

Abstract
Homing endonucleases are sequence-tolerant DNA endonucleases that act as mobile genetic elements. The ability of homing endonucleases to cleave substrates with multiple nucleotide substitutions suggests a high degree of adaptability in that changing or modulating cleavage preference would require relatively few amino acid substitutions. Here, using directed evolution experiments with the GIY-YIG homing endonuclease I-TevI that targets the thymidylate synthase gene of phage T4, we readily isolated variants that dramatically broadened I-TevI cleavage preference, as well as variants that fine-tuned cleavage preference. By combining substitutions, we observed an ∼10 000-fold improvement in cleavage on some substrates not cleaved by the wild-type enzyme, correlating with a decrease in readout of information content at the cleavage site. Strikingly, we were able to change the cleavage preference of I-TevI to that of the isoschizomer I-BmoI which targets a different cleavage site in the thymidylate synthase gene, recapitulating the evolution of cleavage preference in this family of homing endonucleases. Our results define a strategy to isolate GIY-YIG nuclease domains with distinct cleavage preferences, and provide insight into how homing endonucleases may escape a dead-end life cycle in a population of saturated target sites by promoting transposition to different target sites.
INTRODUCTION
Self-splicing group I introns and inteins are found in the genomes of representatives of all domains of life and their associated viruses (1–5). Many self-splicing introns and inteins are also mobile genetic elements due to the presence of a homing endonuclease gene (HEG) encoded within the intron or intein. The homing endonuclease confers mobility on its host intron (or intein) by binding to and cleaving a defined target site in homologous genes that lack the intron, generating a double-strand break (DSB) that is repaired using the intron-containing gene as a template (6). This intron homing pathway results in an extremely high frequency of mobility, and in experimental situations a mobile intron is rapidly incorporated into >95% of naïve genes (7–9). These experiments, coupled with phylogenetic analyses of intron/HEG distribution, led to the proposal that mobile introns undergo a cyclical birth and death process that is driven by the super-Mendelian inheritance of the intron/HEG pair (10). Spread of the intron/HEG rapidly saturates a population of intronless sites. Consequently, the HEG suffers a progressive loss of activity leading to eventual degeneration and deletion unless a population of new target sites becomes available to re-initiate the homing process. The intron/HEG could escape inevitable death by transposing to a different site, and initiating a new lifecycle in a population of intronless alleles. Various mechanisms have been proposed for intron transposition, including reverse splicing of the intron RNA into an RNA template, followed by reverse transcription and integration into the genome (11). An alternate pathway involves rare cleavage by the homing endonuclease at non-allelic sites, triggering DNA repair pathways that result in intron transposition to a new site (12).
Six families of homing endonucleases have been identified to date: the LAGLIDADG, HNH, His-Cys Box, PD-(D/E)xK, EDxHD and GIY-YIG families (13). The GIY-YIG endonucleases are modular two-domain enzymes, with the class-defining amino acid residues of the GIY-YIG motif found in the small (∼100 residue) N-terminal nuclease domain (14–16). The nuclease domain is connected via an inter-domain linker to a C-terminal DNA-binding domain. The best-studied GIY-YIG endonuclease is I-TevI, encoded within a self-splicing group I intron that interrupts the thymidylate synthase gene (td) of bacteriophage T4 (17). I-TevI binds its target site as a monomer (18), and cleavage by the nuclease domain occurs at a 5′-CA↑AC↓G-3′ cleavage site upstream from the DNA-binding site (with ↑ and ↓ representing the bottom- and top-strand nicking sites, respectively) (19,20). Positions C1 and G5 of the cleavage site are critical for I-TevI activity in vitro and in vivo (21,22). I-TevI can tolerate some substitutions within the central three bases, but many 5′-CNNNG-3′ substrates are not cleaved (21,23). I-BmoI, an isoschizomer of I-TevI, binds and cleaves the homologous site in the thymidylate synthase A gene (thyA) of Bacillus mojavensis (24). Interestingly, the I-BmoI cleavage site is 5′-GCCCG-3′, but only the G at position 5 is critical for activity, with no nucleotide preference observed at positions 1–4 (5′-NNNNG-3′) (25,26). The different cleavage preferences at position 1 correlate with different conserved amino acids, and therefore different codons and nucleotide content, at the homologous positions in td and thyA sequences (22,25). From an evolutionary perspective, I-TevI and I-BmoI appear to have distinct evolutionary histories, as the coding region for each endonuclease is located in a different position of their respective group I introns, and the introns themselves are inserted in different positions of the thymidylate synthase genes. Moreover, I-BmoI lacks the zinc-finger found in the I-TevI linker domain. Thus, it appears that each enzyme has independently co-evolved with its DNA substrate to maximize information readout from the conserved components of their respective cleavage sites.
A common feature of homing endonucleases is their ability to tolerate nucleotide variation within their target sites (27,28), which is counterbalanced by the number of nucleotides bound by these enzymes to maintain target specificity. The preferred target site of a homing endonuclease is not a single sequence, but a population of sequences defined by the diversity of nucleotide substitutions tolerated by the homing endonuclease. Sequence-tolerant recognition raises the intriguing question of whether homing endonucleases are highly evolvable enzymes. Since these enzymes are sequence tolerant, they are unlikely to saturate possible base-specific contacts across the length of the target site, as is observed with some restriction enzymes (29). Thus, a change or modulation of DNA preference would be expected to require relatively few amino acid substitutions. The ability to rapidly modulate cleavage preference would provide a mechanism for homing endonucleases to co-evolve with their target sites that have accumulated nucleotide substitutions through genetic drift or other evolutionary processes, perpetuating the homing endonuclease life cycle. Rapid changes in cleavage preference could also generate homing endonuclease variants that target sequences outside of the native (current) target sites, providing a mechanism for endonuclease-mediated invasion of a new population of intronless sites by the intron/HEG pair (30,31).
