

Abstract
Background: Type 2 diabetes mellitus (T2DM) increases the risk of developing Alzheimer’s disease. Most recently, GLP-1 analogs have been shown to have a significant neuroprotective role in several neurodegenerative diseases. However, few are known on its potential mechanism. Objective: In this study, we report the effect of exendin-4 (Ex-4), a GLP-1 receptor agonist, on amyloid-β(1-42) peptide oligomer-induced apoptosis in a PC12 neuronal cell model. Methods: MTT, DAPI and Annexin-V/PI assays revealed that the viability of PC12 cells decreased in a dose- and time-dependent manner after exposure to amyloid-β(1-42) oligomers. This apoptotic effect could be attenuated by Ex-4 (100-300 nM) pre-treatment, compared with the PC12 cells treated with amyloid-β(1-42) oligomers alone. Moreover, treatment with amyloid-β(1-42) oligomers (10 μM) resulted in a decrease in active- and pro-caspase-3 expression, as well as in Bcl-2 protein expression; suggesting that amyloid-β(1-42) oligomers impaired neuronal cells via the apoptosis signaling pathway. A further study of this mechanism revealed that amyloid-β oligomers (AβOs) decreased the phosphorylation of Akt and CREB. As expected, pre-treatment with Ex-4 (300 nM) increased the expression of anti-apoptotic protein Bcl-2 and reduced active caspase-3 expression levels. In addition, Ex-4 upregulated the phosphorylation levels of Akt and CREB. Conclusions: These findings indicate that GLP-1 analogue Ex-4 has a neuroprotective effect against AβO-induced PC12 cell apoptosis through reversing the impairment of the neuronal survival signaling pathway. This strongly suggests that Ex-4 is a potential therapeutic option for ameliorating AβO-induced neurotoxicity in the clinical application of Ex-4 for AD treatment, particularly when associated with diabetes.
Keywords: Exendin-4, amyloid-β(1-42) oligomers, neuronal cells, apoptosis, neuronal survival signaling
Introduction
As one of the most common endocrine and metabolic disease at present, type 2 diabetes mellitus (T2DM) has a gradually increased morbidity rate, as well as Alzheimer’s disease (AD). With the progress of T2DM, patients have experienced symptoms such as learning and memory disorders, cognitive impairment and emotional disturbance. There is a significant correlation between T2DM and cognitive impairment caused by neurodegeneration. T2DM is a risk factor of neuronal dysfunction and progressive degeneration [1]. It causes nervous system diseases, increases the risk of AD, and is considered as an independent risk factor of AD [2]. Many scholars have described AD as ‘type 3 diabetes mellitus’ (2005, Suzanne de la Monte). T2DM is connected to AD through oxidative stress, the formation of advanced glycation end products, and insulin signaling disorders in the central nervous system.
There is increasing interest in developing strategies for reducing cognitive impairment risk caused by T2DM, in addition to blood glucose control. Neuronal survival factors (NTs, IGF-IRa, etc.) → PI3K → Akt → BAD-14-3-3 → CREB → Bc1-2 is an important pathway for neuronal survival signaling, in which key components include Akt/PKB and cAMP-response element binding protein (CREB). Dysfunction of neuronal survival signaling pathway (PI-3K-Akt-CREB) occurs in neurons in T2DM patients. In the T2DM rat model, T2DM was induced by streptozotocin (STZ), and rats were given a high-calorie, high-sugar diet. Results revealed that IGF-IRa, Akt/PKB and CREB expression decreased in the hippocampus of rats [1]. Neurotrophic factors increase the expression of Bcl-2, an anti-apoptotic protein, by the activation of Akt/PKB and CREB, and inhibit neuronal apoptosis signaling through the Bcl-2 family of proteins [3]. Therefore, we assumed that neuronal apoptosis can be prevented by improving dysregulation in neuronal survival signaling.
Amyloid-β oligomers (AβOs) are small and diffusible aggregates that accumulate in the brains of individuals with AD, and are recognized as potent synaptotoxins.
