

Abstract
Methionine aminopeptidases (MetAPs) are metalloenzymes that cleave the N-terminal methionine from newly synthesized peptides and proteins. These MetAP enzymes are present in bacteria, and knockout experiments have shown that MetAP activity is essential for cell life, suggesting that MetAPs are good antibacterial drug targets. MetAP enzymes are also present in the human host and selectivity is essential. There have been significant structural biology efforts and over 65 protein crystal structures of bacterial MetAPs are deposited into the PDB. This review highlights the available crystallographic data for bacterial MetAPs. Structural comparison of bacterial MetAPs with human MetAPs highlights differences that can lead to selectivity. In addition, this review includes the chemical diversity of molecules that bind and inhibit the bacterial MetAP enzymes. Analysis of the structural biology and chemical space of known bacterial MetAP inhibitors leads to a greater understanding of this antibacterial target and the likely development of potential antibacterial agents.
Keywords: Antibacterial, Metalloenzyme, Methionine aminopeptidase (MetAP), Protein crystal structure
1. INTRODUCTION
Methionine aminopeptidase (MetAP) is a dinuclear metalloprotease that removes N-terminal methionine initiators from nascent proteins [1,2]. MetAPs initiate co- and post-translational modifications that are essential for the translocation, activation, regulation and degradation of proteins in eukaryotic cells [3]. Highly conserved substrate specificity results from recognition of the cleavage site by MetAP enzymes. The adjacent residue to N-terminal methionine initiators in nascent proteins indicates the cleavage site, triggering methionine removal via MetAP activity [4]. Site-directed mutagenesis found that substrates for MetAP enzymes contain one of seven amino acids bearing the smallest radii of gyration, while adjacent residues with larger side chains do not interact with MetAPs [5,6]. Additionally, the effect of downstream residues on MetAP substrate recognition was determined to be insignificant. Because amino acid degradation is controlled by the ubiquitin-dependent pathway, other proteins will be recognized by ubiquitin ligase and targeted towards polyubiquitination [7]. Interestingly, MetAPs maintain homeostasis for certain proteins, allowing methionine cleavage only under specific conditions appropriate for the substrate protein's function, preventing premature degradation.
1.1. Different Classes of Methionine Aminopeptidases
Data from partial amino acid sequences of porcine methionine aminopeptidase revealed a catalytic domain similar to that observed for rat p67, but which differs from MetAPs of various prokaryotes and yeast species [8]. However, these proteins were found to be highly conserved, all of which contain a novel protease fold termed the “pita bread” fold [9]. MetAPs originally isolated from eukaryotic sources contain a 60 residue insertion into the catalytic domain providing the distinct characteristic of these enzymes, termed type 2 (MetAP2) (Fig. 1), while MetAPs isolated from prokaryotes are differentiated as type 1 (MetAP1). Both classes of MetAP are known to be dinuclear metallohydrolases requiring divalent metals to hydrolyze substrates [10,11]. Additionally, five specific residues found to coordinate to the metals in the active site are conserved across all species [12]. Concerning natural prevalence, it was later discovered that eukaryotic organisms contain both types of MetAP, with additional N-terminal domains present in each enzyme (MetAP1b: zinc-finger; MetAP2b: polybasic and polyacidic residues) [13], while MetAPs with the absence of these N-terminal domains are denoted as MetAP1a and MetAP2a.
Fig. (1).

Comparison of MetAP1a and MetAP2b isoforms. Left: EcMetAP1a (PDB: 1MAT); Center: HsMetAP2b (PDB: 1BN5). The additional N-terminal domain indicative of type b enzymes is shown in red and the additional C-terminal domain indicative of type 2 enzymes is shown in green. Metal atoms (cobalt) are shown in blue. Right: C-alpha ribbon trace of representative members of 7 bacterial Type1a MetAP structures. Manganese ions are shown as purple spheres.
1.2. Structures of MetAP Available in the PDB
There are 111 deposited MetAP structures from 14 species available in the Protein Data Bank (PDB). The species represented span the spectrum of life and at least one coordinate set is available of a MetAP from each Kingdom of life, Bacteria, Archea and Eukarya. There are 65 structures from bacterial species, 6 structures from a single Archea species, and 40 structures from 4 Eurkaryote species (Table 1). Although there is a broad spectrum of MetAP structures from different species available, the majority of the structures are from only 3 species, E. coli (40 structures), M. tuberculosis (14 structures) and H. sapiens (35 structures). Structures containing methionine-bound or substrate/product mimic bound structures are available for additional species R. prowazekii, P. aeruginosa, S. aureus, P. furiosus, and E. cuniculi.
Table 1.
MetAP structures available in the Protein Data Bank as of Feb 2015. www.pdb.org
| Species | Type | Domain | Kingdom | Gram Stain | # PDBs |
|---|---|---|---|---|---|
| Enterococcus faecalis | 1a | Bacteria | Eubacteria | Gram-pos | 1 |
| Escherichia coli | 1a | Bacteria | Proteobacteria | Gram-neg | 40 |
| Mycobacterium abscessus | 1a | Bacteria | Actinobacteria | Acid Fast | 1 |
| Pseudomonas aeruginosa | 1a | Bacteria | Proteobacteria | Gram-neg | 2 |
| Rickettsia prowazekii | 1a | Bacteria | Proteobacteria | Gram-neg | 2 |
| Staphylococcus aureus | 1a | Bacteria | Eubacteria | Gram-pos | 3 |
| Streptococcus pneumoniae | 1a | Bacteria | Firmicutes | Gram-pos | 1 |
| Thermotoga maritima | 1a | Bacteria | Thermotogae | Gram-neg | 1 |
| Homo sapiens | 1b | Eukarya | Animalia | 17 | |
| Plasmodium falciparum | 1b | Eukarya | Chromalveolata | 1 | |
| Trypanosoma brucei brucei | 1b | Eukarya | Protista | 1 | |
| Mycobacterium tuberculosis | 1c | Bacteria | Actinobacteria | Acid Fast | 14 |
| Pyrococcus furiosus | 2a | Archae | Euryarchaeota | 6 | |
| Homo sapiens | 2b | Eukarya | Animalia | 18 | |
| Encephalitozoon cuniculi | 2c | Eukarya | Fungi | 3 |
The bacterial Typela MetAPs represent the smallest core domain of the pita bread fold composed of about 240 residues. Superposition of the 7 bacterial Typela structures shows high structural similarity with rmsd's between 0.755 and 1.36 Angstrom when compared to the E. coli structure. A noticeable outlier of bacterial type1a structure are the streptococci which contain an insert of 27 amino acids similar in position as seen for the 60 residue insert of type2 MetAPs. An additional observation of the S. pneumoniae structure was crystallization in an apparent closed or inactive conformation with a beta hairpin loop blocking the active site. See Fig. (2).
1.3. Comparison of Human Versus Bacterial Type1 MetAPs
A comparison of the bacterial type1a E. coli MetAP to the human type1b MetAP structure shows conservation of the core pita bread fold with a r.m.s.d. of 0.835 Angstrom across 249 residues (Fig. 1, right). An N-terminal extension of 60 residues of the human type1b structure wraps away from the active site of the enzyme. A similar N-terminal extension is present in other eukaryotic type1 structures as well as the M. tuberculosis type1c structure (Fig. 1, center). These N-terminal extensions sit above the active site pocket and may regulate access to the active site.
The sequence conservation between E. coli MetAP1a (EcMetAP1a) and H. sapiens MetAP1b (HsMetAP1b) is 47% identical (Fig. 2, top). Alignment of the two sequences shows strong conservation within the ligand binding pocket. However, non-conserved residues line the outside of the binding pocket and are noticeable at the bottom of binding pocket, specifically, E. coli residues Cys59, His63 and Val69 (Fig. 2, bottom left).
Fig. (2).

