

Abstract
Abnormal blood rheological properties seldom occur in isolation and instead are accompanied by other complications, often designated as co-morbidities. In the metabolic syndrome with complications like hypertension, diabetes and lack of normal microvascular blood flow, the underlying molecular mechanisms that simultaneously lead to elevated blood pressure and diabetes as well as abnormal microvascular rheology and other cell dysfunctions have remained largely unknown. In this review, we propose a new hypothesis for the origin of abnormal cell functions as well as multiple co-morbidities. Utilizing experimental models for the metabolic disease with diverse co-morbidities we summarize evidence for the presence of an uncontrolled extracellular proteolytic activity that causes ectodomain receptor cleavage and loss of their associated cell function. We summarize evidence for unchecked degrading proteinase activity, e.g. due to matrix metalloproteases, in patients with hypertension, Type II diabetes and obesity, in addition to evidence for receptor cleavage in the form of receptor fragments and decreased extracellular membrane expression levels. The evidence suggest that a shift in blood rheological properties and other co-morbidities may in fact be derived from a common mechanism that is due to uncontrolled proteolytic activity, i.e. an early form of autodigestion. Identification of the particular proteases involved and the mechanisms of their activation may open the door to treatment that simultaneously targets multiple co-morbidities in the metabolic syndrome.
Keywords: Matrix metalloproteinase, serine protease, pancreatic digestive enzyme, cell rheology, mechanotransduction, hypertension, insulin resistance, rarefaction, autodigestion, spontaneously hypertensive rat
1. Introduction
The study of blood rheology is a foundation for analysis and understanding of abnormal microvascular perfusion. Cell mechanical properties, mechanotransduction mechanisms, adhesive properties, and cell interactions have served to understand diseased perfusion states. Frequently the conceptual link between abnormal properties of blood and organ dysfunctions has been assumed to be the result of a shift in the rheological properties of blood with abnormal cellular interactions resulting in a reduction of blood flow. Consequently the observation of abnormal blood rheological properties has lead to the concept that manipulation of blood cell properties may be a means to improve organ dysfunction by restoration of nutrient supply. Yet, after decades of blood rheological research, few new treatment modalities have arisen that are purely based on rheological principles. There is a need to understand the root cause of a shift in rheological properties of blood cells. It may be necessary to view a shift in rheological properties as secondary to a more fundamental pathophysiological event. While some suggestions have been advanced in this regard, e.g. oxygen free radicals, environmental stress, metabolic dysfunctions [1–3], to date no mechanism has been universally accepted. A particular opportunity to further understand the pathophysiology lies in the observation that often a shift in rheological properties of blood is accompanied by co-morbidities that are not necessarily directly derived from blood rheological mechanisms and a compromised blood perfusion that may be linked to them. A case is the metabolic syndrome X in which abnormal blood rheological properties have been identified [4,5].
One of the important and interesting characteristics inherent to the metabolic syndrome X is the simultaneous presence of diverse risk factors. For example, obesity and elevated levels of triglycerides are accompanied by elevated arterial blood pressure and diabetes [6,7] as well as a clustering of unrelated microvascular dysfunctions [8], including cell dysfunctions in endothelium, erythrocytes, leukocytes and platelets [9,10]. Cell dysfunctions appear in the same patient to different degrees and involve different membrane receptors and molecular signaling pathways. To date no hypothesis places the multifaceted dysfunctions encountered in the metabolic syndrome X under one concept. Development of such an overarching concept is of major importance for more effective treatments of patients. Without knowledge of the basic mechanisms governing cell dysfunctions in the metabolic syndrome, current treatment modalities are merely aimed at reducing symptoms of the disease. Such an approach leads to multi-drug treatment in the same patient, often with side effects. The situation can be contrasted with broader interventional opportunities against co-morbidities of the metabolic syndrome, e.g. calorie restriction [11].
We propose a common mechanism that may promote symptoms associated with diabetes, hypertension, microvascular and rheological cell dysfunctions. Our hypothesis will explain the origin of these dysfunctions and co-morbidities under one conceptual roof.
2. Inflammation as a tissue repair mechanism
An important starting point is inflammation, a phenomenon common to many diseases [12]. Clinical studies in the past two decades have served to demonstrate that inflammatory markers accompany a wide range of diseases and tissue complications, from acute injuries to chronic diseases, aging and organ failure [13,14]. The particular inflammatory markers used in such clinical studies include a variety of genetic and biochemical signals (e.g. oxygen free radicals, nuclear factor-kappa B, cytokines, C-reactive protein) [15–17], and include observations about rheological abnormalities of blood cells and vessels. While documented for decades in experimental studies, especially at the microvascular level, and now confirmed in clinical trials, the presence of inflammatory markers in such diverse organs and diseases is remarkable. What do markers of inflammation indicate about a disease?
Inflammation is a biological repair mechanism. Molecules like transcription factors, cytokines, C-reactive protein, growth factors etc. are synthesized by intact cells surrounding an injury sites that serves to restore the tissue. The presence of inflammatory cells, like neutrophils and macrophages, is part of the cascade that facilitates removal of cell debris after an injury and induce new growth of tissues and blood vessels. The entire inflammatory cascade is mediated by multiple families of proteinases, signaling molecules, adhesion molecules, growth factors, stem cells and others tissue growth mechanisms, all of them serve restoration of tissues.
If the inflammatory cascade is a repair process, what are the mechanisms leading to the initial tissue injury? This important issue comes into central focus. There are well-recognized mechanisms that cause injury to a tissue, from different types of trauma (punctures, abrations, tears, mechanical stress waves), to viral, bacterial and fungal infections, abnormal temperature or chemical exposures. All of these can cause changes to the rheological properties of blood. But this review is focused on a form of tissue injury that is different from these classical mechanisms. Our evidence supports the existence of an alternative mechanism designated as autodigestion [18].
3. Digestive enzymes – Friend or foe
Digestive enzymes are an essential component of the digestive track required to extract energy and molecular building blocks for an organism. They are optimized to break down macromolecules into smaller building blocks in order to facilitate absorption by the intestinal mucosa. Digestive enzymes are diverse and are found in the saliva, in the stomach, in the pancreatic juice secreted by pancreatic exocrine cells, and in the intestinal (small and large) secretions, or as part of the lining of the gastrointestinal tract. Based upon their substrate, digestive enzymes are divided into four major families: proteases (hydrolyze proteins into peptides and amino acids); lipases (break down fat into glycerol and fatty acids); carbohydrases (cleave carbohydrates into glucose) and nucleases, which split nucleic acids into nucleotides. The pancreas, as a major contributor to digestive enzymes in the intestine, produces (among others) highly active proteases such as trypsin, chymotrypsin, elastase, carboxypeptidase, lipase, amylase and several nucleases. These enzymes can digest proteins, fats and carbohydrates from many sources, including autologous tissues.
The following discussion is mostly focused on proteases and lipase as contributors to acute cell dysfunction in the circulation [19]. It should be noted that while digestive protease are able to clip the extracellular domain of membrane proteins, such as membrane receptors, and thereby undermine cell function, proteases do not necessarily cause cell death. Any experiment with cell cultures using trypsin to detach cells supports this idea. In contrast, lipases are able to generate free fatty acids which if unbound are highly cytotoxic irrespective of the cell type involved [20,21].
Beside proteases in the digestive tract, other families of circulating and membrane bound proteases are known to cleave proteins and thus may affect cellular functions. Among them are the matrix metalloproteinases, cathepsins, and serine proteases secreted both by the innate immunity cells and endothelial cells lining blood vessels. The following discussion will summarize rheological and other cardiovascular complications possibly caused by uncontrolled protease activity.