Present clinical treatments for AD could improve symptoms other than preventing the development of the disease. As a diabetic drug that has continuously gained attention, glucagon-like peptide-1 (GLP-1) may be a new therapeutic target for AD. GLP-1 receptors are expressed in the pancreas, gastrointestinal tract, muscles and the cardiovascular tissues. It is also expressed in the brain of humans and rodents. Evidences from numerous studies have revealed the influence of GLP-1 in the nervous system. In some studies on neurocytology and electrophysiology, GLP-1 has been found to play an important role as an anti-amyloid; promoting long-term potentiation (LTP) and preventing LTP from amyloid injury [4-6]. In addition, researchers have reported that activating GLP-1 receptors can activate protein kinases such as PKB, PKC, PI3K, MAPK and ERK1/2. Exendin-4 (Ex-4) suppresses high glucose-induced apoptosis in PC12 cells through the PI3K pathway [7].
However, there are no definite reports on whether Ex-4 has a protective effect against amyloid-β(1-42) oligomer-induced PC12 cell apoptosis, and whether this effect is associated to the involvement of the neuronal survival pathway. Thus, we chose amyloid-β(1-42) oligomers to induce PC12 cells and establish a model of neuronal apoptosis. Amyloid-β(1-42) oligomers are of stronger toxicity, and it is easy to form cores of amyloid precipitation [8]; which is closely related to AD. On this basis, Ex-4 (a GLP-1 receptor agonist) was used to intervene in PC12 cells treated with amyloid-β(1-42) oligomers, and to study its effects and mechanism.
Experimental procedures
PC12 cells were obtained from the Shanghai Institute of Cellular Biology of Chinese Academy of Sciences. Dulbecco’s Modified Eagle’s medium (DMEM), Fetal bovine serum (FBS) and 0.25% pancreatic enzymes were purchased from Gibco (USA). Amyloid-β, exendin-4 and 3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) were obtained from Sigma (USA). Dimethyl sulfoxide (DMSO), radio immunoprecipitation assay (RIPA) and 4’, 6-diamidino-2-phenylindole (DAPI) were purchased from Beyotime Institute of Biotechnology (PRC). The following chemicals were purchased from Cell Signaling (USA): Akt antibodies, phosphor-Akt antibodies (Ser473), CREB antibodies, phosphor-CREB antibodies, Bcl-2 antibodies, and caspase-3 antibodies. All other chemicals were purchased from local sources.
Cell culture
Using flasks or culture plates, PC12 cells were cultured in DMEM medium (4,500 mg/L of D-glucose with 10% FBS) in a cell culture incubator at 37°C with 95% air and 5% CO2. The culture medium was replaced every two or three days. PC12 cells were seeded at specified densities before the experiment. On the day of the experiment, the culture medium was replaced with fresh FBS DMEM medium. Amyloid-β(1-42) oligomers were added to cell cultures at a final concentration of 0.1-10 μM. Ex-4 was added to cell cultures at a final concentration of 100-500 nM. Optimum concentrations were determined according to previous experiments and concentrations used by other investigators.
Detection of cell viability by MTT
PC12 cells (3,000 cells/well) were grown in 96-well plates. Cells were separated into three groups: (1) Blank control group, no treatment was conducted; (2) Amyloid-β(1-42) oligomer group, solely treated with amyloid-β(1-42) oligomers (10 μM) for 24 hours; (3) Amyloid-β(1-42) oligomers + exendin-4 group, treated with amyloid-β(1-42) oligomers (10 μM) for 24 hours after pre-treatment with exendin-4 (100-500 nM) for one hour. Ten μl of MTT (5 mg/ml) was added into each hole and incubated for four hours at 37°C. Next, all mediums were replaced with 100 μl of DMSO and oscillated for 10 minutes. Measurement was conducted by a microplate reader at an OD value of 490, and cell viability was calculated in different holes.
Detection of apoptosis by DAPI staining
PC12 cells (0.5×105 cells/well) were grown in 24-well plates. Cells were separated into four groups: (1) Blank control group, no treatment was conducted; (2) Exendin-4 group, treated with exendin-4 (300 nM) for 25 hours; (3) Amyloid-β(1-42) oligomer + exendin-4 group, treated with amyloid-β(1-42) oligomers (10 μM) for 24 hours after pre-treatment with exendin-4 (300 nM) for one hour; (4) Amyloid-β(1-42) oligomer group, solely treated with amyloid-β(1-42) oligomers (10 μM) for 24 hours. Next, after washing with cold phosphate-buffered saline (PBS) for three times, cells were stained with 1 μg/ml of DAPI for 15 minutes at room temperature in the dark. Then, cells were observed and counted using a fluorescence microscope (Olympus). In DAPI staining, apoptotic cells were observed to be smaller and shinier than normal cells. Apoptotic cells had small vesicles and a cleaved nucleus. Images were taken and results were saved.