Here, we have used directed evolution experiments to explore the adaptability of cleavage preference in I-TevI. We find that single substitutions in the nuclease and linker domains can fine-tune cleavage preference, and that combinations of substitutions can generate variants with cleavage preferences dramatically different from the wild-type nuclease domain. One combination of substitutions generates an I-TevI variant with a cleavage profile almost identical to that of I-BmoI. Our data define plausible mutational pathways that recapitulate the evolution of cleavage preference in GIY-YIG isoschizomers, and provide insight into endonuclease-mediated transposition mechanisms of mobile group I introns to distinct populations of intronless target sites.
MATERIALS AND METHODS
Bacterial strains and plasmid construction
Escherichia coli DH5α from New England Biolabs (NEB) was used for plasmid manipulations, ER2566 (NEB) for protein expression and BW25141(λDE3) for bacterial two-plasmid selections (32). All bacterial strains, plasmids and oligonucleotide primers are listed in Supplementary Tables S1, S2 and S3, respectively.
Construction of mutagenized I-TevI nuclease domain libraries
All restriction enzymes were acquired from NEB. Unless stated otherwise, small molecule reagents were acquired from EMD Millipore. I-TevI nuclease domain mutant libraries were generated using Mutazyme II (Agilent). The I-TevI nuclease domain coding region corresponding to amino acids 10–95 was amplified with primers DE-840 and DE-1912 under manufacturer-defined mutagenic conditions for 30 cycles. The polymerase chain reaction (PCR) product was isolated, diluted and mutagenic PCR repeated for another 30 cycles. Taq DNA polymerase (NEB) was used to amplify the mutant nuclease domain sequences under standard conditions for cloning. The average substitution rate was 2.4 amino acid substitutions per clone, as determined by Sanger sequencing of individual clones. A truncated I-TevI linker region (residues 96–169) was amplified using Taq and primers DE-1424 and DE-1045, and then combined with the I-TevI nuclease domain mutant library using splicing by overlap extension (SOEing) PCR with Phusion DNA polymerase (Thermo Scientific) and primers DE-840 and DE-1045. The nuclease domain mutant library with wild-type linker was digested with NcoI-HF and BamHI-HF and ligated using T4 DNA ligase (NEB) into the PciI and BamHI sites of a plasmid encoding the I-OnuI E1 variant (33). The I-OnuI E1 variant is optimized for cleavage against the human monoamine oxidase B gene, and has the following substitutions relative to the wild-type protein; N32S, S35R, S40A, T48C, N51I, K80R, K189N, K229R. The I-OnuI E1 variant used here also possesses a E22Q substitution that knocks out nuclease activity. The I-OnuI E1 variant was cloned with a hexahistidine tag on the 3′ end (C-terminus) to aid purification.
Directed evolution
E. coli BW25141(λDE3) were transformed with a plasmid (pTox) carrying the ccdB gene (27), which encodes a DNA gyrase toxin under control of the arabinose-inducible araBAD promoter, corresponding to different target sites as described previously (34). These individual target cells were made electrocompetent, and 50 μl was transformed with 10 ng of I-TevI nuclease domain (ND) mutant plasmid library, immediately diluted with 500 μl of SOC recovery media and incubated at 37°C with shaking. For the first round of selection, cultures were incubated at 37°C for 6 h, while subsequent rounds were incubated for 1 h. To estimate survival of the library at each round of selection, 100 μl of the culture was diluted and plated on selective Luria Broth (LB) media containing 25 μg/ml chloramphenicol and 10 mM arabinose, and on non-selective LB media containing 25 μg/ml chloramphenicol. Survival was calculated as the ratio of colonies on selective media versus colonies on non-selective media, taking into account dilution factors. Another 200 μl of transformed culture was removed and diluted into 5 ml selective media with 25 μg/ml chloramphenicol and 10 mM arabinose. The diluted cultures were incubated at 30°C with shaking for 18 h before plasmid isolation (Bio Basic) for subsequent rounds of selection. After two rounds of selection, those populations of mutant NDs that showed a measurable increase in survival were PCR amplified with primers DE-840 and DE-1045. The amplified DNA was treated with DpnI (NEB) to destroy any remaining round 2 plasmids, digested with NcoI-HF and BamHI-HF and ligated into pACYCDuet-1. Five colonies from the round 4 selective plates were picked for sequencing and further analyses.
Overexpression and purification of MegaTevs
E. coli ER2566 (NEB) was transformed with the MegaTev expression plasmids, and the MegaTev was purified as previously described (23). MegaTev concentration was determined by measuring the UV absorbance at 280 nm and comparing it to the predicted extinction coefficient of the MegaTev (67 380 M−1·cm−1), assuming no disulfide bonds (35).
Barcode cleavage assays
The kinetics of MegaTev substrate cleavage were assessed using a variation on the barcode assay described by Ulge et al. (36). Barcode assay substrates were prepared by using various pTox plasmids as templates with a pair of primers equidistant from the cleavage motif (Supplementary Table S3). Substrates of 2200, 1900, 1600 or 1320 bp were made, and combined into a single reaction. The 42 bp MegaTev target site (Figure 1) is placed such that two equally-sized products would be generated regardless of the substrate length. Reaction pools were prepared on ice, and comprised 5 nM native substrate and 5 nM of three non-native substrates, 250 nM enzyme and cleavage buffer (50 mM Tris·HCl pH 8.0, 100 mM NaCl, 1 mM DTT, 5% glycerol). An aliquot was removed immediately prior to starting the reaction by adding 2 mM MgCl2, and incubating at 5°C for 30 min. Aliquots were removed at 1, 2, 4, 10 and 30 min time points, and stopped by the introduction of ethylenediaminetetraacetic acid (EDTA) and sodium dodecyl sulphate (SDS) (final concentrations of 33 mM and 0.033%, respectively). Time-points were resolved using agarose gel electrophoresis in TBE (100 mM Tris base, 100 mM boric acid, 2 mM EDTA, pH 8.0) and spot densitometry was used to measure the quantity of substrate remaining in the reaction, and the quantity of product formed. The intensity of the corresponding substrate and product bands at each time-point were summed, and normalized to the intensity of the substrate band at t = 0. The fraction of substrate remaining (fS) is the ratio of the normalized substrate band intensity to the initial intensity. Triplicate values were plotted as fractions of substrate remaining at each time-point, and fit by non-linear regression to a first-order decay curve;
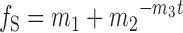 |
(1) |
where fS is the fraction of remaining substrate, m1 corrects for a non-zero baseline, m2 corrects for an initial fS < 1, m3 is the apparent first-order rate constant (kapp) in min−1 and t is time in minutes. The rate constant for decay of each substrate was normalized to kapp for the native cleavage motif decay curve, and reported as krel. Relative cleavage efficiency data for were converted to proportions, and information content in bits was calculated using the seqLogo package available through the Bioconductor R project (37).
