

Abstract
Nuclear receptors (NRs) are ligand-activated transcriptional regulators that play vital roles in key biological processes such as growth, differentiation, metabolism, reproduction, and morphogenesis. Disruption of NRs can result in adverse health effects such as NR-mediated endocrine disruption. A comprehensive understanding of core transcriptional targets regulated by NRs helps to elucidate their key biological processes in both toxicological and therapeutic aspects. In this study, we applied a probabilistic graphical model to identify the transcriptional targets of NRs and the biological processes they govern. The Tox21 program profiled a collection of approximate 10 000 environmental chemicals and drugs against a panel of human NRs in a quantitative high-throughput screening format for their NR disruption potential. The Japanese Toxicogenomics Project, one of the most comprehensive efforts in the field of toxicogenomics, generated large-scale gene expression profiles on the effect of 131 compounds (in its first phase of study) at various doses, and different durations, and their combinations. We applied author-topic model to these 2 toxicological datasets, which consists of 11 NRs run in either agonist and/or antagonist mode (18 assays total) and 203 in vitro human gene expression profiles connected by 52 shared drugs. As a result, a set of clusters (topics), which consists of a set of NRs and their associated target genes were determined. Various transcriptional targets of the NRs were identified by assays run in either agonist or antagonist mode. Our results were validated by functional analysis and compared with TRANSFAC data. In summary, our approach resulted in effective identification of associated/affected NRs and their target genes, providing biologically meaningful hypothesis embedded in their relationships.
Keywords: nuclear receptor, transcriptional regulation, Tox21, toxicogenomics project, author-topic model, integrative analysis
Nuclear receptors (NRs) are a superfamily of multifunctional ligand-activated, DNA-binding transcription factors which play a critical role in a variety of important biological functions such as growth, differentiation, metabolism, and reproduction (Tata, 2002). NR ligands encompass endogenous hormones (eg, 17β-estradiol), lipids and bile acids as well as exogenous chemicals like drugs and toxins. NRs play key roles not only in normal physiology but also in many pathological processes (Tenbaum and Baniahmad, 1997). Due to their significant contribution to pathophysiology, the mechanisms of transcriptional regulation by ligand-bound NRs have been extensively studied over the past several decades. Transcriptional regulation by NRs includes a multistep process involving: binding of NRs to regulatory sites in the genome, ligand-dependent recruitment and function of co-regulators to modify chromatin and associated factors, regulation of Pol II binding and activity at target genes’ promoter resulting in increased gene expression. Disruption of NRs can result in adverse health effects such as estrogen receptor mediated endocrine disruption. A comprehensive understanding of core transcriptional targets regulated by NRs helps elucidate their key biological processes in both toxicological and therapeutic aspects. Consequently, the U.S. Tox21 program conducted many in vitro NR assays. Tox21 is a collaboration between the National Institute of Environmental Health Sciences (NIEHS)/National Toxicology Program (NTP), the U.S. Environmental Protection Agency’s (EPA) National Center for Computational Toxicology (NCCT), the National Institutes of Health (NIH) Chemical Genomics Center (NCGC) (now within the National Center for Advancing Translational Sciences), and the U.S. Food and Drug Administration (FDA). The program profiled a collection of approximately 10 000 compounds (including both industrial chemicals and drugs) against a panel of 11 human NRs in a quantitative high-throughput screening (qHTS) format (Judson et al., 2013). The assays were run in both agonist and antagonist modes at 15 different concentrations in triplicate with concentration-response curves for each chemical. Meanwhile, in the toxicogenomics field, the Japanese Toxicogenomics Project (TGP) in its first phase of the study generated large-scale gene expression profiles for 131 chemicals/drugs on rat liver and primary hepatocytes as well as human primary hepatocytes with varying both doses and treatment durations (Uehara et al., 2010). The integrated analysis of these 2 large datasets offers a unique opportunity to investigate the relationship of drug-induced biological processes and targets from the toxicogenomics study with NR regulatory roles profiled by the Tox21 assays.
In this study, we used a probabilistic graphical model namely author topic model (ATM) (Rosen-Zvi et al., 2004) to investigate biological processes regulated by NRs by combining these 2 different data sources, NR assay data from Tox21 and in vitro human gene expression profiles from TGP. ATM is a text mining approach to investigate the relationship between topics and authors. Specifically, ATM models authors’ interest by inferring topics authors write about and to the extension on which group of authors produce similar work. In many ways, the 2 datasets resemble document collections. Specifically, the TGP expression profiles can be considered as a set of documents, where each gene expression profile consists of mixtures of biological processes that can be thought of as topics, and a biological process consists of a set of genes that can be thought of as the words used to present a topic. In addition, each TGP expression profile has ‘authorship’ information—each expression profile is resulted from a chemical treatment and its authors are a set of NRs activated by the chemical in the Tox21 assays. Using these analogies of the data structure, we applied ATM to examine the relationship between NRs and their biological process with these 2 different data sources.
