

Abstract
A distinctive pattern of recurrent rapid falls in fetal heart rate, called decelerations, are commonly associated with uterine contractions during labour. These brief decelerations are mediated by vagal activation. The reflex triggering this vagal response has been variably attributed to a mechanoreceptor response to fetal head compression, to baroreflex activation following increased blood pressure during umbilical cord compression, and/or a Bezold–Jarisch reflex response to reduced venous return from the placenta. Although these complex explanations are still widespread today, there is no consistent evidence that they are common during labour. Instead, the only mechanism that has been systematically investigated, proven to be reliably active during labour and, crucially, capable of producing rapid decelerations is the peripheral chemoreflex. The peripheral chemoreflex is triggered by transient periods of asphyxia that are a normal phenomenon associated with all uterine contractions. This should not cause concern as the healthy fetus has a remarkable ability to adapt to these repeated but short periods of asphyxia. This means that the healthy fetus is typically not at risk of hypotension and injury during uncomplicated labour even during repeated brief decelerations. The physiologically incorrect theories surrounding decelerations that ignore the natural occurrence of repeated asphyxia probably gained widespread support to help explain why many babies are born healthy despite repeated decelerations during labour. We propose that a unified and physiological understanding of intrapartum decelerations that accepts the true nature of labour is critical to improve interpretation of intrapartum fetal heart rate patterns.

Introduction
Fetal heart rate (FHR) recordings are the only non‐invasive tool that we have to continuously monitor the wellbeing of fetuses during labour. Recordings are most commonly derived from a Doppler ultrasound transducer on the maternal abdomen that measures the contractions of the fetal heart. Alternatively the fetal electrocardiogram can be recorded from an electrode attached directly to the fetal scalp. During labour, the clinician will assess all aspects of the FHR trace, including baseline FHR, FHR variability and the presence of accelerations and decelerations of FHR. Uterine contractions are recorded alongside the FHR trace and thus the FHR features are interpreted in relation to uterine activity to provide a broad, composite indication of fetal wellbeing. Many of these aspects, and the overall interpretation of intrapartum FHR traces have been discussed in detail elsewhere (Parer & Ikeda, 2007; Westgate et al. 2007; Lear et al. 2016) and are beyond the scope of this review.
The most contentious aspect of FHR monitoring is the significance of the rapid but brief falls in FHR, called decelerations, which almost always occur in association with uterine contractions. It is unequivocally established that these decelerations are mediated by vagal activation, and can be blocked by atropine in both humans (Mendez‐Bauer et al. 1963; Caldeyro‐Barcia et al. 1966) and animals (Barcroft, 1946; Itskovitz et al.
1983 b; Westgate et al. 2007). However, the specific reflex that mediates FHR decelerations has been surprisingly controversial. The central scientific and pragmatic clinical question is what, if anything, does a deceleration tell us about the condition of the unborn child?
Before we can assess the scientific evidence we must first address the vexed issue of the terminology used to describe FHR decelerations and commonly held clinical beliefs about causation. Although three categories of deceleration are described (early, late and variable), unfortunately the exact definitions of each type and their clinical significance have varied, both with time and geography. For example, from the 1970s to the 90s most standard British textbooks classified decelerations only on the basis of whether they started and finished within the time frame of (i.e. in time with) a contraction (‘early’ decelerations) or whether there was a lag‐time between the onset of the contraction and the onset of the deceleration (‘late’ decelerations) (Sholapurkar, 2012). This classification was applied irrespective of the deceleration shape. Indeed one of the authors (J.A.W.) recalls being taught only about early and late decelerations when training in the 1980s and only discovered the category of variable decelerations (characterised by their rapid fall in FHR to nadir, and a variable timing relationship with contractions) when commencing a research project in 1990. Even more alarming in retrospect was the associated clinical teaching that early decelerations were always due to head compression and thus were always innocuous whereas late decelerations were always due to fetal hypoxia.
Unfortunately, these clinical associations are still held (even if subconsciously) by many generations of obstetricians. Failure to consider the key variables, depth, duration and frequency of ‘early’ decelerations, has led to many tragic outcomes. There have been noble and useful attempts on both sides of the Atlantic to standardise both the descriptions of FHR decelerations and integrate these into advice on management of overall FHR changes since the late 1990s (Parer et al. 2009; Ugwumadu, 2014). The focus on variable decelerations, which historically were attributed to cord compression, is slowly catching on in the UK but is bemoaned by some (Sholapurkar, 2012). Unfortunately, differences in paper speed (1 cm min−1 in the UK, Australasia and much of Europe compared with 3 cm min−1 in North America) cause geography‐related discrepancies in the appearance and, thus, the interpretation of early and late decelerations (Westgate et al. 2007). The non‐obstetric reader may feel somewhat confused at this point and perhaps amazed that such a critical aspect of intrapartum obstetrics (and one which is responsible for high litigation costs; Williams & Arulkumaran, 2004) remains unresolved.
There is now a wealth of experimental evidence, some old, some new, exploring the causes of FHR decelerations, which enable us both to evaluate commonly held clinical associations and provide an updated scientific basis for understanding decelerations. In contrast to the historically proposed ‘origins’ of decelerations, in this review we argue that the common, brief decelerations in labour are simply peripheral chemoreflex responses to transient but repeated asphyxia.
Are FHR decelerations due to fetal head compression?
During labour the fetal head presses on the uterine wall, leading to increased pressure on the skull, as measured with a strain gauge (Schwarcz et al. 1969). Pressure on the head increases further in the second stage of labour as the head is engaged in the birth canal, and after rupture of the chorio‐amniotic membranes. Not long after the introduction of obstetric auscultation, this pressure on the fetal head was suggested to result in a slowing of the fetal heart rate (variably attributed to Kennedy, 1833 or Naegele, 1839). More recently, it has been further suggested that early decelerations, which often roughly parallel the increase in uterine tone, are consistently the result of this increased intracranial pressure and trigger a non‐hypoxic reflex deceleration (Hon, 1958; Chung & Hon, 1959; Hon & Quilligan, 1968; Ott, 1976; Ball & Parer, 1992; Sholapurkar, 2012; Nageotte, 2015).
The neural pathways controlling such a reflex have never been described. Intuitively, they might be initiated by local pressure receptors in the scalp. However, if head compression was a consistent trigger of early decelerations we might reasonably expect all fetuses to show decelerations either as soon as their head became engaged in the birth canal or during the second stage of labour, as the head descends through the vagina. This is not the case. Clinically, decelerations can equally occur or not occur during all stages of labour (Hon & Quilligan, 1967).
