

Abstract
There has been an escalating interest in the medicinal use of Cannabis sativa in recent years. Cannabis is often administered orally with fat-containing foods, or in lipid-based pharmaceutical preparations. However, the impact of lipids on the exposure of patients to cannabis components has not been explored. Therefore, the aim of this study is to elucidate the effect of oral co-administration of lipids on the exposure to two main active cannabinoids, Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD). In this study, oral co-administration of lipids enhanced the systemic exposure of rats to THC and CBD by 2.5-fold and 3-fold, respectively, compared to lipid-free formulations. In vitro lipolysis was conducted to explore the effect of lipids on the intestinal solubilisation of cannabinoids. More than 30% of THC and CBD were distributed into micellar fraction following lipolysis, suggesting that at least one-third of the administered dose will be available for absorption following co-administration with lipids. Both cannabinoids showed very high affinity for artificial CM-like particles, as well as for rat and human CM, suggesting high potential for intestinal lymphatic transport. Moreover, comparable affinity of cannabinoids for rat and human CM suggests that similar increased exposure effects may be expected in humans. In conclusion, co-administration of dietary lipids or pharmaceutical lipid excipients has the potential to substantially increase the exposure to orally administered cannabis and cannabis-based medicines. The increase in patient exposure to cannabinoids is of high clinical importance as it could affect the therapeutic effect, but also toxicity, of orally administered cannabis or cannabis-based medicines.
Keywords: Δ9-tetrahydrocannabinol, cannabidiol, lymphatic transport, bioavailability, medicinal cannabis, dietary lipids
Introduction
Cannabis sativa, commonly called hemp, has thousands of years-long history of medical use. Cannabis extracts were widely used in Europe and North America for their therapeutic value as sedatives, hypnotics, analgesics, muscle relaxants, and anticonvulsant agents [1-3]. However, cannabis was removed from British and American Pharmacopoeias in 20th century, partially due to politic bias [4]. Although prohibited, many patients were nevertheless self-medicating to obtain therapeutic benefits from cannabis for various conditions, including AIDS wasting syndrome, multiple sclerosis (MS) and spinal injuries [1,4]. More recently, a growing interest in the therapeutic effects of cannabis has developed following the isolation of cannabinoids, the principal chemical compounds of cannabis, as well as the discovery of endocannabinoids and their cognate receptors in humans [5-7]. These advances supported legalisation and wide-spread use of cannabis for therapeutic purposes in many countries. Currently, the use of cannabis for medicinal purposes is legalised in 23 states of the US, as well as in Canada, the Netherlands, and Israel. In addition, the legalisation is currently under consideration in some other US states, as well as in Australia and New Zealand [8]. In Canada, the number of patients enrolled in the federal cannabis for therapeutic purposes program (28,115, as per Dec 2012) represents fewer than 5% of the estimated total users of medical cannabis in the country [9]. In the US, it is estimated that there are currently more than one million legal medicinal cannabis users; the number of non-registered users, however, could be significantly higher (http://medicalmarijuana.procon.org/).
Cannabis is typically consumed by either smoking or oral ingestion. For many people, smoking is the preferred way of consuming medical cannabis as it allows tailoring of the dose to achieve rapid therapeutic effects [1]. However, this method of delivery is not appropriate in considerable number of patients due to the irritant effects of some components in the smoke, the difficulty of consuming cannabis in smoke-free places, and other potential risks and difficulties associated with the smoking process [10]. Oral ingestion of cannabis or cannabis-based medicines is therefore the preferred route of administration in many cases [1,11]. When patients self-medicate with cannabis, it is frequently added to cookies or cakes. The vast majority of cannabis-cooking recipes involve the use of dietary lipids (whole milk, butter, or vegetable oil) for the preparation of these cannabis-containing foods. This was attributed to the fact that therapeutically-active cannabinoids are lipid-soluble and therefore easily extracted from cannabis upon preparation with dietary fats [12]. It has also been proposed that the longer the fatty-acid chains in the lipids, the more potent cannabis-effect is expected following oral administration [12,13].
The two main natural cannabinoids, the psychoactive Δ9-tetrahydrocannabinol (THC) and the non-psychoactive cannabidiol (CBD), have been the focus of research over the last few decades for their potential multiple therapeutic effects [14]. Both cannabinoids are currently available as pharmaceutical formulations. Nabiximols (Sativex®) is a commercially available oromucosal spray that contains a mixture of THC and CBD. It is used to alleviate spasticity in MS patients [15]. Dronabinol (Marinol®), the first oral preparation of synthetic THC, is approved to treat nausea and vomiting associated with cancer chemotherapy and to enhance appetite in AIDS patients suffering from weight loss. In addition, the FDA has recently approved Epidiolex® (an oral solution of CBD) as an orphan antiepileptic drug in the treatment of Dravet syndrome in children [16]. Oral formulations of THC and CBD (Marinol® and Epidiolex®, respectively) contain sesame oil, which is mostly composed of long-chain triglycerides (LCT). It has been stated that the rationale for adding sesame oil to the formulations is to dissolve the lipid-soluble cannabinoids, THC and CBD [1,17]. Moreover, many clinical trials have also reported the use of vegetable oils as vehicles to prepare capsules containing cannabis extracts [18-21].
Thus, the available evidence suggests that the use of dietary fats and pharmaceutical lipid-based excipients is common practice in the preparation of cannabis-containing foods and cannabis-based medicinal formulations. However, despite the widespread use of lipids in cannabis formulations, to our knowledge the effect of lipid excipients on the exposure of patients to orally administered cannabis or cannabinoids has not been investigated. Therefore, the aim of this study is to elucidate the effect of oral co-administration of lipids on the exposure to the main cannabinoids, and hence on the therapeutic effect or potential toxicity of cannabis-based treatments. The possible mechanisms underlying the impact of lipids on the systemic exposure to orally administered cannabinoids have also been investigated in this work.