MATERIALS AND METHODS
Probabilistic graphical model
Our probabilistic graphical model is based on ATM, which is an extension of Latent Dirichlet Allocation (LDA) to include authorship information for document collections. LDA is a text mining approach developed by Blei et al. (2003), to organize and classify a collection of documents. Its underlying concept is that a document has a mixture of topics and that each word is selected with a probability given one of the document topics. ATM is developed for extracting information about authors and topics from large text collections where an author writes a mixture of topics. Therefore, whereas LDA does not require author information for each document, ATM requires additional input indicating about which documents are written by which authors. The ATM analysis produces a set of topics (latent variables) and to the extension of revealing which topics are preferably written by which authors. As a result, each author is represented by a probability distribution over topics whereas each topic is represented as a probability distribution over words. To estimate these 2 matrix parameters, ATM assumes a probabilistically generative model in which each document is generated by 3 sampling processes. First, each word in a document by an author is chosen at random. Next, a topic is chosen from a distribution over topics specific to that author. Lastly, the word is generated from the chosen topic. In this study, the open-source Matlab Topic Modeling Toolbox package from the University of California was applied (http://psiexp.ss.uci.edu/research/programs_data/toolbox.htm) where a Gibbs sampling process was implemented to maximize the posterior probability of 2 observed matrices, authors-documents and documents-words based on the calculated author-topic and topic-word distribution matrix (Rosen-Zvi et al., 2004). As mentioned above, the modeling produces 2 distributions: probability distribution over topics for each author and probability distribution over words associated with each topic. Figure 1 shows an overview of the methodology, where 2 resulting matrices are colored with orange in the middle. Specifically, the former matrix (Θ) is formatted as A × T (authors by topics), with each cell indicating probability of assigning topic t to a word generated by author a. The latter matrix (Θ) is formatted as T × W (topics by words), with each cell indicating the probability of generating word w from topic t. In the context of our study design, Ɵ (NRs by topics) includes the topic distributions for each NR whereas Ф (topics by genes) contains the gene distributions for each topic. By consolidating the 2 matrices, we uncovered hidden biological relationships in terms of target genes regulated by NRs. Our method requires input of several parameters. The number of topics was heuristically determined as 18 by prior knowledge based on the number of NRs to avoid extreme generalization of the model and maximize an informative discovery. Two parameters, α and β were defined as 0.01 and 50/T (number of topics), respectively where α and β is the Dirichlet hyperparameters for author-topic distribution and topic-word distribution, respectively.
FIG. 1.

Overview of the workflow. First, 2 datasets were transformed into document-based form, on which author-topic model was constructed. As a result, 2 matrices, NRs-topic and topic-genes probabilities, were estimated. Through shared topics, the associations between NRs and genes were investigated. (‘TP’ denotes a TGP expression profile; ‘NR’ denotes a nuclear receptor; ‘T’ denotes a topic; ‘G’ denotes a gene).
Dataset
We used the compound-assay activity data from the Tox21 qHTS assays. Briefly, half-maximal activity (AC50) and maximal response (efficacy) values were calculated from the concentration response curves and each curve was assigned a curve class of 1 to 5 based on potency, efficacy, and the quality of curve fit (Inglese et al., 2006). The final activity outcome of a compound in an assay was then determined based on assigned class, reproducibility, and activity in control readouts and counter screens. A detailed description of the compound activity assignment scheme can be found in (Huang et al., 2014). Detailed descriptions of these assays including experimental conditions and the Tox21 qHTS data and activity assignment results are available in PubChem (http://www.ncbi.nlm.nih.gov/pcassay?term=tox21) (Supplementary Table 1). For androgen receptor (AR) and estrogen receptor (ER) assays, 2 types of cell lines were utilized. In this case, a chemical active in at least 1 cell line was considered as active. The final input dataset for analysis was a binary matrix for NR-chemical pairs, with 1 and zero representing a chemical active or inactive against a NR, respectively (Figure 1’s right side green table, Supplementary Table 2).
The TGP dataset was downloaded from CAMDA 2013 (http://dokuwiki.bioinf.jku.at/doku.php/start). The gene expression data was generated using Affymetrix Human Genome U133 Plus 2.0 (in vitro human). After comparing the Tox21 and TGP datasets, we found that 18 NRs in either agonist or antagonist mode are activated by at least 1 compound (Table 1) and 52 chemicals/drugs are shared between Tox21 and TGP (Supplementary Table 2). The in vitro human gene expression profiles from TGP comprise 4 different conditions, the combinations of 2 durations (8 and 24 h) and 2 doses (medium and high). A total of 203 drug-treated gene expression profiles were compiled for further analysis by including 4 combinations of 2 durations and 2 doses. To measure expression level of each gene, the probe-level data of the microarrays were quantile normalized followed by mapping of probe sets into corresponding genes (Dai et al., 2005). Multiple probes for a gene were aggregated using the FARMS method (Hochreiter et al., 2006).
TABLE 1.
Eighteen Nuclear Receptors in Either Agonist or Antagonist Mode used for Analysis
| No. | Nuclear receptor | Nuclear receptor full name |
|---|---|---|
| 1 | RAR agonist | Retinoic acid receptor agonist |
| 2 | RAR antagonist | Retinoic acid receptor antagonist |
| 3 | ROR antagonist | Retinoic acid receptor-related orphan receptor antagonist |
| 4 | RXR agonist | Retinoid × receptor agonist |
| 5 | AhR | Aryl hydrocarbon receptor |
| 6 | AR agonist | Androgen receptor agonist |
| 7 | AR antagonist | Androgen receptor antagonist |
| 8 | ER agonist | Estrogen receptor agonist |
| 9 | ER antagonist | Estrogen receptor antagonist |
| 10 | FXR antagonist | Farnesoid × receptor antagonist |
| 11 | GR agonist | Glucocorticoid receptor agonist |
| 12 | GR antagonist | Glucocorticoid receptor antagonist |
| 13 | PPARδ agonist | Proliferator activated receptor delta agonist |
| 14 | PPARδ antagonist | Proliferator activated receptor delta antagonist |
| 15 | PPARγ agonist | Proliferator activated receptor gamma agonist |
| 16 | PPARγ antagonist | Proliferator activated receptor gamma antagonist |
| 17 | VDR agonist | Vitamin D receptor agonist |
| 18 | VDR antagonist | Vitamin D receptor antagonist |
We generated a ‘document’ for each TGP gene expression profile, which contained ‘words’, ie, genes differentially expressed when compared with the matched control. In the CAMDA dataset, a total of 18 988 genes remained after the preprocessing procedure. We considered the same gene with different transcriptional directions (ie, up or down) as 2 different words. After eliminating genes with low expression in either direction using the criteria |Fold Change|>1.2, a total of 13 006 words were used as the final corpus for our documents. The frequency of a word appearing in each document was determined by multiplying the fold change of the treated samples compared with the time-matched controls by 10 times and rounded to the nearest integer as described in Figure 1’s left side green table. In summary, each TGP expression profile (TP) was represented by 2 vectors; a binary vector consisting of NR activity information whereas another is integer vector indicating summarized expression level of each gene as presented in 2 green tables of Figure 1.