Conservation between human and bacterial (E.coli) Type1 MetAPs. Top: Sequence alignment showing identity (dark shading) and similarity (grey shading) between E.coli type1a and human type1b MetAP. Residues from the substrate binding site are marked with an x. Residues involved in metal binding are marked with an m [1]. Bottom Left: Surface diagram of E.coli structure (PDB: 4A6W) with bound inhibitor. Surface is shaded according to sequence identity and similarity as shown in the top image. The color scheme is as follows: green = identity, yellow = similarity, magenta = non-conserved. Bottom Center and Right: Two poses showing overlaid active site residues of HsMetAP1 (PDB: 2B3K), HsMetAP2 (PDB code: 1BN5) and EcMetAP1 (PDB: 1MAT). Each enzyme is color-coded as follows: cobalt atoms: blue spheres, HsMetAP1 (yellow), HsMetAP2 (orange), EcMetAp1 (green).
1.4. Methionine Aminopeptidase as an Antibacterial Target
The cellular reliance on MetAP enzymatic activity is well established, with inactivation of the pepM gene in S. typhimurium, the gene coding for StMetAP, resulting in cell death [14]. Additionally, the methionine aminopeptidase (map) gene of E. coli, which also codes for MetAP, was substituted for an altered map gene fragment, and cell growth was only observed in the presence of lac operon inducer isopropyl-β-thiogalactoside, again suggesting MetAP knockout results in cell death [15]. Thus, the removal of MetAP activity from single cellular organisms results in growth inhibition and implicates MetAP inhibition as an antibacterial target. Enzymatic inhibition by small organic molecules is a known, tested and utilized therapeutic method. Therefore, the inhibition of MetAP was suggested as a novel druggable target.
Similarities within the core structure of HsMetAP1, HsMetAP2 and EcMetAP1 are found when examining the overlaid structures of these three enzymes (Fig. 2, bottom center and right), demonstrating the conservation of the five residues that bind the metal cofactors [12]. Due to the high conservation of the active sites between various isoforms of these enzymes, the discovery of selective inhibitors of a single species of MetAP is paramount and provides a difficult challenge to groups researching such inhibitors. Additionally, most chemical classes capable of inhibiting these enzymes act via coordination the metal cofactors. This results in the inherent promiscuous inhibition of various enzymes by MetAP inhibitors, as metalloenzymes are extremely prevalent in nature. The discovery of potent, isoform selective inhibitors of MetAPs from various species is therefore a formidable task.
Currently, most accounts detailing classes of bacterial MetAP inhibitors failed to co-screen against human isoforms of the enzyme. If any of these classes of compounds are to be advanced beyond laboratory applications, a detailed understanding of enzymatic selectivity must be documented. Human inhibitory data are therefore shown and discussed as available within published reports.
1.5 Variability of Metals Activating MetAPs
The design of selective inhibitors of bacterial MetAPs is complicated by the lack of information regarding the identity of the native metal cofactors found within the active sites of these enzymes. MetAPs have been shown to exhibit enzymatic activity when utilizing a variety of metals as cofactors, including Co(II), Fe(II), Mn(II), Zn(II) and Ni(II) [16]. However, the native cofactors for most species of MetAP are unknown, although various groups have implicated one or more of these metallic species. For example, D'souza and Holtz have implicated Fe(II) as the native cofactor of E. coli MetAP1 by examining the metal content of the whole cells with inductively coupled plasma (ICP) emission analysis; additionally, MetAP was isolated under both aerobic and anaerobic conditions and screened for activity [17]. Because some metals are oxidative under aerobic conditions, these may be lost under typical enzymatic purification processes, further demonstrating the difficulty in cofactor determination. Finally, many protein purification methods employ the use of affinity columns and cation exchange resins, affording the possibility of MetAP activation by metallic artifacts encountered within purification processes. Therefore, many published reports detailing MetAP inhibitors screen against enzymes containing the various metal cofactors shown to afford enzymatic function. As shown in Section 2, inhibitory values are largely dependent upon the identity of the cofactors, further demonstrating the importance of native cofactor determination.
2. CLASSES OF METAP INHIBITORS
2.1. 1,2,4-Triazole Based Inhibitors
Various 1,2,4-triazole motifs have been identified as bacterial MetAP inhibitors. The compounds do not appear to be species specific inhibitors, with activity demonstrated against MetAPs from numerous bacterial strains. The 1,2,4-triazole pharmacophore is also active against human MetAP2, as demonstrated in a study at GlaxoSmithKline exploring more than 80 3-anilino-5-arylthio-1,2,4-triazole derivatives against HsMetAP2 [18]. The compounds exhibit potent activity, with binding constants (Ki) ranging from >10,000 to 0.04 nM against the Co(II) form of HsMetAP2. The same study also found preferential binding to Co(II) inclusive HsMetAP2 over the Mn(II) derivative, and discovered selectivity of HsMetAP2 binding (Ki = 0.5 nM) over HsMetAP1 (Ki = 3900 nM) when screening the original MetAP2 hit compound (5-(benzylthio)-N-phenyl-4H-1,2,4-triazol-3-amine) against both enzymes. The compounds are competitive inhibitors with bridging coordination to both metals in the enzymatic active site, as demonstrated in the crystal structure of N-(2-isopropylphenyl)-5-((thiophen-2-ylmethyl)thio)-4H-1,2,4-triazol-3-amine bound to HsMetAP2 (PDB: 2OAZ).
A similar binding mode is observed for bacterial MetAPs as demonstrated by the crystalline co-structures of triazole species (1) and (2) with M. tuberculosis MetAP (MtMetAP1) (Fig. 3) [19]. The compounds chelate to the divalent Ni(II) cofactors through the triazole 1 and 2-N atoms at distances around 2.0 Å for both crystal structures. Additionally, compound (1) exhibits parallel-displaced π -π stacking with Phe-211 at a distance of 3.4 Å, possibly exposing a secondary pocket which can be manipulated for stronger binding interactions. Similar interactions with Phe-211 are not observed for the binding of compound (2), presumably due to the inclusion of an additional Ni(II) metal within the crystalline structure. This third metal is bound to the inhibitor through the triazole 4-N (although this nitrogen should be protonated) and coordinates with His-114 through the imidazole N-atom. The coordination sphere of the third metal is filled via coordination to a chloride ion and a water molecule co-crystallized within the enzyme active site. Lu also makes note of the difference in the coordination number of the metals for each structure; the bis-Ni(II) active site metals present in both crystal structures are hexacoordinate for compound (1) and pentacoordinate for compound (2), with the difference in coordination likely arising from the inclusion of the third metal in the case of inhibition by (2) [19].
Fig. (3).

Top: Compound (1) bound to the active site of MtMetAP1c (PDB: 3IU9). Bottom: Compound (2) bound to the active site of MtMetAP1c (PDB: 3IU8). The color scheme is as follows for both figures: red spheres = nickel metals; green sphere = chlorine ion; light green compounds = triazole species; yellow dashes represent coordination to the metals from residues or the inhibitors.
Most triazole-based chemical series that have been synthesized and screened against a variety of bacterial MetAPs are of the general structure shown in Table 2. The compounds exhibit selective binding based upon the identity of the divalent metals in the active site, with the best activity observed for enzymes with Ni(II) and Co(II) cofactors, followed by Mn(II) and Fe(II) respectively. This trend is present for all of the triazole inhibitors, regardless of MetAP species reported. Compound (1) was found to exhibit the most potent inhibition across all metals and MetAP species. The source of the activity of (1) is not readily apparent when examining the crystal structures shown in Fig. (3). No interactions are observed between any residues within the active site and the chlorine atoms at positions 2 and 4 of derivative (1). With compounds (1) and (7) being the only triazole species screened containing ortho-substitution on the benzyl ring, any conclusions drawn concerning this substitution pattern are premature. However, for some bacterial strains, 4-fluoro-benzyl derivative (3) demonstrated comparable activity to that of 2,4-dichloro-benzyl derivative (1). For example, the difference in the inhibition of Co(II) bound A. baumannii MetAPx (AbMetAPx) by (1) and (3) is only 0.1 μM, while the difference for the Co(II) form of MtMetAP1c is 0.48 μM, with (1) being more active against both forms. Additionally, the inclusion of amines at the 2-position of the triazole ring appears to be beneficial, with a 2.5 fold increase in activity for compounds (2) and (3) against MtMetAP1c. Alkylated-phenyl derivatives (4)-(6) exhibit weak inhibition of B. pseudomallei MetAP1 (BpMetAP1) (IC50 = >250 – 3.1 μM, Co(II) cofactors) as compared with the other screened inhibitors suggesting that aryl alkylation in the 4-position produces detrimental effects to binding. Although p-methyl-benzyl derivitive (4) is more active than p-fluoro-benzyl derivative (3) against BpMetAP1 (IC50 = 3.1 and 7.0 μM, respectively), this trend is not observed across different bacterial strains.
Table 2.
Activity of various triazole based inhibitors against bacterial MetAPs.
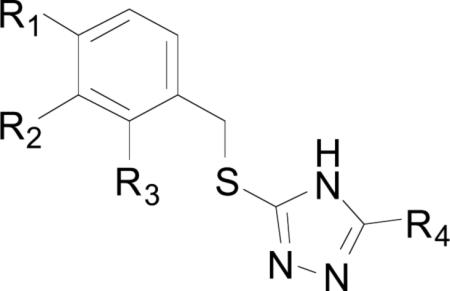
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound Number | R1 | R2 | R3 | R4 | Enzymea | Co(II)b,c | Mn(II)b,c | Fe(II)b,c | Ni(II)b,c | Refd |
| (1) | Cl | H | Cl | NH2 | MtMetAP1a | >250 | >250 | >250 | >250 | [19] |
| MtMetAP1c | 0.26 | 2.0 | 40 | 0.24 | [19,20] | |||||
| AbMetAPxe | 0.78 | 64 | 128 | 4.9 | [21] | |||||
| AbMetAPye | 0.38 | 14 | 94 | 0.40 | [21] | |||||
| BpMetAP1 | 1.0 ± 0.1 | - | - | - | [22] | |||||
| (2) | F | H | H | H | MtMetAP1a | >250 | >250 | >250 | >250 | [19] |
| MtMetAP1c | 2.0 | 143 | >500 | 0.58 | [19,20] | |||||
| (3) | F | H | H | NH2 | MtMetAP1a | >250 | >250 | >250 | >250 | [19] |
| MtMetAP1c | 0.74 | 18 | >500 | 1.3 | [19,20] | |||||
| AbMetAPx | 0.88 | 303 | >500 | 23 | [21] | |||||
| AbMetAPy | 0.64 | 34 | >500 | 2.2 | [21] | |||||
| BpMetAP1 | 7 ± 3 | - | - | - | [22] | |||||
| (4) | Me | H | H | MtMetAP1a | >250 | >250 | >250 | >250 | [19] | |
| MtMetAP1c | 0.69 | 26 | >500 | 2.5 | [19,20] | |||||
| AbMetAPx | 2.1 | 145 | >500 | 44 | [21] | |||||
| AbMetAPy | 3.8 | 14 | 94 | 0.40 | [21] | |||||
| BpMetAP1 | 3.1 ± 0.26 | - | - | - | [22] | |||||
| (5) | i-Pr | H | H | NH2 | BpMetAP1 | >250 | - | - | - | [22] |
| (6) | t-Bu | H | H | NH2 | BpMetAP1 | >250 | - | - | - | [22] |
| (7) | H | F | F | NH2 | BpMetAP1 | >250 | - | - | - | [22] |
MtMetAP1a/ MtMetAPc = Mycobacterium tuberculosis, AbMetAPx/ AbMetAPy = Acinetobacter baumannii, BpMetAP1 = Burkholderia pseudomallei
The different metal headings correspond to the various MetAP cofactors
IC50 values are expressed in units of μM
Standard deviations are shown as available.
The designation of x or y for MetAPs from A. baumannii differentiates MetAPs based upon which gene the enzymes was cloned and expressed form.
Oefner explored the effect of differing 3-substituted triazoles on the activity of S. typhimurium MetAP1 (StMetAP1) [23]. They found that compounds (8) and (9) exhibited strong binding (StMetAP1 IC50 = 0.599 and 0.044 μM, respectively; Co(II) cofactors), where the increase in activity for compound (9) is attributed to the inclusion of the 3-(methylthio)-1-propanamine substituent. The authors present crystal structures of (8) and (9) bound to the StMetAP1 active site and report a similar binding mechanism to that reported earlier in Fig. (3), with the triazole 1 and 2-N atoms bridging the Co(II) cofactors. This remains the only study to date exploring the effect of substitution at the 3-position of the triazole ring on bacterial MetAP1 activity.

The exploration of this class of compounds on bacterial MetAP inhibition is currently in its infancy, with only a few publications reporting activity, and with most screening the same compounds against various species of MetAP. The effect of chain length between the benzyl and triazole rings should be probed in the future in an attempt to promote favorable interactions with the various lipophilic side chains (such as Phe-211 for MtMetAP1c) residing within the active site of these enzymes. Additionally, the amino moiety at the 3-position of the triazole ring should be derivitized and screened against bacterial MetAPs, as demonstrated for compound (9). These compounds should also be co-screened against HsMetAP1 and HsMetAP2 to determine the selectivity for enzymatic inhibition. Although some triazole species exhibit potent activity against bacterial MetAPs, similar triazole species have demonstrated preferential binding to HsMetAP2 over HsMetAP1, possibly foreshadowing selectivity challenges for bacterial targets.
2.2. Biaryl Chelating Inhibitors
Various groups have implicated compounds consisting of biaryl ring systems capable of metal chelation as inhibitors of MetAPs from both human and bacterial sources, with the most common scaffolding including 2,2’-bipyridyl [24-28], 2-(2-pyridine)-benzimidazole [29-31] or thiabendazole [29-32] motifs. Thiabendazole (10) was crystallized with EcMetAP1 containing three Co(II) cofactors, with the inhibitor chelating to the auxiliary Co atom closest to the entrance of the active site (Fig. 4, bottom). Interestingly, this third Co(II) cofactor is not present in the crystal structure of EcMetAP1 with no bound inhibitors (PDB: 2MAT) (Fig. 4, top). A series of similar inhibitors (pyridinylpyrimidines and pyridinylquinazolines) have been crystallized with HsMetAP1, with the chelation of a third Co(II) cofactor as the mode of binding for each structure (PDB: 2G6P, 4IU6, 4HXX, 4IKR, 4IKS, 4IKT). Zhang provides an excellent discussion of these crystal structures and the source of the auxiliary Co(II) ion present in the active site [27]. The crystal structures were generated by first crystallizing the enzyme, then soaking the formed crystals in a solution containing inhibitor and cobalt(II) chloride. The inclusion of the auxiliary Co(II) cofactor, which is not suggested to be present in the native enzyme as demonstrated by the E. coli MetAP1 (EcMetAP1) crystalline structure (PDB: 2MAT) (Fig. 4, top), is therefore thought to be a result of the method of crystallization. However, these compounds exhibit structure-based activity and selectivity of HsMetAP1 over HsMetAP2, possibly revealing a binding mechanism differing from that observed for these crystal structures [27,28].
Fig. (4).

