

Abstract
Key points
Progression of hypoxic pulmonary hypertension is thought to be due, in part, to suppression of voltage‐gated potassium channels (Kv) in pulmonary arterial smooth muscle by hypoxia, although the precise molecular mechanisms have been unclear.
AMP‐activated protein kinase (AMPK) has been proposed to couple inhibition of mitochondrial metabolism by hypoxia to acute hypoxic pulmonary vasoconstriction and progression of pulmonary hypertension.
Inhibition of complex I of the mitochondrial electron transport chain activated AMPK and inhibited Kv1.5 channels in pulmonary arterial myocytes.
AMPK activation by 5‐aminoimidazole‐4‐carboxamide riboside, A769662 or C13 attenuated Kv1.5 currents in pulmonary arterial myocytes, and this effect was non‐additive with respect to Kv1.5 inhibition by hypoxia and mitochondrial poisons.
Recombinant AMPK phosphorylated recombinant human Kv1.5 channels in cell‐free assays, and inhibited K+ currents when introduced into HEK 293 cells stably expressing Kv1.5.
These results suggest that AMPK is the primary mediator of reductions in Kv1.5 channels following inhibition of mitochondrial oxidative phosphorylation during hypoxia and by mitochondrial poisons.
Abstract
Progression of hypoxic pulmonary hypertension is thought to be due, in part, to suppression of voltage‐gated potassium channels (Kv) in pulmonary arterial smooth muscle cells that is mediated by the inhibition of mitochondrial oxidative phosphorylation. We sought to determine the role in this process of the AMP‐activated protein kinase (AMPK), which is intimately coupled to mitochondrial function due to its activation by LKB1‐dependent phosphorylation in response to increases in the cellular AMP:ATP and/or ADP:ATP ratios. Inhibition of complex I of the mitochondrial electron transport chain using phenformin activated AMPK and inhibited Kv currents in pulmonary arterial myocytes, consistent with previously reported effects of mitochondrial inhibitors. Myocyte Kv currents were also markedly inhibited upon AMPK activation by A769662, 5‐aminoimidazole‐4‐carboxamide riboside and C13 and by intracellular dialysis from a patch‐pipette of activated (thiophosphorylated) recombinant AMPK heterotrimers (α2β2γ1 or α1β1γ1). Hypoxia and inhibitors of mitochondrial oxidative phosphorylation reduced AMPK‐sensitive K+ currents, which were also blocked by the selective Kv1.5 channel inhibitor diphenyl phosphine oxide‐1 but unaffected by the presence of the BKCa channel blocker paxilline. Moreover, recombinant human Kv1.5 channels were phosphorylated by AMPK in cell‐free assays, and K+ currents carried by Kv1.5 stably expressed in HEK 293 cells were inhibited by intracellular dialysis of AMPK heterotrimers and by A769662, the effects of which were blocked by compound C. We conclude that AMPK mediates Kv channel inhibition by hypoxia in pulmonary arterial myocytes, at least in part, through phosphorylation of Kv1.5 and/or an associated protein.
Key points
Progression of hypoxic pulmonary hypertension is thought to be due, in part, to suppression of voltage‐gated potassium channels (Kv) in pulmonary arterial smooth muscle by hypoxia, although the precise molecular mechanisms have been unclear.
AMP‐activated protein kinase (AMPK) has been proposed to couple inhibition of mitochondrial metabolism by hypoxia to acute hypoxic pulmonary vasoconstriction and progression of pulmonary hypertension.
Inhibition of complex I of the mitochondrial electron transport chain activated AMPK and inhibited Kv1.5 channels in pulmonary arterial myocytes.
AMPK activation by 5‐aminoimidazole‐4‐carboxamide riboside, A769662 or C13 attenuated Kv1.5 currents in pulmonary arterial myocytes, and this effect was non‐additive with respect to Kv1.5 inhibition by hypoxia and mitochondrial poisons.
Recombinant AMPK phosphorylated recombinant human Kv1.5 channels in cell‐free assays, and inhibited K+ currents when introduced into HEK 293 cells stably expressing Kv1.5.
These results suggest that AMPK is the primary mediator of reductions in Kv1.5 channels following inhibition of mitochondrial oxidative phosphorylation during hypoxia and by mitochondrial poisons.
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide riboside
- AMPK
AMP‐activated protein kinase
- BKCa
large conductance voltage‐ and calcium‐activated K+ channel
- DPO‐1
diphenyl phosphine oxide‐1
- HEK 293
human embryonic kidney 293 cells
- HPV
hypoxic pulmonary vasoconstriction
- Kv
voltage‐gated K+ channel
- LKB1
liver kinase B1
Introduction
Hypoxia without hypercapnia induces pulmonary vasoconstriction, and thus assists ventilation–perfusion matching in the lung (von Euler & Liljestrand, 1946). However, hypoxia may trigger pulmonary hypertension when it is widespread, for example during ascent to altitude (Bartsch et al. 2005) or due to disorders such as cystic fibrosis (Lahm et al. 2014). While current therapies have been shown to prolong survival, pulmonary hypertension remains a life‐threatening disorder (Lahm et al. 2014) and the precise molecular mechanisms underlying it remain unclear. Therefore, greater understanding is critical to the development of effective therapies.
Initially, hypoxic pulmonary vasoconstriction (HPV) is driven by calcium release via ryanodine receptors from the sarcoplasmic reticulum of pulmonary arterial smooth muscle cells (Dipp et al. 2001), but is also associated with concomitant inhibition of voltage‐gated potassium channels (Kv) (Post et al. 1992; Yuan et al. 1993; Archer et al. 2004). The role of Kv channel inhibition in acute HPV remains open to debate (Wilson et al. 2002; Wang et al. 2004; Evans et al. 2005; Lu et al. 2008), but it has been proposed that loss of Kv function contributes to smooth muscle proliferation and thus to the progression of pulmonary hypertension (Sweeney & Yuan, 2000; Moudgil et al. 2006) by promoting cell survival (Ekhterae et al. 2001, 2003).
Kv current suppression during hypoxia (Post et al. 1992; Yuan et al. 1993; Firth et al. 2008) occurs as a consequence of inhibition of mitochondrial oxidative phosphorylation (Firth et al. 2008, 2009). However, the nature of the signalling pathway that couples mitochondrial function to Kv channels has been unclear. In this respect, little attention has been paid to the role of the AMP‐activated protein kinase (AMPK), although we have previously proposed that it couples inhibition of mitochondrial metabolism by hypoxia to acute HPV (Evans et al. 2005; Evans, 2006) and may also contribute to the progression of pulmonary hypertension (Evans et al. 2005; Evans, 2006; Ibe et al. 2013; Goncharov et al. 2014). AMPK, an energy sensor that acts to maintain cellular energy homeostasis, exists as heterotrimers comprising catalytic α subunits and regulatory β and γ subunits (Hardie, 2014a,b,c). AMPK is coupled to mitochondrial metabolism through changes in the cellular AMP:ATP and ADP:ATP ratios. Binding of AMP to the γ subunit causes a 10‐fold increase in AMPK activity by allosteric activation, but a further activation of up to 100‐fold can be generated by binding of either AMP or ADP, which promotes phosphorylation and inhibits dephosphorylation of Thr172 on the α subunit; these effects are antagonised by ATP (Gowans et al. 2013; Ross et al. 2016). Thr172 is primarily phosphorylated by the tumour suppressor kinase LKB1 (liver kinase B1), which appears to be constitutively active (Sakamoto et al. 2004), but which phosphorylates AMPK more rapidly when AMP is bound to the γ subunit. In an alternative Ca2+‐dependent activation mechanism, the calmodulin‐dependent protein kinase CaMKKβ can also phosphorylate Thr172 and hence activate AMPK in an AMP‐independent manner (Hardie, 2014a,b,c). The classical role of AMPK is to maintain energy homeostasis under conditions of metabolic stress, by activating catabolic processes that generate ATP and inhibiting non‐essential anabolic processes that consume ATP. However, AMPK has also been shown to regulate a wide variety of ion channels and membrane transport proteins (Evans et al. 2009; Lang & Foller, 2014), including Kv2.1 (Ikematsu et al. 2011), KCa3.1 (Ross et al. 2011), and Kir 2.1 and Kv7.1 (Lang & Foller, 2014).
Of the various known Kv channel types, it has been established that both Kv2.1 and Kv1.5 contribute to voltage‐gated potassium currents in pulmonary arterial myocytes (Smirnov et al. 2002; Archer et al. 2004; Firth et al. 2011; Olschewski et al. 2014). Their relative contributions vary in a manner related to arterial diameter, with the greatest level of Kv1.5 expression (and contribution to Kv currents) occurring in myocytes from near‐resistance‐sized arteries (Archer et al. 1998, 2004; Smirnov et al. 2002; Moral‐Sanz et al. 2011), the response of which to hypoxia is critical to acute increases in pulmonary arterial perfusion pressure. Moreover, selective down‐regulation of Kv1.5 has been identified as a hallmark of pulmonary hypertension (Yuan et al. 1998; Michelakis et al. 2002; Bonnet et al. 2006; Remillard et al. 2007; Burg et al. 2010; Morales‐Cano et al. 2014). Consistent with this view, overexpression of Kv1.5 enhances apoptosis (Brevnova et al. 2004), while adenoviral expression of a Kv1.5 transgene in vivo reduces pulmonary hypertension and restores HPV (Pozeg et al. 2003).