Materials and methods
Materials
THC (CAS: 1972-08-3) and CBD (CAS: 13956-29-1) were donated by GW Pharmaceuticals (Cambridge, UK). 4,4-Dichlorodiphenyltrichloroethane (DDT, CAS: 50-29-3), probucol (CAS: 23288-49-5), tris maleate, porcine pancreatin powder (8 × USP specifications), L-α-phosphatidylcholine, sodium hydroxide, potassium bromide (KBr), tetrahydrofuran (THF), and Intralipid® were purchased from Sigma Aldrich (Dorset, UK). Sesame oil, peanut oil, taurocholic acid salt hydrate, sodium chloride, acetonitrile (ACN), n-hexane, and water were purchased from Fisher Scientific (Leicestershire, UK). Vitamin D3 (CAS: 67-97-0) and calcium chloride were purchased from Alfa Aesar (Lancashire, UK). All reagents were of analytical grade or higher and used without further purification.
Pharmacokinetic experiments
The protocol for this study was approved by the UK Home Office in accordance with the Animals [Scientific Procedures] Act 1986. Male Sprague Dawley rats (Charles River Laboratories, UK) weighing 300-349 g were used in this study. The rats were housed in the University of Nottingham Bio Support Unit, and kept in a temperature-controlled, 12 hours light-dark cycle environment with free access to water and food.
The right external jugular vein was cannulated with a two-part catheter consisting of polyethylene (PE-50) connected to silastic tubing. Following an average recovery period of 36 hours, animals were divided into the following 6 treatment groups: IV bolus of THC or CBD at a dose of 4 mg/kg (8 mg/mL solution in propylene glycol-ethanol-sterile water (80:10:10, v/v/v)), oral gavage of THC or CBD at a dose of 12 mg/kg in lipid-free formulation (12 mg/mL solution in propylene glycol-ethanol-sterile water (80:10:10, v/v/v)), and oral gavage of THC or CBD in lipid (LCT)-based formulation at a dose of 12 mg/kg (12 mg/mL solution in sesame oil). Blood samples (0.25 mL) were then withdrawn from the cannulae at 5, 15, 30, 60, 120, 240, 360, 480, and 720 minutes after IV injections or 30, 60, 120, 180, 240, 300, 360, 480, and 720 minutes after oral administrations. Plasma was separated by centrifugation (3,000 g, 10 minutes, 15°C) and stored at -80°C until analysis. Phoenix WinNonlin 6.3 (Pharsight, Mountain View, CA, USA) software was used for pharmacokinetic analysis of the data using a non-compartmental approach.
In vitro lipolysis
The effect of LCT on intestinal processing of lipophilic cannabinoids was assessed using an in vitro lipolysis model. This model simulates physiological lipid digestion processes in the small intestine, and is commonly used in the design and development of oral lipid-based drug delivery systems [22-26].
The in vitro lipolysis experiments used in this study were based on previously used and validated conditions [25]. The reaction vessel contained 35.5 mL of the digestion buffer (50 mM tris maleate, 150 mM sodium chloride, and 5 mM calcium chloride, pH = 6.8). Taurocholic acid salt hydrate and L-α-phosphatidylcholine were added to the buffer at concentrations of 5 and 1.25 mM, respectively, to mimic a fasting gastrointestinal state. The vessel was attached to a pH-stat titrator (T50 Graphix, Mettler Toledo Inc,) and placed in a 37°C water bath.
Cannabinoids were dissolved in LCT (sesame oil) to prepare 20 mg/mL solutions. A volume of 160 µL of freshly prepared THC or CBD solution was dispersed in the reaction vessel and mixed for 15 minutes. Lipolysis was then initiated by the addition of 3.5 mL of pancreatin extract. Sodium hydroxide solution (1 M) was used as a titrant to maintain the pH of the reaction medium at 6.8 (Electrode, DG111-SC pH).
After completion of the lipolysis process, the resulting reaction medium was ultracentrifuged at 268,350 g (SORVALL® TH-641 Rotor, Thermo Fisher Scientific, UK) for 90 minutes at 37°C. Upper lipid, middle micellar, and lower sediment fractions were separated after centrifugation and stored at -80°C until analysis.
Association of cannabinoids with artificial chylomicron-like lipid particles and natural rat and human chylomicrons
Preparation of artificial lipid particle emulsions: Intralipid® 20% was used as a source of lipid particles as previously described [27]. Intralipid® is an emulsion of lipid particles that are composed of lecithin, soybean triglycerides and glycerin. Although natural chylomicrons (CM) have more complex structure, the uptake of lipophilic compounds by artificial emulsions has been shown to provide a reasonably close estimate for the degree of association with CM before proceeding with experiments that require materials from animals or humans [27,28].
Intralipid® was diluted with phosphate-buffered saline with a density of 1.006 g/mL and pH of 7.4 to achieve a triglyceride (TG) concentration of 100 mg/dL. A TG enzymatic kit was used to assess TG concentration according to the manufacturer’s protocol (Sigma Aldrich, Dorset, UK) using a BIO-TEK FL600™ plate reader (BIO-TEK INSTRUMENTS, INC. Vermont, USA). The lipid particle emulsion was then used to assess the uptake of THC and CBD as described below in the section on uptake experiments.
Preparation of rat plasma-derived chylomicrons
The protocol for this study was approved by the Home Office in accordance with the Animals [Scientific Procedures] Act 1986. Four male Sprague Dawley rats (Charles River Laboratories, UK) weighing 275-300 g were used in this experiment. The rats were housed in the University of Nottingham Bio Support Unit, and kept in a temperature controlled, 12 hours light-dark cycle environment with free access to water and food.