4. Compromise of blood rheology in the presence of degrading enzymes
The properties of blood cells may be strongly compromised by degrading proteases. When red blood cells are exposed temporarily to SHR plasma or MMPs they loose the glycocalyx and swell. When red blood cells are exposed to matrix metalloproteinases (MMPs) or serine proteases (trypsin and chymotrypsin) they aggregate in dextran (70 kDa at 15 gm/l without plasma) but less in fibrinogen (6 gm/l without plasma) [22]. In contrast, treatment of red cells with amylases produces fibrinogen-induced aggregation. MMP cleavage of red cell glycocalyx reduces their adhesion to macrophages as a mechanism to remove old red blood cells from the circulation [22].
Mechanosensing of neutrophils or macrophages and retraction of pseudopods under fluid shear stress depends on the formyl peptide receptor [23]. When this receptor is cleaved, neutrophils become non-responsive not only to formyl peptides (e.g. F-Met-Leu-Phe) but also to fluid shear stress [24]. Consequently one finds enhanced levels of pseudopod formation by leukocytes in the circulation and they are subject not only to entrapment in capillaries but also they cause increased capillary hemodynamic resistance by disturbing the motion of red cells in single file capillaries [25,26].
A similar compromise of mechanotransduction is observed in vitro in endothelial cells in the presence of serine protease, e.g. trypsin, due to cleavage of the glycocalyx and the vascular endothelial growth factor receptor (VEGFR-2) [27]. The presence of trypsin impairs the endothelial cells’ ability to align in the direction of fluid flow after 12 h of continuous shear stress. But endothelial cells realigned after an additional 12 h of shear stress with protease inhibition. Trypsin reduces the density of the glycocalyx and the insulin receptor under no shear and shear conditions, as well as the ectodomain of VEGFR-2 under fluid shear.
Platelets also change their function in response to degrading enzymes. Serine proteases such as thrombin, mediate platelet activation [28]. Treatment of platelets with MMPs cleaves their membrane surface glycoproteins and they exhibit a lag in response to stimuli such as thrombin [29]. Moreover, proteases secreted from other inflammatory cells, such as neutrophils, also have the capability to induce shape changes in platelets [30].
The question arise whether blood rheological alterations occur in isolation or whether they may arise in conjunction with other cell dysfunctions. To investigate this question it is necessary to examine models with multiple co-morbidities, like the spontaneously hypertensive rat (SHR).
5. Proteolytic receptor cleavage and co-morbidities in the spontaneously hypertensive rat
While extensively investigated for its elevated arterial blood pressure, the SHR is a model that has numerous microvascular dysfunctions [31–33], as well as insulin resistance [34,35]. Besides elevated white cell count, indices of inflammation and high levels of oxygen free radicals [36], this rat strain exhibits elevated levels of protease activity in plasma, including MMP-2, MMP-9 (gelatinase A and B), MMP-8, and MMP-7 (matrilysin) [35,37]. While the elevations of plasma MMP activity are modest (about 10 and 25% above the low pressure control strain) they are permanently elevated and accompanied by reduced levels of tissue inhibitor of metalloproteases 1 (TIMP-1,2) [38]. The enhanced MMP activity is accompanied by elevated MMP protein levels, as detected with immunohistochemistry [35]. In addition, there is evidence for elevated serine proteases in the plasma of the SHR [39].
MMPs were first characterized by their ability to cleave and remodel the extracellular matrix. Yet, studies have shown that other proteins including plasma membrane receptors are substrates to these enzymes [40]. Should the extracellular domain of a membrane receptor be cleaved by an extracellular protease, its function is compromised. Systematic analysis of this concept in our recent investigations points towards an array of receptors that are associated with the characteristic cell dysfunctions seen in hypertension and in the metabolic syndrome. Examples are as follows.
The SHR has constricted arterioles, which leads to elevation of its central arterial blood pressure [41,42]. It also has an attenuated response to β2 adrenergic receptor agonists and antagonists. The defective β2 adrenergic receptor signaling [43] was traced towards extracellular cleavage of the receptor. Exposure of the aorta and the heart muscle of control Wistar rats to plasma from SHRs, but not plasma from normotensive rats, reduces the number of extracellular domains detectable by immunolabeling, but not intracellular domains, of the β2 adrenergic receptor. If the SHR is chronically treated with MMP inhibitors (e.g. doxycycline at a sub-antibiotic dose level, CGS27023A), its plasma and endothelial MMP activity is reduced to levels of the normotensive WKY rats, its elevated blood pressure is normalized and its β2 adrenergic receptor population is restored [35,38].
Insulin resistance is a co-morbidity in the SHR [34], just like seen in many hypertensive patients [44]. In the SHR, insulin resistance is accompanied by reduced density of the extracellular domain of the insulin receptor [35]. Plasma of SHR rats cleaves the ectodomain of the insulin receptor on naive cells, suggestive of an active protease/s in the SHR plasma that is absent/inhibited in WKY plasma. In the SHR, the hyperglycemia and impaired transport of glucose into the cell, as a key marker for insulin resistance, is restored after MMP inhibition and so is the density of the extracellular insulin receptor domains [35].
The SHR also exhibits extensive apoptosis in its vasculature [45] including its capillary network, an effect that leads to loss of capillaries (rarefaction) [46]. In addition, the SHR has attenuated angiogenesis, which is associated with impaired VEGFR-2 signaling [47]. Labeling of the SHR extracellular, but not the intracellular, domain of the VEGFR-2 shows a reduced density in cardiac microvessels (arterioles and capillaries) and on the aortic endothelium [48]. SHR plasma also cleaves the extracellular domain of VEGFR-2 on naive control cells, a process also achieved by application of purified forms of MMP-7 and MMP-9. Chronic inhibition of MMP activity with doxycycline in the SHR abolishes the ability of its plasma to cleave the receptor, restores blood pressure and reduces endothelial apoptosis and facilitates capillary network growth [37].
Other membrane receptors are cleaved in the SHR. The SHR leukocytes exhibit an attenuated ability to roll on the endothelium in postcapillary venules [49–51]. This phenomenon is accompanied by proteolytic cleavage of P-selectin on endothelium and its counter receptor (P-selectin glycoprotein ligand-1) on leukocyte membranes [52]. The reduced density of endothelial P-selectin and ability for leukocytes to roll on the endothelium leads to its elevated leukocyte count in the circulation [53], just like seen in P-selectin knockout animals [54,55] or in human hypertension [56,57]. Normal receptor densities and rolling of leukocytes on the endothelium are restored by chronic MMP inhibition [52].
The resulting immune compromise due to reduced leukocyte-endothelial interaction and reduced leukocyte migration into interstitial tissue in acute inflammation are further enhanced by a lower density of β2 integrins on SHR neutrophils [35,49]. The reduced density of the β2 integrins and increased leukocyte counts in the circulation of the SHR are also normalized after chronic MMP inhibition [35]. The counter receptor to the β2 integrins, Intercellular Adhesion Molecule 1 (ICAM-1), is however upregulated in the SHR in an organ-specific fashion, although accumulation of ICAM-1 fragments was documented in the renal glomeruli indicating cleavage of the receptor in other tissues [58]. The accumulation of cleaved (i.e. soluble) receptors in the glomeruli may play a role in renal failure.
The shear stress response in form of pseudopod retraction is mediated by the f-Met-Leu-Phe receptor (FPR) [23]. It is impaired in SHR leukocytes and also in naïve control leukocytes exposed to SHR plasma [24,25]. The ectodomain of the FPR is cleaved in these cases and restored by chronic MMP inhibition together with its ability to respond to fluid shear stress [24].
The ectodomain of the scavenger receptor and free fatty acid transporter, CD36, is clipped in the SHR. One of the consequences is that the SHR has a reduced ability to adhere to and to remove swollen red cells from the circulation. Chronic MMP inhibition restores the receptor density and its ability to bind to the red cells [38].