Next, we address studies showing that head compression can (if inconsistently; Chung & Hon, 1959) cause decelerations, but that this is only observed if the compression is severe enough to impair cerebral blood flow – hardly a benign event. It is questionable whether such severe increases in intracranial pressure are common during unobstructed labour. In fetal sheep, when intracranial pressure was increased through intracerebroventricular infusions to a level where systemic hypertension was induced but cerebral blood flow was maintained, decelerations were not observed (Harris et al. 1989). In a clinical study, manual pressure on the maternal abdomen over the fetal head variably provoked either decelerations or tachycardia (Walker et al. 1973). The inconsistent FHR response to head compression may be partly related to which particular area of the head is compressed (Chung & Hon, 1959). Additionally, it has been reported that pressure over the inferior aspect of the uterus often produced decelerations with slow resolution (Walker et al. 1973). This suggests that the rapid resolution of increased intracranial pressure is not sufficient to restore heart rate, and that slow recovery was more likely to reflect recovery from frank cerebral ischaemia (Walker et al. 1973).
Strongly supporting these data, manual head compression in full term fetal sheep was associated with decelerations and an accompanying 50% reduction in carotid blood flow (Paul et al. 1964). Highly consistent with the clinical observations of Walker and colleagues noted above, FHR took 10–30 s to recover after release of compression, in parallel with delayed recovery of carotid blood flow. This observation strongly suggests that decelerations were secondary to central nervous system hypoxia (Paul et al. 1964). Similarly, head compression in fetal sheep with a modified rib retractor was only associated with decelerations in 37% of cases. When a deceleration did occur, it was again associated with reduced cerebral blood flow and profound suppression of fetal electroencephalographic activity (Mann et al. 1972).
In summary, direct fetal head compression inconsistently causes decelerations and so is very unlikely to be a major contributor to intrapartum decelerations during the majority of labours. The findings discussed in this section illustrate that when fetal head compression does lead to a deceleration, the phenomenon is highly consistent with a Cushing response; a near‐terminal response to reduced cerebral blood flow secondary to increased intracranial pressure (Fodstad et al. 2006). Thus, if decelerations due to head compression do occur, for example during obstructed labour when the fetal head is engaged in the birth canal, these data suggest that such decelerations reflect severe cerebral hypoperfusion and hypoxia (Schifrin et al. 2016), and in contrast with current proposed interpretation (Sholapurkar, 2012; National Institute for Health and Care Excellence, 2014), should not be considered to be benign.
Are FHR decelerations mediated by the baroreflex?
Others have proposed that the rapid variable decelerations (the category that constitutes the vast majority of intrapartum decelerations) could be a reflex response to the abrupt changes in blood volume and arterial blood pressure secondary to compression of the umbilical cord (Lee et al. 1975; Ott, 1976; Ball & Parer, 1992; Nageotte, 2015; Pinas & Chandraharan, 2016). Partial compression of both the placenta and umbilical vein during contractions may initially promote transfer of blood to the fetus, increasing blood volume and thus blood pressure. With a further increase in contraction strength, both the umbilical vein and arteries become completely occluded, increasing total peripheral resistance of the fetus. Both of these events could increase fetal arterial blood pressure, and hypothetically trigger a baroreflex‐mediated deceleration. As the contraction resolves and the umbilical arteries are unoccluded, peripheral resistance would be lost, with a reciprocal fall in fetal blood pressure and reduction in baroreflex activity (Ball & Parer, 1992; Sunderström et al. 2000; Gibbs & Arulkumaran, 2007; Hendler & Seidman, 2009).
Again, there is strikingly little evidence to support this concept. Umbilical cord compression can occur in labour, for example, during entanglements of the cord or cord prolapse, albeit there is no empirical evidence that it is the predominant cause of FHR decelerations in the majority of labours. Nonetheless, we can test this hypothesis by experimental occlusion of the umbilical cord. First, partial occlusion of the umbilical cord allows the effect of umbilical vein compression (or veins, since sheep have two umbilical veins) to be investigated. In near‐term fetal sheep, FHR fell once umbilical blood flow was reduced by 50% or more (Itskovitz et al. 1983 b). Despite this, two separate studies found no significant changes in arterial pressure, and thus the baroreflex cannot have been triggered during partial occlusion of the umbilical cord (Itskovitz et al. 1983 b; Giussani et al. 1997).
In contrast to partial occlusion, rapid complete occlusion of the umbilical cord is associated with two separate increases in fetal arterial pressure, as seen in Figs 1 and 2. The first small increase occurs immediately at the onset of occlusion, suggesting that it reflects mechanical removal of the low resistance placental vascular bed, leading to a net increase in total peripheral resistance. This increase in arterial blood pressure, however, is fleeting and small, well within normal baseline fluctuations in blood pressure. Thus, it is highly improbable that this first increase in blood pressure is sufficient to trigger a baroreflex.
Figure 1. Relationship between mean arterial pressure (MAP, mmHg), fetal heart rate (FHR, beats min −1 (bpm)) and changes in cerebral oxygenated haemoglobin (μmol (100 g) −1 ), as measured by near‐infrared spectroscopy, during a prolonged complete umbilical cord occlusion in a near‐term fetal sheep (0.85 of gestation) .
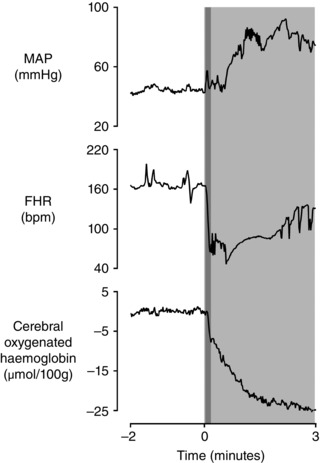
The period of occlusion is shown in light grey. The overlapping dark grey area represents the initial and most rapid portion of the deceleration. Note how both FHR and cerebral oxygenated haemoglobin begin to fall in parallel at 6 s after the start of occlusion when MAP is not markedly higher than baseline. Data are 1 s averages. Cerebral oxygenated haemoglobin is shown relative to the average of the 2 min baseline period.
Figure 2. Fetal heart rate (FHR, beats min −1 (bpm)) and mean arterial pressure (MAP, mmHg) during three successive 1 min complete umbilical cord occlusions, repeated every 5 min in a near‐term fetal sheep (0.85 of gestation) .
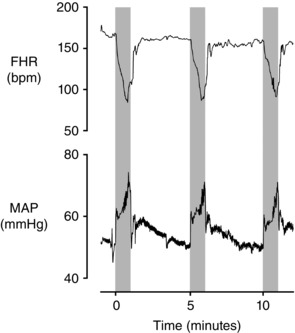
The periods of occlusion are shown in grey. Note that the major actively mediated increase in MAP is delayed until after the onset of decelerations. Additionally the release of occlusion is not associated with the development of hypotension below baseline levels. The first occlusion shown here is the 18th in a series of 49 occlusions; at this time arterial pH was 7.358, with a lactate of 1.6 mmol l−1. Data are 1 s averages.
Supporting this interpretation, Fig. 3 (left panels), shows a spontaneous contraction in a near‐term fetal sheep that was associated with a rapid deep deceleration despite an initial fall in arterial pressure at the onset of deceleration. These data show that hypertension is not necessary to cause spontaneous decelerations. Indeed, only one of the decelerations shown in Fig. 3 was associated with an increase in mean arterial pressure at the start of the deceleration.