CM separation from rat blood was performed as previously described [29]. Briefly, animals were fasted overnight with free access to water. Next morning, animals were administered 0.5 mL peanut oil by oral gavage. Two more doses of peanut oil (0.3 mL each) were administered 1 and 2 hours after the first administration. One hour after the last dose, animals were anesthetised with 2% isoflurane and a total blood volume of 10-12 mL was collected from the posterior vena cava of each animal. Plasma was separated from blood by centrifugation (800 g, 5 minutes, 15°C). KBr (0.57 g) was then added to 4 mL of plasma aliquots to adjust the density to 1.1 g/mL. Standard solutions with densities of 1.006, 1.019, and 1.063 g/mL were prepared and layered on top of plasma aliquots to build a density gradient in polyallomer ultracentrifuge tubes. Samples were ultracentrifuged at 268,350 g (SORVALL® TH-641 Rotor, Thermo Fisher Scientific, UK) for 35 minutes at 15°C. Following ultracentrifugation, the top 1 mL layer containing CM was collected using a glass pipette. TG concentration of CM emulsion was determined using a TG enzymatic kit (Sigma Aldrich, Dorset, UK) and BIO-TEK FL600™ plate reader (BIO-TEK INSTRUMENTS, INC. Vermont, USA). TG concentration was adjusted to 100 mg/dL by dilution with standard solution of 1.006 g/mL density. CM emulsion was kept at 4°C until uptake experiments (< 24 hours).
Preparation of human plasma-derived chylomicron emulsion
The protocol for this experiment was approved by Faculty of Medicine and Health Sciences Research Ethics Committee, Queens Medical Centre, Nottingham University Hospitals, Nottingham, UK (BT12102015 CBS SoP). An exclusion criterion was the use of any medication within one week prior to the study. Three male healthy human volunteers (25-35 years old) were recruited for this study. After 12 hours overnight fasting, participants had a high-fat breakfast. Three to four hours following the meal (expected time of peak plasma-CM level [30,31]) blood samples (30 mL) were collected in heparinised tubes (Vacutainer® Blood Collection Tubes). Plasma was separated from blood by centrifugation (800 g, 10 minutes, 15°C). CM separation was performed as described above for rat CM. The CM emulsion was kept at 4°C pending uptake experiments (< 24 hours).
Uptake experiment
The uptake of THC and CBD by artificial lipid particles emulsion, rat CM emulsion, and human CM emulsion was performed as previously described [29]. Briefly, stock solutions of THC and CBD (110.4 and 110.39 µg/mL, respectively) were prepared in propylene glycol-ethanol (99:1, v/v). A volume of 10 µL of cannabinoid stock solution was added to 2 mL of the emulsion (100 mg/dL TG content) to achieve a molar concentration of 1.75 × 10-6 M. Emulsion, spiked with a cannabinoid, was then incubated at 37°C for 1 hour with continuous mixing. Following incubation, the density of the emulsion was adjusted to 1.1 g/mL using KBr. Artificial lipid particles or natural CM were then separated by density gradient ultracentrifugation (SORVALL® TH-641 Rotor, Thermo Fisher Scientific, 268,350 g, 35 minutes, 15°C). The top 1 mL layer was collected following ultracentrifugation using a glass pipette and kept at -80°C for analysis. The cannabinoid content of this layer represents the fraction of the spiked dose associated with lipid artificial particles, rat CM, or human CM.
Analytical methods
The concentrations of THC and CBD in rat plasma, in vitro lipolysis fractions, artificial emulsion and CM samples were determined using a high performance liquid chromatography (HPLC) system (Waters Alliance 2695 separations module) equipped with photodiode array ultra-violet (UV) detector (Waters 996). Data processing was carried out using EmpowerTM 2 software.
Plasma samples were analysed for cannabinoids concentrations using a previously developed and validated method [32]. Samples from in vitro lipolysis fractions (lipid, micellar, and sediment), artificial emulsion or CM association experiments were prepared for HPLC-UV analysis by a liquid-liquid extraction method which was a slight modification of previously reported method for synthetic lipophilic cannabinoids (Table 1) [33]. Chromatographic conditions for the detection of THC and CBD in plasma, in vitro lipolysis fractions, artificial emulsion, and CM association samples are summarized in Table 1.
Table 1.
Chromatographic conditions for the detection of Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD) in rat plasma, in vitro lipolysis medium fractions, artificial lipid particles emulsion, and chylomicrons (CM) emulsion using high performance liquid chromatography (HPLC)
| Medium | Mobile phase | Stationary phase | Flow rate (mL/min) | Oven temperature (°C) | IS | Detection wavelength (nm) | |
|---|---|---|---|---|---|---|---|
| THC | Plasma | ACN and Water (62:38, v/v) | ACE C18-PFP 150 × 4.6 mm, 3 µm particle size | 1 | 55 | DDT | 220 |
| Lipolysis | Methanol and Water (90:10, v/v) | Phenomenex Luna C18(2) 150 × 2.1 mm, 5 µm | 0.2 | 43 | Vit D3 | 220 | |
| Lipid particles | Methanol and Water (90:10, v/v) | Phenomenex Luna C18(2) 150 × 2.1 mm, 5 µm | 0.2 | 50 | PB | 220 | |
| Rat-CM | Methanol and Water (90:10, v/v) | Phenomenex Luna C18(2) 150 × 2.1 mm, 5 µm | 0.2 | 50 | PB | 220 | |
| Human-CM | ACN and Water (90:10, v/v) | ACE Excel Super C18 100 × 4.6 mm, 5 µm | 0.6 | 43 | PB | 220 | |
| CBD | Plasma | ACN and Water (62:38, v/v) | ACE C18-PFP 150 × 4.6 mm, 3 µm particle size | 1 | 55 | DDT | 220 |
| Lipolysis | ACN and Water (92:08, v/v) | ACE Excel Super C18 100 × 4.6 mm, 5 µm | 0.6 | 43 | PB | 210 | |
| Lipid particles | ACN and Water (75:25, v/v) | ACE Excel Super C18 100 × 4.6 mm, 5 µm | 0.8 | 43 | DDT | 210 | |
| Rat-CM | ACN and Water (75:25, v/v) | ACE Excel Super C18 100 × 4.6 mm, 5 µm | 0.8 | 43 | DDT | 210 | |
| Human-CM | ACN and Water (75:25, v/v) | ACE Excel Super C18 100 × 4.6 mm, 5 µm | 0.8 | 43 | DDT | 210 |
IS, internal standard; DDT, 4,4-dichlorodiphenyltrichloroethane; PB, probucol.