6. Hypertension and cardiovascular remodeling
These studies in the SHR are in line with studies in humans, in which it was shown that elevated activity of MMP-2, MMP-9 and elastase is associated with elevated blood pressure and arterial stiffness [59,60] as well as with a prehypertensive state in obese women [61]. The focus on MMP activity is not only justified due to the possibility for receptor cleavage discussed above, but also since MMP activity promotes vascular smooth muscle cell migration and proliferation and was shown to be a player in the development of endothelial dysfunction [62]. These processes result in increased cardiovascular stiffness and hypertrophy. The MMP activity promotes long-lasting cardiovascular structural and functional alterations in both experimental and clinical hypertension, including activation of the renin-angiotensin-aldosterone system and oxidative stress. Therefore, MMP inhibition may prevent the deleterious effects of hypertension on the cardiovascular system [62].
An elevation of plasma MMP-9 in essential hypertension and a positive correlation between plasma MMP-9 and systolic blood pressure was observed [60]. Plasma levels of MMP-2 and MMP-10 are elevated in subjects with hypertensive-end stage renal disease with a positive correlation between plasma MMP-2 and plasma MMP-10. End stage renal disease patients (in many cases as a consequence of the metabolic syndrome) also have elevation of plasma serine proteases and cleavage of vascular endothelial cadherins (VE-Cadherins) [63]. Inhibition of MMPs reduces the over-expression of contractile proteins (myosin light chain kinase and myosin light chain II) and their transcriptional activators in insulin resistant vascular smooth muscle cells [64]. In a model of hypertension produced by high fructose diet fed rats, MMP-2 produces endothelial dysfunction and acquired systolic hypertension with insulin resistance that is inhibited by MMP blockade [65].
MMPs may not be the only unchecked proteases encountered in hypertension. In the salt-sensitive form of hypertension (Dahl salt sensitive rat), oral administration of a synthetic serine inhibitor (CM) decreases blood pressure with an elevation of the urinary sodium/potassium ratio [24]. It should also be noted that angiotensin-converting-enzyme (ACE) inhibitors, designed to block the conversion of angiotensin I to angiotensin II and used to treat hypertensive and diabetic conditions, also have MMP inhibitory activity [66], and therefore it is not certain which aspect of protease inhibition is relevant with respect to the pathogenesis in hypertension and diabetes. It may be the MMP inhibitory activity that makes ACE inhibitors effective in both of these two different diseases, in line with the concept of proteolytic receptor cleavage as the underlying mechanisms for these two co-morbidities, as seen in the SHR.
It is also interesting to note that oral administration of synthetic serine protease inhibitors (camostat mesilate and an active metabolite of camostat mesilate) significantly decreases blood pressure in Dahl salt-sensitive rats fed a high-salt diet. It result in a decrease in serum creatinine, reduction in urinary protein excretion, and improvement of renal injury markers such as collagen 1, collagen 3, transforming growth factor-β1, and nephrin. Thus the serine protease inhibitor has beneficial effects on both blood pressure and kidney injury in Dahl salt-sensitive rats [67].
Collectively this evidence suggest that hypertension may be mediated by several unchecked degrading protease causing not only an elevated central blood pressure but also a multitude of cell dysfunctions, including microvascular and blood rheological abnormalities.
7. Protease activity in obesity
Like hypertension, obesity is also accompanied by elevated protease activity in plasma and other organs. There are a number of observations on patients in the literature. The development of adipose tissue is mostly mediated by MMPs. MMP-2, MMP-3, MMP-12, MMP-14, MMP-19, and a tissue inhibitor of metalloproteinases (TIMP-1), especially in obese adipose tissues as compared to lean tissues. In contrast, MMP-7 and TIMP-3 mRNAs are markedly decreased in obesity. It was also found that in adipose tissue, the overall balance of MMPs and their inhibitors favors tissue remodeling [68]. Other than MMPs, serine proteases positively regulate adipocyte differentiation in vitro, as well as in vivo [69]. While different reports support MMP activity in mediating adipose tissue development in obesity, not all studies utilizing MMP inhibitors were able to show the anticipated results; e.g. treating adipocytes with MMP inhibitors by neutralizing antibodies decreased differentiation, suggesting that MMP activities were required for adipocyte conversion [70]. The addition of TIMP-1 or GM6001 (a general MMP inhibitor) accelerates adipocytes differentiation [71].
In plasma of obese patients, elevated levels of MMP-2 and MMP-9 may reflect abnormal extracellular matrix metabolism and an inflammatory state [72]. In contrast, MMP-8 levels in obese women are low despite neutrophil activation measured by myeloperoxidase activity [73]. Beside MMPs, plasma of obese individuals has elevated levels of other enzymes, such as serine proteases, originating from activated leukocytes. In that regard, elastase secreted from activated neutrophils was shown to mediate insulin resistance in a high fat diet-induced obesity mouse model [74].
A role for degrading enzymes was also suggested in the brain of obese individuals. MMP-9 was shown to promote brain microvascular disruption and permeability of the blood brain barrier [75]. The enzyme prolylcarboxypeptidase (PRCP) degrades α-melanocyte-stimulating hormone (α-MSH) to an inactive form that is unable to inhibit food intake thus promoting obesity [76].
Various studies show the contribution of proteolytic enzymes to the progression of obesity. A functional role for MMP-2 was described and mice that are lacking this enzyme gain less weight after high fat diet compared to wild type controls [77]. Moreover, inhibition of this protease in an insulin resistance model was shown to improve endothelial dysfunction and prevent hypertension [65].
The observations for proteolytic receptor cleavage are supported also by our preliminary experimental studies with a high fat diet. Rats kept on high fat diet for a period of 12 weeks exhibit protease activity in their small intestine and also in peripheral organs, e.g. skeletal muscle (Fig. 1). This is associated with a reduced level of the ectodomain insulin receptor, e.g. on blood vessels or on circulating cells (Figs 2, 3(A)), and leads to reduced transport of fluorescent glucose analogs into the cell cytoplasm (Fig. 3(B), (C)). It is associated with the appearance of insulin receptor fragments detectable by Western blot analysis (Fig. 4) in spite of the possibility that insulin fragments in plasma (sometimes designated as soluble insulin receptors) [78–80] may be fragmented to a degree that antibody binding is not possible and they remain undetected.
Fig. 1.
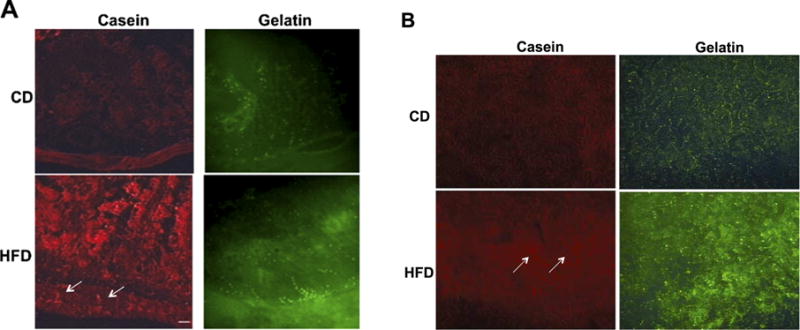
(A) In situ zymography (with fluorescently quenched casein and gelatin in red and green fluorescence, respectively) of the small intestine of rats fed high fat diet (HFD, with 60% calorie intake from fat; Harlan, USA) and a normal control diet (CD) for 12 weeks. Sections are oriented with the muscularis at the bottom of each panel and villi pointing upwards. Casein degrading enzymes activity (left) was low in villi of animals on CD and is enhanced after HFD with penetration to the muscular layer (arrows). Gelatinases activity (right) was present along the villi in CD, but in HFD, elevated activity is recorded at the base of the villi, next to the muscular layer. (B) In-situ zymography on skeletal muscle (gastrocnemius) of rats on CD and HFD for 12 weeks. Conditions are the same as in Fig. 1(A).
Fig. 2.