Figure 3. Cardiovascular responses during spontaneous uterine contractions in a near‐term fetal sheep (0.85 of gestation) .
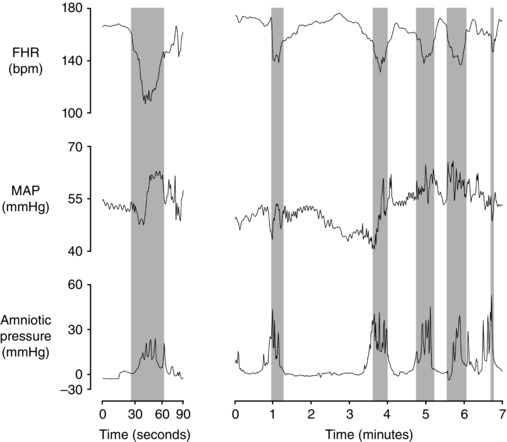
Fetal heart rate (FHR beats min−1 (bpm)), mean arterial pressure (MAP, mmHg) and amniotic pressure (mmHg) during uterine contractions resulting in a deep deceleration (left panels) and multiple moderate decelerations (right panels). Labour was induced by the administration of betamethasone (11.4 mg, i.m. to the ewe x2, 24 h apart) as previously described (Liggins, 1969). Contractions began approximately 48 h after the first dose of betamethasone. Note in the left panels that the deep deceleration occurred despite an initial mild fall in MAP at the start of contraction while delayed hypertension develops late in the deceleration, consistent with the data presented in Figs 1 and 2 during complete umbilical cord occlusion. Only the second to last contraction in the right panels was associated with an increase in MAP at the start of the deceleration. Thus the baroreflex cannot be a consistent trigger of decelerations. Data are 1 s averages.
Next, as shown in Fig. 1 and multiple previous studies, the second, much larger and more sustained, rise in pressure occurs after the onset of the fall in FHR and takes between 2 and 3 min to develop fully (Itskovitz et al. 1983 b; Bennet et al. 2005; Booth et al. 2012). Given the time taken for this increase in pressure, it cannot possibly contribute to the initial fall in FHR and, importantly, may not develop fully during shorter periods of umbilical cord occlusion (Fig. 2). Critically, this second large increase in pressure is actively mediated by increased sympathetic nervous system (SNS) activity leading to fetal peripheral vasoconstriction (Itskovitz et al. 1983 b; Giussani et al. 1993; Galinsky et al. 2014). In contrast, the baroreflex response is sympathoinhibitory and leads to peripheral vasodilatation (Charkoudian & Wallin, 2014), and so cannot explain the intense peripheral vasoconstriction during acute umbilical cord occlusion. This is highly consistent with early work by Lee and Hon in human fetuses, where decelerations were reported to occur after both partial and complete umbilical cord occlusion in association with either increased, decreased or no change in blood pressure. The authors concluded that the baroreflex could not be the common mechanism for decelerations (Lee & Hon, 1963).
Finally, the reader should also note preclinical evidence that the baroreflex is relatively immature before birth. In near‐term fetal sheep, hypertension induced by phenylephrine did indeed result in a deceleration, but this was a very slow deceleration that took up to 30 s to develop fully (Booth et al. 2011). Phenylephrine‐induced hypertension developed faster and was greater than the greatest increase in blood pressure observed after umbilical cord occlusion (Figs 1 and 2). Despite this brisk increase in blood pressure, the baroreflex‐mediated changes in FHR were still much slower than typical decelerations in labour, which in many cases reach a nadir within 5–10 s (Hon & Quilligan, 1967). In conclusion, although the baroreflex is undoubtedly functional and active during fetal life (Segar et al. 1992; Booth et al. 2009, 2011), it is mechanistically unable to contribute to the typical brief intrapartum deceleration.
Hypovolaemia: the Bezold–Jarisch reflex?
Alternatively, selective compression of the umbilical vein could reduce venous return from the placenta to the fetus sufficiently to reduce central blood volume (Sunderström et al. 2000). Potentially, this could trigger the Bezold–Jarisch reflex, whereby reduced blood volume and subsequently reduced cardiac filling pressures activate cardiac C‐fibres, and so trigger a vagal deceleration. This reflex allows a longer filling time, restoring stroke volume and so restoring cardiac output (Mark, 1983). However, there is no evidence of reduced atrial (or ventricular) pressures during partial cord occlusion in fetal sheep that would activate this reflex (Itskovitz et al. 1987). In contrast, microsphere based measurements during partial occlusion demonstrated an increase in blood flow in the abdominal inferior vena cava and in the proportion of umbilical venous blood passing through the ductus venosus. This illustrates that compensatory preferential shunting maintains venous return to the fetal heart and thus atrial filling pressures are maintained (Itskovitz et al. 1987). Although there is evidence that during partial cord occlusion a secondary delayed reduction in FHR occurs in parallel with loss of fetal femoral vascular resistance, which might be consistent with this reflex, this fall in FHR did not occur until after approximately 2 min, well after the initial FHR deceleration (Giussani et al. 1997). Finally, the Bezold–Jarisch reflex is also known to be very immature at birth (Gootman et al. 1983). Critically, just as with the baroreflex, it is sympathoinhibitory (Mark, 1983) and so wholly inconsistent with the rapid increase in SNS activity during umbilical cord occlusion described above. Thus, it is highly improbable that its activation could contribute to the brief decelerations characteristic of labour.
The peripheral chemoreflex?
The above hypotheses are inviting to both families and clinicians because they suggest that fetuses are not being exposed to asphyxia during decelerations in labour. Aside from head compression, which in systematic studies was only associated with decelerations when it was sufficiently severe as to cause frank cerebral hypoperfusion, it is important to appreciate that if the hypothesised events described above did occur during labour, they must all be confounded in time with a rapid fall in fetal arterial oxygen saturation (Kunzel et al. 1980). As shown in Fig. 1, cerebral oxygenated haemoglobin measured by near‐infrared spectroscopy fell within 6 s of complete umbilical cord occlusion in near‐term fetal sheep. This fall parallels the simultaneous fall in FHR. This refutes suggestions that hypoxia only occurs once a deceleration is prolonged (Gibbs & Arulkumaran, 2007; Sholapurkar, 2012). In contrast to the questionable mechanisms described above, the peripheral chemoreflex is reliably and rapidly activated during fetal life and so can readily explain the vast majority of intrapartum decelerations.