Figure 1.

Selection of I-TevI nuclease domain variants on cleavage site sequences non-permissive to the wild-type enzyme. (A) Schematic of the MegaTev endonuclease and DNA substrate. The individual domains of the MegaTev endonuclease are indicated, as are the corresponding regions of DNA substrate contacted by each domain. The I-TevI nuclease domain bottom- and top-strand nicking sites are indicated by black triangles, and the critical C1 and G5 positions of the cleavage site are in bold-type font. (B) The E. coli-based selection relies on a plasmid (pTox) carrying the ccdB gene that encodes a DNA gyrase toxin, and a MegaTev substrate. The MegaTev is expressed from a separate plasmid (pEndo). Selection outcomes with an active or inactive MegaTev when cells are plated on selective (+ara) or non-selective (-ara) media. (C) Results of initial screening and enrichment of 16 populations of MegaTev variants on the indicated CNNNG triplet substrates through four rounds of enrichment. Open circles indicate survival of the wild-type MegaTev (WT).
RESULTS
Identification of I-TevI variants that cleave sub-optimal substrates
To isolate I-TevI cleavage variants, we screened partially randomized libraries of the I-TevI nuclease domain in the MegaTev architecture against variants of the CAACG cleavage motif using an E. coli two-plasmid survival assay (Figure 1A and B). The MegaTev protein is a fusion of residues 1–169 of I-TevI to the N-terminus of the I-OnuI LAGLIDADG homing endonuclease (also called a meganuclease). In this study, the MegaTev constructs contain an active I-TevI nuclease domain, whereas the I-OnuI nuclease was inactivated by a E22Q substitution. This architecture facilitates cloning and overexpression of the otherwise toxic I-TevI nuclease domain in E. coli, and possesses the same cleavage requirements as wild-type I-TevI. (23). In the two-plasmid assay, survival is promoted by cleavage of a target site cloned into a toxic plasmid by a MegaTev expressed from a second plasmid. Previous studies showed that certain substitutions of the central three bases within the CAACG cleavage motif drastically reduced or abolished I-TevI nuclease domain activity (central three bases are underlined) (23). We chose 16 CNNNG substrates (hereafter, ‘triplet’ substrates) for directed evolution studies. After two rounds of selection with the MegaTev nuclease domain library, we generated 16 populations of variants with varying degrees of survival against the triplet substrates (Figure 1C). Survival of the wild-type nuclease domain ranged from 0% to 13.5% on the same 16 triplet substrates, whereas 100% survival was observed on the native CAACG cleavage motif (Supplementary Table S4). The variant populations enriched on the CAAGG, CCCCG, CGAAG, CGCCG, CGGAG and CTGGG substrates were pursued further. To eliminate expression or plasmid stability effects, the MegaTev ORFs from each population were re-cloned, followed by two further rounds of enrichment. After four total round of enrichment, these six populations showed an improvement in survival ranging from 4- to >370-fold relative to the wild-type enzyme on the same substrates (Table 1). All of the mutant populations survived ∼100% on the wild-type CAACG substrate. Sequences of the sampled round 4 survivors revealed a number of mutant genotypes, none of which were fully wild-type. Mapping the position of each amino acid substitution onto the crystal structure of the I-TevI nuclease domain revealed that most lie on the same face of the domain as the active site (Figure 2A) (6). Only two of the identified positions, K26 and I86, lie in conserved blocks of amino acids previously identified in GIY-YIG domains (Figure 2B) (38). Notably, position K26 lies immediately adjacent to R27, implicated in stabilizing the phosphoanion intermediate generated from hydrolysis of the DNA backbone. One exception to this observation was the finding of a Q158R substitution in clones selected against the CAAGG and CGAAG substrates. Residue Q158 lies in the I-TevI zinc-finger that is a component of the inter-domain linker that connects the nuclease and DNA-binding domains (39).
Table 1. Summary of directed evolution experiments.
| Survival rate | ||||
|---|---|---|---|---|
| CNNNG target | MegaTev-WT (n = 3) | R4 population | Mutants | # clones |
| CAAGG | 1.6 ± 0.8 | 16 | Q158R | 4 |
| K26R/Q158R | 1 | |||
| CCCCG | <0.1 | 36 | T95S | 5 |
| CGAAG | 13.5 ± 6.1 | 61 | Q158R | 5 |
| CGCCG | <0.1 | 20 | I86V/T95S | 1 |
| C39R/T95S | 2 | |||
| C39R/I86V/T95S | 3 | |||
| CGGAG | <0.1 | 29 | T95S | 3 |
| K26R/T95S | 2 | |||
| CTGGG | <0.1 | 37 | T95S | 3 |
| K26R/T95S | 2 | |||
Figure 2.