Top: EcMetAP1 with no inhibitors (Co(II) cofactors) (PDB: 2MAT). Bottom: Compound (10) bound to the active site of EcMetAP1 (PDB: 1YVM). The color scheme is as follows for both images: blue spheres = nickel metals; light green compound = thiabendazole; yellow dashes represent coordination to the metals from residues or the inhibitors.
Kishor reports a series of pyridinylpyrimidine based inhibitors of E. faecalis MetAP1a (EfMetAP1a) and MtMetAP1c, where compounds were co-screened against HsMetAP1b to elucidate selectivity for the bacterial forms of the enzyme [24]. The compounds are all of the general structure shown below, where a wide variety of substituents were employed at the positions indicated. Generally, the most active compounds utilized small donating or withdrawing groups at R1 (Me, CF3, NH2, OH), R2 (H, Cl, CN, n-pentyl, CH2-cyclopropyl, CH2CO2Et) and R3 (H, CH3, OH, SH, SMe). For example, compound (11) was determined to be the most active against EfMetAP1a (IC50 = 1.56 μM, Co(II) cofactors), while compound (12) was determined to be the most active against MtMetAP1c with (IC50 = 1.67 μM, Co(II) cofactors). However, these compounds are also active against HsMetAP1b (IC50 = 0.95 and 0.66 μM for (11) and (12), respectively, Co(II) cofactors), suggesting selectivity shortcomings.

2-(2-Pyridine)-benzimidazole scaffolds have been utilized in the design of bacterial MetAP inhibitors [29-31]. The compounds are of the general structure shown above where R1 and R2 are generally H, Alkyl, Cl, F, NH2, CN, NO2 or phenyl ketones [30,31]. Of these possible derivatives, compound (13) was the most active compound exhibiting activity better than that of the parent compound 2-(2-pyridyl)benzimidazole (R1 = R2 = H) with IC50 values of 0.162 and 0.574 μM, respectively, against EcMetAP1 with Co(II) cofactors [31]. Additionally, converting the imidazole NH to an N-benzyl derivative resulted in a slight decrease in activity (IC50 = 0.992 μM), while utilizing a 2-pyrazine instead of the 2-pyridyl biaryl system resulted in a drastic decrease in activity (2-(2-pyrazine)-benzimidazole EcMetAP1 IC50 = 4.59 μM, Co(II) cofactors). The effect of introducing fused imidazole-heterocycle rings was also explored and resulted in a notable increase in activity. For example, 4-azabenzimidazole derivative (14) exhibits a 5-fold increase in activity (EcMetAP1 IC50 = 0.105 μM, Co(II) cofactors) over the parent (2-(2-pyridyl)benzimidazole) compound [31]. For other azabenzimidazole derivatives (2-(2-pyridine)-5-azabenzimidazole, 2-(2-pyridine)-4,6-diazaben-zimidazole and 2-(2-pyridine)-4,7-diazabenzimidazole) the increase in activity is to a lesser extent, although all exhibit better activity than the benzimidazole parent compound. Concerning selectivity, compounds (13) and (14) were co-screened against HsMetAP types 1 and 2 and were found to be more potent against the bacterial enzyme than both human enzymes ((13), IC50 = 28.8 μM, HsMetAP1; (14), IC50 = 27.6 and 26.7 μM, HsMetAP 1 and 2, respectively, Co(II) cofactors) [30].

The most active biaryl chelating inhibitors of bacterial MetAPs are species bearing a thiabendazole (10) motif [29-32]. The general structure of thiabendazole derivatives is shown below, where R1 and R2 were H, Alkyl, Cl, F, NH2, CN, NO2 or phenyl ketones [31]. Unlike the similar 2-(2-pyridine)-benzimidazole derivatives, where some substitution patterns produced compounds of activity greater than that of the 2-(2-pyridine)-benzimidazole parent compound, all substitution patterns explored at R1 and R2 for thiabendazole derivatives produced species exhibiting less potent activity than thiabendazole (10) (EcMetAP1 IC50 = 0.472 μM, Co(II) cofactors). However, N-benzyl derivative (15) and N-methyl-thiabendazole both exhibitited activity comparable to that of thiabendole (EcMetAP1 IC50 = 0.461 and 0.497 μM, respectively, Co(II) cofactors) [31]. Like the 2-(2-pyridine)-benzimidazole derivatives, the introduction of fused imidazole-heterocycle rings was explored for thiabendole derivatives [31]. Interestingly, 4-azabenzimidazole derivative (16) was found to be the most active compound of this series (EcMetAP1 IC50 = 0.078 μM, Co(II) cofactors), while the related 5-azabenzimidazole derivative exhibited less potent activity than the thiabendazole parent compound (EcMetAP1 IC50 = 1.724 μM, Co(II) cofactors). Finally, compounds (15) and (16) were screened against HsMetAP types 1 and 2. Compound (15) exhibited no activity against HsMetAPs at a stock concentration of 25 μM, while compound (16) was determined to exhibit selectivity for the bacterial enzyme over HsMetAP1 (IC50 = 39.8 μM, Co(II) cofactors) [30].

2.3. Aryl Carboxylic Acid Inhibitors
Aryl carboxylic acid derivatives, usually 5-aryl-2-furoic acid or 5-aryl-2-thiophenic acids, have been demonstrated as potent inhibitors of bacterial MetAPs [19-22,33-43]. The compounds bind via chelation to one of the active site metals, as shown in the numerous crystal structures of these compounds bound to MetAPs from bacterial sources (PDB: 1XNZ, 2EVM, 2Q92, 2Q93, 2Q94, 2Q95, 2Q96 and 3IU7). For example, 5-(2-chlorophenyl)-2-furoic acid (17) was crystallized with EcMetAP1 containing Mn(II) cofactors (PDB: 1XNZ) (Fig. 5). The crystal structure clearly demonstrates (17) chelating to one of the Mn(II) cofactors, with additional binding interactions via long distance H-bonding to His-178 (2.7 Å) and offset T- shaped π-π stacking with Tyr-62. Similar modes of binding are observed for the crystal structures of other aryl-furoic acid inhibitors, with the PDB codes listed earlier.
Fig. (5).

5-(2-Chlorophenyl)-2-furoic acid (17) bound to the active site of EcMetAP1 (PDB: 1XNZ). The color scheme is as follows: purple spheres = manganese metals; light green compound = (17); yellow dashes represent coordination to the metals from residues or the inhibitors and H-bonding interactions.
The general structure of these furoic acid and thiophenic acid derivatives is shown below. This class of inhibitors has been demonstrated to bind preferentially to MetAPs containing Mn(II) cofactors over other cofactors, as demonstrated by the differences in activity for compound (17) when screened against various metalloforms of EcMetAP1 (IC50 = 0.511, 138, 141 and 116 μM; Mn(II), Co(II), Ni(II) and Fe(II), respectively) [33]. Additionally, (17) was screened against AbMetAPx and AbMetAPy, with preferential binding again observed for the Mn(II) loaded forms of the enzymes (AbMetAPx IC50 = 13, 394, 188, 358 μM; Mn(II), Co(II), Ni(II) and Fe(II), respectively; AbMetAPy IC50 = 0.92, 381, 186, 288; Mn(II), Co(II), Ni(II) and Fe(II), respectively) [21]. Compound (17) was also screened against MtMetAP1c and was found to be active against the Mn(II) loaded form of the enzyme only (MtMetAP1c IC50 = 14 μM; Mn(II); IC50 = >500 μM; Co(II), Ni(II) and Fe(II)) [40] where inhibition of BpMetAP1 with Co(II) cofactors by (17) was not observed (IC50 = >250 μM) [22].
Furoic acid derivatives appear to be more active than thiophenic acid derivatives, as demonstrated by the activity of 5-(2-chlorophenyl)thiophenic acid derivative (18) (EcMetAP1 IC50 = 3.5, 45.3, 50.0 and 50.0 μM, Mn(II), Co(II), Ni(II) and Fe(II), respectively). Concerning phenyl furoic acid derivatives, substitution at the 2-position of the phenyl substituent generally resulted in the most active compounds, with motifs usually including Cl, Me, MeO, EtO and CF3 substituents [33,34]. For example, 5-(2-trifluoromethylphenyl)-2-furoic acid (19) was found to exhibit the most potent activity against EcMetAP1 (IC50 = 0.15 μM, Mn(II) cofactors) of any test compounds [34]. Substitution at two positions of the phenyl ring increases activity, except in the case of 2,3-disubstituted derivatives, with 2,5-disubstituted motifs generally having the best increase in activity. For example, 5-(2,3-dichlorophenyl)-2-furoic acid (X = O, R1 = R2 = Cl, R3 = R4 = H), 5-(2,4-dichlorophenyl)-2-furoic acid (X = O, R1 = R3 = Cl, R2 = R4 = H) and 5-(2,5-dichlorophenyl)-2-furoic acid (X = O, R1 = R4 = Cl, R2 = R3 = H) all demonstrated superior activity to that of (17) against EcMetAP1 with Mn(II) cofactors (IC50 = 0.94, 0.44, and 0.25 μM, respectively) [34]. This trend is observed for all derivatives with these substitution patterns, regardless of the substituent identity. Additionally, derivatives with aryl substituents other than substituted phenyl rings are also potent inhibitors, as in the case of 5-(2-1H-indolyl)-2-furoic acid (20) (EcMetAP1 IC50 = 0.46 μM, Mn(II) cofactors).