Flow cytometry
PC12 cells (1×105 cells/well) were grown in 6-well plates. Cells were grouped as above. Cells were collected and incubated with Annexin-V/PI staining. After washing, cells were analyzed using a FACSCano II flow cytometer.
Western blot analysis
PC12 cells (1×105 cells/well) were grown in 6-well plates. Cells were grouped as above. Cells were lysed with RIPA buffer for 20 minutes on ice. Cells were harvested by scraping, centrifuged at 14,000 rpm for 15 minutes, and the supernatant was used for detection in western blot analysis. Equal volumes of 5× sample buffer and RIPA buffer were added to PC12 cell lysates for balancing protein. Samples were boiled for five minutes, separated on 5% stacking acrylamide gels and 10% separating acrylamide gels, and transferred onto nitrocellulose membranes. The membranes were individually incubated with anti-caspase-3, anti-active caspase-3, anti-Akt, anti-phospho-Akt, anti-CREB, anti-phospho-CREB, or anti-Bcl-2. Secondary antibodies corresponded to the respective primary antibodies. Chemiluminescence assay was performed with an ECL Western Blotting Detection Reagent according to manufacturer’s instructions. Each membrane was stripped and probed for β-actin to verify equal protein loading.
Statistical analysis
Results were expressed as mean ± standard error of the mean (SEM). Data were analyzed using one-way analysis of variance (ANOVA) for multiple comparisons. Mean comparisons between two samples were carried out by t-test. P < 0.05 was considered statistically significant. All analyses were performed using SPSS version 19.0 J for Windows.
Results
Effect of amyloid-β(1-42) oligomers on PC12 cell viability
Previous studies have demonstrated that oligomeric amyloid-β(1-42) induced neuronal cell death in vitro. In this study, PC12 cell viability was observed to gradually decrease in a dose- and time-dependent manner when exposed to amyloid-β(1-42) oligomers. Thus, amyloid-β(1-42) oligomers could inhibit PC12 cell growth (Figure 1). Cells were treated with 10 μM of amyloid-β(1-42) oligomers for 24 hours to establish the neuronal apoptosis model in PC12 cells.
Figure 1.
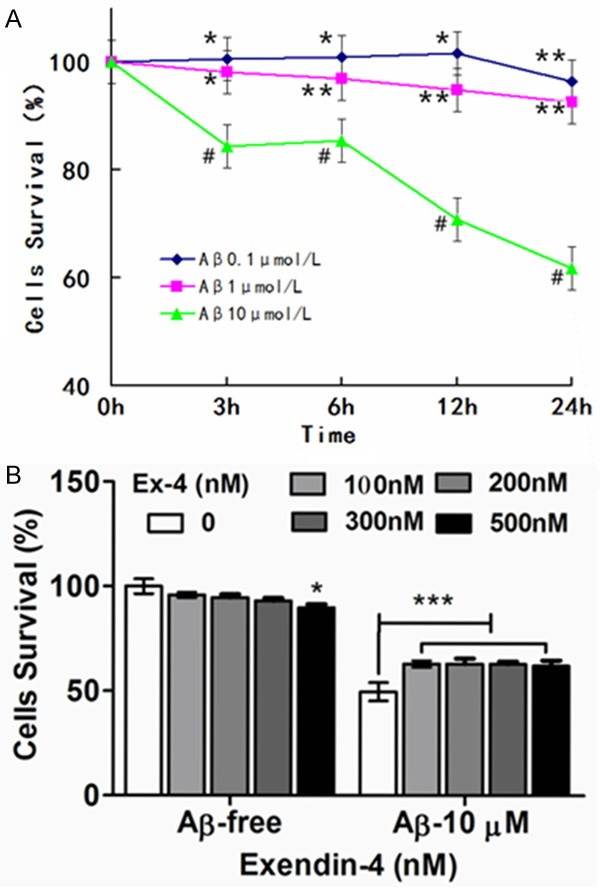
Effects of exendin-4 on the PC12 cell viability after exposure to Aβ(1-42) oligomers. A. The effect of Aβ(1-42) oligomers on the viability of PC12 cells. Cells were treated with Aβ(1-42) oligomers (0.1, 1.0, 10 μmol/L) for 3~24 h. B. Cells were pre-incubated with exendin-4 (0~500 nM) for 1 h, and then exposed to Aβ(1-42) oligomers for 24 h at 10 μmol/L. [***P < 0.001 vs Aβ(1-42) oligomers (10 μmol/L)]. Cell viability was assessed by MTT reduction assay. The values were expressed as mean ± S.E. (n = 5) of at least three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001 is compared with control group (0 μmol/L). [*P > 0.05].