We show here that AMPK selectively inhibits Kv1.5 in pulmonary arterial myocytes, and also phosphorylates and inhibits recombinant Kv1.5 channels expressed in HEK 293 cells.
Methods
Ethical approval
All experiments were performed under the United Kingdom Animals (Scientific Procedures) Act 1986. The animals used in this study were male Sprague Dawley rats that underwent no experimental procedures as recognised under UK Law. They were killed using a Schedule 1 method for collection of tissues only, which does not require formal ethical approval in the UK. Frozen canine tissue was left over from a previous project where surgical procedures and protocols were approved by the Cleveland Clinic Foundation Institutional Animal Care and Use Committee (Cleveland, OH, USA).
Smooth muscle cell isolation
Resistance pulmonary arteries (<200 μm inner diameter) from male Sprague Dawley rats (250–350 g) were dissected into a physiological bath solution of composition (in mm): NaCl 135, KCl 5, MgCl2 1, CaCl2 1, glucose 10, Hepes 10 (pH 7.4). For cell isolation, endothelium denuded arteries were transferred into a nominally calcium‐free bath solution containing (in mg ml−1): 1 papain, 0.8 dithiothreitol and 0.7 BSA. The tissue was incubated in the latter solution for 10 min at 37°C and gently triturated using a fire polished glass pipette to get dispersed pulmonary arterial smooth muscle cells.
Electrophysiological recordings
Pulmonary arterial myocytes or HEK 293 cells that stably expressed Kv1.5 were transferred to a recording chamber and perfused at 1 ml min−1 with bath solution. K+ currents were recorded by whole‐cell patch clamp and a pipette solution of the following composition (mm): KCl 140, MgCl2 1, EGTA 10, Hepes 10, Na2ATP 4, Na3GTP 0.1 (pH 7.2). Cells were superfused (3 ml min−1) at 37°C with bath solution steadily bubbled with either room air (normoxia) or 95% N2/5% CO2 [hypoxia, 4.4 ± 0.3% O2 in the experimental chamber; as measured with an optical oxygen meter (FireStingO2, Pyro Science, Aachen, Germany)]. For some experiments recombinant thiophosphorylated AMPK heterotrimers (α2β2γ1, α1β1γ1 or D157A kinase dead mutant) were added to the pipette solution. Kv currents were assessed by voltage ramps (−100 to +40 mV), single voltage steps (−80 to +40 mV) and by acquisition of full I–V relationships for steady state activation (200 ms steps from −80 to +40 mV in 10 mV increments) or inactivation (2 s inactivation steps from −80 to +40 mV in 10 mV increments, a 10 ms pre‐pulse at −80 mV followed by a single voltage step to +60 mV). Current magnitude was normalised to cell capacitance as required. Conductance values (G) were calculated from the equation G = I/(V − EK), where the Nernst equilibrium potential (EK) was calculated as −89 mV at 37°C. Normalised conductance/voltage profiles for Kv currents were fitted to a single Boltzmann function with the form G = G max/(1 + exp[− (V − V mid)/k]), where G max is the maximal conductance, V mid is the test potential for half‐maximal conductance (G 0.5) and k represents the slope of the activation curve. Patch pipettes had resistances of 4–6 MΩ. Series resistance was compensated for (60–80%) after achieving the whole‐cell configuration. Signals were sampled at 10 kHz and low‐pass filtered at 2 kHz. Voltage‐clamp acquisition and analysis protocols were performed using an Axopatch 200A amplifier/Digidata 1200 interface controlled by Clampex 10.0 software (Molecular Devices, Sunnyvale, CA, USA). Off‐line analysis was performed using Clampfit 10.0 (Molecular Devices). Data are expressed as current density (pA pF–1) or I/I zero, where I zero is the current magnitude recorded at the onset of a given experimental intervention.
Cell culture and transfection
HEK 293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin. Cells were transfected following the manufacturer's instructions with 12 μg of pcDNA3.1 encoding an HA‐tagged human Kv1.5 (KCNA5) using Fugene 6 (Promega, Madison, WI, USA) and lysed 48 h later.
RT‐PCR
Total RNA was isolated from frozen canine tissues and from frozen pelleted HEK 293 cells stably expressing Kv1.5, using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) as per the manufacturer's instructions. Reverse transcription PCRs (RT‐PCRs) were carried out on 200 ng of total RNA using the One‐Step RT‐PCR Kit (ABM, Richmond, Canada) as recommended in the manual. Then, 20 μl of the 50 μl reaction was run on a 1% agarose gel and visualised using Safe‐White (ABM) and a GelDoc equipped with Quantity One software (BioRad, Hercules, CA, USA). Kvβ primer sequences were as previously published (Platoshyn et al. 2004) and for the reference gene ReadyMade GAPDH primers (Integrated DNA Technologies, Coralville, IA, USA) were used with sequences as follows: GAPDH‐For, ACCACAGTCCATGCCATCAC; GAPDH‐Rev, TCCACCACCCTGTTGCTGTA. RT‐PCR was repeated with 250 ng of template and 35 cycles of PCR after very faint bands were observed in the Kv1.5 stable cell line for Kvβ1 and Kvβ3. Canine ventricle was also repeated for comparison.
Kv1.5 phosphorylation assays
Phosphorylation assays were performed as described previously (Ross et al. 2011) using AMPK purified from bacteria and phosphorylated with CAMKKβ (10 units ml−1) in the presence of 200 μm AMP for 30 min at 30°C.
Expression, purification and activation of bacterial AMPK
These were performed as described previously (Ross et al. 2011).
Isoform‐specific AMPK activities
Isoform‐specific AMPK activity was determined by immunoprecipitating tissue lysate with antibodies raised against α1 or α2 subunits bound to protein G‐Sepharose beads and quantified using the AMARA peptide and [γ‐32P]ATP substrates (Cheung et al. 2000).
Statistics
Data are expressed as means ± SEM or means ± SD, as indicated; n represents the number of cells tested from at least four different animals. Statistical analysis was performed using Student's t test for paired observations or one‐way ANOVA followed by a Dunnett's post hoc test. Differences were considered statistically significant at P < 0.05.
Results
Inhibition of mitochondrial oxidative phosphorylation activates AMPK and reduces Kv1.5 current density in pulmonary arterial myocytes
Biguanide drugs such as phenformin inhibit complex I of the mitochondrial respiratory chain (El‐Mir et al. 2000; Owen et al. 2000; Evans et al. 2005) and elicit consequent increases in the cellular AMP:ATP ratio and AMPK activation (Hawley et al. 2010). Consistent with this, pre‐incubation of second‐ and third‐order pulmonary arteries with phenformin (4 h) increased AMPK‐α1‐associated activity from (mean ± S.D.) 0.025 ± 0.001 to 0.403 ± 0.012 nmol min−1 mg−1 protein and AMPK‐α2‐associated activity from 0.0096 ± 0.001 to 0.126 ± 0.006 nmol min−1 mg−1 protein (Fig. 1 A, n = 3; 32 arteries, 8 rats). Furthermore, and in accordance with previously reported effects of mitochondrial inhibitors (Firth et al. 2008) and hypoxia (Platoshyn et al. 2001), pre‐incubation of acutely isolated pulmonary arterial myocytes with 1 mm phenformin (2–4 h) also caused pronounced reductions in Kv current density, from 126 ± 17 pA pF–1 in time‐matched controls to 55 ± 6 pA pF–1 at +40 mV (Fig. 1 Ca–b, n = 9–11, P < 0.001). Consistent with the effects of phenformin and previous investigations by others (Firth et al. 2008), acute application of antimycin A (1 μm), a rapidly acting inhibitor of complex III, caused equivalent reductions in Kv current density, from 131.3 ± 10.4 to 62.6 ± 11.1 pA pF–1 at +40 mV (Fig. 1 Da–b, n = 6, P < 0.001). Unless stated, in these and all subsequent experiments on pulmonary arterial myocytes, potassium currents were recorded in the presence of paxilline (1 μm) to block the large conductance voltage‐ and calcium‐activated K+ (BKCa) channel.
Figure 1. Inhibition of mitochondrial oxidative phosphorylation activates AMPK and reduces Kv1.5 current density in pulmonary arterial myocytes .