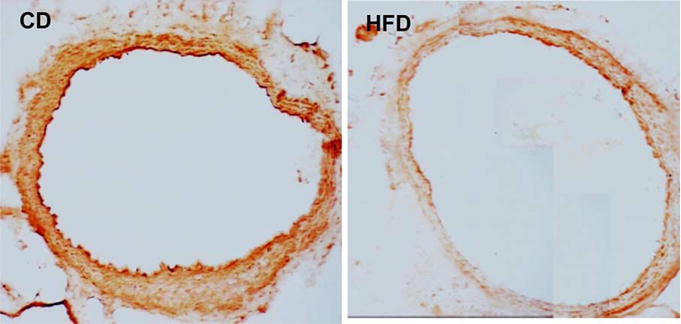
Immunohistochemical sections with primary antibody against the extracellular domain of the insulin receptor (InRα, N-20, sc-710 polyclonal antibody mapping to the N terminus, Santa Cruz Biotechnology) followed by biotin-avidin label [35] showing the expression density of the insulin receptor ectodomain in aortic rings from rats fed HFD (left) or CD (right) for 12 weeks.
Fig. 3.
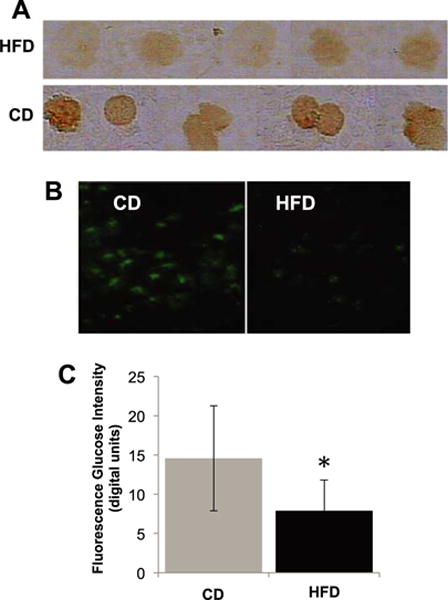
(A) Extracellular domain immunolabel density of the insulin receptor (same techniques as in Fig. 2). (B) Glucose uptake (non-hydrolyzable glucose analog 6-NBD-deoxyglucose; according to Methods described in [35]) in fresh isolated leukocytes after lysis of red blood cells from rats fed HFD and CD for 12 weeks with (C) digital fluorescent intensity measurements. The leukocytes were exposed to the fluorescent glucose analog for a period of 30 min. Note the reduced insulin receptor density and attenuated transmembrane glucose transport into the cells after exposure to plasma of rats on HFD compared to CD. *p < 0.05 HFD vs. CD by student t test. N = 4 rats/group.
Fig. 4.
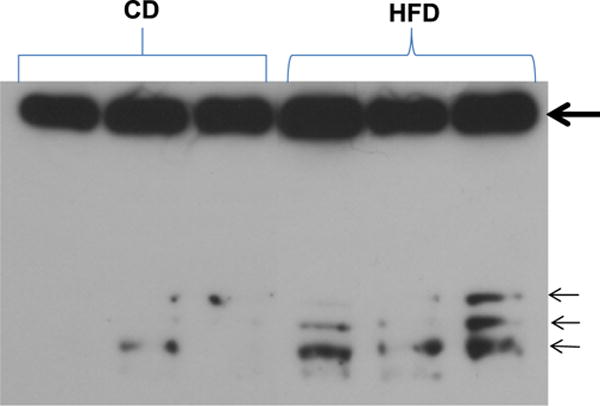
Western blot showing insulin receptor alpha in plasma of rats fed HFD and CD for 12 weeks. In addition to the expected upper band (thick arrow, 130 KDa), three additional bands (small arrows) are present in plasma of HFD rats and low or absent in the plasma of rats on CD. The bands indicate additional cleavage products of the insulin receptor alpha and require further mass-spec analysis. (Colors are visible in the online version of the article; http://dx.doi.org/10.3233/BIR-15045.)
8. Receptor cleavage in diabetes and insulin resistance
The fat sand rat (Psammomys obesus) is an animal that naturally consumes low levels of calories and upon feeding normal Western diet, will develop Type II diabetes, insulin receptor cleavage, elevated plasma glucose levels and glycated hemoglobin levels. In these rats, MMP inhibition (doxycycline) serves to rescue the extracellular domain of the insulin receptor, and reduces the blood glucose and glycated hemoglobin levels. This evidence supports the hypothesis that calorie overconsumption in this model causes ectodomain cleavage of the insulin receptor as seen in the SHR [39]. The observation is especially interesting in light of the extensive body of evidence that calorie restriction has a beneficial effect on cardiovascular risk factors; hypertension as well as diabetes [81–83].
In an alternative transgenic model of diabetes with a leptin receptor deficiency, which shows hyperglycemia and obesity [84,85], we found on average a 19% reduction of the insulin receptor labeling density compared to control. Thus genetic defects associated with insulin resistance may also involve cleavage of the extracellular domain of the insulin receptor [39].
The underlying mechanism for the lack of insulin response in these two models and in the SHR may be an uncontrolled degrading protease activity in the circulation and in the tissue parenchyma. Significantly different models of Type II diabetes and patients have evidence for cleavage of the insulin receptor [78] as well as VEGFR-2, leptin receptor and others [86–89].
Supporting the notion that MMPs are upregulated in many forms of the metabolic syndrome, recent studies in humans have shown that short-term MMP inhibition in diabetic patients serves to improve insulin receptor expression and signaling concomitantly decreasing the level of inflammatory markers [90].
Other potentially degrading proteases may also be involved in metabolic and cardiovascular disease. Neutrophils are major carriers of proteases. Treatment of hepatocytes with neutrophil elastase causes cellular insulin resistance [74]. Tryptase and chymase are serine proteinase ubiquitously stored in cytoplasmic granules of tissue mast cells. Mast cells participate in atherogenesis [91]. These enzymes are locally activated and released into the microenvironment, where they can act on the various extracellular targets, i.e. activate pro-MMPs, cleave extracellular peptides and degrade lipoproteins and fibronectin [92]. Circulating tryptase levels are positively correlated with subclinical atherosclerosis and have a positive correlation with obesity and insulin resistance [93]. Moreover, their levels are an independent risk factor for pre-diabetes and diabetes mellitus [94].
While some specific pathways for MMP activation in the metabolic syndrome have been suggested [95], the in-vivo mechanisms for MMP activations are still uncertain. In vitro studies show that MMP-1, MMP-2, and MMP-9 are highly expressed by diabetic cultured endothelial cells and monocyte-derived macrophages after exposure to an elevated glucose concentration [96]. Yet, this evidence does not explain the primary cause for elevated blood glucose values in diabetes, which requires a mechanism that independently impairs the glucose transport. Our proposed mechanism which include proteolytic cleavage of receptors ectodomains leads inevitably to formation of soluble receptor fragments for which there is ample evidence in diabetic patients [78].
There are other degrading protease subfamilies in the plasma and in the tissue that may be involved in receptor shedding and the cell dysfunctions characteristic for the metabolic syndrome. The type I transmembrane metalloproteinase ADAM17 (A Disintegrin and A Metalloproteinase 17) plays a role in atherosclerosis and insulin resistance [97], cleaves the ectodomain transmembrane proteins and causes receptor shedding [98]. Plasma levels of granzyme B, a serine protease in T-lymphocytes, macrophages, mast cells and others, are elevated in type 2 diabetics with abdominal obesity and significantly correlate with the levels of the soluble insulin receptor fragments, supporting the idea that the receptor may be proteolytically clipped in these patients [97]. Neutrophils, that are primed in the metabolic syndrome, have been shown to undergo degranulation, releasing degrading enzymes to the surrounding milieu and promoting protein cleavage and progression of cell dysfunctions and disease [74].
It is interesting to note that hyperglycemia generates only low or undetectable levels or soluble insulin receptors in vitro in several cell lines, suggesting that glucose per se has low ability to promote cleavage of the insulin receptor [80]. Instead a protease appears to be present in-vivo that clips the receptor ectodomain and elevation of the blood glucose is a consequence.