There is considerable evidence that repeated periods of asphyxia are common during labour, independent of any umbilical cord occlusion (Westgate et al. 2007). The fetus is dependent on continuous placental and umbilical blood flow for oxygen delivery. Normal, uncomplicated labours are associated with a small but consistent fall in pH and oxygen tension, and a rise in carbon dioxide tensions, base deficit and plasma lactate, showing that intermittent interruption of placental exchange occurs during all normal labours (Modanlou et al. 1974; Huch et al. 1977; Wiberg et al. 2006). This in turn is consistent with findings that intrauterine pressure increases during contractions and is associated with a linear fall in maternal uterine artery blood velocity of up to 73% during individual contractions (Janbu & Nesheim, 1987). Similar reductions in uterine blood flow have been found in pregnant sheep and dogs (Assali et al. 1958). Further, decreased umbilical artery blood flow was observed during variable decelerations in humans (Sakai et al. 1997), and during spontaneous second stage contractions in the bovine fetus (Bleul et al. 2007). Clinically, variable, late and prolonged FHR decelerations are universally associated with a rapid reduction in fetal cerebral oxygenation on near‐infrared spectroscopy (Aldrich et al. 1996). Thus, uterine contractions result in impaired fetal arterial oxygen tensions, regardless of whether or not direct umbilical cord compression occurs.
The peripheral chemoreflex is the response to an acute fall in oxygen tension mediated by the peripheral chemoreceptors. The key efferent arms of the peripheral chemoreflex are the rapid vagal‐mediated fall in heart rate, i.e. the deceleration in labour, and SNS mediated peripheral vasoconstriction (Giussani et al. 1993). Presumptively, the fall in FHR reduces myocardial work and so helps to conserve cardiac glycogen levels (Bennet et al. 2009). Although the peripheral chemoreflex matures during the course of normal gestation (Fletcher et al. 2006), it is a robust response throughout pregnancy (Kiserud et al. 2001; Wassink et al. 2007). The immediate fall in cardiac output due to decreased FHR is offset by SNS‐mediated peripheral vasoconstriction. The combined effect of these responses maintains or increases arterial pressure, which in turn maintains or increases blood flow to the ‘central organs’ such as the brain, heart and adrenals, as recently reviewed (Giussani, 2016). After the initial neural‐mediated fall in peripheral blood flow, vasoconstriction is maintained by release of vasoactive mediators (Booth et al. 2012; Giussani, 2016), including adrenal catecholamines (Giussani et al. 1993; Galinsky et al. 2014), vasopressin (Perez et al. 1989) and neuropeptide Y (Fletcher et al. 2000).
When is the chemoreflex triggered during labour?
The vast majority of decelerations occur in association with uterine contractions; the majority of these are unlikely to result in complete cessation of placental and umbilical blood flow. Considering this, fetal oxygenation probably changes dynamically over the course of a contraction, reflecting a balance between the fall in oxygen supply related to contraction strength and duration, and continuing consumption of oxygen by the fetus. The fetus is not a passive player and can decrease oxygen expenditure, increase oxygen extraction and augment shunting of blood to crucial organs to defend itself (Itskovitz et al. 1983 a; Wilkening & Meschia, 1983; Bocking & Harding, 1986). Experimentally, rapid decelerations are not triggered until umbilical blood flow is reduced by at least 50% from baseline (Itskovitz et al. 1983 b; Giussani et al. 1997). Thus, the healthy fetus is often able to adapt to mild reductions in oxygen supply without triggering a deceleration and so not all uterine contractions are associated with decelerations. Broadly, as the reduction in utero‐placental or umbilical flow becomes greater, the FHR deceleration also becomes deeper (Itskovitz et al. 1983 b; Baan et al. 1993).
In well controlled experimental studies in healthy animals, the magnitude of the peripheral chemoreflex‐mediated deceleration is closely correlated with the degree of reduction in utero‐placental flow (Peeters et al. 1979; Itskovitz et al. 1982, 1983 a; Baan et al. 1993). In labour, many other factors contribute to fetal arterial oxygen saturation over the course of a contraction. As illustrated in Fig. 4, the balance between these factors is crucial in determining the delay before oxygenation is impaired sufficiently to trigger a rapid deceleration, as well as the magnitude and duration of the deceleration. An important factor that modulates the magnitude of the fall in fetal oxygen tension is fetal oxygenation status before the contraction. This in turn will be determined by the underlying placental exchange capacity and the duration of placental reperfusion between contractions. Consistent with this, pre‐existing hypoxia in fetal sheep has been associated with deeper and longer decelerations, that began earlier, during maternal aortic occlusion (Itskovitz et al. 1982).
Figure 4. Factors that influence fetal oxygenation over the course of an intrapartum uterine contraction .
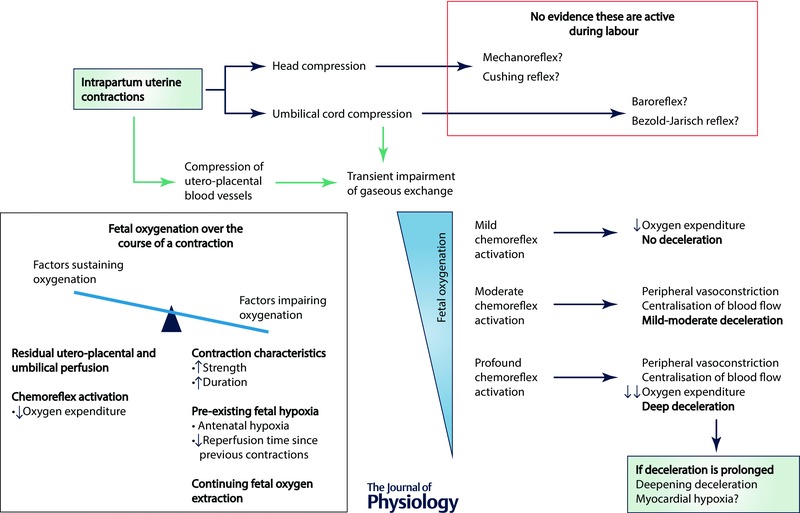
During the course of an individual contraction, there is a critical balance between factors promoting a fall in fetal arterial oxygenation and those that sustain it – the relative strengths of these factors will determine the extent to which fetal arterial oxygenation falls during an individual contraction. Greater strength and duration of a uterine contraction leads to greater impairment of gaseous exchange, while antenatal factors such as impaired utero‐placental perfusion can exacerbate the severity of fetal deoxygenation. Fetal oxygen extraction must continue during a uterine contraction; however, the fetus can adapt to this reduced gas exchange by reducing non‐vital metabolic and behavioural processes, and so reduce its oxygen consumption. The degree to which fetal oxygenation falls during a contraction will determine the magnitude of the peripheral chemoreflex response, and, in turn, the depth of any resulting deceleration.
Does the peripheral chemoreflex evolve over the course of labour?
Labour can last for many hours and it is thus important to consider how this prolonged exposure time affects the fetus’ ability to respond to repeated interruptions in oxygen supply. During severe asphyxia the neural components of the chemoreflex are relatively short lived, and thus it represents a rapid response system. During a prolonged and severe deceleration (i.e. in response to severe asphyxia) both the vagal and SNS efferent components of the peripheral chemoreflex start to become attenuated after 90–120 s (Barcroft, 1946; Harris et al. 1982; Westgate et al. 2007; Booth et al. 2012).