The intra- and inter-day precision and accuracy for the detection of THC and CBD in lipolysis fractions, artificial lipid particles emulsion and CM were within acceptable limits in accordance with the FDA Guidance for Bioanalytical Method Validation (standard deviation (RSD) and relative error (RE) < 15% and within ± 15%, respectively) [34].
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Statistical differences between data sets were assessed using either one-way ANOVA or unpaired two-tailed Student’s t-test, as appropriate. A p value < 0.05 was considered to represent a significant difference.
Results
Effect of lipids on systemic exposure to orally administered cannabinoids
The plasma concentration-time profiles following oral administration of THC, the main psychoactive natural cannabinoid, in lipid-free vehicle and lipid-based formulation are presented in Figure 1. IV bolus administration was used to calculate the absolute bioavailability. The pharmacokinetic parameters derived from these concentration-time profiles are summarised in Table 2. The absolute bioavailability of THC was increased by more than 2.5-fold following oral administration in the lipid-based formulation compared to lipid-free vehicle.
Figure 1.
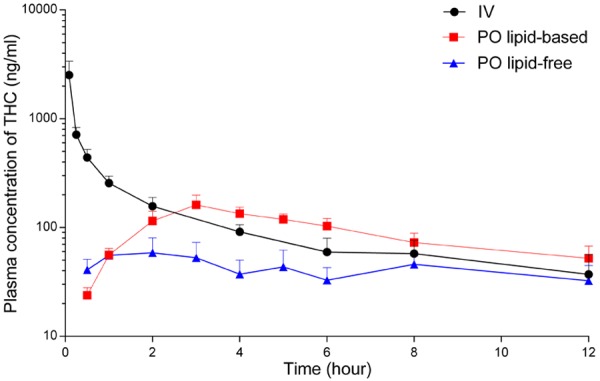
Plasma concentration-time profiles of Δ9-tetrahydrocannabinol (THC) following IV bolus (4 mg/kg, n = 5), oral lipid-free formulation (12 mg/kg, n = 6), and oral long-chain triglyceride (LCT)-based formulation (12 mg/kg, n = 5) to rats. The data are shown as mean ± SEM.
Table 2.
Pharmacokinetic parameters (mean ± SEM) derived from plasma concentration-time profiles following the administration of IV bolus (4 mg/kg), oral lipid-free formulation (12 mg/kg), and long chain triglyceride (LCT)-based formulation (12 mg/kg) of Δ9-tetrahydrocannabinol (THC) to rats
| Administration/formulation | AUC0-t (h.ng/mL) | Vd (mL/kg) | CL (mL/h/kg) | t 1/2 (h) | Cmax (ng/mL) | Tmax (h) | F (%) | n |
|---|---|---|---|---|---|---|---|---|
| IV bolus | 1624 ± 334 | 7921 ± 462 | 2671 ± 680 | 4.6 ± 2.0 | - | - | - | 5 |
| Oral lipid-free | 414 ± 130 | - | - | 6.9 ± 2.0 | 65 ± 17 | 2 | 8.5 ± 2.6 | 6 |
| Oral LCT-based | 1050 ± 169* | - | - | 7.4 ± 2.6 | 172 ± 34 | 3 | 21.5 ± 3.5* | 5 |
Unpaired t-test was used for statistical analysis.
Statistically different from oral lipid-free formulation (P < 0.05).
The plasma concentration-time profiles following the oral administration of CBD, the main non-psychoactive natural cannabinoid, in lipid-free vehicle and lipid-based formulation are presented in Figure 2. The pharmacokinetic parameters derived from these concentration-time profiles are summarised in Table 3. The absolute bioavailability of CBD was increased by almost 3-fold following oral administration in lipid-based formulation compared to lipid-free vehicle.
Figure 2.
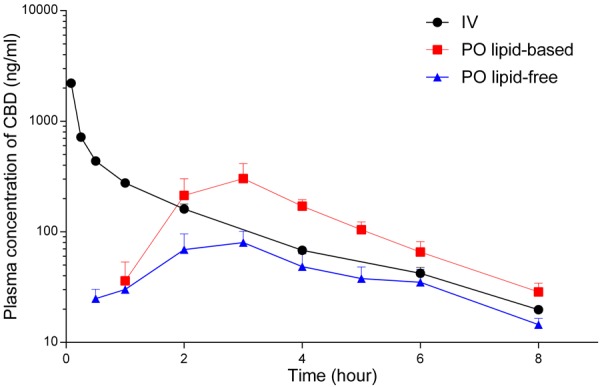
Plasma concentration-time profiles of cannabidiol (CBD) following IV bolus (4 mg/kg, n = 4), oral lipid-free formulation (12 mg/kg, n = 4), and long-chain triglyceride (LCT)-based formulation (12 mg/kg, n = 4) to rats. The data are shown as mean ± SEM.
Table 3.
Pharmacokinetic parameters (mean ± SEM) derived from plasma concentration-time profiles following the administration of IV bolus (4 mg/kg), oral lipid-free formulation (12 mg/kg), and long chain triglyceride (LCT)-based formulation (12 mg/kg) of cannabidiol (CBD) to rats
| Administration/formulation | AUC0-t (h.ng/mL) | Vd (mL/kg) | CL (mL/h/kg) | t 1/2 (h) | Cmax (ng/mL) | Tmax (h) | F (%) | n |
|---|---|---|---|---|---|---|---|---|
| IV bolus | 1380 ± 43 | 12495 ± 2607 | 2794 ± 85 | 2.0 ± 0.1 | - | - | - | 4 |
| Oral lipid-free | 327 ± 91 | - | - | 2.5 ± 0.4 | 87 ± 25 | 3 | 7.9 ± 2.2 | 4 |
| Oral LCT-based | 932 ± 188* | - | - | 1.6 ± 0.1 | 308 ± 109 | 3 | 22.3 ± 4.6* | 4 |
Unpaired t-test was used for statistical analysis.
Statistically different from oral lipid-free formulation (P < 0.05).