The conversion of the acid chelating group to a hydroxamic acid has been shown to increase activity against EcMetAP1, with the most pronounced increases resulting for Co(II) and Fe(II) cofactors. For example, conversion of (17) to hydroxamic acid derivative (21) results in dramatic activity increases when screened against these cofactors (EcMetAP IC50 for (17) = >200, 94 and 2.9 μM; Co(II), Fe (II), Mn(II) cofactors, respectively; EcMetAP IC50 for (21) = 3.5, 1, 1.3 μM; Co(II), Fe (II), Mn(II) cofactors, respectively) [41]. Additionally, (21) was crystallized with EcMetAP1 bearing Mn(II) cofactors (PDB: 4A6W); the crystal structure demonstrates bridging interactions to both active site metals as opposed to chelation demonstrated for the crystal structure of (17) (Fig. 5). Finally, other 5-aryl-2-heterocyclic carboxylic acids (i.e. oxazole, 1,2,4-oxadiazole, 1,3,4-oxadiazole, and imidazole) were screened against EcMetAP1 and were found to exhibit comparable or worse inhibitory activity [41].
This class of inhibitors has also been screened for bacterial growth inhibition against E. coli cells with recombinant MtMetAP1c [40]. Although the in vitro activity of these compounds against bacterial MetAPs has been established, compound (17) was ineffective at inhibiting E. coli growth. Because these furoic acid derivatives have only demonstrated activity for Mn(II) forms of MetAPs, this lack of activity may suggest the native form of MtMetAP1c does not contain Mn(II) cofactors when excluding physicochemical parameter considerations [40]. Unfortunately, compounds of this class have not been screened against HsMetAPs and selectivity data is therefore not discussed.
2.4. Quinoline Inhibitors
Compounds bearing 8-hydroxy or 8-amino quinoline motifs have been demonstrated to inhibit bacterial MetAPs in vitro. As demonstrated by the crystalline structure of N-(8-quinolinyl)methanesulfonamide (22) bound to EcMetAP1 with Mn(II) cofactors (PDB: 2BB7), the compounds bind to an auxiliary metal atom imbedded within the active site via chelation from the quinoline N and sulfonamide N atoms, with hydrophobic interactions to Tyr-62, His-79 and His-178 (Fig. 6) [44]. This binding mechanism is similar to that demonstrated by the biaryl chelating inhibitors discussed previously. Again, because the crystal structure was obtained by mixing the enzyme and inhibitor in a buffered solution containing MnCl2, the inclusion of the auxiliary Mn(II) is likely the result of the method of crystallization and the compounds may act in a mechanism not observed within this crystal structure.
Fig. (6).

N-(quinolin-8-yl)methanesulfonamide (22) bound to the active site of EcMetAP1 (PDB: 2BB7). The color scheme is as follows: purple spheres = manganese metals; light green compound = (22); yellow dashes represent coordination to the metals from residues or the inhibitors and H-bonding interactions.
The structures and bacterial MetAP inhibitory activity for various quinoline based inhibitors are shown in Table 3. Species bearing sulfonamide groups at the 8-position (R1) were found to be selective for MetAPs with Co(II) or Ni(II) cofactors, and are less potent for enzymes bearing Mn(II), Fe(II) and Zn(II) metals. Generally, nitroxoline (27) derivatives (R1 = OH, R2 = H, R3 = NO2) were found to be more active than compounds bearing chloro or sulfuric acid groups at R2 (compare: (26) and (27); (28) and (29)) against BpMetAP1 bearing Co(II) cofactors. Additionally, morpholine and piperidine Mannich products of 5-chloro-8-quinolinol (28) and nitroxoline ((29) and (31)) have demonstrated activity against BpMetAP1 (IC50 = 9.0, 0.1 and 0.03 μM, respectively). However, these derivatives, specifically (29), do not exhibit superior activity to that of the parent compound nitroxoline (27). Although dimethylamino derivative (30) and piperidine derivative (31) do exhibit slightly better activity than nitroxoline (27) against BpMetAP1, the increases in activity are marginal, indicating that drastic improvements upon the activity of these derivatives will likely not be achieved with Mannich products of the 7-position on the quinoline scaffolding.
Table 3.
General structures and enzymatic inhibitory activity of quinoline inhibitors.
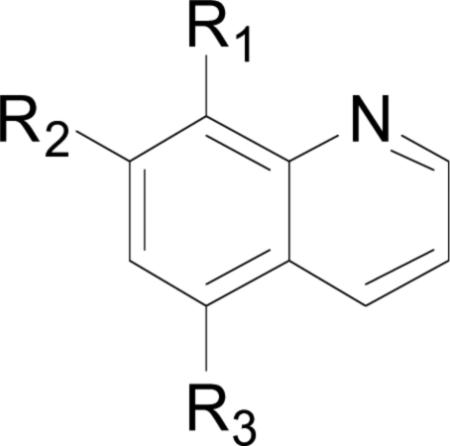
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound Number | R1 | R2 | R3 | Enzyme | Co(II)a | Mn(II)a | Fe(II)a | Ni(II)a | Zn(II)a | Refa |
| (22) |
|
H | H | EcMetAP1 | 0.137 | 2.14 | 3.74 | 0.184 | 1.11 | [44] |
| (23) |
|
H | Cl | EcMetAP1 | 0.154 | 1.64 | 3.93 | 0.171 | 0.806 | [44] |
| (24) |
|
H | H | EcMetAP1 | 1.69 | 18.5 | 13.8 | 7.42 | 11.3 | [44] |
| - | - | - | - | EcMetAP1 | 11.0 | - | - | - | - | [30] |
| - | - | - | - | SaMetAP1 | 22.9 ± 1.9 | - | - | - | - | [30] |
| - | - | - | - | HsMetAP1 | 44.9 ± 1.9 | - | - | - | - | [30] |
| - | - | - | - | HsMetAP2 | 39.8 ± 2.4 | - | - | - | [30] | |
| (25) | OH | H | H | EcMetAP1 | 0.77 | - | - | - | - | [30] |
| - | - | - | - | SaMetAP1 | 18.3 ± 1.3 | - | - | - | - | [30] |
| - | - | - | - | HsMetAP1 | >15 | - | - | - | - | [45] |
| - | - | - | - | HsMetAP2 | - | 2.03 ± 0.3 | [45] | |||
| (26) | OH | H | SO3H | BpMetAP1 | 213 | - | - | - | - | [22] |
| - | - | - | - | HsMetAP1 | >15 | - | - | - | - | [45] |
| - | - | - | - | HsMetAP2 | - | >15 | - | - | - | [45] |
| (27) | OH | H | NO2 | BpMetAP1 | 0.06 | - | - | - | - | [22] |
| - | - | - | - | HsMetAP1 | >15 | - | - | - | - | [45] |
| - | - | - | - | HsMetAP2 | - | 0.055 ± 0.02 | - | - | - | [45] |
| (28) | OH |
|
Cl | BpMetAP1 | 9 ± 6 | - | - | - | - | [22] |
| - | - | - | - | HsMetAP1 | >50 | - | - | - | - | [45] |
| - | - | - | - | HsMetAP2 | - | 2.66 ± 0.2 | - | - | - | [45] |
| (29) | OH |
|
NO2 | BpMetAP1 | 0.10 ± 0.06 | - | - | - | - | [22] |
| (30) | OH | N(CH3)2 | NO2 | BpMetAP1 | 0.04 ± 0.02 | - | - | - | - | [22] |
| (31) | OH |
|
NO2 | BpMetAP1 | 0.03 ± 0.08 | - | - | - | - | [22] |
| (32) | OH | Br | Cl | MtMetAP1a | 5.44 | - | - | - | - | [46] |
| - | - | - | - | MtMetAP1c | 4.92 | - | - | - | - | [46] |
| - | - | - | - | HsMetAP1 | 105.29 | - | - | - | - | [46] |
| - | - | - | - | HsMetAP2 | 112.2 | - | - | - | - | [46] |
| (33) | OH | I | Cl | MtMetAP1a | 9.25 | - | - | - | - | [46] |
| - | - | - | - | MtMetAP1c | 11.16 | - | - | - | - | [46] |
| - | - | - | - | HsMetAP1 | 84.70 | - | - | - | - | [46] |
| - | - | - | - | HsMetAP2 | 80.40 | - | - | - | - | [46] |
Values are expressed as IC50 with units of μM
b. Standard deviations are shown as available
Some of these compounds have shown activity against HsMetAP1 and HsMetAP2 ((24) IC50 = 44.9 and 39.8 μM, HsMetAP1 and HsMetAP2, respectively), demonstrating potential selectivity shortcomings (Table 3) [30]. Nitroxoline (27) also exhibits potent activity against HsMetAP2 (IC50 = 0.055 μM) that is comparable to the activity observed for BpMetAP1 (IC50 = 0.06 μM), providing further evidence of promiscuous MetAP inhibition. Additionally, compounds of this class have been identified as pan-assay interference compounds (PAINS) for their ability to break down under normal assay conditions to species capable of affording false hit signals [47]. The compounds have been identified as redox cyclers and protein covalent modifiers, suggesting selectivity issues against various MetAP isoforms and other host (human) proteins [47]. While some compounds were potent inhibitors of various bacterial MetAPs, this chemical class is unlikely to progress further than in vitro studies against MetAPs and inhibitory studies against bacterial colonies.
2.5. Fumagillin Derivatives
A landmark in the inhibition of HsMetAP came in 1997 with the discovery that the anti-angiogenic properties observed for fumagillin (34) were due to MetAP2 inhibition [48]. A later report details the mechanism of binding, with the epoxide covalently binding to an active site histidine (His-79) residue of EcMetAP1[49]. Additionally, the difference of a single residue in the active sites of HsMetAP1 as compared with HsMetAP2 is responsible for the selectivity of ovalicin (35), a fumagillin derivative, for the latter enzyme [50].
Concerning the inhibition of bacterial MetAPs by fumagillin and its derivatives, only the parent compound fumagillin (34) has been screened (EcMetAP1 IC50 = 9.15 μM; Co(II) cofactors) [31]. Additionally, fumagillin (34) was shown to covalently bind to HsMetAP2 [48] and a derivative (Ovalicin) exhibits selectivity between HsMetAP1 and HsMetAP2 [50]. Although this class of inhibitors has exhibited potent activity against HsMetAP2, a diverse collection of derivatives has not been screened against bacterial targets. More research is necessary to determine MetAP potency against bacterial targets.
2.6. Peptide Based Inhibitors
Peptide based inhibitors of bacterial MetAPs usually include one or both of two motifs: modified methionine moie-ties [51-53] and 3-amino-2-hydroxy amino acid (bestatin) derivatives [49,51,54-56]. Compounds of this class have been crystallized with MetAP (PDB: 2GG0, 2GG9, 2GGB, 2GG8). As demonstrated in Fig. (7), hydroxy amino acid derivatives bind to the MetAP active site via chelation to both metal centers from the primary amine, secondary alcohol and adjacent amide carbonyl to form a tridentate complex [54]. Additionally, the p-(trifluoromethyl)phenyl substituent of (36) exhibits hydrophobic interactions with Tyr62 and His-63 (not shown), maximizing binding to the catalytic domain.
Fig. (7).