From this point on, the deceleration is thought to be maintained by the direct effects of ‘myocardial hypoxia’ (Barcroft, 1946; Westgate et al. 2007), while peripheral vasoconstriction is sustained by humoral factors (Galinsky et al. 2014). Fortunately, most decelerations (and episodes of asphyxia) during labour are brief, lasting for a minute or less. Indeed, we have shown that both parasympathetic and sympathetic responses rapidly recover between brief repeated complete umbilical occlusions in near‐term fetal sheep that were continued for many hours, despite evolving severe metabolic acidosis (Bennet et al. 2005; Galinsky et al. 2014). Thus, the fetus’ crucial autonomic responses to asphyxia can continue to be reactivated by repeated uterine contractions even during prolonged labour.
The next question is whether the responsiveness or magnitude of the peripheral chemoreflex changes over the course of repeated episodes of asphyxia. In fetal sheep, repetition of mild (partial) occlusions that did not result in hypotension or severe acidosis has been associated with attenuation of both the vagal and sympathetic efferents of the peripheral chemoreflex, resulting in a shallower deceleration and less peripheral vasoconstriction with repeated occlusions (Giussani et al. 1997; Green et al. 1999). In contrast, during repeated complete occlusions of the umbilical cord, leading to worsening hypotension and acidosis, the initial vagal‐mediated deceleration became more rapid as fetuses developed progressive cardiovascular compromise with hypotension during occlusions (Bennet et al. 2005). Further, there is recent evidence that although the initial sympathetic neural response to support arterial blood pressure and redistribute cardiac output is transient, it can be repeatedly reactivated between episodes of brief asphyxia over many hours, without becoming attenuated (Galinsky et al. 2014; Lear et al. 2016).
These data strongly suggest that the peripheral chemoreflex response is not only sustained but the vagal response is augmented by repeated exposure to severe asphyxia. It is likely that the progressive metabolic acidosis itself contributes to augmenting the chemoreflex responses, as shown by the finding that mild acidosis induced by infusion of acidified saline was associated with a greater fall in FHR and increased vasoconstriction of the femoral artery in near‐term fetal sheep subjected to moderate hypoxaemia (Thakor & Giussani, 2009).
The contrasting experimental data examined in this section show that the peripheral chemoreflex responses probably do evolve during the course of labour, becoming more rapid (or at least maintained) when fetal survival is threatened, whereas they become relatively attenuated during milder insults that do not lead to significant fetal acidaemia. Thus, during mild homeostatic challenges, the fetus probably partially adapts through other mechanisms.
Experimental studies of brief repeated asphyxia – what can be learned about the fetal adaptation to labour?
The above sections illustrate that during labour every fetus is exposed to brief but repeated asphyxia. This statement should not cause alarm. Indeed this is offset by the striking ability of the fetus to adapt to repeated asphyxia, which needs to be appreciated to understand how the clinician should interpret decelerations. Systematic studies in fetal sheep have illustrated that the healthy fetus can adapt almost indefinitely to severe repeated asphyxia without risk of injury, provided that the time between decelerations is sufficient to allow recovery of the oxygen debt. This is strikingly exemplified by the findings that healthy near‐term fetal sheep did not develop hypotension despite being exposed to 1 min complete umbilical cord occlusions repeated continuously every 5 min for 4 h. These fetuses showed stable variable decelerations and developed only mild acidosis (nadir of pH 7.34 ± 0.07, base deficit 1.3 ± 3.9 mmol l−1 and lactate 4.5 ± 1.3 mmol l−1) (Westgate et al. 1999 a). In contrast, fetuses with pre‐existing spontaneous hypoxia developed progressive cardiovascular compromise during repeated umbilical cord occlusions of the same frequency, with severe, progressive metabolic acidosis (nadir of pH 7.07 ± 0.14, base deficit 14 ± 1.8 mmol l−1 and lactate 10.9 ± 1.9 mmol l−1) and hypotension (nadir of 24 ± 2 mmHg vs. 45.5 ± 3 mmHg in normoxic fetuses; P < 0.001) (Westgate et al. 2005; Wassink et al. 2013).
When the period of time between 1 min umbilical cord occlusions was reduced to 90 s, and so was insufficient to allow complete recovery between occlusions, even normoxic fetuses ultimately developed profound acidosis (pH 6.92 ± 0.03, base deficit 19.2 ± 1.5 mmol l−1 and lactate 14.6 ± 0.8 mmol l−1 at the end of the occlusion series) (Westgate et al. 2001 b). These fetuses initially showed a sustained increase in blood pressure during occlusions, but eventually developed a biphasic response to each occlusion, with continuing initial hypertension that was then followed by a rapid fall in blood pressure (de Haan et al. 1997; Westgate et al. 1999 a, 2001 a,b).
Despite these dramatic changes in fetal blood pressure, fetuses continued to show almost identical variable decelerations for many hours of repeated occlusions (de Haan et al. 1997). Delayed recovery of the FHR after the end of occlusion was seen only as a near‐terminal event, in a minority of fetuses. Despite multiple detailed analyses there is no single consistent FHR marker of fetal compromise (de Haan et al. 1997; Westgate et al. 1999 a, 2001 a,b, 2005, 2007; Bennet et al. 2005; Lear et al. 2016). Overall, developing fetal acidosis and hypotension appears to be associated with relatively modest changes to the FHR response such as an increase in FHR between decelerations, leading to a greater depth and rate of fall in FHR during each deceleration (Bennet et al. 2005). The lack of a strong marker of fetal compromise is consistent with a 4‐year retrospective cohort study of 5388 consecutive term singleton labours, which found that the strongest factor associated with acidaemia was the total deceleration area during repetitive decelerations, whereas many features suggested by current clinical guidelines provided no additional predictive power (Cahill et al. 2012).
What do intrapartum decelerations tell us about the fetus during labour?
The observation of a brief deceleration tells us that the fetus has experienced a brief period of asphyxia; however, this in itself does not tell us anything about the condition of the fetus. Contrary to one common clinical teaching, whether a pattern of repeated intrapartum decelerations is benign or not does not depend on whether the decelerations are ‘reflex’. All brief (<∼1–1.5 min) decelerations are reflex in origin. Rather, the risk that hypotension and profound acidosis will develop depends on the balance between the fetus’ ability to adapt to repeated hypoxic challenges (largely determined by its placental exchange capacity relative to oxygen consumption) and the time available to recover between occlusions. It is this balance which determines the magnitude of the progressive oxygen debt associated with repeated uterine contractions, as illustrated in Fig. 5.
Figure 5. Factors that influence fetal anaerobic reserve and fetal oxygen debt during labour .