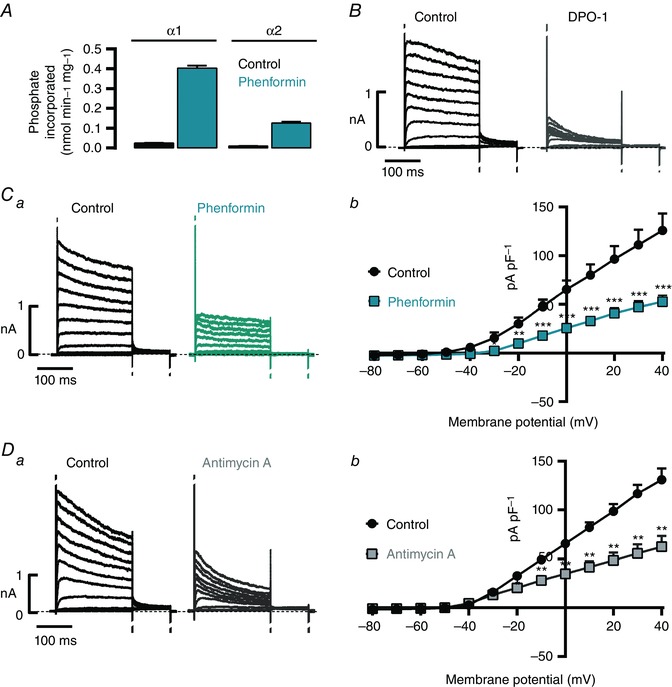
A, bar chart showing the effect of 10 mm phenformin on the activity of AMPK‐α1 and AMPK‐α2 containing heterotrimers, as determined by immunoprecipitate kinase assay (n = 3; 32 arteries, 8 rats). B, representative records (200 ms pulses from −80 to +40 mV in 10 mV increments, holding potential −80 mV) in pulmonary arterial myocytes before (control) and after extracellular application of 1 μm DPO‐1. C and D, representative records (a) and associated I–V relationships (b; 200 ms depolarization pulses from −80 to +40 mV in 10 mV increments, holding potential −80 mV) from myocytes pre‐incubated with 1 mm phenformin versus time‐matched controls (C, green, n = 9–11), or before and after 5–8 min extracellular application of 1 μm antimycin A (D, n = 6). * P < 0.05, ** P < 0.01 and *** P < 0.001.
Given that the contribution to native Kv currents of Kv2.1 and Kv1.5 varies, in a manner related to both the size and the regional location within the lung of the arteries from which myocytes are derived (Smirnov et al. 2002), we assessed the nature of the channels that underpin the Kv current within the cells under study. Application of the Kv1.5 blocker diphenyl phosphine oxide‐1 (DPO‐1, 1 μm, Fig. 1 B) in the absence of paxilline caused almost total inhibition of the Kv currents (96 ± 1% at +40 mV; n = 6, P < 0.0001), consistent with the view that Kv1.5 drives the majority of voltage‐gated K+ currents in myocytes of the near‐resistance‐sized pulmonary arteries studied here, which contribute most to the increase in pulmonary vascular resistance during hypoxia (Kato & Staub, 1966; Archer et al. 2004).
AMPK activation inhibits Kv1.5 currents in pulmonary arterial myocytes
We next assessed the effect on Kv1.5 current amplitude of extracellular application of three AMPK activators with distinct mechanisms of action, i.e. A769662, 5‐aminoimidazole‐4‐carboxamide riboside (AICAR) and C13. Analysis of the time course for Kv1.5 inhibition at steady‐state activation (100 ms at +40 mV) showed the time to onset of effect for A769662 (100 μm), AICAR (1 mm) and C13 (30 μm) to be around 2 min, with apparent maxima for inhibition achieved after 8–10 min (Fig. 2 B). After 10 min, Kv1.5 currents had declined (Fig. 2 A) from 144 ± 12 to 101 ± 9 pA pF–1 in the presence of A769662 (n = 14), from 186 ± 23 to 136 ± 17 pA pF–1 in the presence of AICAR (n = 6) and from 164 ± 8 to 104 ± 10 pA pF–1 in the presence of C13 (n = 8). Note, however, that in 3 of 9 cells superfused with AICAR we observed no effect (excluded from analysis).
Figure 2. Time course for reduction in Kv current in response to AMPK activators .
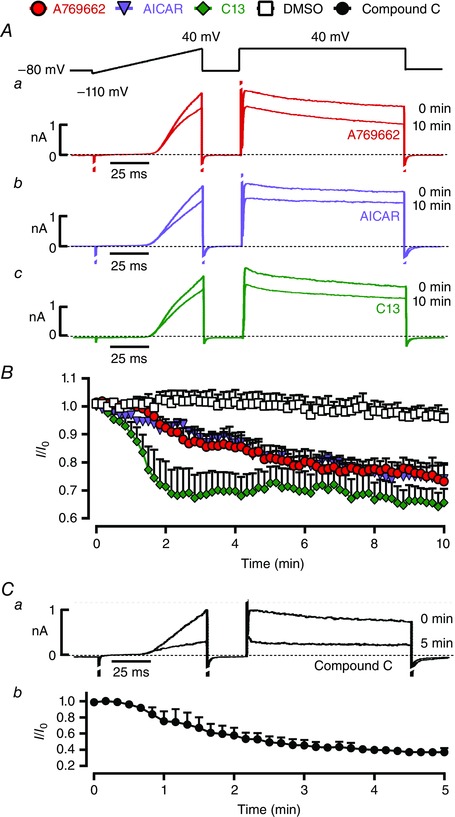
A, representative current records for voltage ramp and step protocol at 0 and 10 min after extracellular application of 100 μm A769662 (a), 1 mm AICAR (b) or 30 μm C13 (c). B, time course for reductions in Kv current during 10 min of extracellular application of DMSO (1:1000, vehicle control), 100 μm A769662, 1 mm AICAR and 30 μm C13. C, the effect on Kv currents of pulmonary arterial myocytes of 5 min extracellular application of 30 μm compound C (a), and the time course for inhibition (b). Results are expressed as mean ± SEM, n = 3–8.
Surprisingly, we also observed inhibition of Kv1.5 currents upon application of the non‐selective AMPK antagonist compound C (30 μm), from 164 ± 62 to 59 ± 24 pA pF–1 (n = 3; Fig. 2 C); this confounding effect precluded the use of this agent in further studies on myocytes. Previous studies have shown that compound C has little or no effect on resting pulmonary arterial tone, but inhibits acute HPV in a concentration‐dependent manner (Robertson et al. 2008), which must therefore be induced independently of the inhibition by hypoxia of Kv1.5 inhibition (Dipp et al. 2001). It is worth noting that in a screen of 70 protein kinases, at least 10 were inhibited by compound C more potently than AMPK (Bain et al. 2007). Thus, it is not a specific inhibitor of AMPK, a point reinforced by our findings that it markedly attenuates Kv1.5 function. This should be considered when interpreting outcomes of other cell‐based assays that have employed compound C, not least with respect to myocyte proliferation and survival (Ibe et al. 2013).
Importantly, upon equilibration of pulmonary arterial myocytes with AICAR, A769662 and C13, reductions in current density were evident throughout the I–V range over which Kv1.5 currents were activated (Fig. 3 A, B). This was confirmed by the fact that A769662 was without effect on residual currents observed following pre‐incubation of cells with DPO‐1 even in the absence of paxilline (Fig. 3 C), current density measuring 10 ± 2 pA pF–1 in the presence of DPO‐1 alone and 11 ± 3 pA pF–1 in the presence of DPO‐1 and A769662 (n = 6).
Figure 3. AMPK activation inhibits Kv1.5 currents in pulmonary arterial myocytes .
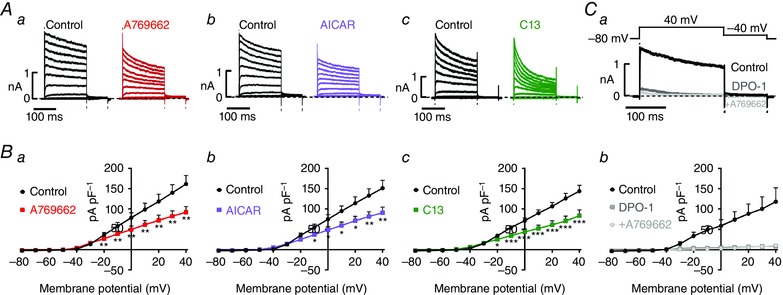
A, representative current traces (200 ms depolarization pulses from −80 to +40 mV in 10 mV increments, holding potential −80 mV); and B, I–V relationships for Kv current recorded before (control) and after extracellular application of A769662 (a, 100 μm), AICAR (b, 1 mm) or C13 (c, 30 μm); measurements taken at end of pulse. C, representative current trances (a) and I–V relationships (b) for Kv currents in the absence and presence of 1 μm DPO‐1 and the effect of 100 μm A769662 in the continued presence of DPO‐1. Results are expressed as mean ± SEM, n = 5–7. * P < 0.05, ** P < 0.01 and *** P < 0.001.
Intracellular application of active AMPK heterotrimers inhibits Kv1.5 in pulmonary arterial myocytes
Although A769662, AICAR and C13 activate AMPK by different mechanisms and are therefore unlikely to have the same off‐target effects, we also analysed the effect on endogenous Kv1.5 channel function of bacterially expressed human AMPK heterotrimers (α2β2γ1 or α1β1γ1 complexes). These had been thiophosphorylated at Thr172 using CaMKKβ to yield active, recombinant AMPK that is also resistant to phosphatases (Ross et al. 2011). Intracellular dialysis of either of the active α2β2γ1 or α1β1γ1 heterotrimers (Fig. 4 A) evoked Kv1.5 current inhibitions that were similar in magnitude (−33 ± 5% for α2β2γ1 and −36 ± 7% for α1β1γ1 at +40 mV; n = 5–7) to the reductions induced by pharmacological activation of AMPK. Importantly, current inhibition was not observed upon intracellular dialysis of an inactive AMPK heterotrimer [Fig. 4 B; α2β2γ1 complex with D157A mutation in α2 (Ross et al. 2011)]. Based on the use both of pharmacological activation of endogenous AMPK and of exogenous recombinant AMPK, we can conclude that AMPK mediates, either directly or indirectly, inhibition of Kv1.5 currents in pulmonary arterial myocytes.
Figure 4. Intracellular application of active AMPK heterotrimers inhibits Kv1.5 in pulmonary arterial myocytes .