Bestatin derivative (36) bound to the active site of EcMetAP1 (PDB: 2GG0). The color scheme is as follows: blue spheres = cobalt metals; light green compound = (36); yellow dashes represent coordination to the metals from residues or the inhibitors and H-bonding interactions.
Generally, compounds of this class do not exhibit binding specificity based upon the identity of the active site metals. For example, (2S,3R)-3-amino-2-hydroxyheptanoic acid (37) does not exhibit drastic changes in activity for EcMetAP1 with Co(II), Mn(II), Zn(II), or Ni(II) cofactors (IC50 = 12.3, 11.5, 9.3 and 22.3 μM, respectively) [51]. Changing the free acid to a short peptide chain, as in the case of Ala-Leu-OMe derivative (38), results in compounds with similar activity that still bear the same nonselective character as free acid (37) against the various EcMetAP1 metalloforms (IC50 = 22.9, 25.2, 16.7 and 12.5 μM; Co(II), Mn(II), Zn(II), and Ni(II), respectively) [51]. Although (38) demonstrates better inhibitory activity against EcMetAP1 with Ni(II) than free acid (37), the activities of these two derivatives are comparable for all metalloforms, suggesting binding contacts involving nonspecific hydrophobic interactions to auxiliary binding pockets play a larger role in enzymatic inhibition than chelation to the active site metals. Conversion of the n-butyl chain to p-(trifluoro)phenyl substituent, as in the case of (36), results in an increase in activity against EcMetAP1 with Co(II) cofactors (IC50 = 1.7 μM) [54] as compared with similar derivative (38).

Some peptide based inhibitors of bacterial MetAPs incorporate methionine residues for enzymatic recognition of the natural substrate. For example, pyridine derivative (39) was found to inhibit EcMetAP1 (IC50 = 2.5 μM; Co(II) cofactors) and even displayed selectivity for the bacterial iso-form over HsMetAP1 and HsMetAP2 (IC50 = 48 and 91 μM, respectively), although the source of the isoform selectivity is not discussed [53]. Additionally, conversion of the methionine sulfur to a methylene to form an n-butyl chain results in a decrease in activity against EcMetAP1 (IC50 = 59 μM; Co(II) cofactors) demonstrating the advantage of substrate recognition. Some peptide based inhibitors exhibiting activity against bacterial MetAP isoforms incorporate both methionine and amino alcohol motifs. For example, compound (40) was shown to potently inhibit EcMetAP1 (IC50 = 2.5 μM; Co(II) cofactors) while incorporating both methionine recognition and the chelating ability of amino-alcohol motifs. Interestingly, conversion of the N-terminal cysteine of (40) to a valine residue results in a slightly less active species (EcMetAP1 IC50 = 4.3 μM; Co(II) cofactors) [53].

2.7. Thiazole Containing Inhibitors
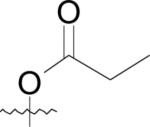
Inhibitors containing thiazole rings are generally composed of N-(2-thiazolyl)picolinamide [57-60], N-(2-thiazolyl)oxamide [36,38,42,43], or N-(2-thiazolyl)thiazole-4-carboxamide motifs [61,62]. The sole crystal structure of EcMetAP1 utilizing thiazole inhibitors contains N1-cyclopentyl-N2-(2-thiazolyl)oxalamide (41) bound to an auxiliary Co(II) atom crystallized near the active site of EcMetAP1 (PDB: 2EVO) (Fig. 8). Like similar crystal structures containing auxiliary metal atoms, (41) coordinates in a bidentate fashion, with the thiazole amide nitrogen and cyclopentyl amide carbonyl groups coordinating to the metal center. The coordination sphere of the auxiliary cobalt atom is filled by coordination to His-79 and three water atoms. Additionally, (41) exhibits π-π stacking with Tyr-65 via the thiazole ring.
Fig. (8).

Oxalamide derivative (41) bound to the active site of EcMetAP1 (PDB: 2EVO). The color scheme is as follows: blue spheres = cobalt metals; light green compound = (41); yellow sashes represent coordination to the metals from residues or the inhibitors and H-bonding interactions.
Compounds bearing a (2-thiazolyl)picolinamide scaffolding are of the general structure shown below. Components of this series contain the (2-thiazolyl)picolinamide core (42) with substitution at various positions around the pyridine ring, with the most active compounds utilizing amides or esters at the 3-position[34d, 60].
Additionally, any substitution pattern appears to enhance activity as compared with the picolinamide core (42), which was found to inhibit EcMetAP1 (IC50 = 5.0; Co(II) cofactors). For example, 3-propionamido (R1 = propionamide, R2 = R3 = R4 = H) derivative exhibits potent activity against EcMetAP1 with Co(II) cofactors (IC50 = 0.26), while similar 4-acetylamido (R2 = acetylamide, R1 = R3 = R4 = H) and 5-but-3-eneamido (R3 = but-3-eneamide, R1 = R2 = R4 = H) derivatives exhibited less potent activity [60]. Interestingly, all compounds bearing substitution at the 6-position (R4) of the pyridine ring were found to be inactive against both EcMetAP1, except in the case of 6-hydroxymethyl substitution (R1 = R2 = R3 = H, R4 = CH2OH; IC50 = 5.82 μM; Co(II) cofactors) [60].
Compounds containing ester or amide motifs at the 3-position (R1) exhibit the best activity of this class, with amides generally exhibiting superior activity than the corresponding esters. For example, phenyl ester derivative (43) exhibits weaker acitivity than the corresponding amide (44) against EcMetAP1 (IC50 = 1.22 and 0.89 μM, respectively; Co(II) cofactors) [57]. Interestingly, phenyl ester (43) is significantly more active than amide (44) against S. cerevisiae MetAP1 (IC50 = 1.03 and >100 μM, respectively; Co(II) cofactors), demonstrating the selectivity of amide derivatives for bacterial MetAPs over fungal MetAPs. Concerning phenyl esters and amides, only compounds bearing 2-substituted phenyl esters were found to be active against ScMetAP1, although the source of this activity is not discussed [57]. However, this is an important observation; the most difficult problem to overcome in the search for bacterial MetAP inhibitors is enzymatic selectivity. Because these enzymes are highly conserved across numerous lifeforms, the selective inhibition of isoforms from a single kingdom is paramount.
As stated previously, compounds bearing alkyl esters and amides at the 3-position (R1) of the pyridyl ring generally exhibit more potent activity than aryl derivatives against various MetAPs. Additionally, the incorporation of alkyl amines greatly improves MetAP inhibitory activity; for example, 3-(2-(diethylamino)acetamido derivative (45) exhibits potent activity against EcMetAP1 (IC50 = 0.049 μM; Co(II) cofactors) [59]. Various other alkyl amine derivatives (dimethyl, pyrrolidinyl, piperadinyl, and morpholino) were screened against EcMetAP1, although none were as active as (45).

Compounds of this class have been screened in bacterial whole-cell assays and the activities can be found in Table 4 [57]. While compound (42) exhibits activity against both EcMetAP1 and was shown to be active against some bacterial cell lines (S. aureus and E. coli), all ester and amide derivatives screened were not active bacterial growth inhibitors. Considering the fact that derivatives of (42) exhibit more potent activity against EcMetAP1 than parent compound (42), it is surprising to see that the compounds are essentially ineffective as antibacterial agents. The authors of the study provide two hypotheses to explain this observation:
Table 4.
Whole-cell inhibition of various bacteria by (2-thiazolyl)picolinamide derivatives.
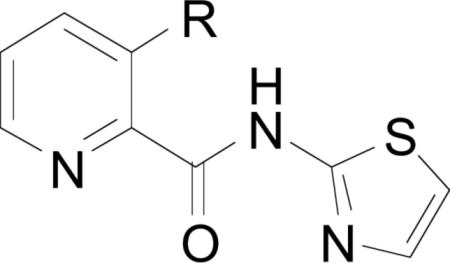
First, the compounds may not be able to penetrate cell walls as effectively as (42) and second, the physiological active site metals for MetAPs from the various bacterial species may not be cobalt. Because the physicochemical parameters of this class of compounds have not been determined and due to the fact that the natural cofactors of bacterial MetAPs are unknown, either of these hypotheses could be responsible for the observed lack of cell growth abatement for these derivatives.
Finally, compounds of this class have been screened against HsMetAPs to elucidate isoform selectivity. For example, compound (42) was found to be less active against HsMetAP1 (IC50 = 10.4 μM; Co(II) cofactors), than EcMetAP1 (Table 4) [63]. Although (42) only exhibits a two-fold increase in activity favoring the bacterial isoform, the incorporation of lipophilic amide functionalities at the 3 position of the picolinamide scaffold affords compounds inactive against HsMetAPs. When employing a benzamide at postion 3, a potent inhibitor of EcMetAP1 with selectivity over HsMetAP1 results (IC50 = 0.89 and >100 μM; Co(II) cofactors, respectively) [63].
In addition to (2-thiazolyl)picolinamide derivatives, N-(2-thiazolyl)oxamide derivatives have been shown to inhibit bacterial MetAPs. Based upon the crystal structure of EcMetAP1 containing cyclopentylamide derivative (41), the compounds inhibit the enzyme via coordination to an auxiliary Co(II) atom not found within the native enzyme. As stated previously, the incorporation of this third metal atom in the active site of MetAPs is likely the result of the method of crystallization; because metal salts are included in the crystallization mother liquor with both the enzyme and the inhibitors present, it is impossible to know if the inhibitor is chelating to the auxiliary metal atom prior to enzyme coordination.
Only three oxamide derivatives have been screened against bacterial MetAPs and are shown in Table 5. These compounds were screened against EcMetAP1 containing the various cofactors shown in the table and were found to be selective for MetAPs containing Co(II) cofactors. Slight activity was observed for EcMetAP1 with Ni(II) cofactors, while the compounds are essentially inactive against both the Mn(II) and Fe(II) forms of the enzyme. Interestingly, the compounds were found to be inactive as bacterial growth inhibitors of E. coli [43]. Such observed lack of activity in the bacterial whole-cell assay is noteworthy, as the compounds clearly inhibit some metalloforms of the enzyme. However, because the native cofactor has not been assigned (although Chai suggests the native cofactor for E. coli is Fe(II) [42]), the compounds may not be active against the natural form of the enzyme. Additionally, the physicochemical parameters of these compounds have not been evaluated and any conclusions drawn regarding the observed lack of activity against E. coli in the whole-cell assay are premature. Finally, a wider test set of these compounds must be screened to determine if progression from hit to lead is feasible. To date, species bearing an oxamide scaffold have not been screened against HsMetAPs to determine isoform selectivity.
Table 5.
Oxamide Based MetAP Inhibitors.
| Compound | Co(II)a | Mn(II)a | Ni(II)a | Fe(II)a | E. coli AS19 Cell-Growth Inhibitionb | Ref.c |
|---|---|---|---|---|---|---|
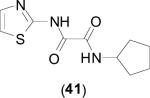
|
0.067 | 53 | 1.0 | 46 | >1000 | [38,43] |
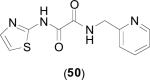
|
0.073 | 54 | 2.0 | 65 | >1000 | [38,43] |
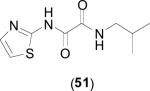
|
0.28 | 108 | 3.4 | 118 | - | [38] |
Inhibition of EcMetAP1 with indicated cofactors; expressed as IC50 (μM); Expressed as IC50 (μM); No standard deviations reported
Species bearing a N-(2-thiazolyl)thiazole-4-carboxamide core are active against EcMetAP1, although their activity has not been verified in whole-cell assays [61,62]. Derivatives of this class of inhibitors utilize functionalization at the 4-position of the central thiazole ring. They are isosteric with the (2-thiazolyl)picolinamide derivatives discussed previously and exhibit similar activity, although the selectivity for bacterial (E. coli) MetAPs over fungi (S. cerevisiae) MetAPs is not as pronounced. Generally, these compounds utilize alkyl, aryl and amide functionalization at the 4-position of the thaizole ring [64,65]. Essentially any substitution at this position results in potent inhibitors of EcMetAP1 (IC50 = <1.0 μM), while the parent compound, N-(2-thiazolyl)thiazole-4-carboxamide (R = H), was found to be less potent (IC50 = 1.97 μM, Co(II) cofactors) [62]. Because drastic changes in activity are not observed for these inhibitors, a true structure-activity relationship is difficult to deduce. However, the activity of piperidine derivatives (52) – (54) appears to follow a logical SAR, with activity increasing with decreasing length between the piperidine and thiazole rings. For example, amide (52) was found to be a potent inhibitor of EcMetAP1 (IC50 = 0.14 μM, Co(II) cofactors) [62], while removal of the amide N and carbonyl to afford (53) results in a 3-fold increase in activity (IC50 = 0.045 μM, Co(II) cofactors) [61], and shortening of the alkyl chain by a methylene, (54), results in an additional 4-fold increase in activity (IC50 = 0.010 μM, Co(II) cofactors) [62] to afford the most potent compound of this class screened against EcMetAP1 to date. Although compound (54) is a potent inhibitor of EcMetAP1, it has also been demonstrated as a potent inhibitor of ScMetAP1 (IC50 = 0.075 μM, Co(II) cofactors), demonstrating possible selectivity issues not observed for similar (2-thiazolyl)picolinamide derivatives. Finally, compounds of this class have not been co-screened against HsMetAPs and isoform selectivity against human enzymes is therefore unknown.
2.8. Other Inhibitors
In addition to the classes of bacterial MetAP inhibitors discussed, other classes of inhibitors have been identified, such as barbiturates [66], salicylate derivatives [33,67], catechols [43,64,65,68] and bengamide derivatives [69]. In general, the barbiturate based inhibitors exhibit moderate activity against EcMetAP1 (Ki = 4 – 517 μM, Co(II) cofactors) [66], with only one compound exhibiting potent activity (Ki = 0.05 μM) and were found to be nonselective inhibitors of MetAPs, as demonstrated by comparable activity with HsMetAP1. Because the compounds are not very active against MetAPs, exhibit nonselective inhibition and only one report exists detailing their activity, we have chosen to exclude a detailed overview.