9. Mechanisms for protease activation
A critical issue that comes into focus in this discussion about uncontrolled protease activity is the mechanisms for generation of active degrading enzymes in the circulation. Understanding these mechanisms will be a guide towards prevention and early interventions. The mechanisms likely depend on the specific medical conditions and are largely unknown.
In the case of the SHR preliminary evidence suggests that serine protease may leak directly out of the pancreas into the central circulation (A. Chan, G.W. Schmid-Schönbein, unpublished results). Serine proteases, like trypsin, are expected to be blocked in part by serpins (e.g. anti-alpha trypsin), but more refined measurements suggest that such native inhibition may be not entirely complete and may also be transient over the course of a day [99]. Even low concentrations of trypsin can convert proenzymes into active enzymes (like in the case for MMPs activation). Thus, digestive proteases leaking from the pancreas may serve as a mechanism for elevation of MMP activity in the SHR, although this hypothesis remains to be investigated.
It is interesting to note in this context the case of physiological shock and sepsis, a life threatening condition, in which elevated protease activity can be detected in the circulation. In this case a key source of serine proteases in the circulation and in peripheral organs is the pancreatic digestive enzymes [100, 101]. They are derived predominantly from the small intestine where they are fully activated and present in high concentrations as essential part of normal digestion. Their escape into the central circulation is facilitated by a breakdown of the mucosal barrier in the small intestine [102,103] under conditions that compromise the barrier properties of the mucins and the epithelial cells lining the intestinal villi and cause autodigestion and insulin resistance [104]. The dramatic unchecked proteolytic activity in shock with severe autodigestion is but an extreme case of the more subtle lower level of receptor cleavage in chronic conditions, suggesting that the metabolic syndrome is accompanied by a milder and early form of autodigestion.
10. Conclusion
There is increasing evidence for enhanced proteolytic activity in the metabolic syndrome that is associated with receptor cleavage and loss of cell functions. The mechanism may serve as a step towards understanding the root cause of the spectrum of patho-physiologies involving blood rheology and diversity of cell dysfunction in chronic diseases, including hypertension, diabetes, the metabolic syndrome. Protease activation and receptor cleavage will have different consequences based on the tissue and specific receptors/proteases involved. Receptor cleavage is followed by reconstitution of receptors in the cell membrane after biosynthesis in the cytoplasm and therefore is a dynamic process involving autophagy and neobiosythnthesis. The dynamics of such receptor turnover remain to be studied in specific diseases and cell types. Identifying the exact proteases that are responsible for receptor cleavage and the sequence of activation of these proteases is of great importance and needs to be in the center of metabolic disease research.
Acknowledgments
Supported in part by NIH grants HL 10881 and GM 67825 and grants from the American Heart Association.
Footnotes
Conflict of interest
Dr. Schmid-Schönbein owns stock in Leading Bioscience, Inc., San Diego, CA, which is involved in development of shock treatments.
References
- 1.Zappulla D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? Journal of the Cardiometabolic Syndrome. 2008;3(1):30–4. doi: 10.1111/j.1559-4572.2008.07263.x. [DOI] [PubMed] [Google Scholar]
- 2.Brun JF. Hormones, metabolism and body composition as major determinants of blood rheology: Potential pathophysiological meaning. Clinical Hemorheology and Microcirculation. 2002;26(2):63–79. [PubMed] [Google Scholar]
- 3.Mozsik G, Fiegler M, Juricskay I, Mezey B, Toth K. Oxygen free radicals, lipid metabolism, and whole blood and plasma viscosity in the prevention and treatment of human cardiovascular diseases. Bibliotheca Nutritio et Dieta. 1992;49:111–24. doi: 10.1159/000421440. [DOI] [PubMed] [Google Scholar]
- 4.Lowe GD. Blood viscosity and cardiovascular disease. Thrombosis and Haemostasis. 1992;67(5):494–8. [PubMed] [Google Scholar]
- 5.Sugimori H, Tomoda F, Koike T, Kinuno H, Kurosaki H, Masutani T, Inoue H. Blood rheology and platelet function in untreated early-stage essential hypertensives complicated with metabolic syndrome. International Journal of Hypertension. 2012;2012:109830. doi: 10.1155/2012/109830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizzo M, Rizvi AA, Rini GB, Berneis K. The therapeutic modulation of atherogenic dyslipidemia and inflammatory markers in the metabolic syndrome: What is the clinical relevance? Acta Diabetol. 2009;46(1):1–11. doi: 10.1007/s00592-008-0057-4. [DOI] [PubMed] [Google Scholar]
- 7.Mule G, Cottone S, Nardi E, Andronico G, Cerasola G. Metabolic syndrome in subjects with essential hypertension: Relationships with subclinical cardiovascular and renal damage. Minerva Cardioangiol. 2006;54(2):173–94. [PubMed] [Google Scholar]
- 8.Serne EH, de Jongh RT, Eringa EC, Ijzerman RG, de Boer MP, Stehouwer CD. Microvascular dysfunction: Causative role in the association between hypertension, insulin resistance and the metabolic syndrome? Essays in Biochemistry. 2006;42:163–76. doi: 10.1042/bse0420163. [DOI] [PubMed] [Google Scholar]
- 9.Chien S. Blood rheology in myocardial infarction and hypertension. Biorheology. 1986;23(6):633–53. doi: 10.3233/bir-1986-23614. [DOI] [PubMed] [Google Scholar]
- 10.Levy Y, Elias N, Cogan U, Yeshurun D. Abnormal erythrocyte rheology in patients with morbid obesity. Angiology. 1993;44(9):713–7. doi: 10.1177/000331979304400907. [DOI] [PubMed] [Google Scholar]
- 11.Fontana L. Calorie restriction and cardiometabolic health. Eur J Cardiovasc Prev Rehabil. 2008;15(1):3–9. doi: 10.1097/HJR.0b013e3282f17bd4. [DOI] [PubMed] [Google Scholar]
- 12.Schmid-Schönbein GW. Analysis of inflammation. Annual Review of Biomedical Engineering. 2006;8:93–131. doi: 10.1146/annurev.bioeng.8.061505.095708. [DOI] [PubMed] [Google Scholar]
- 13.Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Research and Clinical Practice. 2014;105(2):141–50. doi: 10.1016/j.diabres.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 2014;69(Suppl 1):S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 15.Jung UJ, Choi MS. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. International Journal of Molecular Sciences. 2014;15(4):6184–223. doi: 10.3390/ijms15046184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metabolism. 2011;13(1):11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haffner SM. The metabolic syndrome: Inflammation, diabetes mellitus, and cardiovascular disease. American Journal of Cardiology. 2006;97(2A):3A–11A. doi: 10.1016/j.amjcard.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 18.Schmid-Schönbein GW, Chang M. The autodigestion hypothesis for shock and multi-organ failure. Annals of Biomedical Engineering. 2014;42(2):405–14. doi: 10.1007/s10439-013-0891-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kramp WJ, Waldo S, Schmid-Schönbein GW, Hoyt D, Coimbra R, Hugli TE. Characterization of two classes of pancreatic shock factors: Functional differences exhibited by hydrophilic and hydrophobic shock factors. Shock. 2003;20(4):356–62. doi: 10.1097/01.shk.0000082442.66379.90. [DOI] [PubMed] [Google Scholar]
