer Res. 2006;66:3071–3077. doi: 10.1158/0008-5472.CAN-05-2871. [DOI] [PubMed] [Google Scholar]
- 30.Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A, Kleer CG, Essner JJ, Nasevicius A, Luker GD. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci U S A. 2007;104:15735–15740. doi: 10.1073/pnas.0610444104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, Singh RK. Small-molecule antagonists for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing tumor cell proliferation, survival, and angiogenesis. Clin Cancer Res. 2009;15:2380–2386. doi: 10.1158/1078-0432.CCR-08-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Varney ML, Singh S, Li A, Mayer-Ezell R, Bond R, Singh RK. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011;300:180–188. doi: 10.1016/j.canlet.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wente MN, Keane MP, Burdick MD, Friess H, Büchler MW, Ceyhan GO, Reber HA, Strieter RM, Hines OJ. Blockade of the chemokine receptor CXCR2 inhibits pancreatic cancer cell-induced angiogenesis. Cancer Lett. 2006;241:221–227. doi: 10.1016/j.canlet.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 34.Hertzer KM, Donald GW, Hines OJ. CXCR2: a target for pancreatic cancer treatment? Expert Opin Ther Targets. 2013;17:667–680. doi: 10.1517/14728222.2013.772137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu ZD, Li JY, Li M, Gu J, Shi XT, Ke Y, Chen KN. Matrix metalloproteinases expression correlates with survival in patients with esophageal squamous cell carcinoma. Am J Gastroenterol. 2005;100:1835–1843. doi: 10.1111/j.1572-0241.2005.50018.x. [DOI] [PubMed] [Google Scholar]
- 36.El-Shahat M, Lotfy M, Fahmy L, Abouel-Nour M, El-Kenawy Ael M. Prognostic value of microvessel density, matrix metalloproteinase-9 and p53 protein expression in esophageal cancer. J Egypt Natl Canc Inst. 2004;16:224–230. [PubMed] [Google Scholar]
- 37.Samantaray S, Sharma R, Chattopadhyaya T, Gupta SD, Ralhan R. Increased expression of MMP-2 and MMP-9 in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2004;130:37–44. doi: 10.1007/s00432-003-0500-4. [DOI] [PubMed] [Google Scholar]
- 38.Gu ZD, Chen KN, Li M, Gu J, Li JY. Clinical significance of matrix metalloproteinase-9 expression in esophageal squamous cell carcinoma. World J Gastroenterol. 2005;11:871–874. doi: 10.3748/wjg.v11.i6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakrabarti S, Patel KD. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J Leukoc Biol. 2005;78:279–288. doi: 10.1189/jlb.1004612. [DOI] [PubMed] [Google Scholar]
- 40.Juffermans NP, Dekkers PE, Peppelenbosch MP, Speelman P, van Deventer SJ, Van der Poll T. Expression of the Chemokine Receptors CXCR1 and CXCR2 on Granulocytes in Human Endotoxemia and Tuberculosis: Involvement of the p38 Mitogen-Activated Protein Kinase Pathway. J Infect Dis. 2000;182:888–894. doi: 10.1086/315750. [DOI] [PubMed] [Google Scholar]
- 41.Singh S, Nannuru K, Sadanandam A, Varney M, Singh R. CXCR1 and CXCR2 enhances human melanoma tumourigenesis, growth and invasion. Br J Cancer. 2009;100:1638–1646. doi: 10.1038/sj.bjc.6605055. [DOI] [PMC free article] [PubMed] [Google Scholar]