Mutations that modulate cleavage preference map to non-conserved positions of the nuclease domain. (A) Shown is the crystal structure of the I-TevI nuclease domain (PDB 1LN0) shaded according to amino acid conversation, with cyan representing low conservation and magenta representing high conservation. Positions implicated in nuclease domain catalysis are shown in sticks with black labels. Positions identified in the genetic selections are shown as sticks overlaid with spheres with red labels. (B) Amino acid alignment of residues 1–100 of I-TevI and I-BmoI with the homologous positions of Eco29kI. Residues are colored according to physiochemical properties. The five conserved motifs (A–E) previously identified by Kowalski et al. (38) are indicated above the alignment. Amino acid positions identified in the directed evolution experiments are indicated by red stars and rectangles.
Determining the functional impact of individual amino acid substitutions
To de-convolute the importance of each amino acid substitution, MegaTev single or double mutant variants were constructed and tested against the native CAACG and triplet substrates using the bacterial two-plasmid assay. As shown in heat map format for the variants with the strongest phenotypes, each substitution conferred improvements in survival rate against a different subset of the triplet substrates, and no individual substitution reduced survival against the native CAACG substrate (Figure 3 and Supplementary Table S4). Further, survival rate was generally enhanced when substitutions were combined. A K26R mutation conferred ∼31% survival against the CAAGG substrate, while Q158R conferred ∼53% survival against the same substrate. When combined into the double mutant K26R/Q158R, we observed ∼86% survival against the CAAGG substrate, an approximately additive effect. Additionally, neither the K26R nor Q158R mutation conferred more than 2% survival against the CCAGG substrate individually, but the K26R/Q158R double mutant conferred ∼26% survival against CCAGG, an apparent cooperative effect. We also made a triple mutant consisting of substitutions that individually conferred an improvement in survival rate on a wide range of substrates. This triple mutant (K26R/T95S/Q158R, hereafter MegaTev-T3) conferred the highest survival rates against the broadest range of substrates tested.
Figure 3.

De-convoluting the impact of individual substitutions on MegaTev activity. Shown is a heat map of MegaTev survival on the indicated CNNNG triplet or C1 substrates. The mean survival percentage from a minimum of three experimental replicates was used to generate the heat map. The complete list of all MegaTev variants tested is in Supplementary Table S4.
We also tested the ability of the MegaTev variants to confer survival against substrates with substitutions at positions C1 and G5. Previous studies showed that substitutions in either or both of these positions drastically reduced in vivo survival and in vitro cleavage by I-TevI and the wild-type MegaTev (MegaTev-WT)(21–23). Surprisingly, we observed 100% survival of the MegaTev-T3 triple mutant against the C1T substrate (TAACG), representing an ≥10 000-fold improvement in survival relative to the wild-type nuclease domain (Figure 3). Lower levels of survival were observed against the C1G and C1A substrates (∼4% and ∼3%, respectively). The Q158R and K26R/Q158R mutants also displayed weak survival against the C1T substrates. No survival was observed against substrates with mutations in position G5 (Supplementary Table S4). Collectively, these data demonstrate that combinations of amino acid substitutions in the I-TevI nuclease and linker domains can dramatically increase survival on substrates that are non-permissive for the wild-type enzyme.
The Q158R linker substitution does not affect the position of DNA cleavage
The Q158R mutation lies in the zinc-finger motif of the I-TevI inter-domain linker, and broadens the cleavage range of MegaTev variants (Figure 3). The I-TevI zinc finger functions to position the nuclease domain on substrate to cleave at the CAACG motif that is correctly spaced from the DNA-binding site (40,41). Knocking out zinc-coordination relaxes the distance constraint, allowing the I-TevI nuclease domain to cleave at CNNNG sequences closer to, or farther from, the DNA-binding site (15,41). Thus, it is possible that variants containing the Q158R mutation were isolated because they are cleaving at an alternative TCTAG or CTCAG motif that is present in the DNA spacer of all of the substrates adjacent to the native CAACG motif. We tested this possibility by mapping the top- and bottom-strand nicking sites of MegaTev-T3 on plasmids that contained the CAACG native cleavage motif, or one of the three C1 substitutions. The MegaTev-T3 mutant cleaved at the correct site and distance from the DNA-binding site, regardless of the substrate used (Supplementary Figure S1). We conclude that the Q158R substitution in the context of MegaTev-T3 faithfully maintains the distance constraint of the wild-type enzyme, and that enrichment of Q158R was not due to cleavage at an alternative motif.
In vitro barcode assays
To determine if the observed survival on the non-permissive substrates by MegaTev-T3 mutant was due to differences in DNA cleavage rather than factors such as in vivo protein stability, off-target cleavage of pTox, or expression levels, we purified both the MegaTev-WT and MegaTev-T3 enzymes for in vitro barcode cleavage assays. In this assay, three substrates that differ in CAACG cleavage motif, and that are of different lengths, are incubated with purified protein and the native CAACG substrate in a competitive cleavage reaction. Because each substrate is a different length, and because the MegaTev cleavage site is in the middle of each substrate, cleavage generates a unique length product that can be resolved from the other products on an agarose gel (Figure 4A and Supplementary Figure S2). One of the substrates contains the native cleavage motif, allowing determination of an apparent reaction rate (kapp, Supplementary Figure S3) for each substrate relative to the native CAACG substrate (krel, Figure 4B). Control experiments showed that cleavage is non-cooperative and is unaffected by substrate length (Supplementary Figure S3).
Figure 4.

An in vitro barcode assay reveals that the MegaTev-T3 variant has a broader range of cleavage preference than the wild-type protein. (A) Schematic of the assay, where each substrate and product has a defined length that can be resolved on agarose gel. (B) Representative cleavage assay over time with MegaTev-WT and four substrates, including the wild type (indicated by an asterisk) and three CNNNG triplet substrates. The corresponding kinetic analysis to determine krel reaction rates is shown on the right. (C) Plot of MegaTev-WT versus the MegaTev-T3 krel rates for substrates used in the directed evolution experiments. Each krel rate is indicated by a dot, with the boundary of the ellipse corresponding to the standard error of the mean for each MegaTev. (D) krel rates for additional CNNNG and C1 substrates, plotted as in panel (C).