- 20.Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. 2007;27(3):296–304. doi: 10.1097/01.shk.0000235139.20775.7f. [DOI] [PubMed] [Google Scholar]
- 21.Penn AH, Altshuler AE, Small JW, Taylor SF, Dobkins KR, Schmid-Schönbein GW. Digested formula but not digested fresh human milk causes death of intestinal cells in vitro: Implications for necrotizing enterocolitis. Pediatric Research. 2012;72(6):560–7. doi: 10.1038/pr.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pot C, Chen AY, Ha JN, Schmid-Schönbein GW. Proteolytic cleavage of the red blood cell glycocalyx in a genetic form of hypertension. Cellular and Molecular Bioengineering. 2011;4(4):678–92. doi: 10.1007/s12195-011-0180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makino A, Prossnitz ER, Bünemann M, Wang JM, Yao W, Schmid-Schönbein GW. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. American Journal of Physiology. Cell Physiology. 2006;290:C1633–9. doi: 10.1152/ajpcell.00576.2005. [DOI] [PubMed] [Google Scholar]
- 24.Chen AY, DeLano FA, Valdez SR, Ha JN, Shin HY, Schmid-Schönbein GW. Receptor cleavage reduces the fluid shear response in neutrophils of the spontaneously hypertensive rat. American Journal of Physiology. Cell Physiology. 2010;299(6):C1441–9. doi: 10.1152/ajpcell.00157.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukuda S, Yasu T, Kobayashi N, Ikeda N, Schmid-Schönbein GW. Contribution of fluid shear response in leukocytes to hemodynamic resistance in the spontaneously hypertensive rat. Circulation Research. 2004;95(1):100–8. doi: 10.1161/01.RES.0000133677.77465.38. [DOI] [PubMed] [Google Scholar]
- 26.Helmke BP, Bremner SN, Zweifach BW, Skalak R, Schmid-Schönbein GW. Mechanisms for increased blood flow resistance due to leukocytes. American Journal of Physiology. 1997;273(6 Pt 2):H2884–90. doi: 10.1152/ajpheart.1997.273.6.H2884. [DOI] [PubMed] [Google Scholar]
- 27.Altshuler AE, Morgan MJ, Chien S, Schmid-Schönbein GW. Proteolytic activity attenuates the response of endothelial cells to fluid shear stress. Cellular and Molecular Bioengineering. 2012;5(1):82–91. doi: 10.1007/s12195-011-0207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nylander S, Mattsson C. Thrombin-induced platelet activation and its inhibition by anticoagulants with different modes of action. Blood Coagulation & Fibrinolysis: An International Journal in Haemostasis and Thrombosis. 2003;14(2):159–67. doi: 10.1097/00001721-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 29.McGowan EB, Detwiler TC. Differential effect of Serratia protease on platelet surface glycoproteins Ib and V. Blood. 1985;65(4):1033–5. [PubMed] [Google Scholar]
- 30.Peng X, Ramstrom S, Kurz T, Grenegard M, Segelmark M. The neutrophil serine protease PR3 induces shape change of platelets via the Rho/Rho kinase and Ca(2+) signaling pathways. Thrombosis Research. 2014;134(2):418–25. doi: 10.1016/j.thromres.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki H, Zweifach BW, Schmid-Schönbein GW. The multifaceted contribution of microvascular abnormalities to the pathophysiology of the hypertensive syndrome. In: Zanchetti A, Mancia G, editors. Handbook of hypertension, Vol 17, Pathopysiology of hypertension. Amsterdam: Elsevier Science B.V.; 1997. pp. 482–523. [Google Scholar]
- 32.Gunduz F, Baskurt OK, Meiselman HJ. Vascular dilation responses of rat small mesenteric arteries at high intravascular pressure in spontaneously hypertensive rats. Circulation Journal: Official Journal of the Japanese Circulation Society. 2009;73(11):2091–7. doi: 10.1253/circj.cj-09-0392. [DOI] [PubMed] [Google Scholar]
- 33.Lominadze D, Joshua IG, Schuschke DA. Blood flow shear rates in arterioles of spontaneously hypertensive rats at early and established stages of hypertension. Clin Exp Hypertens. 2001;23(4):317–28. doi: 10.1081/ceh-100102670. [DOI] [PubMed] [Google Scholar]
- 34.Hulman S, Falkner B, Freyvogel N. Insulin resistance in the conscious spontaneously hypertensive rat: Euglycemic hyperinsulinemic clamp study. Metabolism: Clinical and Experimental. 1993;42(1):14–8. doi: 10.1016/0026-0495(93)90165-k. [DOI] [PubMed] [Google Scholar]
- 35.DeLano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage: Mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. 2008;52(2):415–23. doi: 10.1161/HYPERTENSIONAHA.107.104356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi N, DeLano FA, Schmid-Schönbein GW. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(10):2114–21. doi: 10.1161/01.ATV.0000178993.13222.f2. [DOI] [PubMed] [Google Scholar]
- 37.Tran ED, DeLano FA, Schmid-Schönbein GW. Enhanced matrix metalloproteinase activity in the spontaneously hypertensive rat: VEGFR-2 cleavage, endothelial apoptosis, and capillary rarefaction. Journal of Vascular Research. 2010;47(5):423–31. doi: 10.1159/000281582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santamaria MH, Chen AY, Chow J, Muñoz DC, Schmid-Schönbein GW. Cleavage and reduced CD36 ectodomain density on heart and spleen macrophages in the spontaneously hypertensive rat. Microvascular Research. 2014;95:131–42. doi: 10.1016/j.mvr.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delano FA, Zhang H, Tran EE, Zhang C, Schmid-Schönbein GW. A new hypothesis for insulin resistance in hypertension due to receptor cleavage. Expert Review of Endocrinology & Metabolism. 2010;5(1):149–58. doi: 10.1586/eem.09.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Research. 2006;69(3):562–73. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Schmid-Schönbein GW, Zweifach BW, DeLano FA, Chen P. Microvascular tone in a skeletal muscle of spontaneously hypertensive rats. Hypertension. 1987;9:164–71. doi: 10.1161/01.hyp.9.2.164. [DOI] [PubMed] [Google Scholar]
- 42.Zweifach BW, Lipowsky HH. Pressure-flow relations in blood and lymph microcirculation. In: Renkin EM, Michel CC, editors. Handbook of physiology, Section 2: The cardiovascular system. Vol. 4. Microcirculation I. Bethesda, MD: American Physiological Society; 1984. pp. 251–307. [Google Scholar]
- 43.Rodrigues SF, Tran ED, Fortes ZB, Schmid-Schönbein GW. Matrix metalloproteinases cleave the beta2-adrenergic receptor in spontaneously hypertensive rats. American Journal of Physiology. Heart and Circulatory Physiology. 2010;299(1):H25–35. doi: 10.1152/ajpheart.00620.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reaven GM. Relationships among insulin resistance, type 2 diabetes, essential hypertension, and cardiovascular disease: Similarities and differences. J Clin Hypertens (Greenwich) 2011;13(4):238–43. doi: 10.1111/j.1751-7176.2011.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim HH, DeLano FA, Schmid-Schönbein GW. Life and death cell labeling in the microcirculation of the spontaneously hypertensive rat. Journal of Vascular Research. 2001;38(3):228–36. doi: 10.1159/000051051. [DOI] [PubMed] [Google Scholar]
- 46.Tran ED, Schmid-Schönbein GW. An in-vivo analysis of capillary stasis and endothelial apoptosis in a model of hypertension. Microcirculation. 2007;14(8):793–804. doi: 10.1080/10739680701419992. [DOI] [PubMed] [Google Scholar]
- 47.Wang H, Olszewski B, Rosebury W, Wang D, Robertson A, Keiser JA. Impaired angiogenesis in SHR is associated with decreased KDR and MT1-MMP expression. Biochemical and Biophysical Research Communications. 2004;315(2):363–8. doi: 10.1016/j.bbrc.2004.01.059. [DOI] [PubMed] [Google Scholar]