Topic modeling
By learning the parameters of the model, 2 matrices, NR-topic (Θ) and topic-gene (Ф) probabilities, were yielded. The NR-topic matrix identifies which topics are preferably used by which NRs whereas the topic-gene matrix identifies genes associated with corresponding topics. To investigate the relationship between NRs and genes, we used topic variable, which defines a pair of probability distributions over NRs and genes. The highly ranked genes for each topic were defined as target genes regulated by the mostly probable NR from the same topic.
Functional analysis
The second outcome of our model is the probability distribution of genes within a given topic. Specifically, ϕwt is the probability of gene w occurring in topic t, giving a measure of contribution of gene w to topic t. Since our probabilistic graphical model is to cluster genes co-occurring frequently chosen by a certain NR with respect to a collection of gene expression profiles, genes highly ranked in a topic are presumably regulated by the NR that is most likely associated with that topic. To determine the overrepresentation of biological processes governed by a particular NR, we extracted 300 genes for each topic followed by a functional analysis using the Gene Ontology and KEGG. Over-enriched terms were identified using Fisher’s exact test. To construct an interaction network from top 300 genes, we utilized the ‘Significant interactions within set(s)’ feature of MetaCore.
RESULTS
Nuclear Receptors and Topics
One of our modeling results is a probability matrix for each NR over topics, with each element θta representing the probability of assigning topic t to genes regulated by NR a. Some of the NRs were highly relevant to the topics whereas others were less apparent. This is expected because some of the chemicals activate a single NR whereas others activate as many as 11 NRs in either agonist or antagonist modes. For example, acarbose showed agonist activity against ER only whereas griseofulvin showed agonist activity for 11 NRs. Figure 2A shows a heat map of chemical-NR activity matrix generated from the activity profile of the 52 chemicals across the NR assays. Figure 2B shows the distribution of the number of chemicals in the context of the number of active NRs. ROR antagonist shows the most active frequency of 22 whereas VDR agonist, PPARδ agonist and PPARδ antagonist have least active frequency by activating only one single chemical. The average number of chemicals active against an NR in agonist mode is 9 whereas the average number of chemicals for antagonist is 11. Table 2 shows the top 2 NRs assigned to each topic. Some of the NRs like PPARδ agonist, PPARδ antagonist, VDR agonist and VDR antagonist are not enriched with any of the topics whereas several other NRs are enriched with several topics. For example, ER agonist is associated with as many as 3 topics (Topic 3, 13, and 17) as the most probable NR. We found that topic 4, 5, 7, 9 and 13 showed a strong association with only 1 NR such as RXR agonist, RAR agonist, AhR, RAR agonist, and ER agonist, respectively (θta > 0.9).
FIG. 2.

(A) Heatmap for chemical-NR activity matrix generated from the activity profile of the 52 chemicals across the NR assays. (B) Distribution of number of chemicals in the context of the number of active NRs.
TABLE 2.
Author-Topic Probabilities Matrix Includes Topic Distributions for each NR
| Topic | Top NR | Probability | Second NR | Probability |
|---|---|---|---|---|
| Topic 1 | RXR agonist | 0.46178 | AR antagonist | 0.30304 |
| Topic 2 | GR agonist | 0.54651 | AR agonist | 0.42918 |
| Topic 3 | ER agonist | 0.35156 | RAR agonist | 0.33942 |
| Topic 4 | RXR agonist | 0.99838 | AhR | 0.00041 |
| Topic 5 | RAR agonist | 0.9979 | AhR | 0.00025 |
| Topic 6 | ROR antagonist | 0.64334 | RXR agonist | 0.35511 |
| Topic 7 | AhR | 0.94849 | PPARγ antagonist | 0.05068 |
| Topic 8 | AR antagonist | 0.4382 | AhR | 0.27876 |
| Topic 9 | RAR agonist | 0.99784 | RAR antagonist | 0.00029 |
| Topic 10 | PPARγ antagonist | 0.45153 | RXR agonist | 0.23791 |
| Topic 11 | FXR antagonist | 0.55794 | VDR antagonist | 0.43917 |
| Topic 12 | RAR antagonist | 0.66447 | ROR antagonist | 0.33393 |
| Topic 13 | ER agonist | 0.99839 | PPARδ agonist | 0.00019 |
| Topic 14 | AR antagonist | 0.54336 | ER antagonist | 0.4545 |
| Topic 15 | ER antagonist | 0.99758 | ROR antagonist | 0.00042 |
| Topic 16 | GR antagonist | 0.45849 | AR antagonist | 0.21767 |
| Topic 17 | ER agonist | 0.49531 | AhR | 0.38676 |
| Topic 18 | ROR antagonist | 0.46391 | ER agonist | 0.44006 |
NRs are ranked according to the probability of each topic from author-topic matrix. The table shows the top 2 NRs and their probabilities for each topic in the model.