We assayed the MegaTev-WT and MegaTev-T3 enzymes against 34 substrates that included 15 of the substrates used in the initial enrichment experiments and 15 substrates that differed by a single base (CGAAG was excluded due to the consistently high survival rate with all MegaTev variants, Figure 3). As shown in Figure 4B and C, the krel data are consistent with a broadening of substrate preference by MegaTev-T3 relative to MegaTev-WT. The data are also consistent with the in vivo survival results using the two-plasmid selection, as increases in survival were generally correlated with an increased krel rate of the T3 mutant. Notable examples of this correlation include the CGCCG substrate, against which MegaTev-WT did not confer survival and had a krel of 0.32, whereas MegaTev-T3 conferred 71% survival and had a krel of 0.78. The krel data support the conclusion that the combination of the K26R/T95S/Q158R substitutions broaden the cleavage range of the I-TevI nuclease domain.
Single substitutions in the nuclease domain affect information readout at the cleavage motif
To examine more precisely nucleotide preference at the cleavage motif, we used a barcode cleavage assay to profile cleavage on DNA substrates with all possible substitutions at a single position. The cleavage efficiency on each singly substituted substrate is compared to cleavage of the native substrate (which is normalized to 1), providing a relative contribution of each base to cleavage efficiency at each position. The relative cleavage profile at each position is then converted to information content in bits to assess the information readout by the nuclease domain. As shown in Figure 5A and B, we first analyzed the cleavage profile of MegaTev-WT and found a C at position 1 and a G at position 5 were preferred over other bases, in agreement with previous studies (22,23). We next analyzed the cleavage profiles of a number of MegaTev variants, and found two different phenotypes (Figure 5A and B). The first phenotype, found for the K26R, T95S, Q158R, K26R/T95S and K26R/Q158R variants, maintained the preference for C and G at positions 1 and 5 with minimal changes to the information content readout at these positions. The second phenotype, found for the T95S/Q158R and T3 (K26R/T95S/Q158R) variants, was much more dramatic as both variants did not exhibit a strong preference for any nucleotide at position 1. Moreover, the T95S/Q158R variant exhibited no nucleotide preference at position 5, whereas the T3 variant maintained a G preference. Comparison of initial reaction rates indicated that the T95S/Q158R double mutant is ∼4-fold slower than the T3 enzyme that may explain why survival in the two-plasmid assay was lower for the T95S/Q158R than for the MegaTev-T3 variant. Although the effect of combining mutations was likely confounded by epistatic effects, the mutations greatly broadened substrate preference.
Figure 5.

Barcode assay of the cleavage motif reveals higher rates of relative cleavage at the C1 and G5 positions by MegaTev variants. (A) Representative gel images of time course barcode cleavage assays for MegaTev-WT and MegaTev-T3. Each gel image corresponds to a position within the CAACG motif. The nucleotide substitution for each length substrate (S) is indicated on the left side of the gel in black font. The corresponding product (P) is indicated in red font. The images for each substrate were cropped from separate images and assembled into a composite figure. (B) Barplot of relative cleavage at each nucleotide position (on left) and corresponding sequence logos plot representing information content readout by each nuclease domain (on right). At each position, cleavage activity is plotted relative to the wild-type substrate, which is normalized to 1 (grey dashed line). Error bars are standard error of the mean derived from minimum of 3 independent replicates.
DISCUSSION
In this study, we explored the capacity of the I-TevI DNA-cleavage domain to adapt to new target sequences. Using a PCR-based random mutagenesis strategy, we readily selected variants that cleave substrates the wild-type enzyme cannot. We consider these variants to have changed cleavage preference rather than undergone a complete relaxation of specificity, since they cleave a subset of the sequences with which they were challenged. In particular, the I-TevI variants cannot cleave certain CNNNG triplet substrates, or substrates with mutations in position G5 (with the exception of the T95S/Q158R variant). Thus, cleavage preference of the GIY-YIG nuclease domain appears to be an adaptable trait, and we discuss the mechanistic and evolutionary interpretations of our findings.
One common feature of intron-encoded homing endonucleases is their ability to tolerate nucleotide variation within their target sites (27,28). The degree to which individual homing endonucleases tolerate nucleotide substitutions depends on the molecular mechanisms of DNA recognition and cleavage, and is arguably best understood for the LAGLIDADG endonucleases (42). Little is known about the molecular mechanisms that regulate cleavage preference of the nuclease domain in GIY-YIG homing endonucleases. In the modular I-TevI, the N-terminal nuclease and C-terminal binding domains independently interact with different regions of substrate, whereas the interdomain linker communicates with the nuclease domain to position it correctly at the 5′-CNNNG-3′ motif. From an evolutionary perspective, the separation of biochemical activities of the nuclease and binding domain allows each to co-evolve independently with its DNA substrate to accommodate genetic drift at the target site. The specificity of the I-TevI nuclease domain was initially described as 5′-CNNNG-3′ (22), but as is evident in Figure 3, some NNN triplets are very poor substrates for the enzyme. The fact that no single position in the NNN triplet is critical for cleavage suggests that the I-TevI nuclease domain is sensitive to the sum of perturbations arising from the different DNA structures of the NNN sequences, as is also seen with cleavage of the central 4 bases in the LAGLIDADG target sites (43,44). This idea is supported by the correlation between the predicted flexibility of the individual CNNNG sequences and low cleavage activity (Supplementary Figure S4). In contrast, the strict preference of I-TevI for a C and G at positions 1 and 5 of the cleavage site implies a high sensitivity to structural perturbations or major groove contacts at these base pairs individually.