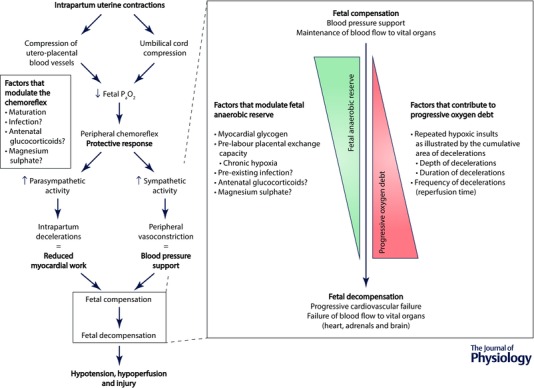
Periods of brief asphyxia are frequent and a normal component of labour. This is offset by the fetus’ ability to adapt to these challenges through peripheral chemoreflex activation and a high anaerobic tolerance. Thus, the vast majority of fetuses adapt effectively to labour and are born healthy. The ability to adapt is finite, however, and can fail in two situations. Firstly, a fetus with good antepartum health and adequate anaerobic reserves can progressively decompensate if exposed to severe repeated asphyxial challenges that are too frequent to allow complete recovery between intense uterine contractions. Secondly, and perhaps of more clinical importance, is the scenario of a fetus entering labour with poor utero‐placental exchange capacity and reduced glycogen reserves. Such fetuses are unable to fully compensate to the repeated asphyxial challenges of a typical labour and will quickly decompensate, leading to hypotension and increasing risk of neural injury.
Decelerations that become deeper and longer over the course of labour reflect more severe and longer periods of interrupted gaseous exchange, respectively. Thus the severity of the fetal oxygen debt is closely associated with the depth, duration and frequency of intrapartum decelerations (i.e. the cumulative deceleration area). The frequency of decelerations is important as it determines the reperfusion time between asphyxial insults. If there is a sufficient duration of recovery the fetus will overcome its acute oxygen debt and begin to replenish its anaerobic reserves. Crucially, these relationships are all heavily modulated by fetal condition, including the amount of stored cardiac glycogen (Dawes et al. 1959) and the adequacy of the pre‐labour placental exchange capacity (Westgate et al. 2005; Wassink et al. 2013).
Supporting these preclinical findings, clinically it is well established that conditions associated with antenatal hypoxia and growth restriction are also associated with increased risk of neonatal encephalopathy and an overall increase in perinatal morbidity and mortality (Nicolaides et al. 1989; Westgate et al. 1999 b; Nelson et al. 2012; McIntyre et al. 2013). Unfortunately, the overall appearance of decelerations does not appear to alter substantially despite the progressive development of fetal compromise – a testament to how robust the peripheral chemoreflex truly is.
What is left unanswered?
Why are late decelerations associated with fetal compromise?
Little is known about the pathophysiology behind late decelerations and why they have been (inconsistently) seen in compromised fetuses (Thomas, 1975; Fleischer et al. 1982; Sameshima & Ikenoue, 2005; Parer & Ikeda, 2007; Cahill et al. 2012). Late decelerations are defined by a lag between the contraction and the beginning of the deceleration. As previously reviewed (Westgate et al. 2007), it is important to distinguish between two types of ‘late’ decelerations.
Firstly, the true late decelerations consistent with Hon's original description occur before labour, and are often accompanied by baseline tachycardia, low to absent variability and absent accelerations. These are usually observed with painless uterine ‘tightenings’ or contractures. The predominant clinical scenario is a mother who reports reduced fetal movements, or a known growth restricted fetus with reduced fetal movements. This setting suggests that the fetus probably has poor utero‐placental perfusion and is thus unable to adapt to even the small decreases in placental exchange associated with mild contractures without triggering a deceleration. If the fetus is still neurologically intact, this deceleration will still represent a peripheral chemoreflex response. This antenatal pattern appears to identify the compromised fetus who will not adapt to the stress of labour, as previously reviewed (Westgate et al. 2007).
In contrast, intrapartum ‘late’ decelerations are almost always simply variable decelerations that are late in timing. There is no evidence that these late decelerations are associated with a greater physiological challenge to the fetus than early or variable decelerations (Westgate et al. 2007; Cahill et al. 2012). It is unknown why there is a lag time between contraction and deceleration. Presumptively, the delay is related to the time taken for fetal oxygenation to become impaired sufficiently to trigger a deceleration and then to recover.
What is ‘myocardial hypoxia’?
It is well known that complete parasympathetic blockade only delays the onset of a FHR deceleration by approximately 90–120 s during sustained, severe asphyxia (Barcroft, 1946; Westgate et al. 2007). This is believed to represent the direct depressant effects of hypoxia on the heart. However, more research is needed to understand the specific mechanisms. For example, accumulation of the cardiodepressant adenosine is reported to contribute in part to bradycardia during prolonged moderate hypoxia (Giussani et al. 2001), but its role during more severe insults is unclear.
As discussed previously, in intact fetuses it is believed that the peripheral chemoreflex is replaced by myocardial hypoxia during deep decelerations if they are prolonged, although there is no specific evidence from measurements of fetal vagal activity to directly confirm inhibition of parasympathetic activity. Nonetheless, the point at which myocardial hypoxia contributes to a deceleration probably reflects the transition from a period of asphyxia that was completely compensated for by the fetus, and one that was not. Thus a marker of such a transition is likely to be clinically useful.
We believe this transition is, albeit imperfectly, reflected by the development of overshoot tachycardia immediately after a deceleration. Overshoot tachycardia refers to a large increase in FHR immediately after a deceleration that can last up to 10–30 s. It should be distinguished from the so‐called ‘shoulders’ that can occasionally be observed clinically. Overshoot can be seen in association with either a normal or increased baseline FHR (Westgate et al. 2007). In fetal sheep, overshoot tachycardia was observed in association with the development of acidosis and hypotension after brief 1 min umbilical cord occlusions repeated every 2.5 min (Westgate et al. 2001 b). In contrast, overshoot tachycardia was observed immediately from the first occlusion onwards during longer, 2 min, repeated occlusions when fetuses were neither acidotic nor hypotensive (Westgate et al. 2001 b, 2007; Lear et al. 2016). Thus overshoot is not a consistent marker of fetal compromise. Nevertheless, it does appear to indicate the loss of vagal tone, and likely accompanying myocardial hypoxia, during an individual deep deceleration.
Where do the afferent chemoreflex signals originate?
In strong contrast to the response to moderate acute hypoxia, where carotid chemodenervation completely prevents any fall in FHR (Itskovitz & Rudolph, 1982; Bartelds et al. 1993; Giussani et al. 1993), isolated carotid chemodenervation only slows the fall in heart rate during severe asphyxia (Jensen & Hanson, 1995). These data suggest that there are substantial unidentified afferent inputs that can stimulate the peripheral chemoreflex. Speculatively, one key additional input might be greater recruitment of aortic chemoreceptors during severe asphyxia (Hanson, 1997).
The role of central chemoreceptors in fetal life is also poorly described but they are probably more sensitive to changes in the partial pressure of carbon dioxide and pH than to oxygen levels, as is the case in adults (Nattie, 2006). In light of this, they probably provide longer‐term feedback on the acid–base status of the fetus and, speculatively, may contribute to the modulation of peripheral chemoreflex responses during repeated insults as discussed above.