Functional Analysis
The second outcome of our model is the probability distribution of genes conditioned on a given particular topic (Ф). Based on the model, genes co-occurring frequently across expression profiles are clustered together. Functional analysis of the highly ranked genes in each topic provides the information about the biological processes regulated by each NR. To determine the overrepresentation of biological processes governed by a particular NR, we performed a functional analysis with the top 300 ranked genes in each topic against Gene Ontology and KEGG (P < .05, Supplementary Table 3). The most frequently observed pathway from KEGG database was p53 signaling, which appeared in 13 topics, followed by cell cycle across 12 topics. Metabolism of xenobiotics by cytochrome P450, retinol metabolism and drug metabolism were enriched in 10 topics. Generally, p53 pathway responds to various stress signals to disrupt cellular homeostatic mechanisms. Specifically, drug-induced DNA damage and oxidative stress cause a stressful state for cells, so that p53 signaling is triggered as a defensive mechanism, ultimately resulting in cell cycle arrest and apoptotic pathway. However, the aggressive pro-oxidative mechanism of p53 was also discovered in instances where a high level of oxidative stress was leading to cell death. We found that PPAR signaling pathway is enriched in 2 topics, topics 3 and 10 which the mostly associated NR is ER agonist and PPARγ antagonist, respectively. Both topics 5 and 9 have the RXR agonist as the mostly related NR. We found that some of the biological processes over-represented in topic 5, such as carboxylic acid metabolic process, organic acid metabolic process, cofactor metabolic process, and oxidation reduction were identified in previous work (He et al., 2013). Topic 14 is associated with ER antagonist (θta = 0.54) and AR antagonist (θta = 0.45). Both ER and AR are steroidal receptors and play important roles in developing prostate and breast cancer. The GO analysis indicated that cell proliferation related biological processes such as cell cycle (P = 7.67E–07) and regulation of cell cycle (P = 7.49E–05) were highly ranked. In the KEGG analysis, DNA replication is top ranked (P = 4.00E–04). Topic 10 is associated with PPARγ antagonist and its apoptotic mechanism in various cancer cells are widely studied (Fajas et al., 2003). Our result showed that apoptotic process was highly enriched in topic 10 (P = .00015) and regulation of apoptotic process (P = .00028). Topic 11 is highly associated with FXR antagonist. The GO analysis showed that topic 11 was significantly associated with cell migration (P = .0029) and cell motility (P = .0042), and abnormal regulation driving cancer metastasis. It is well known that FXR plays a vital role in cancer metastasis and FXR inhibition is an effective approach to diminish tumor growth (Lee et al., 2011). Additionally, FXR is involved in cholesterol homeostasis; in our result cholesterol homeostasis was significantly identified (P = .026). Table 3 shows the unique KEGG pathways over-represented in each topic and not in the other topics. Intriguingly, topic 10 is enriched in pathways for both cancer and apoptosis. ER agonist is top NR in topic 10 (θ = 0.49), which has a critical role in breast cancer as mentioned above. Topic 10 of which top NR is PPARγ antagonist, is enriched with fatty acid metabolism and, their relationship was also studied (Ciaraldi et al., 2002).
TABLE 3.
Functional Analysis was Conducted with 18 topics’ top 300 Genes Against KEGG and GO Database
| Topic | Top NR | No. of pathways | KEGG pathways |
|---|---|---|---|
| 1 | RXR agonist | 1 | Circadian rhythm |
| 3 | ER agonist | 1 | Purine metabolism, Propanoate metabolism |
| 6 | ROR antagonist | 1 | Aminoacyl-tRNA biosynthesis |
| 7 | AhR | 1 | Pentose and glucoronate interconversions |
| 9 | RAR agonist | 1 | Spliceosome |
| 10 | PPARγ antagonist | 1 | Fatty acid metabolism |
| 11 | FXR antagonist | 1 | Nicotinate and nicotinamide metabolism |
| 12 | RAR antagonist | 1 | RNA degradation |
| 13 | ER agonist | 1 | Ascorbate and aldarate metabolism |
| 16 | GR antagonist | 2 | Nitrogen metabolism, Androgen and estrogen metabolism |
| 17 | ER agonist | 2 | Pathways in cancer, Apoptosis |
| 18 | ROR antagonist | 2 | Tyrosine metabolism, Citrate cycle (TCA cycle) |
The table shows unique KEGG pathways over-represented in single topic.
Nuclear Receptor Target Genes
We defined the top 300 genes for each topic as target genes regulated by the mostly probable NR from the corresponding topic. The top 10 genes from 18 topics are presented in Table 4. All 300 genes for each topic are provided in Supplementary Table 4. Each of the 18 topics is unique, as evident by a pairwise similarity assessment of topics using the Tanimoto method based on the top 300 genes (Figure 3) where the largest Tanimoto coefficient was only 0.15 between topic 2 and topic 15. This indicated that each topic represented a unique aspect of biology. A literature search was conducted to validate target genes regulated by NRs. We compared the known 13 AhR target genes with top 300 genes of topic 7 (associated most strongly with AhR). Among the 13 known genes, 5 genes with the same regulation direction appeared such as Cyp1a1, Cyp1b1, Cyp1a2, Nfe2l2, and Tiparp (P = 6.983e–06) (Watson et al., 2014). Cyp3a7 was appearing as most likely gene in 2 topics, topics 2 and 4 of which top NR is GR agonist and RAR agonist, respectively. Cyp3a7 is one of the biomarkers for human fetal liver; its gene expression was induced after treatment of glucocorticoids (Pang et al., 2012). PXR:RXR complex binds to ER6 elements upstream of the Cyp3a4 and Cyp3a7 and activates their gene expression (Pascussi et al., 1999). Topic 9 has Cyp26a1 as a top gene of which top NR is RAR agonist, supported by (Pozzi et al., 2006). In topic 14, both AR and ER antagonist are highly associated. Its most likely gene is GDF15 (upregulated) that is known for association with estrogen resistance and liver injury as a member of the transforming growth factor-beta superfamily.
TABLE 4.