While there is no structural information for I-TevI-substrate interactions at the cleavage site, the dimeric Type IIP restriction enzymes Eco29kI and Hpy188I that contain the GIY-YIG catalytic motif were co-crystallized with their DNA substrates (42,45). In both structures, sequence-recognition is achieved through direct contact with amino acid residues present in structural elements that are largely absent from I-TevI, I-BmoI and other GIY-YIG homing endonucleases. Mapping positions we identified in our genetic selection (K26, C39, I86 and T95) onto homologous positions in the Eco29kI- and Hpy188I-DNA co-crystals (Figure 2A) revealed no obvious candidate(s) that would directly regulate specificity at positions 1 and 5 of the I-TevI cleavage site. In the Hpy188I structure, S87 makes a direct base contact and lies in an α-helix that contains the active site R84 residue proposed to stabilize the phosphoanion intermediate generated from hydrolysis of the DNA backbone; the equivalent residue in I-TevI is R27 (6). The adjacent K26R substitution might enhance this stabilization, resulting in the enhanced cleavage that we observed with the MegaTev-T3 mutant (K26R/T95S/Q158R). Another possibility is that K26R increases the preference for C at position 1 as the K26R variants, except for MegaTev-T3, possessed a slightly elevated preference for C1. Interestingly, two of the substitutions we identified (C39R and T95S) occur naturally in I-BmoI, while an S is found in position 26. We attempted to convert the specificity of the I-TevI nuclease domain to that of I-BmoI by making the K26S substitution alone, and in combination with the C39R and T95S substitutions, anticipating that these variants would show relaxed specificity at position C1. All of the converted variants survived on the native CAACG substrate, but none survived on any of the C1 mutant substrates (Supplementary Table S4), indicating that these combinations of residues in the I-TevI backbone does not recapitulate the tolerance of I-BmoI to substitutions at position C1. The T95S substitution is also found in a number of I-TevI homologs found in group I introns that interrupt the thymidylate synthase genes of related T4-like phage that infect enteric bacteria. The predicted cleavage sites of a number of these endonucleases is 5′-CAACG-3′, identical to that of I-TevI, making it difficult to ascertain if the homologs with the T95S substitution have cleavage preferences similar to those of the T95S variants described here.
One identified substitution with a significant influence on cleavage was Q158R. The Q158 position has immediate structural relevance as it is in a loop that extends from the core of the zinc finger motif that communicates with the nuclease domain to position it for cleavage at the correct 5′-CNNNG-3′ motif within the I-TevI binding site (40,41,39). As noted by Van Roey et al. (39), Q158 is slightly outside of hydrogen-bonding range of the DNA substrate used in that structural study, but the Q158R substitution (with a longer side chain) could create a hydrogen bond contact with a base, or with the DNA backbone, effectively stabilizing the interaction of the zinc finger, and consequently the nuclease domain, with substrate. A longer-lived interaction between the nuclease domain and DNA substrate would increase the probability of cleavage by the nuclease domain on substrates that are unfavorable for cleavage by the wild-type protein, particularly substrates with predicted high flexibility (Supplementary Figure S4).
One of the most striking results from our study was the conversion of I-TevI nuclease domain preference to any base at position 1 from the strict requirement for a C. This preference is very similar to that of the isoschizomer I-BmoI (5′-NNNNG-3′) (25). Interestingly, all of the variants we characterized retained a high level of activity against the native I-TevI 5′-CAACG-3′ target site. This result was somewhat surprising because we naïvely expected that substitutions that changed cleavage preference would do so at the cost of reduced cleavage of the native site. From an evolutionary perspective, retention of activity against the native target site allows homing endonuclease variants with altered cleavage preferences to sample different populations of cleavage sites without affecting the ability to perpetuate the homing cycle within the current population of native sites (Figure 6). Homing endonuclease variants with distinct cleavage preferences could then ‘jump’ to a new population of sites, assuming that the sites were permissive to intron insertion and splicing. Natural selection would favor homing endonuclease variants with optimized target site interactions, as well as optimized interactions between the intron and exon sequences to promote efficient RNA splicing. This scenario could provide a mechanism for an intron/HEG pair to escape death by degeneration and loss of activity in an intron-saturated population, and initiate a new life cycle in a distinct population of intronless target sites. Given the similarities between the paralogous thymidylate synthase genes, and the I-TevI and I-BmoI cleavage site preferences (25), it is tempting to speculate that this mechanism facilitated the evolution of mobile group I introns interrupting thymidylate synthase genes in bacteria and their phages.
Figure 6.

Escape and invasion of new target sites perpetuates the homing endonuclease life cycle. A modified schematic of the homing endonuclease life cycle depicts two populations of target sites, the CNNNG and NNNNG sites. Amino acid substitutions in the I-TevI nuclease domain that perpetuate maintenance and cycling through the CNNNG target site population are shown on the bottom right, while amino acid substitutions that facilitate invasion of the NNNNG target site population are shown on the bottom left.
In conclusion, we have shown that cleavage preference of the I-TevI nuclease domain can be readily modulated by straightforward genetic selections. In addition to providing insight into the evolution of cleavage specificity in sequence-tolerant DNA endonucleases, our data imply that I-TevI nuclease domains with distinct cleavage preferences can be isolated. The modular I-TevI nuclease and linker domains have been fused to various DNA-binding platforms to generate highly specific genome-editing reagents (23,34,46). Targeting of these reagents requires the presence of the native CAACG I-TevI cleavage site positioned ∼15 bp upstream of the DNA binding site. In this regard, the I-TevI variants with altered cleavage profiles that we describe here would broaden the targeting potential of the chimeric reagents by alleviating the requirement for a CAACG cleavage site.
Supplementary Material
Acknowledgments
The authors thank Jason Wolfs for helpful comments on the directed evolution experiments, and on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Sciences and Engineering Research Council of Canada, Discovery Grant [RGPIN-2015-04800 to D.R.E.]; PGS-D scholarship from the National Sciences and Engineering Research Council of Canada [to A.C.R.]. Funding for open access charge: Natural Sciences and Engineering Research Council of Canada, Discovery Grant [RGPIN-2015-04800 to D.R.E] and New England Biolabs.