Exendin-4 protects amyloid-β(1-42) oligomer-induced PC12 cell from apoptosis
Viability increased by 15% in groups pre-treated with Ex-4 (100-300 nM), compared with the group treated with amyloid-β(1-42) oligomers alone (Figure 2). Ex-4 had an obvious protective effect on neuronal apoptosis induced by amyloid-β(1-42) oligomers. Under a light microscope, cells in the control group were observed to be clear. Furthermore, cells had clearer boundaries and longer dendrites after being treated with Ex-4. In contrast, the group treated with amyloid-β(1-42) oligomers alone had low cell density, shorter synapses, and less synapse frequency. Moreover, cells were easy to gather. Therefore, cells were improved after pre-treatment with Ex-4 (Figure 3).
Figure 2.
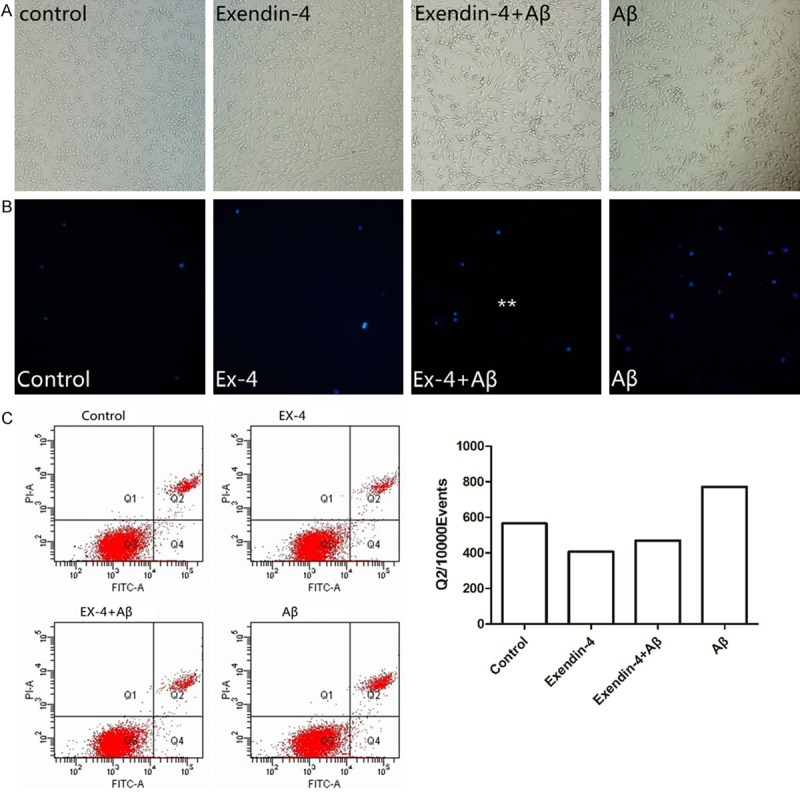
Effects of exendin-4 on PC12 cells apoptosis after exposure to Aβ(1-42) oligomers. A. Pretreatment with Ex-4 cells apoptosis in Aβ(1-42) oligomer-induced PC12 cells under optical microscope (*200). B. Cells were stained with DAPI, then evaluated by fluorescence microscope (*200). C. Effects of Ex-4 on PC12 cell apoptosis after exposure to Aβ(1-42) oligomers. Cells were stained with Annexin-V/PI, and then evaluated by flow cytometry. The values were expressed as mean ± S.E. (n = 4) of at least three independent experiments. *P < 0.05, **P < 0.01, or ***P < 0.001 is compared with control group (0 μmol/L) **P < 0.01 vs. Aβ-treated group.