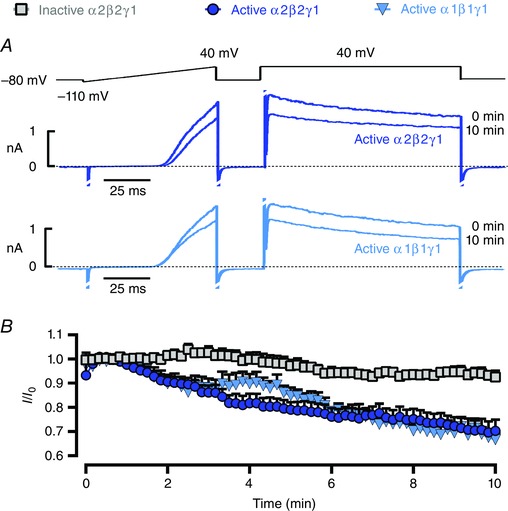
A, voltage ramp and step protocol recorded at 0 and 10 min after intracellular dialysis of the indicated recombinant, thiophosphorylated active AMPK heterotrimers (α2β2γ1 or α1β1γ1). B, time course for reduction in Kv current following intracellular dialysis of either active α2β2γ1 (thiophosphorylated, 5 U ml−1), active α1β1γ1 (thiophosphorylated, 5 U ml−1) or inactive α2β2γ1 (D157A mutant) AMPK heterotrimer. Results are expressed as mean ± SEM, n = 5–7.
Hypoxia and mitochondrial inhibitors attenuate Kv1.5 currents and occlude further current inhibition by AMPK activation
AMPK is intimately coupled to mitochondrial metabolism through changes in the AMP:ATP and ADP:ATP ratios (Gowans et al. 2013), which is evident from the fact that AMPK activity was increased by hypoxia (Evans et al. 2005) and by the mitochondrial inhibitor phenformin (Fig. 1 A). To assess whether AMPK acted as a downstream mediator of Kv1.5 inhibition during hypoxia and inhibition of mitochondrial oxidative phosphorylation, we therefore carried out studies to determine if Kv1.5 inhibition by these stimuli was additive with respect to that induced by AMPK activators.
Superfusion of pulmonary arterial myocytes with a hypoxic solution (∼4% O2, > 10 min) markedly inhibited Kv1.5 currents, with a maximal reduction achieved after ∼10 min (Fig. 5 A, B, 150 ± 9 pA pF–1 during normoxia vs. 63 ± 6 pA pF–1, n = 14–19, after 10–40 min of hypoxia; P < 0.001). Most importantly, continued hypoxia markedly attenuated the degree of Kv1.5 inhibition upon application of 100 μm A769662, which reduced current density from 53 ± 4 pA pF–1 during hypoxia alone to 45 ± 5 pA pF–1 (n = 6, P < 0.05; Figs 5 B, C and 6 A). As described previously, pre‐incubation of acutely isolated myocytes with 1 mm phenformin (2 h) also caused pronounced reductions in Kv current density, from 126 ± 17 to 55 ± 6 pA pF–1 at +40 mV (Fig. 1 Ca–b, n = 9–11, P < 0.001). Moreover, and consistent with the effects of hypoxia, pre‐incubation of cells with phenformin markedly attenuated the degree to which Kv1.5 currents were inhibited upon application of A769662, which reduced current density from 57 ± 12 pA pF–1 in the presence of phenformin alone to 49 ± 10 pA pF–1 (Figs 5 Cb and 6 A; n = 4, P = 0.09). In this respect antimycin A was equally effective, A769662 reducing Kv1.5 current density from 44 ± 4 pA pF–1 in the presence of antimycin A alone to 33 ± 4 pA pF–1 (Figs 5 Cc and 6 A; n = 3, P < 0.05). Therefore, Kv1.5 inhibition by pharmacological AMPK activation is non‐additive with respect to the action of either hypoxia or respiratory poisons. We can therefore conclude that in myocytes from near‐resistance‐sized pulmonary arteries, AMPK probably acts as the primary downstream mediator of Kv1.5 channel inhibition not only during hypoxia, but also in response to the inhibition of mitochondrial oxidative phosphorylation.
Figure 5. Hypoxia and mitochondrial inhibitors attenuate Kv1.5 currents and occlude further current inhibition by AMPK activation .
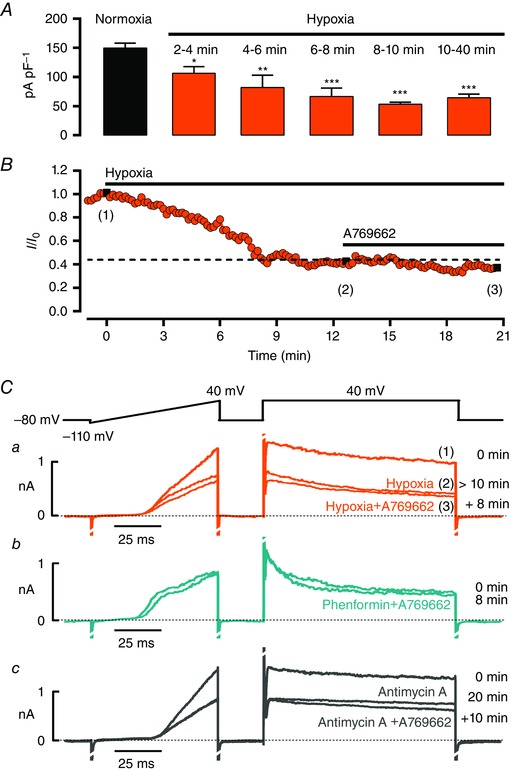
A, bar chart showing the reduction in current density at +40 mV from myocytes superfused with a hypoxic solution (4% O2; n = 4–19). B, time course for reduction in Kv current during extracellular superfusion with hypoxic solution and the effect of subsequent addition of 100 μm A769662; measurements taken at the end of a 100 ms step pulse to +40 mV. Ca, example records for voltage ramp and step protocol under normoxia (1), > 10 min of hypoxia (2), after 8 min of superfusion with hypoxia + 100 μm A769662 (3). Cb‐c, as in a but representative current traces show the effect of 100 μm A769662 on myocytes pre‐incubated with 1 mm phenformin (b) or after 20 min extracellular application of 1 μm antimycin A (c). * P < 0.05, ** P < 0.01 and *** P < 0.001 vs. normoxia.
Figure 6. Comparison of the change in current density and voltage–conductance relationships induced by activated AMPK and AMPK activators in pulmonary arterial myocytes .
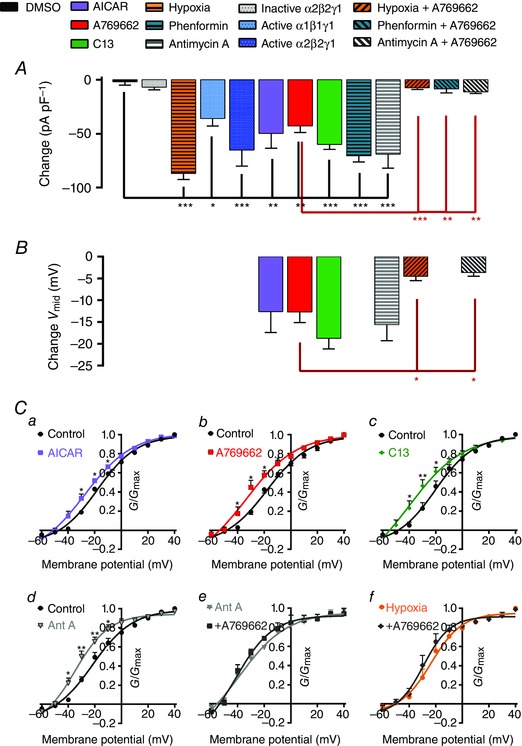
A, bar chart showing mean ± SEM change in current density at the end of each experimental intervention after 6–10 min of DMSO (1:1000), inactive α2β2γ2, hypoxia (∼4% O2), 5 U active α2β2γ2, 5 U active α1β1γ1, 1 mm AICAR, 100 μm A769662, 30 μm C13, 1 mm phenformin (2–4 h), 1 μm antimycin A, 100 μm A769662 in the presence of hypoxia (∼4% O2, > 10 min), phenformin and antimycin A (n = 3–16). B, similar to A but showing the net change in V mid of the voltage–conductance plots for 1 mm AICAR, 100 μm A769662, 30 μm C13, 1 μm antimycin A, 100 μm A769662 in the presence of 1 μm antimycin A and 100 μm A769662 in the presence of hypoxia (∼4% O2, > 10 min); n = 3–7. C, voltage–conductance plots showing effects of 1 mm AICAR (a), 100 μm A769662 (b), 30 μm C13 (c), 1 μm antimycin A (d), 100 μm A769662 in the presence of 1 μm antimycin A (e) and 100 μm A769662 in the presence of hypoxia (f, ∼4% O2, > 10 min). Results are expressed as mean ± SEM, n = 3–7. * P < 0.05, ** P < 0.01 and *** P < 0.001.