Some salicylate derivatives have demonstrated activity in a bacterial whole-cell assay when screened against E. coli AS19. The compounds contain biaryl systems between the salicylate core and thiophene rings, with the most active compounds against bacterial growth utilizing 4-(2-thiophene)-benzoic acid motifs. Unlike most other classes of MetAP inhibitors, salicylate derivatives do not exhibit metal-loform selectivity in the inhibition of MetAPs. For example, compound (55) was shown to inhibit E. coli AS19 growth (IC50 = 34.1 μM; MIC = 28 mg/L) [37], although potent inhibition of EcMetAP1 is not observed (IC50 = 87.7, 62.7 and 73.0; μM; Co(II), Mn(II) and Fe(II), respectively). (55) was the most potent compound screened against E. coli AS19 growth, with all salicylate derivatives exhibiting similar activity (IC50 = 48.5 - >1000 μM; MIC = 30 - >248 mg/L). Species bearing various substitution patterns were screened for both enzymatic and antibacterial activity, although the activities were generally too close to determine SAR effects. However, the authors were able to show the 2-hydroxybenzoic acid substitution pattern is necessary for potent activity, which would facilitate metal chelation.
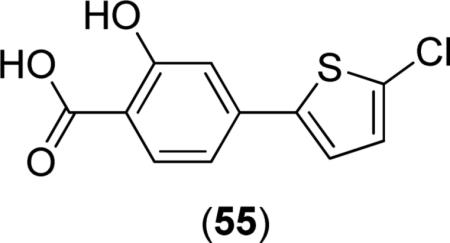
Catechol based inhibitors have been implicated as possible antibacterial agents which target MetAP, as demonstrated by both their potent in vitro enzymatic activity and antibacterial activity [43,64,65,68]. Generally, this class of inhibitors consists of catechol – heterocycle biaryl systems, with the most potent heterocycles usually consisting of thiophene, furan or thiazole rings, and pyridine, pyrimidine, 1,2,3-triazole, imidazole or oxazole rings usually producing less active species. Thiophene derivative (56) has been crystallized with EcMetAP1 and was found to exhibit the expected binding mode, with the catechol moiety chelating one of the active site metals (PDB: 3D27) (Fig. 9). Like the similar aryl carboxylic acid derivatives, the thiophene ring exhibits T-shaped π- π stacking interactions with Tyr-62.
Fig. (9).

Catechol derivative (56) bound to the active site of EcMetAP1 (PDB: 3D27). The color scheme is as follows: blue spheres = cobalt metals; light green compound = (56); yellow dashes represent coordination to the metals from residues or the inhibitors and H-bonding interactions.
The general structure of catechol based inhibitors of bacterial MetAPs is shown in Table 6. Compounds of this class of inhibitor exhibit selectivity for MetAPs with Fe(II) cofactors and have been screened against various bacterial species to determine antibacterial activity. Species utilizing heterocycles other than thiophene have been excluded from the table as they generally exhibit less activity against both the enzyme in vitro and against cell growth inhibition, with the exception of furan derivatives containing alkyl amides, as in the case of compound (62). Generally, 3-catechol thiophene derivatives are more active than 2-catechol thiophenes against both EcMetAP1 and the various bacterial strains, as demonstrated by the difference in activity for compounds (57) and (58). Additionally, functionalization at any position of the thiophene ring results in an increase in antibacterial activity; for example, compounds (59) – (61) exhibit activity comparable or better than parent compound (57) against the various bacterial strains. However, these compounds generally do not exhibit more potent activity than (57) in the enzymatic assay, suggesting a mechanism of action other than MetAP inhibition when excluding cell-permeability considerations.
Table 6.
Catechol based inhibitors.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| R | Co(II)a | Mn(II)a | Fe(II)a | E. coli AS19b | E. coli D22b | B. subtilis b | B. megaterium b | Refc |
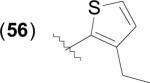
|
62.6 | 55.7 | 12.9 | 15.4 (5) | 11.8 (6) | 21.8 (7) | 27.8 (11) | [43,64] |
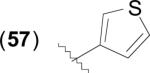
|
40.4 | 27.5 | 3.8 | 29.9 (15) | 2.0 (2) | 15.7 (9) | 19.2 (10) | [43,64] |
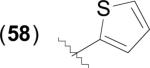
|
23.7 | 15.7 | 5.1 | 32.8 | 4.0 (3) | 16.1 (7) | 21.2 (9) | [43,64] |

|
>100 | >100 | 7.4 | 18.5 (9) | 3.7 (4) | 16.5 (5) | 16.0 (5) | [43,64] |
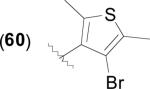
|
2.4 | 2.1 | 3.1 | 10.8 (5) | 7.7 (9) | 12.8 (8) | 17.5 (10) | [43,64] |
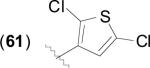
|
62.4 | 48.6 | 20.2 | 6.1 (3) | 7.3 (2) | 6.0 (2) | 5.0 (1) | [43,64] |
|
|
30.1 | 11.8 | 0.9 | 23.4 (19) | - | - | - | [65] |
Inhibition of EcMetAP1 with various cofactors (IC50 = μM)
Inhibition of bacterial growth (IC50 = μM); Numbers in parenthesis are MIC (μg/mL)
No standard deviations reported.
Although these compounds initially appear to be selective inhibitors of bacterial MetAPs, they have been implicated as pan-assay interference compounds [47]. Species bearing catechol rings are known promiscuous inhibitors of metalloenzymes and often exhibit false positive responses in assays. Therefore, any future studies utilizing this class of inhibitors should be diligent in their investigations and rigorously question the result.
Finally, bengamide derivatives have demonstrated activity against MtMetAP1a and MtMetAP1c and have been crystallized with MtMetAP1 (PDB: 3PKC, 3PKD, 3PKE) [69,70]. The general structure of these inhibitors is shown below and the compounds chelate both active site metals via the 1,2,3-triol chain. Other enzyme-inhibitor interactions involve numerous H-bonding and hydrophobic coordination. As demonstrated by compounds (64) and (65), derivatives of this class of inhibitors utilize the same side chain as the natural product (63) with functionalization involving the conversion of the caprolactam ring to other lipophilic ring systems.