Intraluminal processing of cannabinoids co-administered with dietary fats or pharmaceutical lipid excipients
The intraluminal processing of cannabinoids co-administered with dietary fats or pharmaceutical lipid excipients has been assessed in this work using an in vitro lipolysis model. The results are shown in Figure 3. Upon lipolysis of sesame oil, around one-third of THC (31.2%, panel A) and CBD amounts (32.8%, Panel B) was observed to be solubilised in the micellar layer, which is the fraction readily available for absorption. The remaining approximately 70% of both compounds was distributed in the undigested lipid fraction and the sediment layer which are considered to represent the proportion of the drug not readily available for absorption following oral administration with lipids [35,36].
Figure 3.
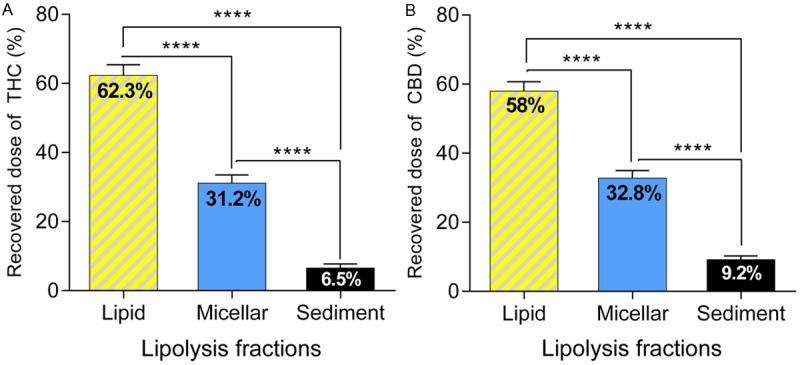
Panel A: Distribution of Δ9-tetrahydrocannabinol (THC) in the micellar, lipid, and sediment layers after lipolysis of 160 μL of the long-chain triglyceride (LCT) sesame oil containing 20 mg of THC (n = 6). Panel B: Distribution of cannabidiol (CBD) in the micellar, lipid, and sediment layers after lipolysis of 160 μL of the LCT sesame oil containing 20 mg of CBD (n = 6). The data are shown as mean ± SEM. One-way ANOVA with Tukeys post-hoc test was used for statistical analysis. ****P < 0.0001.
Intestinal lymphatic transport potential of cannabinoids
The intestinal lymphatic transport potential of THC and CBD was assessed using incubation studies with artificial lipid emulsion and with natural rat and human CM [27,29]. The results of the uptake are shown in Figure 4. The association values of both cannabinoids with artificial lipid particles and natural CM were in the range of 70-80%. No significant differences were seen between the uptake of cannabinoids by artificial lipid particles, rat CM or human CM (Figure 4).
Figure 4.
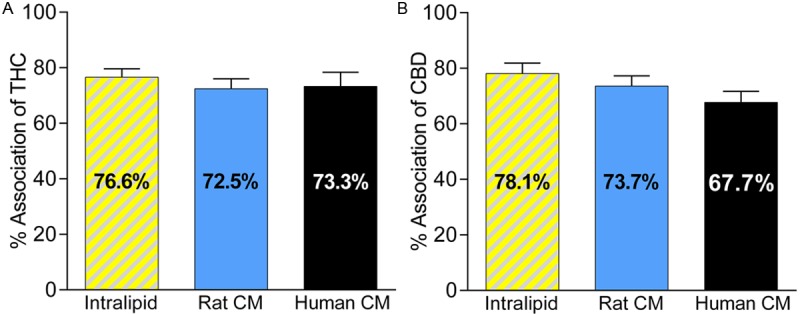
Panel A: Uptake of Δ9-tetrahydrocannabinol (THC) by lipid particles (from Intralipid®, n = 6) and plasma derived chylomicrons (CM) isolated from rats (n = 5) or humans (n = 6). Panel B: Uptake of cannabidiol (CBD) by lipid particles (n = 9) and plasma derived CM isolated from rats (n = 7) or humans (n = 5). The data are shown as mean ± SEM of % association. One-way ANOVA with Tukey’s post-hoc test was used for statistical analysis. Differences between data sets were statistically non-significant.
Discussion
Over the last few years, the medicinal use of cannabis has gained growing interest after a long period of marginalization [9]. The legalisation of medical cannabis programs has noticeably increased the access of patients to cannabis and cannabis-based medicines in many countries [8]. For many patients, orally administered cannabis and cannabis-based medicines are preferred [1,11]. Orally administered cannabis is often consumed with dietary fat-containing food (such as cookies). Lipids are also commonly used in pharmaceutical formulations of cannabis or cannabinoids. The rationale for the use of dietary fats and lipids is to enhance the extraction of the lipid-soluble active constituents [1,12,13]. However, the impact of dietary fats or pharmaceutical lipid excipients on the systemic exposure of patients to the cannabinoids has not previously been explored. This could be of particular importance when it comes to therapeutic efficacy or potential toxicity. In this study we aimed to assess the effect of lipids on the systemic exposure to the main constituents of cannabis, THC and CBD, following oral consumption of cannabis with dietary fats or oral administration of cannabis-based medicines.
Our results show that the co-administration of cannabinoids with lipids enhances the bioavailability of THC in rats by more than 2.5-fold (Figure 1 and Table 2) and of CBD by almost 3-fold (Figure 2 and Table 3). Such a profound increase in systemic exposure can significantly affect the therapeutic effects or toxicity of these cannabinoids.
To the best of our knowledge there are no previously reported studies of absolute oral bioavailability of these cannabinoids in rats. In humans, the reported bioavailabilities of THC and CBD, based on a very limited available number of studies, were less than 10% [37-39]. In our study, oral administration of THC and CBD in lipid-free formulations to rats showed similar range of bioavailability to that reported in humans (Tables 2 and 3).