Each Topic is Composed of a Set of Genes
| Topic 1 | Topic 2 | Topic 3 | Topic 4 | Topic 5 | Topic 6 |
|---|---|---|---|---|---|
| SDS_up | CYP3A7_up | RRM2_down | CYP3A7_up | THRSP_up | HSPA1B_up |
| RRM2_down | THRSP_up | SOX4_down | CYP3A4_up | CYP3A7_up | ADM_up |
| PBK_down | RRM2_down | POR_up | NDRG1_down | NPTX2_up | KRT7_down |
| CDK1_down | CCL2_down | BAAT_down | TRIM22_down | GPAM_up | RRM2_down |
| PPP1R15A_up | CYP3A4_up | CP_down | TYMS_down | AHSG_down | MAD2L1_down |
| CDC20_down | CYP3A5_up | ARL14_down | ZWINT_down | GPR37_up | MAL2_down |
| AJUBA_down | CTGF_down | INSIG1_up | INSC_down | UGT2B15_down | TFDP1_down |
| LDLR_up | INSIG1_up | CAD_down | TSKU_up | ALAS1_up | TRPM8_down |
| TNFRSF10D_down | HMGB2_down | ACSL1_up | PRC1_down | CPEB4_up | HSD17B2_up |
| ANLN_down | ANLN_down | CITED2_down | MCM2_down | BHMT_up | CXCL6_down |
| Topic 7 | Topic 8 | Topic 9 | Topic 10 | Topic 11 | Topic 12 |
| CYP1A1_up | HMOX1_up | CYP26A1_up | FGF21_up | HIST1H4H_up | RASD1_up |
| RRM2_down | IL18_down | CYP1A1_up | CYP1A1_up | LOC100507025_up | PCK1_up |
| CYP1B1_up | ZWINT_down | RGS2_down | ALAS1_up | SLC2A2_down | CYP1A1_up |
| CCL2_down | MAFF_up | H1F0_up | TSKU_up | ALDH1A1_down | TAT_up |
| CYP1A2_up | TYMS_down | DIO1_up | MTHFD2_up | AKR1B10_down | TIPIN_down |
| ANGPTL4_down | PRSS23_down | C8orf4_up | UPP1_up | RRAD_up | RRM2_down |
| CXCL6_down | IFT80_down | CYP3A5_up | UGT2B15_down | BRD2_up | A2M_up |
| CXCL1_down | CITED4_down | NDRG1_down | ALDH8A1_down | GSTA1_down | CYP3A7_up |
| GLDC_down | CLK1_up | ZNF367_down | PPP1R3C_down | SERTAD1_up | TSC22D3_up |
| FGF21_up | SORBS2_down | BBOX1_up | SLC38A4_down | HIST1H2AE_up | NUAK2_down |
| Topic 13 | Topic 14 | Topic 15 | Topic 16 | Topic 17 | Topic 18 |
| RRM2_down | GDF15_up | RRM2_down | TSKU_up | SLC2A2_down | ARG1_up |
| HMOX1_up | CYP3A7_up | INSIG1_up | HSPA1B_up | KRT7_down | PCK1_up |
| CXCL2_down | PCK1_down | ANGPTL4_down | TRPM8_down | PPP1R15A_up | ENC1_down |
| HSPA1B_up | GINS1_down | CCNE2_down | ALDH8A1_down | ID1_down | PPP1R3C_down |
| GLDC_down | CYP3A4_up | PBK_down | UGT2B15_down | ALDH1A1_down | FOS_up |
| GEM_up | LINC00261_down | ACSS2_up | NAT8_down | PBK_down | FST_up |
| MIR22HG_up | SLC2A2_down | MELK_down | SRXN1_up | TYMS_down | SDS_up |
| CLK1_up | BIRC3_down | DTL_down | SLC3A1_down | ADM_up | FIGNL1_down |
| ANGPTL4_down | CDK1_down | TYMS_down | TRIM22_down | GDF15_up | CCL2_down |
| HSPA1A_up | RGCC_up | NR0B2_up | ARL14_down | FSTL1_down | ZWINT_down |
Genes are ranked according to the probability of topic-gene matrix. The table shows the top 10 genes ranked by probability for each topic.
FIG. 3.

Topic similarity calculated from Tanimoto coefficient based on shared genes. Darker represents strong similarity between 2 topics. For an intuitive visualization, the pairs between same topics located on diagonal were colored with white.
For further validation, we compared our target genes with TRANSFAC database which includes transcription factor’s experimentally-confirmed binding sites and regulated genes (Matys et al., 2003). We identified the target genes for 9 NRs as presented in Table 5. We then counted the number of targets that were common to TRANSFAC and our model for a given NR. The largest intersection was 8 target genes of TRANSFAC’s GR target genes, which were identified for Topic 2 (most likely NR was GR, P = 1.946e–07)—Cyp2c9, Fkbp5, Igfbp1, Lpin1, Sepp1, Tat, Tnfaip3, and Tsc22d3. Another example is Topic 13 of which top NR is ER agonist, in which 6 genes are common with TRANSFAC’s ER target genes such as Cdkn1a, Cyp2a6, Hmox1, Nr0b2, Nr5a2, and Ugt2b15 (P = 8.688e−04).
TABLE 5.
Number of Common Target Genes Between 18 Topics’ Top 300 Genes and 9 NRs’ Target Genes from TRANSFAC
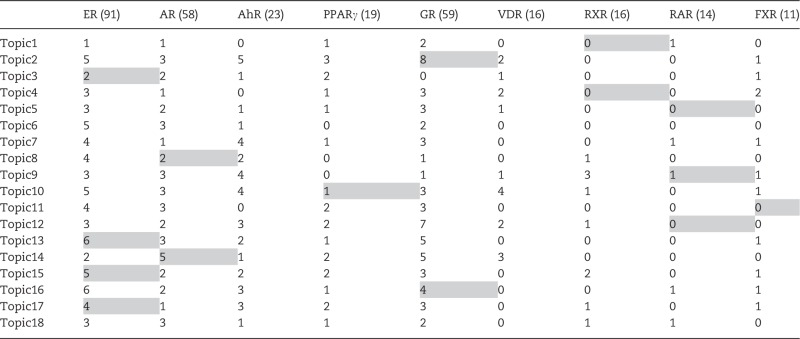 |
The row and column represents 18 topics and TRANSFAC’s 9 NRs, respectively. The number within parenthesis in the column name shows the number of total target genes from TRANSFAC. If the topic in a certain row is most associated with NR in a particular column, that cell is colored with gray.