Figure 3.
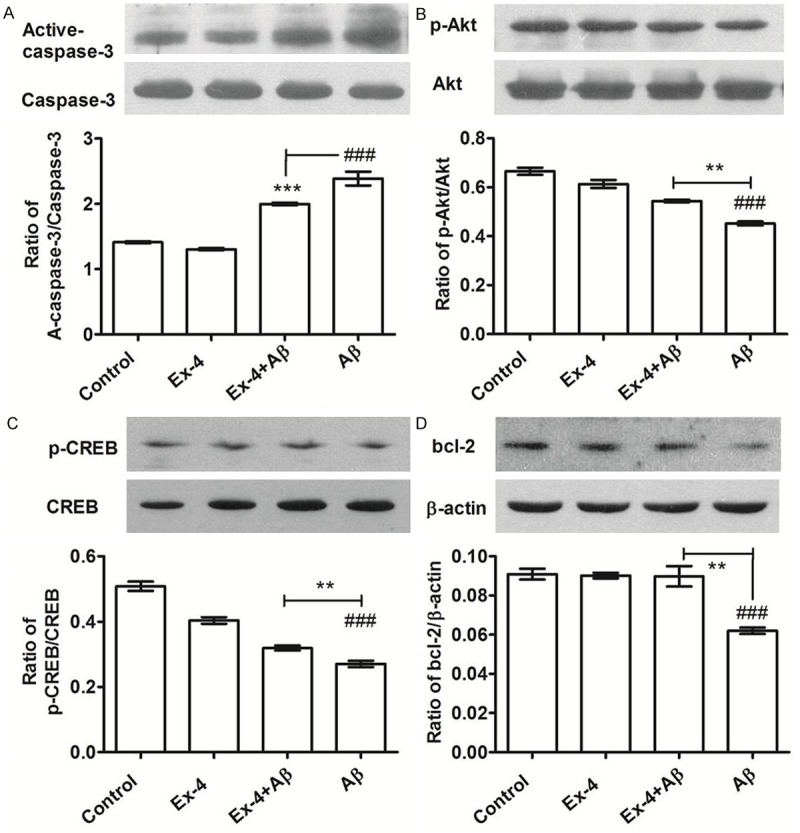
A. Western blot evaluates the effects of exendin-4 on the expression of active-caspase-3 in PC12 cells after exposure to Aβ(1-42) oligomers (10 μmol/L). x̅±s, n = 3, ***P < 0.01 vs control group, ###P < 0.01 vs Aβ group. B-D. Western blot evaluates the effects of exendin-4 on the expression of p-Akt, p-CREB and Bcl-2 in PC12 cells after exposure to Aβ(1-42) oligomers (10 μmol/L). x̅±s, n = 3, ###P < 0.01 vs control group, **P < 0.01 vs Aβ group.
In DAPI staining, apoptotic cells were smaller and shinier than normal cells, and had small vesicles and a cleaved nucleus. Compared with the amyloid-β(1-42) oligomers group, the amount of stained PC12 cell nuclei was significantly reduced after pre-treatment with Ex-4 (Figure 2B, Ex-4+Aβ group). The effect of Ex-4 on PC12 cell apoptosis was examined by AnnexinV/PI flow cytometry analysis (Figure 2C). By assessing the effect of Ex-4 on the activity of caspase-3 using western blot (Figure 3A), we found that pre-treatment with Ex-4 (300 nmol/L) on PC12 cells resulted in the reduction of expression of the active fragment of Caspase-3 (reduced by 35.3%, P < 0.01). Furthermore, it was revealed that Ex-4 has an inhibitory effect on PC12 cell apoptosis induced by amyloid-β(1-42) oligomers.