To explore the mechanism(s) by which lipids could enhance the oral bioavailability of THC and CBD, we first assessed the effect of lipids on intraluminal (intestinal) solubilisation of cannabinoids using in vitro lipolysis experiments. In vitro lipolysis is a commonly used model in pharmaceutics to assess the intraluminal processing of drugs administered orally with lipid-based formulation, or following high-fat meals [26]. The results of our lipolysis experiments showed that around one-third of THC and CBD was solubilised in mixed micelles. These spherical structures are created as a result of lipid digestion process and have the ability to solubilise lipophilic compounds in aqueous medium, and thus could facilitate the diffusion and absorption of lipophilic drugs (Figure 3A and 3B, respectively). The remaining two-thirds of THC and CBD were retained within the undigested lipids and the sediment layer. This suggests that two-thirds of the administered dose of THC and CBD is not readily available for absorption when cannabinoids are administered orally in the presence of lipids. Long intestinal transit times and increased bile salt and phospholipid levels due to concomitant food intake might permit more efficient solubilisation of the drugs in vivo [35].
To assess post-luminal (inside the enterocytes) effects of lipids on the absorption of THC and CBD, we evaluated the role of the intestinal lymphatic transport in the absorption process of cannabinoids. The absorption of dietary lipids (in the form LCT) involves the formation of CM in enterocytes (Figure 5). The association of lipophilic compounds with CM in the enterocyte is a pre-requisite for their intestinal lymphatic transport. The affinity of compounds for CM ex vivo has previously been shown to be predictive for the intestinal lymphatic absorption of drugs [29]. In this study, the lymphatic transport potential was initially investigated by assessing the uptake of THC and CBD by artificial CM-like lipid particles. Both compounds showed remarkable association with lipid particles (> 76%). However, lipid particles lack the surface apoproteins found in natural CM which might affect the process of association [27]. Association experiments were also performed with natural CM isolated from rats and showed association values of 72.5 ± 3.6% and 73.7 ± 3.6% for THC (Figure 4A) and CBD (Figure 4B), respectively. Therefore, the data suggest that CM serve as carriers to transfer THC and CBD to the systemic circulation via the intestinal lymphatic system following oral administration with lipids. Drugs that are transported via the intestinal lymphatic system avoid hepatic first-pass metabolism and therefore achieve significantly higher bioavailability than after administration in lipid-free formulation (Figure 5). It has previously been suggested that THC and CBD exhibit substantial first-pass metabolism [40,41]. Indeed, higher bioavailabilites were reported after administration by routes that avoid first-pass metabolism such as inhalation of THC [37,42] and CBD [39], or rectal administration of THC [40]. Comparable results were reported previously for the synthetic lipophilic cannabinoid PRS-211,220, which had 66% association with rat CM, and showed 3-fold increase in oral bioavailability following oral administration with LCT compared to lipid-free formulation [33]. In addition, it was found in that study that about two-thirds of the absolute bioavailability of PRS-211,220 was solely due to a contribution of the intestinal lymphatic transport. These observations support our proposed mechanism of intestinal lymphatic transport as a primary mechanism underlying the enhanced exposure to THC and CBD when co-administered with LCT in rats. In order to assess if intestinal lymphatic transport might affect bioavailability of cannabinoids in humans, the uptake of THC and CBD by CM isolated from human volunteers was also assessed in our study. Association values observed in these experiments were similar to the uptake profile seen in rat CM (Figure 4). Therefore, it is reasonable to assume that similar effects of increased systemic exposure to orally administered cannabinoids when co-administered with lipids would occur in humans.
Figure 5.
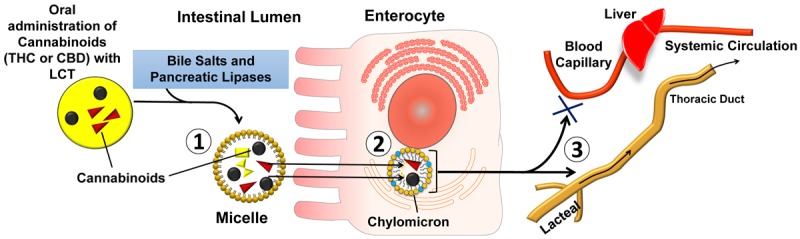
Schematic representation of the proposed mechanism for the intestinal lymphatic transport of Δ9-tetrahydrocannabinol (THC) and cannabidiol (CBD) following oral co-administration with dietary fats of long-chain triglycerides (LCT). (1) Intestinal solubilisation of THC and CBD in the mixed micelles as a result of lipid digestion process. (2) Uptake of cannabinoids by the chylomicrons (CM) inside the enterocytes. (3) Transfer of THC and CBD by CM to the systemic circulation via the intestinal lymphatic system avoiding hepatic first-pass metabolism.
It is unclear if there is a minimal volume of lipids that is required to activate intestinal lymphatic transport mechanism. Some studies show that as little as 1 g of lipid emulsion was sufficient to activate intestinal lymphatic transport of a highly lipophilic compound in dogs [43]. In contrast, it has been demonstrated that the administration of a low dose of lipids to rats (equivalent to 1 g in humans) was not sufficient to enhance intestinal lymph flow. However, a higher lipid dose (equivalent to 10 g in humans) significantly increased lymph flow [44]. These amounts of lipids can easily be obtained from the average meal in Western diet [45]. It currently remains unclear if the administration of a small-volume capsule with lipid-based formulation of cannabinoids (such as Marinol®) in fasting conditions would activate lymphatic transport and increase significantly the bioavailability of cannabinoids. Indeed, low bioavailability of cannabinoids were reported in humans after oral administration in low volumes of lipid-based formulations (0.25-0.5 mL capsules containing the drug dissolved in sesame oil) under fasting conditions [38,46]. However, our results suggest that the same lipid-based formulation-containing capsule administered with a meal, or lipid-rich cannabis-containing cookies, may result in a profound increase in systemic exposure, similar to what has been observed in this study in a rat model.
Conclusions
In conclusion, co-administration of dietary lipids or pharmaceutical lipid excipients may substantially increase the systemic exposure to orally administered cannabis or cannabis-based medicines. Our data suggest that the primary mechanism of the increased absorption of cannabinoids in the presence of lipids is intestinal lymphatic transport. The amount of lipids present in cannabis-containing foods, or following a high-fat meal, is sufficient to activate intestinal lymphatic transport and lead to increased systemic exposure to cannabinoids. The increase in systemic exposure to cannabinoids in humans is of potentially high clinical importance as it could turn a barely effective dose of orally administered cannabis into a highly effective one, or indeed a therapeutic dose into a toxic one. Therefore, it is important for cannabis-prescribing clinicians and those who self-medicate with cannabis to carefully consider the effect of the co-administration of lipids on the therapeutic outcomes of orally administered cannabis or cannabis-based medicines.