AMPK activators induce a leftward shift in the I–V relationship for Kv1.5 current activation that is occluded by hypoxia and mitochondrial inhibitors
AMPK activation not only reduced Kv1.5 current density in pulmonary arterial myocytes throughout the I–V range over which Kv1.5 currents were activated, but also induced a significant 12–14 mV hyperpolarizing shift in the activation curve (Fig. 6 B, C) analysed as G/G max and fitted to a single Boltzmann function. V mid measured: −17.9 ± 1.2 and −30.5 ± 4.5 mV in the absence and presence of AICAR, respectively; −17.4 ± 1.6 and −30.2 ± 3.5 mV in the absence and presence of A769662; −20.9 ± 2.6 and −39.7 ± 4.6 mV in the absence and presence of C13 (n = 5–7, P < 0.01). To allow for direct comparison of the maximal effect of each of these agents, we also expressed the leftward shift as the net change in V mid (Fig. 6 B); ΔV mid was −12.6 ± 4.8, −12.7 ± 2.4 and −18.7 ± 2.3 mV, respectively, for AICAR, A769662 and C13. As previously observed for current inhibition, the leftward shift in the I–V range for Kv1.5 activation that was induced by AMPK activation was non‐additive with respect to the effect of hypoxia and prior inhibition of mitochondrial oxidative phosphorylation, ΔV mid for A769662 measuring −4.5 ± 0.9 mV in the presence of hypoxia and −3.7 ± 0.6 mV in the presence of antimycin A.
To explore further the functional significance of a leftward shift in the I–V relationship we assessed the voltage‐dependence of both Kv1.5 activation and inactivation in the absence and presence A769662, and thus determined the effect of AMPK activation on the window current, i.e. the proportion of current at a given potential that is never inactivated. Figure 7 clearly shows that AMPK activation by A769662 induced a leftward shift in Kv1.5 activation and inactivation curves and thus of the window current, lowering the threshold for activation while reducing the available non‐inactivating current.
Figure 7. A769662 induces a leftward shift in the activation and inactivation curves of Kv1.5 .
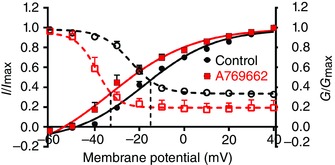
Plot shows the voltage–conductance relationship for Kv1.5 activation and inactivation in the absence (control, black) and presence of 100 μm A769662 (red). Activation is indicated by filled symbols and continuous lines; inactivation is indicated by open symbols and dashed lines. Data points are mean ± SEM (n = 3–6). Curves were obtained by fitting to the sigmoidal Boltzmann equation.
AMPK phosphorylates Kv1.5 and reduces K+ currents carried by recombinant Kv1.5 channels stably expressed in HEK 293 cells
To determine whether AMPK modulates Kv1.5 channel function directly, we examined the effects of AMPK activation on human Kv1.5 channels stably expressed in HEK 293 cells. Application of A769662 (100 μm) reduced K+ currents carried by recombinant human Kv1.5 (Fig. 8 Aa) in a manner that was blocked by the non‐selective AMPK inhibitor compound C (40 μm, Fig. 8 B). Moreover, intracellular dialysis of active AMPK α2β2γ1 or α1β1γ1 heterotrimers also reduced Kv1.5 currents, which remained unaffected in the presence of an inactive (D157A mutant) α2β2γ1 heterotrimer (Fig. 8 Ab–C). Like pulmonary arterial myocytes, therefore, currents carried by human Kv1.5 expressed in HEK 293 cells were similarly inhibited both by AMPK activators and by intracellular dialysis of recombinant active AMPK heterotrimers (Fig. 8 D).
Figure 8. AMPK activation phosphorylates Kv1.5 and reduces K+ currents carried by recombinant Kv1.5 channels stably expressed in HEK 293 cells .
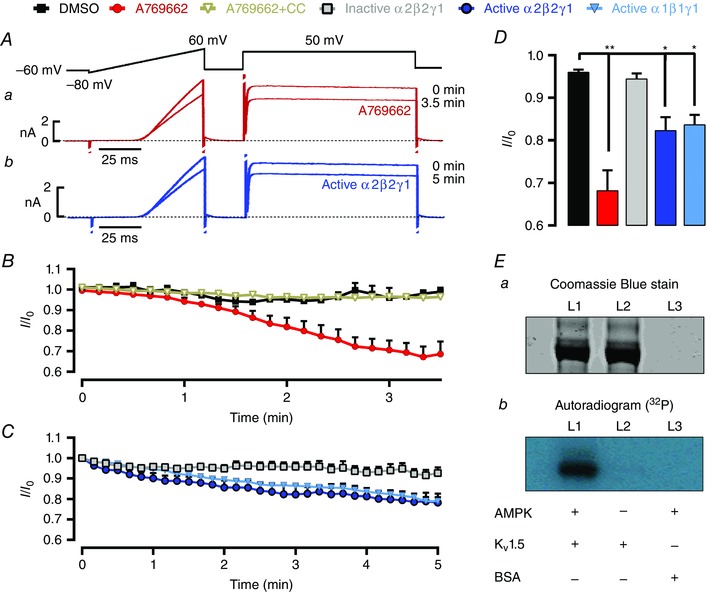
A, K+ currents carried by Kv1.5 stably expressed in HEK 293 cells before (0 min) and 3.5 min after extracellular application of (a) 100 μm A769662 or 5 min following intracellular dialysis of the active AMPK heterotrimer (b) (α2β2γ1, thiophosphorylated, 2 U ml−1). B, time course for reduction of whole‐cell K+ currents during extracellular application of DMSO (1:1000, vehicle control), 100 μm A769662, the combined application of 40 μm compound C and 100 μm A769662; or C, intracellular dialysis of active α2β2γ1 (thiophosphorylated, 2 U ml−1), active α1β1γ1(thiophosphorylated, 2 U ml−1) or inactive α2β2γ1 (D157A mutant). D, bar chart showing mean ± SEM residual current at the end of each experimental intervention (3.5–5 min). Results are means ± SEM, n = 3–7.* P < 0.05 and ** P < 0.01 vs. control. E, Coomassie blue stained SDS‐PAGE (a) and autorad (b) of Kv1.5 protein incubated with okodaic acid, 200 μm AMP, 32P‐ATP in the presence (L1) or absence (L2) of bacterial activated AMPK (α2β2γ1), and (L3) 1 μg of BSA treated as in L1.
We also examined whether AMPK directly phosphorylates Kv1.5, using as substrate the human protein immunoprecipitated from these HEK 293 cells. We first treated the immunoprecipitate with recombinant protein phosphatase (PP1γ) to remove endogenous phosphate groups, then phosphorylated with purified rat liver AMPK (a mixture of α1β1γ1 and α2β1γ1 isoforms) and [γ‐32P]ATP in the presence and absence of 200 μm AMP. The stoichiometry of phosphorylation was estimated by cutting out and counting the 32P‐labelled band and estimating the protein content by comparison with serum albumin standards run on the same gel. We obtained estimates of 0.57 and 0.13 moles of phosphate per mole of protein in the presence and absence of AMP (data not shown). We repeated the experiment using recombinant human α1β1γ1 and α2β2γ1 complexes expressed in bacteria, and obtained stoichiometries of 0.7 and 1.6 moles of phosphate per mole of protein respectively (Fig. 8 E, we did this only in the presence of AMP because, for reasons that remain unclear, the bacterially expressed complexes are much less AMP‐dependent). While we have not yet determined the number and identity of the sites phosphorylated on Kv1.5, these results indicate that different AMPK complexes can catalyse a substantial AMP‐activated phosphorylation of Kv1.5 in cell‐free assays.
Discussion
The present investigation describes, for the first time, evidence that AMPK couples Kv1.5 channel function (defined by the Kv1.5 blocker DPO‐1) to the inhibition by hypoxia of mitochondrial metabolism in pulmonary arterial myocytes. Consistent with this proposal, inhibition by phenformin or hypoxia of the mitochondrial electron transport chain (El‐Mir et al. 2000; Owen et al. 2000) increases NAD(P)H autofluorescence (Evans et al. 2005), activates AMPK and inhibits Kv currents in pulmonary arterial myocytes. That AMPK activation may specifically regulate Kv1.5 in response to metabolic stresses such as hypoxia gained further support from our findings that AMPK activators that are structurally distinct and have different mechanisms of action, namely A769662, AICAR and C13, all markedly inhibited Kv currents in pulmonary arterial myocytes. A769662, which primarily causes allosteric activation (Goransson et al. 2007; Scott et al. 2014), binds in a site located between the α and β subunits of AMPK (Xiao et al. 2013). AICAR (Corton et al. 1995) and C13 (Gomez‐Galeno et al. 2010) act similarly in the sense that they are both taken up into cells and converted to molecules (ZMP and C2, respectively) that bind to the γ subunit, mimicking the effects of AMP. However, C2 is a much more potent activator of AMPK than ZMP and, unlike the latter, does not affect other AMP‐sensitive enzymes such as glycogen phosphorylase or fructose‐1,6‐bisphosphatase (Hunter et al. 2014). Moreover, A769662 is selective for complexes containing the β1 subunit (Scott et al. 2008), while C2 is selective for complexes containing the α1 subunit (Hunter et al. 2014). These data suggest that Kv1.5 current inhibition in pulmonary arterial myocytes may be delivered in whole or in part by AMP‐dependent activation of heterotrimers containing α1 and β1. Furthermore, inhibition of Kv1.5 by hypoxia or by pre‐incubation with inhibitors of mitochondrial oxidative phosphorylation prevented further current reduction by AMPK activators, suggesting that AMPK may act as the primary regulator of Kv1.5 downstream of inhibition of mitochondrial oxidative phosphorylation during hypoxia. This conclusion is also supported by previous findings that hypoxia activates AMPK (Evans et al. 2005) and achieves maximal AMPK phosphorylation within ∼10 min (Ibe et al. 2013). Crucially, given the possible off‐target effects of any pharmacological agents, intracellular dialysis of active and phosphatase‐resistant (thiophosphorylated) recombinant AMPK heterotrimers selectively inhibited Kv currents in pulmonary arterial myocytes, while a kinase‐dead AMPK mutant was without effect. Although our results with C13 and A769662 suggest that an endogenous α1β1‐containing complex may regulate Kv1.5 in pulmonary arterial myocytes, our results with intracellular dialysis of α1β1γ1 and α2β2γ1 heterotrimers suggest that both are capable of regulating Kv1.5.