Signaling pathways involved in exendin-4
Neuronal survival signaling plays an important role in the growth, survival and apoptosis of nerve cells. PC12 cells were treated by amyloid-β(1-42) oligomers (10 μM) for 6-36 hours, and the expression levels of p-Akt, Akt, p-CREB, CREB and Bcl-2 were measured. Amyloid-β(1-42) oligomers induced the expression of p-Akt, Akt, p-CREB, CREB and Bcl-2 to decrease, as shown in Figure 4. Thus, this indicate that PC12 cell apoptosis induced by amyloid-β(1-42) oligomers may be affiliated with the inhibition of neuronal survival signaling. In further experiments, expression levels of p-Akt, Akt, p-CREB, CREB and Bcl-2 were measured after pre-treatment with Ex-4. Results in Figure 3B-D show that exendin-4 induced the expression of p-Akt, Akt, p-CREB, CREB and Bcl-2 to significantly increase; indicating that the activation of neuronal survival signaling may be one of the molecular mechanisms for protecting PC12 cells. Compared with the amyloid-β(1-42) oligomers group, PC12 cells pre-treated with Ex-4 (300 nM) enhanced the phosphorylation of Akt (by 1.20 times, P < 0.01) and CREB (by 1.10 times, P < 0.01), and induced the expression of anti-apoptotic protein Bcl-2 to increase (by 1.68 times, P < 0.01).
Figure 4.
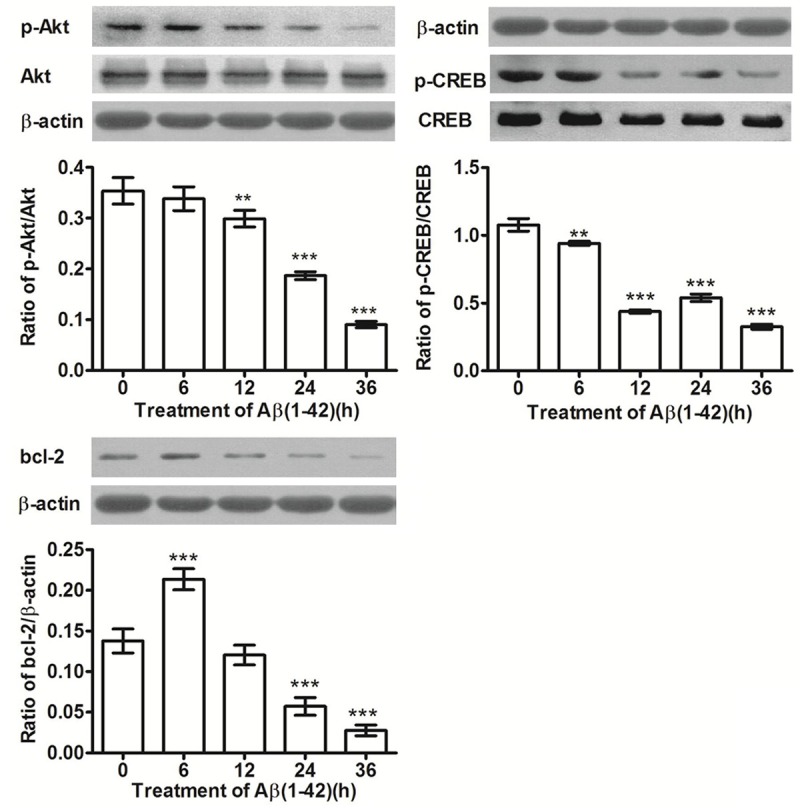
Time courses of p-Akt, p-CREB and Bcl-2 expression in PC12 cells after exposure to Aβ(1-42) oligomers (10 μmol/L). x̅±s, n = 3, **P < 0.01, ***P < 0.001, vs control (0 h).
Conclusion
During the amyloid-β incubation and aging process, a large number of soluble oligomers were formed and the toxicity of amyloid-β increased [9]. Amyloid-β(1-42) oligomers play a key role in the early stage of AD with strong neurotoxicity and ability to induce inflammatory response. Our study has proven that amyloid-β(1-42) oligomers could induce PC12 cell apoptosis. Neuronal apoptosis is an important pathological mechanism in diabetic neuropathy and neurodegeneration that occur during the development process of AD. The means to protect neurons and maintain its functions has profound significance for preventing diabetic neuropathy and the development process of AD. GLP-1 agonists and DPP-4 inhibitors could promote the synthesis and secretion of insulin, suppress glucagon secretion, promote the proliferation and differentiation of β-cells, and inhibit apoptosis. In addition, it also places many other roles.