Acknowledgements
This work was supported by The University of Nottingham School of Pharmacy, the Iraqi Ministry of Higher Education and Scientific Research (MOHESR) and the Centre for Doctoral Training in Targeted Therapeutics and Formulation Sciences at the University of Nottingham (EPSRC Grant EP/L01646X). THC and CBD were generously donated by GW Pharmaceuticals.
Disclosure of conflict of interest
None.
References
- 1.Iversen LL. The Science of Marijuana. Oxford University Press; 2007. [Google Scholar]
- 2.Mikuriya TH. Marijuana in medicine: past, present and future. California Medicine. 1969;110:34. [PMC free article] [PubMed] [Google Scholar]
- 3.Zuardi AW. History of cannabis as a medicine: a review. Revista Brasileira De Psiquiatria. 2006;28:153–157. doi: 10.1590/s1516-44462006000200015. [DOI] [PubMed] [Google Scholar]
- 4.Rasmusson X. History and Policy of Clinical Cannabis versus Medical Marijuana: US History and Policy. Journal of Social Science for Policy Implications. 2014;2:15–30. [Google Scholar]
- 5.Koppel BS, Brust JC, Fife T, Bronstein J, Youssof S, Gronseth G, Gloss D. Systematic review: efficacy and safety of medical marijuana in selected neurologic disorders: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2014;82:1556–1563. doi: 10.1212/WNL.0000000000000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackie K. Cannabinoid receptors: Where they are and what they do. J Neuroendocrinol. 2008;20:10–14. doi: 10.1111/j.1365-2826.2008.01671.x. [DOI] [PubMed] [Google Scholar]
- 7.Seely KA, Prather PL, James LP, Moran JH. Marijuana-based Drugs: Innovative Therapeutics or Designer Drugs of Abuse? Mol Interv. 2011;11:36–51. doi: 10.1124/mi.11.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sznitman SR, Zolotov Y. Cannabis for therapeutic purposes and public health and safety: a systematic and critical review. Int J Drug Policy. 2015;26:20–29. doi: 10.1016/j.drugpo.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Belle-Isle L, Walsh Z, Callaway R, Lucas P, Capler R, Kay R, Holtzman S. Barriers to access for Canadians who use cannabis for therapeutic purposes. Int J Drug Policy. 2014;25:691–699. doi: 10.1016/j.drugpo.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Hall W, Solowij N. Adverse effects of cannabis. Lancet. 1998;352:1611–1616. doi: 10.1016/S0140-6736(98)05021-1. [DOI] [PubMed] [Google Scholar]
- 11.Wang T, Collet JP, Shapiro S, Ware MA. Adverse effects of medical cannabinoids: a systematic review. CMAJ. 2008;178:1669–1678. doi: 10.1503/cmaj.071178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb A. Cooking with Cannabis: The Most Effective Methods of Preparing Food and Drink with Marijuana, Hashish, and Hash Oil Third E. Ronin Publishing; 2009. [Google Scholar]
- 13.Magazine EHT, McDonough E, Remington S. The Official High Times Cannabis Cookbook: More Than 50 Irresistible Recipes That Will Get You High. Chronicle Books LLC; 2012. [Google Scholar]
- 14.Pertwee RG. Pharmacological and therapeutic targets for Δ9 tetrahydrocannabinol and cannabidiol. Euphytica. 2004;140:73–82. [Google Scholar]
- 15.Wright S, Guy G. Licensed Cannabis-Based Medicines: Benefits and Risks. Handbook of Cannabis. 2014:373. [Google Scholar]
- 16.Collins TR. Special Report: What Neurologists Are Doing About Medical Marijuana. Neurology Today. 2014;14:1–28. [Google Scholar]
- 17.Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, Miller I, Flamini R, Wilfong A, Filloux F, Wong M, Tilton N, Bruno P, Bluvstein J, Hedlund J, Kamens R, Maclean J, Nangia S, Singhal NS, Wilson CA, Patel A, Cilio MR. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurology. 2016;15:270–8. doi: 10.1016/S1474-4422(15)00379-8. [DOI] [PubMed] [Google Scholar]
- 18.Consroe P, Laguna J, Allender J, Snider S, Stern L, Sandyk R, Kennedy K, Schram K. Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacol Biochem Behav. 1991;40:701–708. doi: 10.1016/0091-3057(91)90386-g. [DOI] [PubMed] [Google Scholar]
- 19.Zajicek J, Fox P, Sanders H, Wright D, Vickery J, Nunn A, Thompson A. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): multicentre randomised placebo-controlled trial. Lancet. 2003;362:1517–1526. doi: 10.1016/S0140-6736(03)14738-1. [DOI] [PubMed] [Google Scholar]
- 20.Holdcroft A, Smith M, Jacklin A, Hodgson H, Smith B, Newton M, Evans F. Pain relief with oral cannabinoids in familial Mediterranean fever. Anaesthesia. 1997;52:483–486. doi: 10.1111/j.1365-2044.1997.139-az0132.x. [DOI] [PubMed] [Google Scholar]
- 21.Freeman RM, Adekanmi O, Waterfield MR, Waterfield AE, Wright D, Zajicek J. The effect of cannabis on urge incontinence in patients with multiple sclerosis: a multicentre, randomised placebo-controlled trial (CAMS-LUTS) Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:636–641. doi: 10.1007/s00192-006-0086-x. [DOI] [PubMed] [Google Scholar]
- 22.Sek L, Porter CJ, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54:29–41. doi: 10.1211/0022357021771896. [DOI] [PubMed] [Google Scholar]