To assess our results qualitatively, we generated another target gene sets by using fold change. We gathered a collection of gene expression profiles treated with the chemicals that are activated for a certain NR then calculated average fold change for each gene. Then top 300 genes were extracted on the ranking of absolute average fold change. Those target genes were then compared with TRANSFAC database that is presented in Table 6. When GR agonist’s 300 target genes extracted from fold change were compared with GR’s 59 target genes from TRANSFAC, 3 genes such as Cyp2c9, Lpin1, and Tat were found to be common. The list of common target genes for entire pairs is summarized in Supplementary Table 5.
TABLE 6.
Number of Common Target Genes Between Top 300 Genes when using Fold change and 9 NRs’ Target Genes from TRANSFAC
| ER (91) | AR (58) | AhR (23) | PPARγ (19) | GR (59) | VDR (16) | RXR (16) | RAR (14) | FXR (11) | |
|---|---|---|---|---|---|---|---|---|---|
| AhR | 5 | 2 | 4 | 1 | 1 | 2 | 1 | 1 | 1 |
| AR agonist | 5 | 1 | 5 | 2 | 4 | 3 | 0 | 0 | 0 |
| AR antagonist | 5 | 3 | 3 | 1 | 3 | 2 | 1 | 1 | 0 |
| ER agonist | 4 | 2 | 3 | 4 | 2 | 2 | 0 | 0 | 1 |
| ER antagonist | 5 | 3 | 3 | 1 | 3 | 2 | 1 | 1 | 0 |
| FXR antagonist | 4 | 2 | 4 | 3 | 2 | 2 | 0 | 1 | 0 |
| GR agonist | 4 | 2 | 4 | 4 | 3 | 3 | 0 | 1 | 1 |
| GR antagonist | 5 | 1 | 5 | 2 | 1 | 2 | 0 | 1 | 0 |
| PPARδ agonist | 4 | 2 | 1 | 2 | 4 | 2 | 0 | 1 | 0 |
| PPARδ antagonist | 3 | 2 | 4 | 0 | 2 | 4 | 0 | 0 | 1 |
| PPARγ agonist | 4 | 1 | 3 | 3 | 2 | 2 | 1 | 0 | 1 |
| PPARγ antagonist | 4 | 3 | 4 | 2 | 2 | 2 | 1 | 0 | 1 |
| RAR agonist | 4 | 5 | 5 | 2 | 2 | 2 | 2 | 1 | 1 |
| RAR antagonist | 4 | 2 | 4 | 1 | 3 | 2 | 0 | 0 | 0 |
| ROR antagonist | 4 | 2 | 4 | 1 | 1 | 2 | 2 | 0 | 0 |
| RXR agonist | 5 | 3 | 4 | 1 | 2 | 2 | 1 | 0 | 0 |
| VDR agonist | 4 | 2 | 1 | 2 | 4 | 2 | 0 | 1 | 0 |
| VDR antagonist | 4 | 1 | 1 | 4 | 2 | 2 | 0 | 1 | 1 |
The row and column represents 18 NRs in either agonist or antagonist mode and TRANSFAC’S 9 NRs, respectively. The number within parenthesis in the column name shows the number of total target genes from TRANSFAC.
Lastly, to validate further the modularity of our target genes, we examined an interactivity of target genes by counting the number of interaction pairs from top 300 genes using MetaCore’s significant interactions within set(s). As a result, the number of interactions was much higher than one from the randomly selected 300 genes, presented in Figure 4 where the average number of interactions was 169 and 57.7 when it was our discovered 18 topics and randomly selected 50 topics, respectively. The statistical significance was .0001 against the null hypothesis that the means of interaction numbers from topics are randomization are equal. The entire set of interaction pairs is presented in Supplementary Table 6, where topic 12 has the largest number of interactions (389 interacting pairs), of which the most likely NR is RAR antagonist. In the context of a network from topic 12, there were several hub genes such as CDK2, CDK1, and FOS. Their connection to RAR is supported by previous literature reports (Bao et al., 2006; Ødum, 2013; Talmage and Lackey, 1992).
FIG. 4.
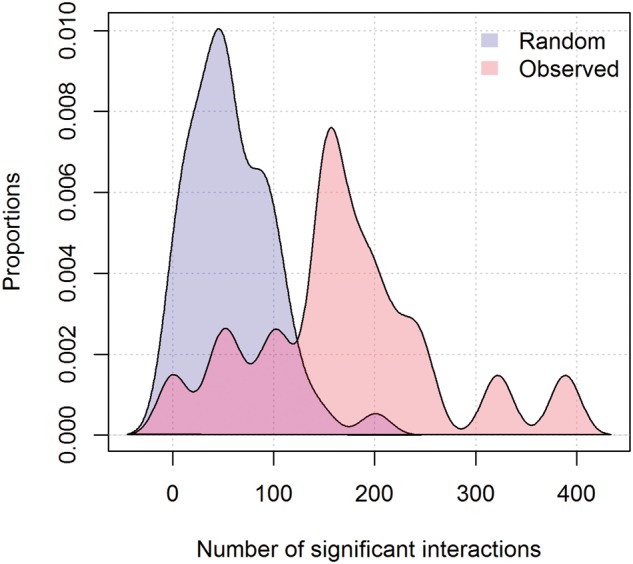
The distribution of the number of interactions between 300 genes. Purple is distribution from 50 times randomly selected 300 genes whereas pink is one from identified top 300 genes for 18 topics.
DISCUSSION
In this study, we performed an integrative analysis of 2 different datasets, small molecule bioassays from Tox21 and in vitro human gene expression profiles from TGP to discover transcriptional targets that are regulated by NRs. We applied ATM that analyzed these 2 different data sources efficiently with latent variables for an enhanced integration. This approach originated from Latent Dirichlet Allocation (LDA) whose objective is to identify the hidden structure embedded in a set of documents. Besides uncovering the hidden structure, ATM examines the relationship between authors (ie, NRs) and words (ie, genes) through latent topics (ie, biological processes). This ultimately leads to modeling the content of documents (ie, gene expression profiles) and the interests of authors (ie, NRs).