In recent studies on GLP-1 and neuroprotection, the therapeutic action of GLP-1 agonists on diabetes-related AD has been confirmed by scholars [10,11]. Many drugs such as liraglutide and exenatide are highly focused on neuroprotective effects, and various clinical trials on these drugs are currently being conducted [12]. Studies have confirmed that liraglutide improves amyloid-β deposition, cognition, glucose uptake and cerebral blood flow in experimental animals [13]; and this result has been confirmed in APP/PS1 mice [14]. In addition, it has been found that this protective effect may be related to the activation of the cAMP signaling pathway in the brain [15]. In this study, MTT, DAPI and flow cytometry assays were performed to determine PC12 neuronal survival. Results revealed that Ex-4 improved amyloid-β(1-42) oligomer-induced PC12 neuronal state and increased survival rate. In further studies on caspase-3 activity, it was found that PC12 cells reflected some ‘resistance’ to the interference of amyloid-β(1-42) oligomers after pre-treatment with Ex-4.
A research has exhibited that Ex-4 can reduce the formation of amyloid in pancreatic islets and curb the damage caused by amyloid [16]. In the study of diabetes-related AD animal models, scholars found that, by adjusting the PI3K pathway, Ex-4 resisted the decease of PC12 cell viability induced by high glucose, and improved damage induced by oxidative stress, as well as the learning and memory ability of ICV-STZ rats [17]. Furthermore, Ex-4 improved cognitive dysfunction caused by hyperglycemia-induced chronic inflammation, reduced the occurrence of diabetic complications caused by NF-κB-related inflammatory reactions, and suppressed the development of these complications [18]. Akt participates in cell survival mainly through three ways: direct regulation of apoptosis-related protein, regulation of transcription factors, and control of the metabolic mode. The PI3K/Akt pathway could regulate cell survival and inhibit apoptosis through growth factors. Studies have found that GLP-1-based therapy could reduce the incidence of AD in the elderly population [19]. GLP-1 plays a role in promoting survival through Akt pathway [20]. Du F et al. found that the intestinal neuronal survival signaling pathway could be activated by the PI3K/Akt pathway [21]. Our experimental results also suggest that there is a certain link between GLP-1 and Akt. Ex-4 can promote and/or extend Akt, activate CREB, and increase the expression of Bcl-2 to reduce amyloid-β(1-42) oligomer-induced PC12 neuronal apoptosis.
Currently, there are reports on the neuroprotective mechanism of GLP-1 and GLP-1 receptor agonists [22,23]. The reason for neuronal apoptosis may be due to nerve cell apoptosis induced by multiple factors such as inflammation, oxidative stress and hypoxia. Therefore, GLP-1 and its receptor agonist can protect nerve cells through diverse mechanisms. In brief, Ex-4 inhibits amyloid-β(1-42) oligomer-induced PC12 cell apoptosis, and this effect may worksthrough nerve cell survival signaling pathways.
Acknowledgements
This study is supported by the Outstanding Young Persons’ Research Program for Chinese diabetes (to Li-Bin Liu), and Young Persons’ funding of Health committee of Fujian Province, China (2011-1-10) (to Yan-Ping Wang). Key clinical specialty Discipline Construction Program of Fujian and Nation, China (to Yan-Ping Wang).
Disclosure of conflict of interest
None.