Although these compounds exhibit potent activity against MetAP in enzymatic assays ((64): MtMetAP1c IC50 = 39, 0.40, 150 and 61 μM; Co(II), Mn(II), Ni(II) and Fe(II) cofactors, respectively), they do not exhibit large differences in activity for the various substitution patterns, as demonstrated by the differences in activity for compounds (64) and (65) (MtMetAP1c IC50 = 52, 0.2, >250 and 68 μM; Co(II), Mn(II), Ni(II) and Fe(II) cofactors, respectively) [69]. Additionally, they are essentially inactive against M. tuberculosis in a microplate Alamar Blue (MABA) cellular growth inhibition assay ((64) % inhibition at 128 μM = 15%; (65) % inhibition at 128 μM = 48%) [69]. Therefore, more research must be conducted to maximize activity if these compounds are to progress from hit identification to lead refinement.
CONCLUSION
MetAPs have been implicated as novel targets for antibacterial applications. Although many compound classes have been screened against MetAPs, a class demonstrating potent, selective inhibition of these enzymes while also exhibiting potent antibacterial activity has not been discovered. Therefore, the study of bacterial MetAP inhibitors is in its infancy, with only a few classes of inhibitors having been confirmed. Furthermore, many crystal structures of MetAPs with bound inhibitors include an auxiliary metal atom not present in the native enzyme. Structures such as these do not accurately describe the catalytic environment of active enzymes and are thus misleading. Future efforts should therefore utilize structures free of such auxiliary metals for inhibitor design.
Additionally, all species currently being explored as bacterial MetAP inhibitors act as metal chelating agents, effectively blocking substrates from the active site. This leads to promiscuous metalloenzyme inhibition and non-selective behavior. Future classes of inhibitors should focus on maximizing interactions with active site residues in addition to cofactor chelation to improve selectivity and potency. Additionally, studies focusing on the development of antibacterial MetAP inhibitors should explore isoform selectivity against HsMetAPs to further maximize binding interactions and reduce promiscuity.
ACKNOWLEDGEMENTS
This project has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No.: HHSN272201200025C.
Biography
 T.J. Hagen
T.J. Hagen
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Addlagatta A, Hu X, Liu JO, Matthews BW. Structural Basis for the Functional Differences between Type I and Type II Human Methionine Aminopeptidases. Biochemistry. 2005;44(45):14741–14749. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 2.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and Nα-acetyl transferase families. Trends Biochem. Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 3.Krishna RG, Wold F. Post-translational modification of proteins. Adv. Enzymol Relat. Areas Mol. Biol. 1993;67:265–298. doi: 10.1002/9780470123133.ch3. [DOI] [PubMed] [Google Scholar]
- 4.Sherman F, Stewart JW, Tsunasawa S. Methionine or not methionine at the beginning of a protein. BioEssay. 1985;3(1):27–31. doi: 10.1002/bies.950030108. [DOI] [PubMed] [Google Scholar]
- 5.Huang S, Elliott RC, Liu PS, Koduri RK, Weickmann JL, Lee JH, Blair LC, Bryan KM, Ghosh-Dastidar P, Einarson B, Kendall RL. Specificity of cotranslational amino-terminal processing of proteins in yeast. Biochemistry. 1987;26(25):8242–2846. doi: 10.1021/bi00399a033. [DOI] [PubMed] [Google Scholar]
- 6.Boissel JP, Kasper TJ, Bunn HF. Cotranslational amino-terminal processing of cytosolic proteins. Cell-free expression of site-directed mutants of human hemoglobin. J. Biol. Chem. 1988;263(17):8443–8449. [PubMed] [Google Scholar]
- 7.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc. Natl. Acad. Sci. 1996;93(22):12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc. Natl. Acad. Sci. 1995;92(17):7714–7718. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bazan JF, Weaver LH, Roderick SL, Huber R, Matthew BW. Sequence and structure comparison suggest that methionine aminopeptidase, prolidase, aminopeptidase P, and creatinase share a common fold. Proc. Natl. Acad. Sci. 1994;91:2473–2477. doi: 10.1073/pnas.91.7.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilcox DE. Binuclear metalloproteases. Chem. Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 11.Lowther WT, Matthews BW. Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem. Rev. 2002;102(12):4581–4608. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 12.Roderick SL, Matthew BW. Structure of cobalt-independent methionine aminopeptidase from Escherichia coli: a new type of proteolytic enzyme. Biochemistry. 1993;32(15):3907–3912. doi: 10.1021/bi00066a009. [DOI] [PubMed] [Google Scholar]
- 13.Walker KW, Arfin SM, Bradshaw RA. Methionyl aminopeptidase type 2. In: Barrett A, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Vol. 1. Elsevier Academic Press; London: 2004. pp. 917–921. [Google Scholar]
- 14.Miller CG, Kukral AM, Miller JL, Movva NR. pepM is an essential gene in Salmonella typhimurium. J. Bacteriol. 1989;171(9):5215–5217. doi: 10.1128/jb.171.9.5215-5217.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang SY, McGary EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989;171(7):4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker KW, Bradshaw RA. Yeast methionine aminopeptidase I can utilize either Zn2+ or Co2+ as a cofactor: a case of mistaken identity? Protein Sci. 1998;7(12):2684–2687. doi: 10.1002/pro.5560071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D'Souza VM, Holz RC. The Methionyl Aminopeptidase from Escherichia coli can Function as an Iron(II) Enzyme. Biochemistry. 1999;38(34):11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- 18.Marino JP, Fisher PW, Hofmann GA, Kirkpatrick RB, Janson CA, Johnson RK, Ma C, Mattern M, Meek TD, Ryan MD, Schulz C, Smith WW, Tew DG, Tomazek TA, Veber DF, Xiong WC, Yamamoto Y, Yamashita K, Yang G, Thompson SK. Highly Potent Inhibitors of Methionine Aminopeptidase-2 Based on a 1,2,4-Triazole Pharmacophore. J. Med. Chem. 2007;50(16):3777–3785. doi: 10.1021/jm061182w. [DOI] [PubMed] [Google Scholar]
- 19.Lu J-P, Ye Q-Z. Expression and characterization of Mycobacterium tuberculosis methionine aminopeptidase type 1a. Bioorg. Med. Chem. Lett. 2010;20(9):2776–2779. doi: 10.1016/j.bmcl.2010.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu J-P, Chai SC, Ye Q-Z. Catalysis and Inhibition of Mycobacterium tuberculosis Methionine Aminopeptidase. J. Med. Chem. 2009;53(3):1329–1337. doi: 10.1021/jm901624n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan H, Chai SC, Lam CK, Howard Xu H, Ye Q-Z. Two methionine aminopeptidases from Acinetobacter baumannii are functional enzymes. Bioorg. Med. Chem. Lett. 2011;21(11):3395–3398. doi: 10.1016/j.bmcl.2011.03.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wangtrakuldee P, Byrd MS, Campos CG, Henderson MW, Zhang Z, Clare M, Masoudi A, Myler PJ, Horn JR, Cotter PA, Hagen TJ. Discovery of Inhibitors of Burkholderia pseudomallei Methionine Aminopeptidase with Antibacterial Activity. ACS Med. Chem. Lett. 2013;4(8):699–703. doi: 10.1021/ml400034m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oefner C, Douangamath A, D'Arcy A, Häfeli S, Mareque D, Mac Sweeney A, Padilla J, Pierau S, Schulz H, Thormann M, Wadman S, Dale GE. The 1.15Å Crystal Structure of the Staphylococcus aureus Methionyl-aminopeptidase and Complexes with Triazole Based Inhibitors. J. Mol. Biol. 2003;33(1):13–21. doi: 10.1016/s0022-2836(03)00862-3. [DOI] [PubMed] [Google Scholar]
- 24.Kishor C, Arya T, Reddi R, Chen X, Saddanapu V, Marapaka AK, Gumpena R, Ma D, Liu JO, Addlagatta A. Identification, Biochemical and Structural Evaluation of Species-Specific Inhibitors against Type I Methionine Aminopeptidases. J. Med. Chem. 2013;56(13):5295–5305. doi: 10.1021/jm400395p. [DOI] [PubMed] [Google Scholar]
- 25.Kishor C, Gumpena R, Reddi R, Addlagatta A. Structural studies of Enterococcus faecalis methionine aminopeptidase and design of microbe specific 2,2[prime or minute] -bipyridine based inhibitors. MedChemComm. 2012;3(11):1406–1412. [Google Scholar]
- 26.Musonda CC, Whitlock GA, Witty MJ, Brun R, Kaiser M. Synthesis and evaluation of 2-pyridyl pyrimidines with in vitro antiplasmodial and antileishmanial activity. Bioorg. Med. Chem. Lett. 2009;19(2):401–405. doi: 10.1016/j.bmcl.2008.11.098. [DOI] [PubMed] [Google Scholar]
- 27.Zhang F, Bhat S, Gabelli SB, Chen X, Miller MS, Nacev BA, Cheng YL, Meyers DJ, Tenney K, Shim JS, Crews P, Amzel LM, Ma D, Liu JO. Pyridinylquinazolines Selectively Inhibit Human Methionine Aminopeptidase-1 in Cells. J. Med. Chem. 2013;56(10):3996–4016. doi: 10.1021/jm400227z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang P, Yang X, Zhang F, Gabelli SB, Wang R, Zhang Y, Bhat S, Chen X, Furlani M, Amzel LM, Liu JO, Ma D. Pyridinylpyrimidines selectively inhibit human methionine aminopeptidase-1. Bioorg. Med. Chem. 2013;21(9):2600–2617. doi: 10.1016/j.bmc.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garkani-Nejad Z, Saneie F. QSAR Study of Benzimidazole Derivatives Inhibition of Escherichia Coli Methionine Aminopeptidase. Bulletin Chem. Soc. Ethiopia. 2010;24(3):317–325. [Google Scholar]
- 30.Altmeyer MA, Marschner A, Schiffmann R, Klein CD. Subtype-selectivity of metal-dependent methionine aminopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2010;20(14):4038–4044. doi: 10.1016/j.bmcl.2010.05.093. [DOI] [PubMed] [Google Scholar]
- 31.Schiffmann R, Neugebauer A, Klein CD. Metal-Mediated Inhibition of Escherichia coli Methionine Aminopeptidase: Structure–Activity Relationships and Development of a Novel Scoring Function for Metal–Ligand Interactions. J. Med. Chem. 2006;49(2):511–522. doi: 10.1021/jm050476z. [DOI] [PubMed] [Google Scholar]
- 32.Schiffmann R, Heine A, Klebe G, Klein CDP. Metal Ions as Cofactors for the Binding of Inhibitors to Methionine Aminopeptidase: A Critical View of the Relevance of In vitro Metalloenzyme Assays. Angew. Chemie. Int. Ed. 2005;44(23):3620–3623. doi: 10.1002/anie.200500592. [DOI] [PubMed] [Google Scholar]
- 33.Huang Q-Q, Huang M, Nan F-J, Ye Q-Z. Metalloform-selective inhibition: Synthesis and structure–activity analysis of Mn(II)-form-selective inhibitors of Escherichia coli methionine aminopeptidase. Bioorg. Med. Chem. Lett. 2005;15(24):5386–5391. doi: 10.1016/j.bmcl.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 34.Vedantham P, Guerra JM, Schoenen F, Huang M, Gor PJ, Georg GI, Wang JL, Neuenswander B, Lushington GH, Mitscher LA, Ye Q-Z, Hanson PR. Ionic Immobilization, Diversification, and Release: Application to the Generation of a Library of Methionine Aminopeptidase Inhibitors. J. Comb. Chem. 2007;10(2):185–194. doi: 10.1021/cc700085c. [DOI] [PubMed] [Google Scholar]