Considering the similar data structure between authors-documents and NRs-gene expression profiles, we successfully applied an ATM to the 2 different data sources. ATM is a text mining approach to interrogate authors’ interest by investigating which topics authors write about and to the extension of revealing which group of authors often collaborate together in writing the same topics. In many ways, differentially expressed genes selectively regulated by NRs in gene expression profiles can be viewed as if words are preferably selected by authors in writing documents. Two matrices were yielded from the ATM analysis: one is NR-topic probabilities matrix and the other is topic-gene probabilities matrix. The NR-topic matrix identifies which topics are preferably used by which NRs whereas the topic-gene matrix identifies which genes are associated with which topics. Here, a ‘topic’ consisted of a set of genes with each having a probabilistic measure of its importance to the topic and it is enriched in the expression profiles for sets of chemicals that activate particular NRs (or pairs, triples, etc. of NRs). The main goal of this analysis is to infer which genes are targets by which NRs. Specifically, by analyzing 2 matrices, the highly probable genes for each topic were defined as target genes regulated by the most probable NR from the same topic.
For each topic, we performed functional analysis, which provides us with an intuitive understanding of the biological processes associated with NRs. Among the functional analysis results, some of the cell death related functions may be associated with potential cytotoxicity rather than NR’s induced effect in the 2 data sets because some of the targets may be regulated as a result of cell stress at high drug cocentration. In the Tox21 dataset, this issue is more relevant for the antagonist mode assays, where an inhibitory effect caused by cytotoxicity could be mistaken for antagonist action. We have tried to minimize the interference from cytotoxic responses by running a cell viability counterscreen for every antagonist mode assay, and only considering a chemcial as active when it showed significantly more potent activity against the NR target than in the cell viability counterscreen. The method may not remove cytotoxicity interference completely, but the target gene/pathway validation results show that with our analysis method we can see true NR related signals that are not merely responses to cytotoxicity. We found that p53 signaling pathway is most frequently over-represented on all the 18 topics. The relationship between NRs and p53 has been widely studied in both structural and functional aspects (Lee et al., 2010; Yang et al., 2012). For example, ER interacts with p53, leading to the suppression of p53-mediated transcriptional repression (Rasti et al., 2012). It is also reported that both ER agonist (estradiol) and the ER antagonist (tamoxifen) can promote p53 inhibition (Bailey et al., 2012). Besides the functional analysis of each topic, we compared top 300 genes in our 18 topics with TRANSFAC’s target genes. As a result, the functions of some topics are verified, for example, among 300 genes in topic 2 where the most probable NR is GR, 8 genes are overlapped with TRANSFAC GR’s target genes. Of note, a relatively large false discovery could be anticipated due to the low hit rate, 0.14 (8/59) and potential false positive targets. Differences in experimental conditions, such as cell type, could account for the relatively low overlap between the target genes identified by our method and the TRANSFAC genes resulting in potential false discoveries. Specifically, the gene expression profiles of TGP are generated in primary human hepatocytes, but most of the NRs-target relationships reported in TRANSFAC are only validated in human cancer cells. Another common approach to identify candidate targets is to compare DEGs (or fold change alone) with TRANSFAC. This approach has only 3 genes overlapped with TRANSFAC’s GR target genes, which is far less than our ATM approach. Thus, our approach is clearly more robust in correctly identifying target genes despite of the seemingly low hit rate for some targets. Lastly, we analyzed the interaction/connection between top 300 genes for each topic, which demonstrated our observed topics are much highly correlated with each other than randomly selected 300 genes.
Uncovering hidden structure embedded in different datasets is a non-trivial problem due to their distinct characteristics such as different variable types and/or their scales. However, utilizing the ATM method we were able to integrate 2 heterogeneous data sources in an efficient manner through latent variables (ie, topics), which connects NRs and associated (or regulated) differentially expressed genes. With that said, we found that certain NRs (such as PPARδ agonist, PPARδ antagonist, VDR agonist, and VDR antagonist) were not associated with any topic. It is likely due to the fact that because the method is model-based, if the hidden structure is not apparent compared with others, it would not appear as detectable pattern. In contrast, we could define target genes for every NR when using fold change. Additionally, even though a few NRs is associated with multiple topics, it does not imply that our topics are redundant as evident by the pairwise similarity assessment of 18 topics where the nearest pair was between topic 2 and 15 with a Tanimoto coefficient of 0.15 (80 genes were shared). These results demonstrate that even though the same NR is activated, the expression pattern could be different according to the biological context. Importantly, our approach was successfully validated with functional analysis in comparison with a curated database, leading to the discovery of transcriptional targets of NRs with interpretable biological insights.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. Food and Drug Administration and the National Center for Advancing Translational Sciences, National Institutes of Health. We would also like to thank Dr. Rajarshi Guha for helpful discussion. The views expressed in this article are those of the authors and do not necessarily reflect the statements, opinions, views, conclusions, or policies of the National Center for Advancing Translational Sciences, National Institutes of Health, U.S. Food and Drug Administration, or the United States government. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