References
- 1.Umegaki H. Neurodegeneration in diabetes mellitus. Adv Exp Med Biol. 2012;724:258–65. doi: 10.1007/978-1-4614-0653-2_19. [DOI] [PubMed] [Google Scholar]
- 2.Profenno LA, Porsteinsson AP, Faraone SV. Meta-Analysis of Alzheimer’s Disease Risk with Obesity, Diabetes, and Related Disorders. Biol Psychiatry. 2010;67:505–512. doi: 10.1016/j.biopsych.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- 4.Subhas CB, Jean B, Lloyd AG. Glucagon-like Pepfide-1(GLP-1) diminishes neuronal degeneration and death caused by NGF deprivation by suppressing bim induction. Neurochem Res. 2008;33:1845–1851. doi: 10.1007/s11064-008-9646-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhen XQ, Zhong WS, Jing H. Mutated recombinant human glucagon-like peptide-1 protects SH-SY5Y cells from apoptosis induced by amyloid-B peptide (1-42) Neurosci Lett. 2008;444:217–221. doi: 10.1016/j.neulet.2008.08.047. [DOI] [PubMed] [Google Scholar]
- 6.Gault VA, Hölscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol. 2008;587:112–117. doi: 10.1016/j.ejphar.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age (Dordr) 2012;34:1211–24. doi: 10.1007/s11357-011-9303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhen MG, Ruan ZG, Liu J. Metabolic Mechanism of Amyloid. Chinese Journal of Neuroantomy. 2011;27:344–348. [Google Scholar]
- 9.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Invitroaging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 10.Candeias EM, Sebastião IC, Cardoso SM, Correia SC, Carvalho CI, Plácido AI, Santos MS, Oliveira CR, Moreira PI, Duarte AI. Gut-brain connection: The neuroprotective effects of the anti-diabetic drug liraglutide. World J Diabetes. 2015;25:807–827. doi: 10.4239/wjd.v6.i6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talbot K, Wang HY. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement. 2014;10:S12–S25. doi: 10.1016/j.jalz.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hölscher C. Potential Role of Glucagon-Like Peptide-1 (GLP-1) in Neuroprotection. CNS Drugs. 2012;26:871–882. doi: 10.2165/11635890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.Egefjord L, Gejl M, Møller A, Brændgaard H, Gottrup H, Antropova O, Møller N, Poulsen HE, Gjedde A, Brock B, Rungby J. Effects of liraglutide on neurodegeneration, blood flow and cognition in Alzheimer’s disease - protocol for a controlled, randomized double-blinded trial. Dan Med. 2012;59:A4519. [PubMed] [Google Scholar]
- 14.Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C, O’Neill C. The Diabetes Drug Liraglutide Ameliorates Aberrant Insulin Receptor Localisation and Signalling in Parallel with Decreasing Both Amyloid-beta Plaque and Glial Pathology in a Mouse Model of Alzheimer’s Disease. Neuromolecular Med. 2013;15:102–14. doi: 10.1007/s12017-012-8199-5. [DOI] [PubMed] [Google Scholar]
- 15.Han WN, Hölscher C, Yuan L, Yang W, Wang XH, Wu MN, Qi JS. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging. 2013;34:576–88. doi: 10.1016/j.neurobiolaging.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Aston-Mourney K, Hull RL, Zraika S, Udayasankar J, Subramanian SL, Kahn SE. Exendin-4 increases islet amyloid deposition but offsets the resultant beta cell toxicity in human islet amyloid polypeptide transgenic mouse islets. Diabetologia. 2011;54:1756–65. doi: 10.1007/s00125-011-2143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S, Liu AR, An FM, Yao WB, Gao XD. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. Age (Dordr) 2012;34:1211–24. doi: 10.1007/s11357-011-9303-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang HJ, Chen YH, Liang KC, Jheng YS, Jhao JJ, Su MT, Lee-Chen GJ, Hsieh-Li HM. Exendin-4 protected against cognitive dysfunction in hyperglycemic mice receiving an intrahippocampal lipopolysaccharide injection. PLoS One. 2012;7:e39656. doi: 10.1371/journal.pone.0039656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Velmurugan K, Bouchard R, Mahaffey G, Pugazhenthi S. Neuroprotective actions of Glucagon-like peptide-1 in differentiated human neuroprogenitor cells. Neurochem. 2012;123:919–31. doi: 10.1111/jnc.12036. [DOI] [PubMed] [Google Scholar]
- 20.Chin PC, D’Mello SR. Survival of cultured cerebellar granule neurons can be maintained by Akt-dependent and Akt-independent signaling pathways. Brain Res Mol Brain Res. 2004;127:140–5. doi: 10.1016/j.molbrainres.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 21.Du F, Wang L, Qian W, Liu S. Loss of enteric neurons accompanied by decreased expression of GDNF and PI3K/Akt pathway in diabetic rats. Neurogastroenterol Motil. 2009;21:1229–e114. doi: 10.1111/j.1365-2982.2009.01379.x. [DOI] [PubMed] [Google Scholar]
- 22.Jolivalt CG, Fineman M, Deacon CF, Carr RD, Calcutt NA. GLP-1 signals via ERK in peripheral nerve and prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab. 2011;13:990–1000. doi: 10.1111/j.1463-1326.2011.01431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salcedo I, Tweedie D, Li Y, Greig NH. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166:1586–1599. doi: 10.1111/j.1476-5381.2012.01971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]