- 23.Christensen JO, Schultz K, Mollgaard B, Kristensen HG, Mullertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23:287–296. doi: 10.1016/j.ejps.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Williams HD, Sassene P, Kleberg K, Bakala-N’Goma JC, Calderone M, Jannin V, Igonin A, Partheil A, Marchaud D, Jule E, Vertommen J, Maio M, Blundell R, Benameur H, Carriere F, Mullertz A, Porter CJ, Pouton CW. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012;101:3360–3380. doi: 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- 25.Benito-Gallo P, Franceschetto A, Wong JC, Marlow M, Zann V, Scholes P, Gershkovich P. Chain length affects pancreatic lipase activity and the extent and pH-time profile of triglyceride lipolysis. Eur J Pharm Biopharm. 2015;93:353–362. doi: 10.1016/j.ejpb.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 26.Dahan A, Hoffman A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm Res. 2006;23:2165–2174. doi: 10.1007/s11095-006-9054-x. [DOI] [PubMed] [Google Scholar]
- 27.Gershkovich P, Fanous J, Qadri B, Yacovan A, Amselem S, Hoffman A. The role of molecular physicochemical properties and apolipoproteins in association of drugs with triglyceride-rich lipoproteins: in-silico prediction of uptake by chylomicrons. J Pharm Pharmacol. 2009;61:31–39. doi: 10.1211/jpp/61.01.0005. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Qiu YT, Qi JP, Feng MQ, Ju DW, Wu W. Biomimetic reassembled chylomicrons as novel association model for the prediction of lymphatic transportation of highly lipophilic drugs via the oral route. Int J Pharm. 2015;483:69–76. doi: 10.1016/j.ijpharm.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 29.Gershkovich P, Hoffman A. Uptake of lipophilic drugs by plasma derived isolated chylomicrons: linear correlation with intestinal lymphatic bioavailability. Eur J Pharm Sci. 2005;26:394–404. doi: 10.1016/j.ejps.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Cohn JS, McNamara JR, Krasinski SD, Russell RM, Schaefer EJ. Role of triglyceride-rich lipoproteins from the liver and intestine in the etiology of postprandial peaks in plasma triglyceride concentration. Metabolism. 1989;38:484–490. doi: 10.1016/0026-0495(89)90203-5. [DOI] [PubMed] [Google Scholar]
- 31.Cohn JS, McNamara JR, Cohn SD, Ordovas JM, Schaefer EJ. Postprandial plasma lipoprotein changes in human subjects of different ages. J Lipid Res. 1988;29:469–479. [PubMed] [Google Scholar]
- 32.Zgair A, Wong JC, Sabri A, Fischer PM, Barrett DA, Constantinescu CS, Gershkovich P. Development of a simple and sensitive HPLC-UV method for the simultaneous determination of cannabidiol and Δ 9-tetrahydrocannabinol in rat plasma. J Pharm Biomed Anal. 2015;114:145–51. doi: 10.1016/j.jpba.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Gershkovich P, Qadri B, Yacovan A, Amselem S, Hoffman A. Different impacts of intestinal lymphatic transport on the oral bioavailability of structurally similar synthetic lipophilic cannabinoids: dexanabinol and PRS-211,220. Eur J Pharm Sci. 2007;31:298–305. doi: 10.1016/j.ejps.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 34.Health UDo and Services H. Guidance for industry, bioanalytical method validation. 2001. http://www.fda.gov/cder/guidance/index.htm.
- 35.Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129:1–10. doi: 10.1016/j.jconrel.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 36.Thomas N, Holm R, Rades T, Mullertz A. Characterising Lipid Lipolysis and Its Implication in Lipid-Based Formulation Development. Aaps Journal. 2012;14:860–871. doi: 10.1208/s12248-012-9398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohlsson A, Lindgren JE, Wahlen A, Agurell S, Hollister LE, Gillespie HK. Plasma delta-9 tetrahydrocannabinol concentrations and clinical effects after oral and intravenous administration and smoking. Clin Pharmacol Ther. 1980;28:409–416. doi: 10.1038/clpt.1980.181. [DOI] [PubMed] [Google Scholar]
- 38.Consroe P, Kennedy K, Schram K. Assay of plasma cannabidiol by capillary gas chromatography/ion trap mass spectroscopy following high-dose repeated daily oral administration in humans. Pharmacol Biochem Behav. 1991;40:517–522. doi: 10.1016/0091-3057(91)90357-8. [DOI] [PubMed] [Google Scholar]
- 39.Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, Hollister LE. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom. 1986;13:77–83. doi: 10.1002/bms.1200130206. [DOI] [PubMed] [Google Scholar]
- 40.Mattes RD, Shaw LM, Edling-Owens J, Engelman K, Elsohly MA. Bypassing the first-pass effect for the therapeutic use of cannabinoids. Pharmacol Biochem Behav. 1993;44:745–747. doi: 10.1016/0091-3057(93)90194-x. [DOI] [PubMed] [Google Scholar]
- 41.Siemens AJ, Walczak D, Buckley FE. Characterization of blood disappearance and tissue distribution of [3H] cannabidiol. Biochem Pharmacol. 1980;29:462–464. doi: 10.1016/0006-2952(80)90532-8. [DOI] [PubMed] [Google Scholar]
- 42.Lindgren JE, Ohlsson A, Agurell S, Hollister L, Gillespie H. Clinical effects and plasma levels of delta 9-tetrahydrocannabinol (delta 9-THC) in heavy and light users of cannabis. Psychopharmacology (Berl) 1981;74:208–212. doi: 10.1007/BF00427095. [DOI] [PubMed] [Google Scholar]
- 43.Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res. 2003;20:1460–1465. doi: 10.1023/a:1025718513246. [DOI] [PubMed] [Google Scholar]
- 44.Trevaskis NL, Charman WN, Porter CJ. Targeted drug delivery to lymphocytes: a route to site-specific immunomodulation? Mol Pharm. 2010;7:2297–2309. doi: 10.1021/mp100259a. [DOI] [PubMed] [Google Scholar]
- 45.Joint F. Fats and fatty acids in human nutrition. Karger; 2009. [Google Scholar]
- 46.Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42:327–360. doi: 10.2165/00003088-200342040-00003. [DOI] [PubMed] [Google Scholar]