How do maturation and additional factors affect peripheral chemoreflex responses?
Although the peripheral chemoreflex is known to be active from very early in gestation, and the preterm fetus is known to have a qualitatively similar response to severe prolonged asphyxia to that seen at term (Gunn et al. 2001; Wassink et al. 2007), there is limited evidence on how preterm fetuses adapt to the repeated asphyxial insults characteristic of labour (Keunen & Hasaart, 1999), or whether fetal responses are modulated by exposure to chronic intrauterine infection or inflammation (Booth et al. 2013). For example, the cortisol surge that occurs later in gestation is associated with further maturation of the peripheral chemoreflex response to moderate hypoxaemia (Fletcher et al. 2006). It is unclear how this alters the peripheral chemoreflex response to the more severe homeostatic challenge of repeated asphyxia. The effects of many common clinical drugs are also not fully understood. In the fetal sheep, exposure to maternal dexamethasone is associated with a more prolonged deceleration during acute moderate hypoxia (Fletcher et al. 2003; Jellyman et al. 2005). Further, maternal treatment with magnesium sulphate can reduce baseline FHR, in addition to its known vasodilatory effects (Nensi et al. 2014; Galinsky et al. 2016). Thus, it is clearly important to determine how these treatments affect the ability of the fetus to sustain peripheral chemoreflex responses to repeated severe asphyxia in labour.
The way forward: physiology over descriptive titles
There is now overwhelming evidence that brief FHR decelerations in labour are mediated by the peripheral chemoreflex, and are related to acute but brief asphyxia. The complex and un‐physiological explanations described at the start of this review probably arose to try to explain why many babies are born without acidosis despite repeated decelerations during labour. The experiments reviewed above show that this apparent discrepancy is the result of the truly remarkable ability of the healthy mammalian fetus to adapt to the short periods of asphyxia characteristic of labour without systemic compromise (Westgate et al. 2001 a, 2005).
Consistent with decelerations being predominantly mediated by peripheral chemoreflex activation, there is considerable evidence that the clinical practice of classifying decelerations based on shape and timing does not improve the obstetrician's ability to identify acidotic fetuses (Parer & Ikeda, 2007; Cahill et al. 2012). Similarly, the recent large retrospective study from Cahill and colleagues found that the cumulative area of decelerations is one of the best available measures of progressive fetal oxygen debt during labour (Cahill et al. 2012). Thus, we believe that when it comes to interpreting decelerations it is better to focus on the frequency, depth and total duration of decelerations during labour rather than on the timing, shape or supposed aetiology of specific decelerations. This of course must take into account the overall clinical context of these decelerations, and would include evaluation of baseline FHR, FHR variability, contraction frequency and, importantly, the evolution of all of these features over the course of labour, coupled with knowledge of relevant clinical factors.
Additional information
Competing interests
None declared.
Funding
This study was supported by grants from the Health Research Council of New Zealand (grant numbers 12/613 and 14/216), the Auckland Medical Research Foundation (grant number 1108004) and the Lotteries Board of New Zealand (grant numbers 209214 and 340855). C. A. Lear was supported by an Auckland Medical Research Foundation Doctoral Scholarship (grant number 1213003).
Biographies
Christopher Lear is a PhD student working with Professors Alistair Gunn and Laura Bennet. His goal is to understand why some fetuses survive oxygen deprivation and infection and grow up to be completely normal, whereas others suffer injury or die after apparently identical exposure. His studies have highlighted both the extraordinary physiological resilience of the unborn child and the critical importance of understanding the physiological mechanisms of the fetal responses to major homeostatic challenges.

Alistair Jan Gunn is a paediatrician‐scientist who has conducted groundbreaking basic research into ways of identifying compromised fetuses in labour, the mechanisms and treatment of asphyxial brain injury and the mechanisms of life threatening events in infancy. He has helped to develop a range of new, clinically relevant chronically instrumented fetal sheep paradigms to support translation of the team's findings to clinical practice. His research helped to establish mild cooling as the first ever technique to reduce brain injury due to low oxygen levels at birth.
Linked articles This article is highlighted by a Perspective by Giussani. To read this Perspective, visit http://dx.doi.org/10.1113/JP272339.
References
- Aldrich CJ, D'Antona D, Spencer JA, Delpy DT, Reynolds EO & Wyatt JS (1996). Fetal heart rate changes and cerebral oxygenation measured by near‐infrared spectroscopy during the first stage of labour. Eur J Obstet Gynecol Reprod Biol 64, 189–195. [DOI] [PubMed] [Google Scholar]
- Assali NS, Dasgupta K, Kolin A & Holms L (1958). Measurement of uterine blood flow and uterine metabolism. V. Changes during spontaneous and induced labor in unanesthetized pregnant sheep and dogs. Am J Physiol 195, 614–620. [DOI] [PubMed] [Google Scholar]
- Baan J Jr, Boekkooi PF, Teitel DF & Rudolph AM (1993). Heart rate fall during acute hypoxemia: a measure of chemoreceptor response in fetal sheep. J Dev Physiol 19, 105–111. [PubMed] [Google Scholar]
- Ball RH & Parer JT (1992). The physiologic mechanisms of variable decelerations. Am J Obstet Gynecol 166, 1683–1688. [DOI] [PubMed] [Google Scholar]
- Barcroft J (1946). Researches on Pre‐natal Life. Blackwell Scientific Publications Ltd, London and Oxford. [Google Scholar]
- Bartelds B, van Bel F, Teitel DF & Rudolph AM (1993). Carotid, not aortic, chemoreceptors mediate the fetal cardiovascular response to acute hypoxemia in lambs. Pediatr Res 34, 51–55. [DOI] [PubMed] [Google Scholar]
- Bennet L, Westgate JA, Gluckman PD & Gunn AJ (2009). Pathophysiology of asphyxia In Fetal and Neonatal Neurology and Neurosurgery, 4th edn, eds Levene M. & Chervenack FA, pp. 472–490. Churchill Livingstone Elsevier, Philadelphia. [Google Scholar]
- Bennet L, Westgate JA, Lui YC, Wassink G & Gunn AJ (2005). Fetal acidosis and hypotension during repeated umbilical cord occlusions are associated with enhanced chemoreflex responses in near‐term fetal sheep. J Appl Physiol (1985) 99, 1477–1482. [DOI] [PubMed] [Google Scholar]