Conflict of interest statement. None declared.
REFERENCES
- 1.Stoddard B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 2.Michel F., Westhof E. Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J. Mol. Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- 3.Cech T.R. Conserved sequences and structures of group I introns: Building an active site for RNA catalysis–a review. Gene. 1988;73:259–271. doi: 10.1016/0378-1119(88)90492-1. [DOI] [PubMed] [Google Scholar]
- 4.Gimble F.S., Thorner J. Homing of a DNA endonuclease gene by meiotic gene conversion in saccharomyces cerevisiae. Nature. 1992;357:301–306. doi: 10.1038/357301a0. [DOI] [PubMed] [Google Scholar]
- 5.Belfort M., Derbyshire V., Stoddard B.L., Wood D. Homing Endonucleases and Inteins. Berlin: Springer; 2005. [Google Scholar]
- 6.Van Roey P., Meehan L., Kowalski J.C., Belfort M., Derbyshire V. Catalytic domain structure and hypothesis for function of GIY-YIG intron endonuclease I-TevI. Nat. Struct. Biol. 2002;9:806–811. doi: 10.1038/nsb853. [DOI] [PubMed] [Google Scholar]
- 7.Netter P., Petrochilo E., Slonimski P.P., Bolotin-Fukuhara M., Coen D., Deutsch J., Dujon B. Mitochondrial genetics. VII. allelism and mapping studies of ribosomal mutants resistant to chloramphenicol, erythromycin and spiramycin in S. cerevisiae. Genetics. 1974;78:1063–1100. doi: 10.1093/genetics/78.4.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacquier A., Dujon B. An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell. 1985;41:383–394. doi: 10.1016/s0092-8674(85)80011-8. [DOI] [PubMed] [Google Scholar]
- 9.Gimble F.S., Thorner J. Homing of a DNA endonuclease gene by meiotic gene conversion in saccharomyces cerevisiae. Nature. 1992;357:301–306. doi: 10.1038/357301a0. [DOI] [PubMed] [Google Scholar]
- 10.Goddard M.R., Burt A. Recurrent invasion and extinction of a selfish gene. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13880–13885. doi: 10.1073/pnas.96.24.13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodson S.A., Cech T.R. Reverse self-splicing of the tetrahymena group I intron: Implication for the directionality of splicing and for intron transposition. Cell. 1989;57:335–345. doi: 10.1016/0092-8674(89)90971-9. [DOI] [PubMed] [Google Scholar]
- 12.Belfort M., Perlman P.S. Mechanisms of intron mobility. J. Biol. Chem. 1995;270:30237–30240. doi: 10.1074/jbc.270.51.30237. [DOI] [PubMed] [Google Scholar]
- 13.Belfort M., Bonocora R.P. Homing endonucleases: From genetic anomalies to programmable genomic clippers. Methods Mol. Biol. 2014;1123:1–26. doi: 10.1007/978-1-62703-968-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas M., Davis R.W. Studies on the cleavage of bacteriophage lambda DNA with EcoRI restriction endonuclease. J. Mol. Biol. 1975;91:315–328. doi: 10.1016/0022-2836(75)90383-6. [DOI] [PubMed] [Google Scholar]
- 15.Liu Q., Dansereau J.T., Puttamadappa S.S., Shekhtman A., Derbyshire V., Belfort M. Role of the interdomain linker in distance determination for remote cleavage by homing endonuclease I-TevI. J. Mol. Biol. 2008;379:1094–1106. doi: 10.1016/j.jmb.2008.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Q., Derbyshire V., Belfort M., Edgell D.R. Distance determination by GIY-YIG intron endonucleases: Discrimination between repression and cleavage functions. Nucleic Acids Res. 2006;34:1755–1764. doi: 10.1093/nar/gkl079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shub D.A., Gott J.M., Xu M.Q., Lang B.F., Michel F., Tomaschewski J., Pedersen-Lane J., Belfort M. Structural conservation among three homologous introns of bacteriophage T4 and the group I introns of eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 1988;85:1151–1155. doi: 10.1073/pnas.85.4.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller J.E., Smith D., Bryk M., Belfort M. Intron-encoded endonuclease I-TevI binds as a monomer to effect sequential cleavage via conformational changes in the td homing site. EMBO J. 1995;14:5724–5735. doi: 10.1002/j.1460-2075.1995.tb00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quirk S.M., Bell-Pedersen D., Belfort M. Intron mobility in the T-even phages: High frequency inheritance of group I introns promoted by intron open reading frames. Cell. 1989;56:455–465. doi: 10.1016/0092-8674(89)90248-1. [DOI] [PubMed] [Google Scholar]
- 20.Bell-Pedersen D., Quirk S.M., Bryk M., Belfort M. I-TevI, the endonuclease encoded by the mobile td intron, recognizes binding and cleavage domains on its DNA target. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7719–7723. doi: 10.1073/pnas.88.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryk M., Belisle M., Mueller J.E., Belfort M. Selection of a remote cleavage site by I-tevI, the td intron-encoded endonuclease. J. Mol. Biol. 1995;247:197–210. doi: 10.1006/jmbi.1994.0133. [DOI] [PubMed] [Google Scholar]
- 22.Edgell D.R., Stanger M.J., Belfort M. Coincidence of cleavage sites of intron endonuclease I-TevI and critical sequences of the host thymidylate synthase gene. J. Mol. Biol. 2004;343:1231–1241. doi: 10.1016/j.jmb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Wolfs J.M., DaSilva M., Meister S.E., Wang X., Schild-Poulter C., Edgell D.R. MegaTevs: Single-chain dual nucleases for efficient gene disruption. Nucleic Acids Res. 2014;42:8816–8829. doi: 10.1093/nar/gku573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgell D.R., Shub D.A. Related homing endonucleases I-BmoI and I-TevI use different strategies to cleave homologous recognition sites. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7898–7903. doi: 10.1073/pnas.141222498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgell D.R., Stanger M.J., Belfort M. Importance of a single base pair for discrimination between intron-containing and intronless alleles by endonuclease I-BmoI. Curr. Biol. 2003;13:973–978. doi: 10.1016/s0960-9822(03)00340-3. [DOI] [PubMed] [Google Scholar]