REFERENCES
- Bailey S. T., Shin H., Westerling T., Liu X. S., Brown M. (2012). Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 109, 18060–18065. doi:10.1073/pnas.1018858109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G. C., Wang J.-G., Jong A. (2006). Increased p21 expression and complex formation with cyclin E/CDK2 in retinoid-induced pre-B lymphoma cell apoptosis. FEBS Lett. 580, 3687–3693. doi:10.1016/j.febslet.2006.05.052. [DOI] [PubMed] [Google Scholar]
- Blei D. M., Ng A. Y., Jordan M. I. (2003). Latent dirichlet allocation. J. Mach. Learn. Res. 3, 993–1022. [Google Scholar]
- Ciaraldi T. P., Cha B.-S., Park K.-S., Carter L., Mudaliar S. R., Henry R. R. (2002). Free Fatty Acid Metabolism in Human Skeletal Muscle Is Regulated by PPARγ and RXR Agonists. Ann. N. Y. Acad. Sci. 967, 66–70. doi:10.1111/j.1749-6632.2002.tb04264.x. [DOI] [PubMed] [Google Scholar]
- Dai M., Wang P., Boyd A. D., Kostov G., Athey B., Jones E. G., Bunney W. E., Myers R. M., Speed T. P., Akil H., et al. (2005). Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33, e175 doi:10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L., Egler V., Reiter R., Miard S., Lefebvre A. M., Auwerx J. (2003). PPARgamma controls cell proliferation and apoptosis in an RB-dependent manner. Oncogene 22, 4186–4193, 10.1038/sj.onc.1206530. [DOI] [PubMed] [Google Scholar]
- He Y., Gong L., Fang Y., Zhan Q., Liu H.-X., Lu Y., Guo G., Lehman-McKeeman L., Fang J., Wan Y.-J. (2013). The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling. BMC Genomics 14, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter S., Clevert D.-A., Obermayer K. (2006). A new summarization method for affymetrix probe level data. Bioinformatics 22, 943–949. doi:10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- Huang R., Sakamuru S., Martin M. T., Reif D. M., Judson R. S., Houck K. A., Casey W., Hsieh J. H., Shockley K. R., Ceger P., et al. (2014). Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 4, 5664 doi:10.1038/srep05664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J., Auld D. S., Jadhav A., Johnson R. L., Simeonov A., Yasgar A., Zheng W., Austin C. P. (2006). Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. 103, 11473–11478. doi:10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson R., Kavlock R., Martin M., Reif D., Houck K., Knudsen T., Richard A., Tice R. R., Whelan M., Xia M., et al. (2013). Perspectives on validation of high-throughput assays supporting 21(st) century toxicity testing(). ALTEX 30, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. W., Martinez-Yamout M. A., Dyson H. J., Wright P. E. (2010). Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry 49, 9964–9971. doi:10.1021/bi1012996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y., Lee K. T., Lee J. K., Lee K. H., Jang K. T., Heo J. S., Choi S. H., Kim Y., Rhee J. C. (2011). Farnesoid X receptor, overexpressed in pancreatic cancer with lymph node metastasis promotes cell migration and invasion. Br. J. Cancer 104, 1027–1037. doi:10.1038/bjc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V., Fricke E., Geffers R., Gossling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A. E., Kel-Margoulis O. V., et al. (2003). TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ødum N. (2013). CDK1 links to RARγ in treatment response of cancer cells. Cell Cycle 12, 1659 doi:10.4161/cc.25069. [DOI] [PubMed] [Google Scholar]
- Pang X. Y., Cheng J., Kim J. H., Matsubara T., Krausz K. W., Gonzalez F. J. (2012). Expression and regulation of human fetal-specific CYP3A7 in mice. Endocrinology 153, 1453–1463. doi:10.1210/en.2011-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi J. M., Jounaidi Y., Drocourt L., Domergue J., Balabaud C., Maurel P., Vilarem M. J. (1999). Evidence for the presence of a functional pregnane X receptor response element in the CYP3A7 promoter gene. Biochem. Biophys. Res. Commun. 260, 377–381. doi:10.1006/bbrc.1999.0745. [DOI] [PubMed] [Google Scholar]
- Pozzi S., Rossetti S., Bistulfi G., Sacchi N. (2006). RAR-mediated epigenetic control of the cytochrome P450 Cyp26a1 in embryocarcinoma cells. Oncogene 25, 1400–1407. doi:10.1038/sj.onc.1209173. [DOI] [PubMed] [Google Scholar]
- Rasti M., Arabsolghar R., Khatooni Z., Mostafavi-Pour Z. (2012). p53 Binds to estrogen receptor 1 promoter in human breast cancer cells. Pathol. Oncol. Res. 18, 169–175. doi:10.1007/s12253-011-9423-6. [DOI] [PubMed] [Google Scholar]
- Rosen-Zvi M., Griffiths T., Steyvers M., Smyth P. (2004). The author-topic model for authors and documents. In Proceedings of the 20th conference on Uncertainty in artificial intelligence, AUAI Press, Banff, Canada, pp. 487–494. [Google Scholar]
- Talmage D. A., Lackey R. S. (1992). Retinoic acid receptor alpha suppresses polyomavirus transformation and c-fos expression in rat fibroblasts. Oncogene 7, 1837–1845. [PubMed] [Google Scholar]
- Tata J. R. (2002). Signalling through nuclear receptors. Nat. Rev. Mol. Cell Biol. 3, 702–710. [DOI] [PubMed] [Google Scholar]
- Tenbaum S., Baniahmad A. (1997). Nuclear receptors: structure, function and involvement in disease. Int. J. Biochem. Cell Biol. 29, 1325–1341. [DOI] [PubMed] [Google Scholar]
- Uehara T., Ono A., Maruyama T., Kato I., Yamada H., Ohno Y., Urushidani T. (2010). The Japanese toxicogenomics project: Application of toxicogenomics. Mol. Nutr. Food Res. 54, 218–227. doi:10.1002/mnfr.200900169. [DOI] [PubMed] [Google Scholar]
- Watson J. D., Prokopec S. D., Smith A. B., Okey A. B., Pohjanvirta R., Boutros P. C. (2014). TCDD dysregulation of 13 AHR-target genes in rat liver. Toxicol. Appl. Pharmacol. 274, 445–454. doi:10.1016/j.taap.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Yang Z., Zhang Y., Kemper J. K., Wang L. (2012). Cross-regulation of protein stability by p53 and nuclear receptor SHP. PLoS One 7, e39789 doi:10.1371/journal.pone.0039789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.