It is also interesting to note that AMPK activation not only induced a leftward shift in half maximal activation of Kv currents, as previously reported for effects of mitochondrial inhibitors (Firth et al. 2008), but also a leftward shift in half maximal inactivation and thus reduced the available non‐inactivating current, i.e. the window current. The overall effect would be to lower the threshold for Kv1.5 activation and thus increase the threshold for membrane depolarization, while reducing the opposition to membrane depolarization once initiated. These outcomes argue against a role for Kv1.5 inhibition in the initiation, through membrane depolarisation, of acute HPV. This view is supported by the fact that compound C inhibits Kv1.5, but has little or no effect on resting pulmonary arterial tone despite the fact that it inhibits acute HPV in a concentration‐dependent manner (Robertson et al. 2008). When considered together, our study therefore provides further support for the view that acute HPV is induced in a manner independent of Kv1.5 inhibition (Dipp et al. 2001; Prieto‐Lloret et al. 2015). That aside, our findings suggest that care must be taken when assessing studies that have employed compound C to examine the role of AMPK in pulmonary vascular function, given that the marked attenuation of Kv1.5 by compound C presents an important confounding variable with respect to investigations on myocyte proliferation and the progression of pulmonary hypertension (Ibe et al. 2013). Moreover in a screen of 70 protein kinases, at least 10 were inhibited by compound C more potently than AMPK (Bain et al. 2007). Compound C cannot, therefore, be considered to be a selective inhibitor of AMPK, a point reinforced by our findings.
Our conclusions from studies on native Kv currents in pulmonary arterial myocytes were confirmed by further investigations on the regulation of hKv1.5 stably expressed in HEK 293 cells. Native rat liver AMPK and two combinations (α1β1γ1 and α2β2γ1) of bacterially expressed human AMPK isoforms were found to incorporate near‐stoichiometric amounts of phosphate into immunoprecipitated hKv1.5 channels, and with rat liver AMPK this was stimulated by AMP, making it very unlikely that the phosphorylation was catalysed by a contaminating kinase. AMPK activators and recombinant heterotrimers also inhibited currents carried by recombinant Kv1.5 channels in intact HEK 293 cells. This suggests that AMPK may directly regulate the channel protein even though Kvβ1, Kvβ2 and Kvβ3 may be expressed to varying degrees in HEK 293 cells that stably express Kv1.5 (Fig. 9), as has been reported previously (Platoshyn et al. 2004).
Figure 9. Transcripts for all three Kvβ genes are expressed in the HEKKv1.5 cell line .
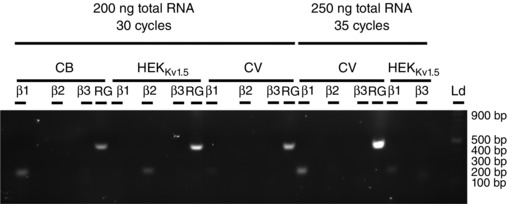
Gel showing RT‐PCR amplicons for Kvβ1, Kvβ2, Kvβ3 and the reference gene (RG), GAPDH, from canine brain (CB), canine ventricle (CV) and the HEKKv1.5 stable line used in this study. The 500 base pair band of the ladder represents ∼1200 ng of DNA (2‐Log DNA Ladder, New England Biolabs, Ipswich, MA, USA).
Our results support a model in which inhibition of mitochondrial oxidative phosphorylation by hypoxia in pulmonary arterial myocytes triggers AMPK‐dependent inhibition of Kv1.5 channels, in line with the observation that co‐expression of Kv1.5 and AMPK reduced Kv current and Kv1.5 channel abundance in the cell membrane of oocytes (Mia et al. 2012). Our finding that AMPK phosphorylates and inhibits Kv1.5 is also entirely consistent with previous evidence that AMPK mediates acute HPV (Evans et al. 2005; Robertson et al. 2008), and the proposal that AMPK may also contribute to smooth muscle proliferation and the development of pulmonary arterial hypertension (Ibe et al. 2013; Goncharov et al. 2014). This is evident from the fact that down‐regulation of Kv1.5 expression and activity is a hallmark not only of HPV but also of pulmonary hypertension (Yuan et al. 1998; Michelakis et al. 2002; Bonnet et al. 2006; Remillard et al. 2007; Burg et al. 2010; Morales‐Cano et al. 2014). This down‐regulation may lead to increased survival of smooth muscle cells due to attenuation of K+ channel‐dependent apoptosis (Krick et al. 2001; Brevnova et al. 2004; Moudgil et al. 2006), and facilitate the phenotypic switch from a contractile to a proliferative state (Cidad et al. 2012, 2015). Further support for this view may be taken from the finding that over‐expression of Kv1.5 enhances apoptosis (Brevnova et al. 2004), while adenoviral transgene expression of Kv1.5 in vivo reduces pulmonary hypertension and restores HPV (Pozeg et al. 2003). Therefore, it is possible that dysfunction of the mitochondrial–AMPK signalling pathway may predispose individuals to hypoxia and other forms of pulmonary arterial hypertension (Bonnet et al. 2006). In this respect, it is interesting to note that single nucleotide polymorphisms (SNPs) in the gene encoding Kv1.5 predispose to pulmonary hypertension and reduce Kv1.5 channel availability in pulmonary arterial myocytes (Remillard et al. 2007), raising the intriguing possibility that this may be due, at least in part, to alterations in AMPK‐dependent regulation of Kv1.5.
AMPK phosphorylates target proteins containing a Φ(X,β)XXS/TXXXΦ (Φ, hydrophobic; β, basic) recognition motif (Hardie et al. 2016). The protein sequence for Kv1.5 presents 15 serines and 4 threonines susceptible to phosphorylation by serine–threonine kinases (Blom et al. 1999). However, none of these represents good matches to the consensus recognition sites for AMPK (http://scansite3.mit.edu), despite the fact that our studies on 32P phosphorylation indicate that the immunoprecipitated channel protein might be a direct substrate for AMPK. This raises two distinct possibilities, (1) AMPK recognises non‐canonical sites within the Kv1.5 sequence, as has been shown for other proteins (Jones et al. 2005; Chang et al. 2009; Egan et al. 2011); or (2) AMPK phosphorylates one or more associated protein(s), such as the regulatory β subunits. The fact that AMPK phosphorylates and regulates Kv1.5 suggests that its effects are mediated, at least in part, independently of such interactions. Nevertheless, we cannot rule out the possibility that outcomes may be modulated by the β subunits, given that rat pulmonary arterial myocytes express Kvβ1, Kvβ2 and Kvβ3 (Platoshyn et al. 2006) and phosphorylation of either Kv1.5 or regulatory Kvβ subunits may modulate not only channel gating and inactivation kinetics (Holmes et al. 1996; Williams et al. 2002) but also the sensitivity of Kv1.5 to regulation by Kvβ subunits (Kwak et al. 1999; David et al. 2012; Macias et al. 2014). It is equally plausible that AMPK‐dependent phosphorylation of Kv1.5 in pulmonary arterial myocytes may alter Kvα−Kvβ interactions, sensitivity to metabolic stress, channel trafficking (Martens et al. 1999; Tipparaju et al. 2012) and/or degradation via ubiquitin ligases (Mia et al. 2012; Andersen et al. 2015). Further studies will be aimed at identifying the AMPK phosphorylation sites on Kv1.5 and on associated β subunits.
In conclusion, we propose that AMPK couples the inhibition of mitochondrial oxidative phosphorylation to Kv1.5 channel inhibition in pulmonary arterial myocytes, which may contribute to the regulation by AMPK of smooth muscle proliferation and thus to the development of pulmonary hypertension. In addition, AMPK‐dependent modulation of Kv1.5 channel availability may also contribute to proliferative potential associated with other diseases, such as cancer (Bonnet et al. 2007; Comes et al. 2013; Vallejo‐Gracia et al. 2013).
Additional information
Competing interests
None of the authors have any disclosures.
Author contributions
This manuscript was written by J.M‐S. and A.M.E. All authors contributed to the conception and design or analysis and interpretation of data, and the drafting of the article or revising it critically for important intellectual content. All authors provided final approval of the version to be published. In particular, the idea for the study was conceived by A.M.E. Tissue collection and isolation of pulmonary arterial smooth muscle cells were done by J.M‐S. J.M‐S. completed all electrophysiology on pulmonary arterial myocytes. A.M.D. completed electrophysiology on HEK293 cells. AMPK trimers were provided by D.G.H. Immunoprecipitate kinase assays and phosphorylation assays were done by F.A.R. D.F and J.E. generated the HEK293 cell line that stably expressed hKv1.5 and analysed the expression of Kvβ in these cells. Studies were conducted at the Centre of Integrative Physiology (University of Edinburgh, Edinburgh, UK), College of Life Sciences (University of Dundee, Dundee, UK) and Department of Anaesthesiology. Pharmacology and Therapeutics, University of British Columbia (Life Sciences Centre, Vancouver, Canada). All authors revised and approved the final version of the manuscript.