- 48.Tran ED, Yang M, Chen A, Delano FA, Murfee WL, Schmid-Schönbein GW. Matrix metalloproteinase activity causes VEGFR-2 cleavage and microvascular rarefaction in rat mesentery. Microcirculation. 2011;18(3):228–37. doi: 10.1111/j.1549-8719.2011.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arndt H, Smith CW, Granger DN. Leukocyte-endothelial cell adhesion in spontaneously hypertensive and normotensive rats. Hypertension. 1993;21(5):667–73. doi: 10.1161/01.hyp.21.5.667. [DOI] [PubMed] [Google Scholar]
- 50.Suematsu M, Suzuki H, Delano FA, Schmid-Schönbein GW. The inflammatory aspect of the microcirculation in hypertension: Oxidative stress, leukocytes/endothelial interaction, apoptosis. Microcirculation. 2002;9(4):259–76. doi: 10.1038/sj.mn.7800141. [DOI] [PubMed] [Google Scholar]
- 51.Suematsu M, Suzuki H, Tamatani T, Iigou Y, DeLano FA, Miyasaka M, Forrest MJ, Kannagi R, Zweifach BW, Ishimura Y, et al. Impairment of selectin-mediated leukocyte adhesion to venular endothelium in spontaneously hypertensive rats. The Journal of Clinical Investigation. 1995;96(4):2009–16. doi: 10.1172/JCI118248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen AY, Ha JN, Delano FA, Schmid-Schönbein GW. Receptor cleavage and P-selectin-dependent reduction of leukocyte adhesion in the spontaneously hypertensive rat. Journal of Leukocyte Biology. 2012;92(1):183–94. doi: 10.1189/jlb.0112010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmid-Schönbein GW, Seiffge D, DeLano FA, Shen K, Zweifach BW. Leukocyte counts and activation in spontaneously hypertensive and normotensive rats. Hypertension. 1991;17:323–30. doi: 10.1161/01.hyp.17.3.323. [DOI] [PubMed] [Google Scholar]
- 54.Mayadas TN. Gene knockout on P-selectin: Its biology and function. Trends in Cardiovascular Medicine. 1995;5(4):149–57. doi: 10.1016/1050-1738(95)00057-G. [DOI] [PubMed] [Google Scholar]
- 55.Wagner DD. P-selectin knockout: A mouse model for various human diseases. Ciba Foundation Symposium. 1995;189:2–10. doi: 10.1002/9780470514719.ch2. discussion 10–16, 77–18. [DOI] [PubMed] [Google Scholar]
- 56.Friedman GD, Selby JV, Quesenberry CP., Jr The leukocyte count: A predictor of hypertension. Journal of Clinical Epidemiology. 1990;43:907–11. doi: 10.1016/0895-4356(90)90074-y. [DOI] [PubMed] [Google Scholar]
- 57.Gillum RF, Mussolino ME. White blood cell count and hypertension incidence. The NHANES I epidemiologic followup study. Journal of Clinical Epidemiology. 1994;47(8):911–9. doi: 10.1016/0895-4356(94)90195-3. [DOI] [PubMed] [Google Scholar]
- 58.Tong S, Neboori HJ, Tran ED, Schmid-Schönbein GW. Constitutive expression and enzymatic cleavage of ICAM-1 in the spontaneously hypertensive rat. Journal of Vascular Research. 2011;48(5):386–96. doi: 10.1159/000323474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yasmin, McEniery CM, Wallace S, Dakham Z, Pulsalkar P, Maki-Petaja K, Ashby MJ, Cockcroft JR, Wilkinson IB. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(2):372. doi: 10.1161/01.ATV.0000151373.33830.41. [DOI] [PubMed] [Google Scholar]
- 60.Friese RS, Rao F, Khandrika S, Thomas B, Ziegler MG, Schmid-Schönbein GW, O’Connor DT. Matrix metalloproteinases: Discrete elevations in essential hypertension and hypertensive end-stage renal disease. Clin Exp Hypertens. 2009;31(7):521–33. doi: 10.3109/10641960802668730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.El-Eshmawy MM, El-Adawy EH, Mousa AA, Zeidan AE, El-Baiomy AA, Abdel-Samie ER, Saleh OM. Elevated serum neutrophil elastase is related to prehypertension and airflow limitation in obese women. BMC Women’s Health. 2011;11:1. doi: 10.1186/1472-6874-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castro MM, Tanus-Santos JE. Inhibition of matrix metalloproteinases (MMPs) as a potential strategy to ameliorate hypertension-induced cardiovascular alterations. Current Drug Targets. 2013;14(3):335–43. doi: 10.2174/1389450111314030005. [DOI] [PubMed] [Google Scholar]
- 63.Cohen-Mazor M, Mazor R, Kristal B, Sela S. Elastase and cathepsin G from primed leukocytes cleave vascular endothelial cadherin in hemodialysis patients. BioMed Research International. 2014;2014:459–640. doi: 10.1155/2014/459640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagareddy PR, MacLeod KM, McNeill JH. GPCR agonist-induced transactivation of the EGFR upregulates MLC II expression and promotes hypertension in insulin-resistant rats. Cardiovascular Research. 2010;87(1):177–86. doi: 10.1093/cvr/cvq030. [DOI] [PubMed] [Google Scholar]
- 65.Nagareddy PR, Rajput PS, Vasudevan H, McClure B, Kumar U, Macleod KM, McNeill JH. Inhibition of matrix metalloproteinase-2 improves endothelial function and prevents hypertension in insulin-resistant rats. British Journal of Pharmacology. 2012;165(3):705–15. doi: 10.1111/j.1476-5381.2011.01583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin Y, Han HC, Lindsey ML. ACE inhibitors to block MMP-9 activity: New functions for old inhibitors. Journal of Molecular and Cellular Cardiology. 2007;43(6):664–6. doi: 10.1016/j.yjmcc.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maekawa A, Kakizoe Y, Miyoshi T, Wakida N, Ko T, Shiraishi N, Adachi M, Tomita K, Kitamura K. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt-sensitive hypertension. J Hypertens. 2009;27(1):181–9. doi: 10.1097/hjh.0b013e328317a762. [DOI] [PubMed] [Google Scholar]
- 68.Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, Tartare-Deckert S. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. The Journal of Biological Chemistry. 2003;278(14):11888–96. doi: 10.1074/jbc.M209196200. [DOI] [PubMed] [Google Scholar]
- 69.Selvarajan S, Lund LR, Takeuchi T, Craik CS, Werb Z. A plasma kallikrein-dependent plasminogen cascade required for adipocyte differentiation. Nature Cell Biology. 2001;3(3):267–75. doi: 10.1038/35060059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bouloumie A, Sengenes C, Portolan G, Galitzky J, Lafontan M. Adipocyte produces matrix metalloproteinases 2 and 9: Involvement in adipose differentiation. Diabetes. 2001;50(9):2080–6. doi: 10.2337/diabetes.50.9.2080. [DOI] [PubMed] [Google Scholar]
- 71.Alexander CM, Selvarajan S, Mudgett J, Werb Z. Stromelysin-1 regulates adipogenesis during mammary gland involution. The Journal of Cell Biology. 2001;152(4):693–703. doi: 10.1083/jcb.152.4.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Derosa G, Ferrari I, D’Angelo A, Tinelli C, Salvadeo SA, Ciccarelli L, Piccinni MN, Gravina A, Ramondetti F, Maffioli P, et al. Matrix metalloproteinase-2 and -9 levels in obese patients. Endothelium: Journal of Endothelial Cell Research. 2008;15(4):219–24. doi: 10.1080/10623320802228815. [DOI] [PubMed] [Google Scholar]
- 73.Andrade VL, Petruceli E, Belo VA, Andrade-Fernandes CM, Caetano Russi CV, Bosco AA, Tanus-Santos JE, Sandrim VC. Evaluation of plasmatic MMP-8, MMP-9, TIMP-1 and MPO levels in obese and lean women. Clinical Biochemistry. 2012;45(6):412–5. doi: 10.1016/j.clinbiochem.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 74.Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nature Medicine. 2012;18(9):1407–12. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McColl BW, Rose N, Robson FH, Rothwell NJ, Lawrence CB. Increased brain microvascular MMP-9 and incidence of haemorrhagic transformation in obese mice after experimental stroke. Journal of Cerebral Blood Flow and Metabolism. 2010;30(2):267–72. doi: 10.1038/jcbfm.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu ZW, Gao XB, Diament A, Haus KA, Shariat-Madar Z, et al. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of Clinical Investigation. 2009;119(8):2291–303. doi: 10.1172/JCI37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Hul M, Lijnen HR. A functional role of gelatinase A in the development of nutritionally induced obesity in mice. Journal of Thrombosis and Haemostasis. 2008;6(7):1198–206. doi: 10.1111/j.1538-7836.2008.02988.x. [DOI] [PubMed] [Google Scholar]