- 26.Kleinstiver B.P., Wolfs J.M., Edgell D.R. The monomeric GIY-YIG homing endonuclease I-BmoI uses a molecular anchor and a flexible tether to sequentially nick DNA. Nucleic Acids Res. 2013;41:5413–5427. doi: 10.1093/nar/gkt186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argast G.M., Stephens K.M., Emond M.J., Monnat R.J., Jr I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J. Mol. Biol. 1998;280:345–353. doi: 10.1006/jmbi.1998.1886. [DOI] [PubMed] [Google Scholar]
- 28.Bryk M., Quirk S.M., Mueller J.E., Loizos N., Lawrence C., Belfort M. The td intron endonuclease I-TevI makes extensive sequence-tolerant contacts across the minor groove of its DNA target. EMBO J. 1993;2:2141–2149. doi: 10.1002/j.1460-2075.1993.tb05862.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pingoud A., Wilson G.G., Wende W. Type II restriction endonucleases–a historical perspective and more. Nucleic Acids Res. 2014;42:7489–7527. doi: 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loizos N., Tillier E.R., Belfort M. Evolution of mobile group I introns: Recognition of intron sequences by an intron-encoded endonuclease. Proc. Natl. Acad. Sci. U.S.A. 1994;91:11983–11987. doi: 10.1073/pnas.91.25.11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgell D.R., Belfort M., Shub D.A. Barriers to intron promiscuity in bacteria. J. Bacteriol. 2000;182:5281–5289. doi: 10.1128/jb.182.19.5281-5289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleinstiver B.P., Fernandes A.D., Gloor G.B., Edgell D.R. A unified genetic, computational and experimental framework identifies functionally relevant residues of the homing endonuclease I-BmoI. Nucleic Acids Res. 2010;38:2411–2427. doi: 10.1093/nar/gkp1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeuchi R., Lambert A.R., Mak A.N., Jacoby K., Dickson R.J., Gloor G.B., Scharenberg A.M., Edgell D.R., Stoddard B.L. Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13077–13082. doi: 10.1073/pnas.1107719108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleinstiver B.P., Wolfs J.M., Kolaczyk T., Roberts A.K., Hu S.X., Edgell D.R. Monomeric site-specific nucleases for genome editing. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8061–8066. doi: 10.1073/pnas.1117984109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gill S.C., von Hippel P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 36.Ulge U.Y., Baker D.A., Monnat R.J., Jr Comprehensive computational design of mCreI homing endonuclease cleavage specificity for genome engineering. Nucleic Acids Res. 2011;39:330–4339. doi: 10.1093/nar/gkr022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–212. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowalski J.C., Belfort M., Stapleton M.A., Holpert M., Dansereau J.T., Pietrokovski S., Baxter S.M., Derbyshire V. Configuration of the catalytic GIY-YIG domain of intron endonuclease I-TevI: Coincidence of computational and molecular findings. Nucleic Acids Res. 1999;27:2115–2125. doi: 10.1093/nar/27.10.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Roey P., Waddling C.A., Fox K.M., Belfort M., Derbyshire V. Intertwined structure of the DNA-binding domain of intron endonuclease I-TevI with its substrate. EMBO J. 2001;20:3631–3637. doi: 10.1093/emboj/20.14.3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgell D.R., Derbyshire V., Van Roey P., LaBonne S., Stanger M.J., Li Z., Boyd T.M., Shub D.A., Belfort M. Intron-encoded homing endonuclease I-TevI also functions as a transcriptional autorepressor. Nat. Struct. Mol. Biol. 2004;11:936–944. doi: 10.1038/nsmb823. [DOI] [PubMed] [Google Scholar]
- 41.Dean A.B., Stanger M.J., Dansereau J.T., Van Roey P., Derbyshire V., Belfort M. Zinc finger as distance determinant in the flexible linker of intron endonuclease I-TevI. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8554–8561. doi: 10.1073/pnas.082253699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mak A.N., Lambert A.R., Stoddard B.L. Folding, DNA recognition, and function of GIY-YIG endonucleases: Crystal structures of R.Eco29kI. Structure. 2010;18:1321–1331. doi: 10.1016/j.str.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szeto M.D., Boissel S.J., Baker D., Thyme S.B. Mining endonuclease cleavage determinants in genomic sequence data. J. Biol. Chem. 2011;286:32617–32627. doi: 10.1074/jbc.M111.259572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert A.R., Hallinan J.P., Shen B.W., Chik J.K., Bolduc J.M., Kulshina N., Robins L.I., Kaiser B.K., Jarjour J., Havens K., et al. Indirect DNA sequence recognition and its impact on nuclease cleavage activity. Structure. 2016;24:862–873. doi: 10.1016/j.str.2016.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sokolowska M., Czapinska H., Bochtler M. Hpy188I-DNA pre- and post-cleavage complexes–snapshots of the GIY-YIG nuclease mediated catalysis. Nucleic Acids Res. 2011;39:1554–1564. doi: 10.1093/nar/gkq821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kleinstiver B.P., Wang L., Wolfs J.M., Kolaczyk T., McDowell B., Wang X., Schild-Poulter C., Bogdanove A.J., Edgell D.R. The I-TevI nuclease and linker domains contribute to the specificity of monomeric TALENs. G3 (Bethesda) 2014;4:1155–1165. doi: 10.1534/g3.114.011445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.