Funding
This work was primarily funded by the Wellcome Trust (WT081195MA and WT097726) and the British Heart Foundation (RG/12/14/29885), but was also supported by the Canadian Institute for Health Research and the Heart and Stroke Foundation of Canada.
References
- Andersen MN, Skibsbye L, Tang C, Petersen F, MacAulay N, Rasmussen HB & Jespersen T (2015). PKC and AMPK regulation of Kv1.5 potassium channels. Channels (Austin) 9, 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Souil E, Dinh‐Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen‐Huu L, Reeve HL & Hampl V (1998). Molecular identification of the role of voltage‐gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101, 2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer SL, Wu XC, Thebaud B, Nsair A, Bonnet S, Tyrrell B, McMurtry MS, Hashimoto K, Harry G & Michelakis ED (2004). Preferential expression and function of voltage‐gated, O2‐sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells. Circ Res 95, 308–318. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR & Cohen P (2007). The selectivity of protein kinase inhibitors: a further update. Biochem J 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch P, Mairbaurl H, Maggiorini M & Swenson ER (2005). Physiological aspects of high‐altitude pulmonary edema. J Appl Physiol (1985) 98, 1101–1110. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S & Brunak S (1999). Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis‐Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B & Michelakis ED (2007). A mitochondria‐K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Michelakis ED, Porter CJ, Andrade‐Navarro MA, Thebaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK & Archer SL (2006). An abnormal mitochondrial‐hypoxia inducible factor‐1α‐Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113, 2630–2641. [DOI] [PubMed] [Google Scholar]
- Brevnova EE, Platoshyn O, Zhang S & Yuan JX (2004). Overexpression of human KCNA5 increases I K(V) and enhances apoptosis. Am J Physiol Cell Physiol 287, C715‐722. [DOI] [PubMed] [Google Scholar]
- Burg ED, Platoshyn O, Tsigelny IF, Lozano‐Ruiz B, Rana BK & Yuan JX (2010). Tetramerization domain mutations in KCNA5 affect channel kinetics and cause abnormal trafficking patterns. Am J Physiol Cell Physiol 298, C496–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TJ, Chen WP, Yang C, Lu PH, Liang YC, Su MJ, Lee SC & Chuang LM (2009). Serine‐385 phosphorylation of inwardly rectifying K+ channel subunit (Kir6.2) by AMP‐dependent protein kinase plays a key role in rosiglitazone‐induced closure of the KATP channel and insulin secretion in rats. Diabetologia 52, 1112–1121. [DOI] [PubMed] [Google Scholar]
- Cheung PC, Salt IP, Davies SP, Hardie DG & Carling D (2000). Characterization of AMP‐activated protein kinase γ‐subunit isoforms and their role in AMP binding. Biochem J 346, 659–669. [PMC free article] [PubMed] [Google Scholar]
- Cidad P, Jimenez‐Perez L, Garcia‐Arribas D, Miguel‐Velado E, Tajada S, Ruiz‐McDavitt C, Lopez‐Lopez JR & Perez‐Garcia MT (2012). Kv1.3 channels can modulate cell proliferation during phenotypic switch by an ion‐flux independent mechanism. Arterioscler Thromb Vasc Biol 32, 1299–1307. [DOI] [PubMed] [Google Scholar]
- Cidad P, Miguel‐Velado E, Ruiz‐McDavitt C, Alonso E, Jimenez‐Perez L, Asuaje A, Carmona Y, Garcia‐Arribas D, Lopez J, Marroquin Y, Fernandez M, Roque M, Perez‐Garcia MT & Lopez‐Lopez JR (2015). Kv1.3 channels modulate human vascular smooth muscle cells proliferation independently of mTOR signaling pathway. Pflugers Arch 467, 1711–1722. [DOI] [PubMed] [Google Scholar]
- Comes N, Bielanska J, Vallejo‐Gracia A, Serrano‐Albarras A, Marruecos L, Gomez D, Soler C, Condom E, Ramon YCS, Hernandez‐Losa J, Ferreres JC & Felipe A (2013). The voltage‐dependent K+ channels Kv1.3 and Kv1.5 in human cancer. Front Physiol 4, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA & Hardie DG (1995). 5‐Aminoimidazole‐4‐carboxamide ribonucleoside: a specific method for activating AMP‐activated protein kinase in intact cells? Eur J Biochem 229, 558–565. [DOI] [PubMed] [Google Scholar]
- David M, Macias A, Moreno C, Prieto A, Martinez‐Marmol R, Vicente R, Gonzalez T, Felipe A, Tamkun MM & Valenzuela C (2012). Protein kinase C (PKC) activity regulates functional effects of Kvβ1.3 subunit on KV1.5 channels: identification of a cardiac Kv1.5 channelosome. J Biol Chem 287, 21416–21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipp M, Nye PC & Evans AM (2001). Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol 281, L318–325. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M & Shaw RJ (2011). Phosphorylation of ULK1 (hATG1) by AMP‐activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekhterae D, Platoshyn O, Krick S, Yu Y, McDaniel SS & Yuan JX (2001). Bcl‐2 decreases voltage‐gated K+ channel activity and enhances survival in vascular smooth muscle cells. Am J Physiol Cell Physiol 281, C157–165. [DOI] [PubMed] [Google Scholar]
- Ekhterae D, Platoshyn O, Zhang S, Remillard CV & Yuan JX (2003). Apoptosis repressor with caspase domain inhibits cardiomyocyte apoptosis by reducing K+ currents. Am J Physiol Cell Physiol 284, C1405–1410. [DOI] [PubMed] [Google Scholar]
- El‐Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M & Leverve X (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275, 223–228. [DOI] [PubMed] [Google Scholar]
- Evans AM (2006). AMP‐activated protein kinase underpins hypoxic pulmonary vasoconstriction and carotid body excitation by hypoxia in mammals. Exp Physiol 91, 821–827. [DOI] [PubMed] [Google Scholar]
- Evans AM, Hardie DG, Peers C, Wyatt CN, Viollet B, Kumar P, Dallas ML, Ross F, Ikematsu N, Jordan HL, Barr BL, Rafferty JN & Ogunbayo O (2009). Ion channel regulation by AMPK: the route of hypoxia‐response coupling in thecarotid body and pulmonary artery. Ann NY Acad Sci 1177, 89–100. [DOI] [PubMed] [Google Scholar]
- Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, Kinnear NP & Hardie DG (2005). Does AMP‐activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2‐sensing cells? J Biol Chem 280, 41504–41511. [DOI] [PubMed] [Google Scholar]
- Firth AL, Gordienko DV, Yuill KH & Smirnov SV (2009). Cellular localization of mitochondria contributes to Kv channel‐mediated regulation of cellular excitability in pulmonary but not mesenteric circulation. Am J Physiol Lung Cell Mol Physiol 296, L347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AL, Remillard CV, Platoshyn O, Fantozzi I, Ko EA & Yuan JX (2011). Functional ion channels in human pulmonary artery smooth muscle cells: voltage‐dependent cation channels. Pulm Circ 1, 48–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth AL, Yuill KH & Smirnov SV (2008). Mitochondria‐dependent regulation of Kv currents in rat pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 295, L61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Galeno JE, Dang Q, Nguyen TH, Boyer SH, Grote MP, Sun Z, Chen M, Craigo WA, van Poelje PD, MacKenna DA, Cable EE, Rolzin PA, Finn PD, Chi B, Linemeyer DL, Hecker SJ & Erion MD (2010). A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med Chem Lett 1, 478–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharov DA, Kudryashova TV, Ziai H, Ihida‐Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM & Goncharova EA (2014). Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG & Sakamoto K (2007). Mechanism of action of A‐769662, a valuable tool for activation of AMP‐activated protein kinase. J Biol Chem 282, 32549–32560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans GJ, Hawley SA, Ross FA & Hardie DG (2013). AMP is a true physiological regulator of AMP‐activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014. a). AMP‐activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med 276, 543–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014. b). AMPK–sensing energy while talking to other signaling pathways. Cell Metab 20, 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2014. c). AMPK: positive and negative regulation, and its role in whole‐body energy homeostasis. Curr Opin Cell Biol 33C, 1–7. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE & Brunet A (2016). AMPK: an energy‐sensing pathway with multiple inputs and outputs. Trends Cell Biol 26, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM & Hardie DG (2010). Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes TC, Fadool DA, Ren R & Levitan IB (1996). Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science 274, 2089–2091. [DOI] [PubMed] [Google Scholar]
- Hunter RW, Foretz M, Bultot L, Fullerton MD, Deak M, Ross FA, Hawley SA, Shpiro N, Viollet B, Barron D, Kemp BE, Steinberg GR, Hardie DG & Sakamoto K (2014). Mechanism of action of Compound‐13: an α1‐selective small molecule activator of AMPK. Chem Biol 21, 866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibe JC, Zhou Q, Chen T, Tang H, Yuan JX, Raj JU & Zhou G (2013). Adenosine monophosphate‐activated protein kinase is required for pulmonary artery smooth muscle cell survival and the development of hypoxic pulmonary hypertension. Am J Resp Cell Mol Biol 49, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikematsu N, Dallas ML, Ross FA, Lewis RW, Rafferty JN, David JA, Suman R, Peers C, Hardie DG & Evans AM (2011). Phosphorylation of the voltage‐gated potassium channel Kv2.1 by AMP‐activated protein kinase regulates membrane excitability. Proc Natl Acad Sci USA 108, 18132–18137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ & Thompson CB (2005). AMP‐activated protein kinase induces a p53‐dependent metabolic checkpoint. Mol Cell 18, 283–293. [DOI] [PubMed] [Google Scholar]