- 78.Group TSRS. Soluble insulin receptor ectodomain is elevated in the plasma of patients with diabetes. Diabetes. 2007;56(8):2028–35. doi: 10.2337/db07-0394. [DOI] [PubMed] [Google Scholar]
- 79.Hiriart M, Sanchez-Soto C, Diaz-Garcia CM, Castanares DT, Avitia M, Velasco M, Mas-Oliva J, Macias-Silva M, Gonzalez-Villalpando C, Delgado-Coello B, et al. Hyperinsulinemia is associated with increased soluble insulin receptors release from hepatocytes. Frontiers in Neuroendocrinology. 2014;5:95. doi: 10.3389/fendo.2014.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yuasa T, Amo K, Ishikura S, Nagaya H, Uchiyama K, Hashida S, Ebina Y. Development of in vitro model of insulin receptor cleavage induced by high glucose in HepG2 cells. Biochemical and Biophysical Research Communications. 2014;445(1):236–43. doi: 10.1016/j.bbrc.2014.01.187. [DOI] [PubMed] [Google Scholar]
- 81.Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(17):6659–63. doi: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dolinsky VW, Morton JS, Oka T, Robillard-Frayne I, Bagdan M, Lopaschuk GD, Des Rosiers C, Walsh K, Davidge ST, Dyck JR. Calorie restriction prevents hypertension and cardiac hypertrophy in the spontaneously hypertensive rat. Hypertension. 2010;56(3):412–21. doi: 10.1161/HYPERTENSIONAHA.110.154732. [DOI] [PubMed] [Google Scholar]
- 83.Larson-Meyer DE, Heilbronn LK, Redman LM, Newcomer BR, Frisard MI, Anton S, Smith SR, Alfonso A, Ravussin E. Effect of calorie restriction with or without exercise on insulin sensitivity, beta-cell function, fat cell size, and ectopic lipid in overweight subjects. Diabetes Care. 2006;29(6):1337–44. doi: 10.2337/dc05-2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation. 2007;115(2):245–54. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 85.Park Y, Capobianco S, Gao X, Falck JR, Dellsperger KC, Zhang C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. American Journal of Physiology. Heart and Circulatory Physiology. 2008;295(5):H1982–8. doi: 10.1152/ajpheart.01261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wada H, Satoh N, Kitaoka S, Ono K, Morimoto T, Kawamura T, Nakano T, Fujita M, Kita T, Shimatsu A, et al. Soluble VEGF receptor-2 is increased in sera of subjects with metabolic syndrome in association with insulin resistance. Atherosclerosis. 2010;208(2):512–7. doi: 10.1016/j.atherosclerosis.2009.07.045. [DOI] [PubMed] [Google Scholar]
- 87.Maamra M, Bidlingmaier M, Postel-Vinay MC, Wu Z, Strasburger CJ, Ross RJ. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology. 2001;142(10):4389–93. doi: 10.1210/endo.142.10.8442. [DOI] [PubMed] [Google Scholar]
- 88.Filozof C, Gomez-Garre D, Reinares L, Gonzalez-Rubio ML, Munoz-Pacheco P, Rueda A, Alvarez-Arcaya A, Calle-Pascual AL, Fernandez-Cruz A. Relationship between plasma levels of soluble CD40L and insulin sensitivity and insulin secretion status in non-diabetic dyslipidemic patients. Diabetes Research and Clinical Practice. 2008;79(1):48–55. doi: 10.1016/j.diabres.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 89.Kappelmayer J, Nagy B, Jr, Miszti-Blasius K, Hevessy Z, Setiadi H. The emerging value of P-selectin as a disease marker. Clinical Chemistry and Laboratory Medicine: CCLM/FESCC. 2004;42(5):475–86. doi: 10.1515/CCLM.2004.082. [DOI] [PubMed] [Google Scholar]
- 90.Frankwich K, Tibble C, Torres-Gonzalez M, Bonner M, Lefkowitz R, Tyndall M, Schmid-Schönbein GW, Villarreal F, Heller M, Herbst K. Proof of concept: Matrix metalloproteinase inhibitor decreases inflammation and improves muscle insulin sensitivity in people with type 2 diabetes. J Inflamm (Lond) 2012;9(1):35. doi: 10.1186/1476-9255-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92(5):1084–8. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 92.Fukuoka Y, Schwartz LB. Active monomers of human beta-tryptase have expanded substrate specificities. International Immunopharmacology. 2007;7(14):1900–8. doi: 10.1016/j.intimp.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moreno M, Puig J, Serrano M, Moreno-Navarrete JM, Ortega F, Ricart W, Fernandez-Real JM. Circulating tryptase as a marker for subclinical atherosclerosis in obese subjects. PLoS ONE. 2014;9(5):e97014. doi: 10.1371/journal.pone.0097014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Z, Zhang H, Shen XH, Jin KL, Ye GF, Qiu W, Qian L, Li B, Zhang YH, Shi GP. Immunoglobulin E and mast cell proteases are potential risk factors of impaired fasting glucose and impaired glucose tolerance in humans. Annals of Medicine. 2013;45(3):220–9. doi: 10.3109/07853890.2012.732234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Berry E, Bosonea AM, Wang X, Fernandez-Patron C. Insights into the activity, differential expression, mutual regulation, and functions of matrix metalloproteinases and a disintegrin and metalloproteinases in hypertension and cardiac disease. Journal of Vascular Research. 2013;50(1):52–68. doi: 10.1159/000345240. [DOI] [PubMed] [Google Scholar]
- 96.Federici M, Menghini R, Mauriello A, Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G, Lauro R. Insulin-dependent activation of endothelial nitric oxide synthase is impaired by O-linked glycosylation modification of signaling proteins in human coronary endothelial cells. Circulation. 2002;106(4):466–72. doi: 10.1161/01.cir.0000023043.02648.51. [DOI] [PubMed] [Google Scholar]
- 97.Menghini R, Fiorentino L, Casagrande V, Lauro R, Federici M. The role of ADAM17 in metabolic inflammation. Atherosclerosis. 2013;228(1):12–7. doi: 10.1016/j.atherosclerosis.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 98.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282(5392):1281–4. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 99.Lefkowitz RB, Marciniak JY, Hu C-M, Schmid-Schönbein GW, Heller MJ. An electrophoretic method for the detection of chymotrypsin and trypsin activity directly in whole blood. Electrophoresis. 2010;31(2):403–10. doi: 10.1002/elps.200900424. [DOI] [PubMed] [Google Scholar]
- 100.DeLano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Science Translational Medicine. 2013;5(169):169r11. doi: 10.1126/scitranslmed.3005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Altshuler AE, Penn AH, Yang JA, Kim GR, Schmid-Schönbein GW. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS ONE. 2012;7(3):e32672. doi: 10.1371/journal.pone.0032672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. 2012;37(3):297–305. doi: 10.1097/SHK.0b013e318240b59b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Altshuler AE, Lamadrid I, Li D, Ma SR, Kurre L, Schmid-Schönbein GW, Penn AH. Transmural intestinal wall permeability in severe ischemia after enteral protease inhibition. PLoS ONE. 2014;9(5):e96655. doi: 10.1371/journal.pone.0096655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DeLano FA, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the small intestine prevents insulin resistance in hemorrhagic shock. Shock. 2014;41(1):55–61. doi: 10.1097/SHK.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]