- 35.Vedantham P, Zhang M, Gor PJ, Huang M, Georg GI, Lushington GH, Mitscher LA, Ye Q-Z, Hanson PR. Studies Towards the Synthesis of Methionine Aminopeptidase Inhibitors: Diversification Utilizing a ROMP-Derived Coupling Reagent. J. Comb. Chem. 2007;10(2):195–203. doi: 10.1021/cc7000869. [DOI] [PubMed] [Google Scholar]
- 36.Xie SX, Huang WJ, Ma ZQ, Huang M, Hanzlik RP, Ye QZ. Structural Analysis of Metalloform-Selective Inhibition of Methionine Aminopeptidase. Acta Crystallographica Setction D. 2006;D62:425–432. doi: 10.1107/S0907444906003878. [DOI] [PubMed] [Google Scholar]
- 37.Wang W-L, Chai SC, Ye Q-Z. Synthesis and biological evaluation of salicylate-based compounds as a novel class of methionine aminopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2011;21(23):7151–7154. doi: 10.1016/j.bmcl.2011.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye Q-Z, Xie S-X, Huang M, Huang W-J, Lu J-P, Ma Z-Q. Metalloform-Selective Inhibitors of Escherichia coli Methionine Aminopeptidase and X-ray Structure of a Mn(II)-Form Enzyme Complexed with an Inhibitor. J. Am. Chem. Soc. 2004;126(43):13940–13941. doi: 10.1021/ja045864p. [DOI] [PubMed] [Google Scholar]
- 39.Ma Z-Q, Xie S-X, Huang Q-Q, Nan F-J, Hurley TD, Ye Q-Z. Structural analysis of inhibition of E. coli methionine aminopeptidase: implication of loop adaptability in selective inhibition of bacterial enzymes. BMC Struct. Biol. 2007;7(84) doi: 10.1186/1472-6807-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huguet F, Melet A, Alves de Sousa R, Lieutaud A, Chevalier J, Maigre L, Deschamps P, Tomas A, Leulliot N, Pages J-M, Artaud I. Hydroxamic Acids as Potent Inhibitors of FeII and MnIIE. coli Methionine Aminopeptidase: Biological Activities and X-ray Structures of Oxazole Hydroxamate–EcMetAP-Mn Complexes. ChemMedChem. 2012;7(6):1020–1030. doi: 10.1002/cmdc.201200076. [DOI] [PubMed] [Google Scholar]
- 41.Chai SC, Wang W-L, Ye Q-Z. FE(II) Is the Native Cofactor for Escherichia coli Methionine Aminopeptidase. J. Biol. Chem. 2008;283(40):26879–26885. doi: 10.1074/jbc.M804345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chai SC, Ye Q-Z. A cell-based assay that targets methionine aminopeptidase in a physiologically relevant environment. Bioorg. Med. Chem. Lett. 2010;20(7):2129–2132. doi: 10.1016/j.bmcl.2010.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang M, Xie S-X, Ma Z-Q, Huang Q-Q, Nan F-J, Ye Q-Z. Inhibition of Monometalated Methionine Aminopeptidase: Inhibitor Discovery and Crystallographic Analysis. J. Med. Chem. 2007;50(23):5735–5742. doi: 10.1021/jm700930k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang M, Xie S-X, Ma Z-Q, Hanzlik RP, Ye Q-Z. Metal mediated inhibition of methionine aminopeptidase by quinolinyl sulfonamides. Biochem. Biophys. Res. Commun. 2006;339(2):506–513. doi: 10.1016/j.bbrc.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 45.Bhat S, Shim JS, Zhang F, Chong CR, Liu JO. Substituted oxines inhibit endothelial cell proliferation and angiogenesis. Org. Biomol. Chem. 2012;10(15):2979–2992. doi: 10.1039/c2ob06978d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olaleye O, Raghunand TR, Bhat S, Chong C, Gu P, Zhou J, Zhang Y, Bishai WR, Liu JO. Characterization of clioquinol and analogues as novel inhibitors of methionine aminopeptidases from Mycobacterium tuberculosis. Tuberculosis. 2011;91(Supplement 1):S61–S65. doi: 10.1016/j.tube.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baell JB, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 48.Sin N, Meng L, Wang MQW, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. 1997;94(12):6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowther WT, Orville AM, Madden DT, Lim S, Rich DH, Matthews BW. Escherichia coli Methionine Aminopeptidase: Implications of Crystallographic Analyses of the Native, Mutant, and Inhibited Enzymes for the Mechanism of Catalysis. Biochemistry. 1999;38(24):7678–7688. doi: 10.1021/bi990684r. [DOI] [PubMed] [Google Scholar]
- 50.Brdlik CM, Crews CM. A Single Amino Acid Residue Defines the Difference in Ovalicin Sensitivity between Type I and II Methionine Aminopeptidases. J. Biol. Chem. 2004;279(10):9475–9480. doi: 10.1074/jbc.M307246200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J-Y, Chen L-L, Cui Y-M, Luo Q-L, Li J, Nan F-J, Ye Q-Z. Specificity for inhibitors of metal-substituted methionine aminopeptidase. Biochem. Biophys. Res. Commun. 2003;307(1):172–179. doi: 10.1016/s0006-291x(03)01144-6. [DOI] [PubMed] [Google Scholar]
- 52.Douangamath A, Dale GE, D'Arcy A, Almstetter M, Eckl R, Frutos-Hoener A, Henkel B, Illgen K, Nerdinger S, Schulz H, MacSweeney A, Thormann M, Treml A, Pierau S, Wadman S, Oefner C. Crystal Structures of Staphylococcus aureus Methionine Aminopeptidase Complexed with Keto Heterocycle and Aminoketone Inhibitors Reveal the Formation of a Tetrahedral Intermediate. J. Med. Chem. 2004;47(6):1325–1328. doi: 10.1021/jm034188j. [DOI] [PubMed] [Google Scholar]
- 53.Hu X, Zhu J, Srivathsan S, Pei D. Peptidyl hydroxamic acids as methionine aminopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2004;14(1):77–79. doi: 10.1016/j.bmcl.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 54.Evdokimov AG, Pokross M, Walter RL, Mekel M, Barnett BL, Amburgey J, Seibel WL, Soper SJ, Djung JF, Fairweather N, Diven C, Rastogi V, Grinius L, Klanke C, Siehnel R, Twinem T, Andrews R, Curnow A. Serendipitous discovery of novel bacterial methionine aminopeptidase inhibitors. Proteins: Structure, Function, and Bioinformatics. 2007;66(3):538–546. doi: 10.1002/prot.21207. [DOI] [PubMed] [Google Scholar]
- 55.Keding SJ, Dales NA, Lim S, Beaulieu D, Rich DH. Synthesis of (3R)-Amino-(2S)-Hydroxy Amino Acids for Inhibition of Methionine Aminopeptidase-1. Synth. Commun. 1998;28(23):4463–4470. [Google Scholar]
- 56.Mitra S, Sheppard G, Wang J, Bennett B, Holz R. Analyzing the binding of Co(II)-specific inhibitors to the methionyl aminopeptidases from Escherichia coli and Pyrococcus furiosus. J. Biol. Inorg. Chem. 2009;14(4):573–585. doi: 10.1007/s00775-009-0471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo Q-L, Li J-Y, Liu Z-Y, Chen L-L, Li J, Qian Z, Shen Q, Li Y, Lushington GH, Ye Q-Z, Nan F-J. Discovery and Structural Modification of Inhibitors of Methionine Aminopeptidases from Escherichia coli and Saccharomyces cerevisiae. J. Med. Chem. 2003;46(13):2631–2640. doi: 10.1021/jm0300532. [DOI] [PubMed] [Google Scholar]
- 58.Meetei P, Hauser A, Raju P, Rathore RS, Prabhu NP, Vindal V. Investigations and design of pyridine-2-carboxylic acid thiazol-2-ylamide analogs as methionine aminopeptidase inhibitors using 3D-QSAR and molecular docking. Med. Chem. Res. 2014;23(8):3861–3875. [Google Scholar]
- 59.Luo Q-L, Li J-Y, Chen L-L, Li J, Ye Q-Z, Nan F-J. Inhibitors of type I MetAPs containing pyridine-2-carboxylic acid thiazol-2-ylamide. Part 2: SAR studies on the pyridine ring 3-substituent. Bioorg. Med. Chem. Lett. 2005;15(3):639–644. doi: 10.1016/j.bmcl.2004.11.035. [DOI] [PubMed] [Google Scholar]
- 60.Luo Q-L, Li J-Y, Liu Z-Y, Chen L-L, Li J, Ye Q-Z, Nan F-J. Inhibitors of type I MetAPs containing pyridine-2-carboxylic acid thiazol-2-ylamide. Part 1: SAR studies on the determination of the key scaffold. Bioorg. Med. Chem. Lett. 2005;15(3):635–638. doi: 10.1016/j.bmcl.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 61.Cui Y-M, Huang Q-Q, Xu J, Chen L-L, Li J-Y, Ye Q-Z, Li J, Nan F-J. Identification of potent type I MetAPs inhibitors by simple bioisosteric replacement. Part 2: SAR studies of 5-heteroalkyl substituted TCAT derivatives. Bioorg. Med. Chem. Lett. 2005;15(18):4130–4135. doi: 10.1016/j.bmcl.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 62.Cui Y-M, Huang Q-Q, Xu J, Chen L-L, Li J-Y, Ye Q-Z, Li J, Nan F-J. Identification of potent type I MetAP inhibitors by simple bioisosteric replacement. Part 1: Synthesis and preliminary SAR studies of thiazole-4-carboxylic acid thiazol-2-ylamide derivatives. Bioorg. Med. Chem. Lett. 2005;15(16):3732–3736. doi: 10.1016/j.bmcl.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 63.Li J-Y, Chen L-L, Cui Y-M, Luo Q-L, Gu M, Nan F-J, Ye Q-Z. Characterization of Full Length and Truncated Type I Human Methionine Aminopeptidases Expressed from Escherichia coli. Biochemistry. 2004;43(24):7892–7898. doi: 10.1021/bi0360859. [DOI] [PubMed] [Google Scholar]
- 64.Wang W-L, Chai SC, Huang M, He H-Z, Hurley TD, Ye Q-Z. Discovery of Inhibitors of Escherichia coli Methionine Aminopeptidase with the Fe(II)-Form Selectivity and Antibacterial Activity. J. Med. Chem. 2008;51(19):6110–6120. doi: 10.1021/jm8005788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang W-L, Chai SC, Ye Q-Z. Synthesis and structure-function analysis of Fe(II)-form-selective antibacterial inhibitors of Escherichia coli methionine aminopeptidase. Bioorg. Med. Chem. Lett. 2009;19(4):1080–1083. doi: 10.1016/j.bmcl.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haldar MK, Scott MD, Sule N, Srivastava DK, Mallik S. Synthesis of barbiturate-based methionine aminopeptidase-1 inhibitors. Bioorg. Med. Chem. Lett. 2008;18(7):2373–2376. doi: 10.1016/j.bmcl.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krátký M, Vinšová J, Novotná E, Mandíková J, Wsól V, Trejtnar F, Ulmann V, Stolaříková J, Fernandes S, Bhat S, Liu JO. Salicylanilide derivatives block Mycobacterium tuberculosis through inhibition of isocitrate lyase and methionine aminopeptidase. Tuberculosis. 2012;92(5):434–439. doi: 10.1016/j.tube.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 68.Chai SC, Wang W-L, Ding D-R, Ye Q-Z. Growth inhibition of Escherichia coli and methicillin-resistant Staphylococcus aureus by targeting cellular methionine aminopeptidase. Eur. J. Med. Chem. 2011;46(8):3537–3540. doi: 10.1016/j.ejmech.2011.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu J-P, Yuan X-H, Yuan H, Wang W-L, Wan B, Franzblau SG, Ye Q-Z. Inhibition of Mycobacterium tuberculosis Methionine Aminopeptidases by Bengamide Derivatives. ChemMedChem. 2011;6(6):1041–1048. doi: 10.1002/cmdc.201100003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu J-P, Yuan X-H, Ye Q-Z. Structural analysis of inhibition of Mycobacterium tuberculosis methionine aminopeptidase by bengamide derivatives. Eur. J. Med. Chem. 2012;47(0):479–484. doi: 10.1016/j.ejmech.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]