- Kato M & Staub NC (1966). Response of small pulmonary arteries to unilobar hypoxia and hypercapnia. Circ Res 19, 426–440. [DOI] [PubMed] [Google Scholar]
- Krick S, Platoshyn O, Sweeney M, Kim H & Yuan JX (2001). Activation of K+ channels induces apoptosis in vascular smooth muscle cells. Am J Physiol Cell Physiol 280, C970–979. [DOI] [PubMed] [Google Scholar]
- Kwak YG, Navarro‐Polanco RA, Grobaski T, Gallagher DJ & Tamkun MM (1999). Phosphorylation is required for alteration of kv1.5 K+ channel function by the Kvβ1.3 subunit. J Biol Chem 274, 25355–25361. [DOI] [PubMed] [Google Scholar]
- Lahm T, Tuder RM & Petrache I (2014). Progress in solving the sex hormone paradox in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307, L7–26. [DOI] [PubMed] [Google Scholar]
- Lang F & Foller M (2014). Regulation of ion channels and transporters by AMP‐activated kinase (AMPK). Channels (Austin) 8, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wang J, Shimoda LA & Sylvester JT (2008). Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 295, L104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias A, de la Cruz A, Prieto A, Peraza DA, Tamkun MM, Gonzalez T & Valenzuela C (2014). PKC inhibition results in a Kv 1.5 + Kv β1.3 pharmacology closer to Kv 1.5 channels. Br J Pharmacol 171, 4914–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JR, Kwak YG & Tamkun MM (1999). Modulation of Kv channel α/β subunit interactions. Trends Cardiovasc Med 9, 253–258. [DOI] [PubMed] [Google Scholar]
- Mia S, Munoz C, Pakladok T, Siraskar G, Voelkl J, Alesutan I & Lang F (2012). Downregulation of Kv1.5 K channels by the AMP‐activated protein kinase. Cell Physiol Biochem 30, 1039–1050. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R & Archer SL (2002). Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage‐gated potassium channels. Circulation 105, 244–250. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Thebaud B, Weir EK & Archer SL (2004). Hypoxic pulmonary vasoconstriction: redox regulation of O2‐sensitive K+ channels by a mitochondrial O2‐sensor in resistance artery smooth muscle cells. J Mol Cell Cardiol 37, 1119–1136. [DOI] [PubMed] [Google Scholar]
- Moral‐Sanz J, Gonzalez T, Menendez C, David M, Moreno L, Macias A, Cortijo J, Valenzuela C, Perez‐Vizcaino F & Cogolludo A (2011). Ceramide inhibits Kv currents and contributes to TP‐receptor‐induced vasoconstriction in rat and human pulmonary arteries. Am J Physiol Cell Physiol 301, C186–194. [DOI] [PubMed] [Google Scholar]
- Morales‐Cano D, Menendez C, Moreno E, Moral‐Sanz J, Barreira B, Galindo P, Pandolfi R, Jimenez R, Moreno L, Cogolludo A, Duarte J & Perez‐Vizcaino F (2014). The flavonoid quercetin reverses pulmonary hypertension in rats. PloS ONE 9, e114492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudgil R, Michelakis ED & Archer SL (2006). The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13, 615–632. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Papp R, Nagaraj C & Olschewski H (2014). Ion channels and transporters as therapeutic targets in the pulmonary circulation. Pharmacol Ther 144, 349–368. [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E & Halestrap AP (2000). Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348, 607–614. [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Brevnova EE, Burg ED, Yu Y, Remillard CV & Yuan JX (2006). Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 290, C907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E & Yuan JX (2004). Diversity of voltage‐dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 287, L226–238. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Yu Y, Golovina VA, McDaniel SS, Krick S, Li L, Wang JY, Rubin LJ & Yuan JX (2001). Chronic hypoxia decreases KV channel expression and function in pulmonary artery myocytes. Am J Physiol Lung Cell Mol Physiol 280, L801–812. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL & Weir EK (1992). Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 262, C882–890. [DOI] [PubMed] [Google Scholar]
- Pozeg ZI, Michelakis ED, McMurtry MS, Thebaud B, Wu XC, Dyck JR, Hashimoto K, Wang S, Moudgil R, Harry G, Sultanian R, Koshal A & Archer SL (2003). In vivo gene transfer of the O2‐sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 107, 2037–2044. [DOI] [PubMed] [Google Scholar]
- Prieto‐Lloret J, Ramirez M, Olea E, Moral‐Sanz J, Cogolludo A, Castaneda J, Yubero S, Agapito T, Gomez‐Nino A, Rocher A, Rigual R, Obeso A, Perez‐Vizcaino F & Gonzalez C (2015). Hypoxic pulmonary vasoconstriction, carotid body function and erythropoietin production in adult rats perinatally exposed to hyperoxia. J Physiol 593, 2459–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O'Connor D T & Yuan JX (2007). Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292, C1837–1853. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Mustard KJ, Lewis TH, Clark JH, Wyatt CN, Blanco EA, Peers C, Hardie DG & Evans AM (2008). AMP‐activated protein kinase and hypoxic pulmonary vasoconstriction. Eur J Pharmacol 595, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FA, Jensen TE & Hardie DG (2016). Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem J 473, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FA, Rafferty JN, Dallas ML, Ogunbayo O, Ikematsu N, McClafferty H, Tian L, Widmer H, Rowe IC, Wyatt CN, Shipston MJ, Peers C, Hardie DG & Evans AM (2011). Selective expression in carotid body type I cells of a single splice variant of the large conductance calcium‐ and voltage‐activated potassium channel confers regulation by AMP‐activated protein kinase. J Biol Chem 286, 11929–11936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Goransson O, Hardie DG & Alessi DR (2004). Activity of LKB1 and AMPK‐related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol Endocrinol Metab 287, E310–E317. [DOI] [PubMed] [Google Scholar]
- Scott JW, Ling N, Issa SM, Dite TA, O'Brien MT, Chen ZP, Galic S, Langendorf CG, Steinberg GR, Kemp BE & Oakhill JS (2014). Small molecule drug A‐769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol 21, 619–627. [DOI] [PubMed] [Google Scholar]
- Scott JW, van Denderen BJ, Jorgensen SB, Honeyman JE, Steinberg GR, Oakhill JS, Iseli TJ, Koay A, Gooley PR, Stapleton D & Kemp BE (2008). Thienopyridone drugs are selective activators of AMP‐activated protein kinase β1‐containing complexes. Chem Biol 15, 1220–1230. [DOI] [PubMed] [Google Scholar]
- Smirnov SV, Beck R, Tammaro P, Ishii T & Aaronson PI (2002). Electrophysiologically distinct smooth muscle cell subtypes in rat conduit and resistance pulmonary arteries. J Physiol 538, 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney M & Yuan JX (2000). Hypoxic pulmonary vasoconstriction: role of voltage‐gated potassium channels. Respir Res 1, 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipparaju SM, Li XP, Kilfoil PJ, Xue B, Uversky VN, Bhatnagar A & Barski OA (2012). Interactions between the C‐terminus of Kv1.5 and Kvβ regulate pyridine nucleotide‐dependent changes in channel gating. Pflugers Arch 463, 799–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo‐Gracia A, Bielanska J, Hernandez‐Losa J, Castellvi J, Ruiz‐Marcellan MC, Ramon Y, Cajal S, Condom E, Manils J, Soler C, Comes N, Ferreres JC & Felipe A (2013). Emerging role for the voltage‐dependent K+ channel Kv1.5 in B‐lymphocyte physiology: expression associated with human lymphoma malignancy. J Leukocyte Biol 94, 779–789. [DOI] [PubMed] [Google Scholar]
- von Euler US & Liljestrand G (1946). Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 12, 301–320. [Google Scholar]
- Wang J, Shimoda LA & Sylvester JT (2004). Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286, L848–858. [DOI] [PubMed] [Google Scholar]
- Williams CP, Hu N, Shen W, Mashburn AB & Murray KT (2002). Modulation of the human Kv1.5 channel by protein kinase C activation: role of the Kvβ1.2 subunit. J Pharmacol Exp Ther 302, 545–550. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Mason HS, Smith GD, Nicholson N, Johnston L, Janiak R & Hume JR (2002). Comparative capacitative calcium entry mechanisms in canine pulmonary and renal arterial smooth muscle cells. J Physiol 543, 917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, Giordanetto F, Martin SR, Carling D & Gamblin SJ (2013). Structural basis of AMPK regulation by small molecule activators. Nat Commun 4, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV, Jr , Gaine SP, Orens JB & Rubin LJ (1998). Dysfunctional voltage‐gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98, 1400–1406. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Goldman WF, Tod ML, Rubin LJ & Blaustein MP (1993). Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 264, L116–123. [DOI] [PubMed] [Google Scholar]