- Bleul U, Lejeune B, Schwantag S & Kahn W (2007). Ultrasonic transit‐time measurement of blood flow in the umbilical arteries and veins in the bovine fetus during stage II of labor. Theriogenology 67, 1123–1133. [DOI] [PubMed] [Google Scholar]
- Bocking AD & Harding R (1986). Effects of reduced uterine blood flow on electrocortical activity, breathing, and skeletal muscle activity in fetal sheep. Am J Obstet Gynecol 154, 655–662. [DOI] [PubMed] [Google Scholar]
- Booth LC, Drury PP, Muir C, Jensen E, Gunn AJ & Bennet L (2013). Acute on chronic exposure to endotoxin is associated with enhanced chemoreflex responses in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol 304, R799–R803. [DOI] [PubMed] [Google Scholar]
- Booth LC, Gunn AJ, Malpas SJ, Barrett CJ, Davidson JO, Guild SJ & Bennet L (2011). Baroreflex control of renal sympathetic nerve activity and heart rate in near‐term fetal sheep. Exp Physiol 96, 736–744. [DOI] [PubMed] [Google Scholar]
- Booth LC, Malpas SC, Barrett CJ, Guild SJ, Gunn AJ & Bennet L (2009). Is baroreflex control of sympathetic activity and heart rate active in the preterm fetal sheep? Am J Physiol Regul Integr Comp Physiol 296, R603–R609. [DOI] [PubMed] [Google Scholar]
- Booth LC, Malpas SC, Barrett CJ, Guild SJ, Gunn AJ & Bennet L (2012). Renal sympathetic nerve activity during asphyxia in fetal sheep. Am J Physiol Regul Integr Comp Physiol 303, R30–R38. [DOI] [PubMed] [Google Scholar]
- Cahill AG, Roehl KA, Odibo AO & Macones GA (2012). Association and prediction of neonatal acidemia. Am J Obstet Gynecol 207, 206 e201–208. [DOI] [PubMed] [Google Scholar]
- Caldeyro‐Barcia R, Medez‐Bauer C, Poseiro J, Escarcena L, Pose S, Bieniarz J, Arnt I, Gulin L & Althabe O (1966). Control of the human fetal heart rate during labour In The Heart and Circulation in the Newborn and Infant, ed. Cassels D, pp. 7–36. Grune & Stratton, New York. [Google Scholar]
- Charkoudian N & Wallin BG (2014). Sympathetic neural activity to the cardiovascular system: integrator of systemic physiology and interindividual characteristics. Compr Physiol 4, 825–850. [DOI] [PubMed] [Google Scholar]
- Chung F & Hon EH (1959). The electronic evaluation of fetal heart rate. I. With pressure on the fetal skull. Obstet Gynecol 13, 633–640. [PubMed] [Google Scholar]
- Dawes GS, Mott JC & Shelley HJ (1959). The importance of cardiac glycogen for the maintenance of life in foetal lambs and newborn animals during anoxia. J Physiol 146, 516–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan HH, Gunn AJ & Gluckman PD (1997). Fetal heart rate changes do not reflect cardiovascular deterioration during brief repeated umbilical cord occlusions in near‐term fetal lambs. Am J Obstet Gynecol 176, 8–17. [DOI] [PubMed] [Google Scholar]
- Fleischer A, Schulman H, Jagani N, Mitchell J & Randolph G (1982). The development of fetal acidosis in the presence of an abnormal fetal heart rate tracing. I. The average for gestational age fetus. Am J Obstet Gynecol 144, 55–60. [DOI] [PubMed] [Google Scholar]
- Fletcher AJ, Edwards CM, Gardner DS, Fowden AL & Giussani DA (2000). Neuropeptide Y in the sheep fetus: effects of acute hypoxemia and dexamethasone during late gestation. Endocrinology 141, 3976–3982. [DOI] [PubMed] [Google Scholar]
- Fletcher AJ, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2003). Cardiovascular and endocrine responses to acute hypoxaemia during and following dexamethasone infusion in the ovine fetus. J Physiol 549, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AJ, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2006). Development of the ovine fetal cardiovascular defense to hypoxemia towards full term. Am J Physiol Heart Circ Physiol 291, H3023–H3034. [DOI] [PubMed] [Google Scholar]
- Fodstad H, Kelly PJ & Buchfelder M (2006). History of the Cushing reflex. Neurosurgery 59, 1132–1137. [DOI] [PubMed] [Google Scholar]
- Galinsky R, Davidson JO, Drury PP, Wassink G, Lear CA, van Den Heuij LG, Gunn AJ & Bennet L (2016). Magnesium sulphate and cardiovascular and cerebrovascular adaptations to asphyxia in preterm fetal sheep. J Physiol 594, 1281–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinsky R, Jensen EC, Bennet L, Mitchell CJ, Gunn ER, Wassink G, Fraser M, Westgate JA & Gunn AJ (2014). Sustained sympathetic nervous system support of arterial blood pressure during repeated brief umbilical cord occlusions in near‐term fetal sheep. Am J Physiol Regul Integr Comp Physiol 306, R787–R795. [DOI] [PubMed] [Google Scholar]
- Gibbs D & Arulkumaran S (2007). Control of fetal heart rate and NICE guidelines In Fetal Monitoring in Practice, 3rd edn, pp. 27–44. Churchill Livingstone, London. [Google Scholar]
- Giussani DA (2016). The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol 594, 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Gardner DS, Cox DT & Fletcher AJ (2001). Purinergic contribution to circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep. Am J Physiol Regul Integr Comp Physiol 280, R678–R685. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Spencer JA, Moore PJ, Bennet L & Hanson MA (1993). Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol 461, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Unno N, Jenkins SL, Wentworth RA, Derks JB, Collins JH & Nathanielsz PW (1997). Dynamics of cardiovascular responses to repeated partial umbilical cord compression in late‐gestation sheep fetus. Am J Physiol 273, H2351–H2360. [DOI] [PubMed] [Google Scholar]
- Gootman PM, Gootman N & Buckley BJ (1983). Maturation of central autonomic control of the circulation. Fed Proc 42, 1648–1655. [PubMed] [Google Scholar]
- Green LR, Homan J, White SE & Richardson BS (1999). Cardiovascular and metabolic responses to intermittent umbilical cord occlusion in the preterm ovine fetus. J Soc Gynecol Investig 6, 56–63. [DOI] [PubMed] [Google Scholar]
- Gunn AJ, Quaedackers JS, Guan J, Heineman E & Bennet L (2001). The premature fetus: not as defenseless as we thought, but still paradoxically vulnerable? Dev Neurosci 23, 175–179. [DOI] [PubMed] [Google Scholar]
- Hanson MA (1997). Do we now understand the control of the fetal circulation? Eur J Obstet Gynecol Reprod Biol 75, 55–61. [DOI] [PubMed] [Google Scholar]
- Harris AP, Koehler RC, Gleason CA, Jones MD Jr & Traystman RJ (1989). Cerebral and peripheral circulatory responses to intracranial hypertension in fetal sheep. Circ Res 64, 991–1000. [DOI] [PubMed] [Google Scholar]
- Harris JL, Krueger TR & Parer JT (1982). Mechanisms of late decelerations of the fetal heart rate during hypoxia. Am J Obstet Gynecol 144, 491–496. [DOI] [PubMed] [Google Scholar]
- Hendler I & Seidman DS (2009). Intrapartum evaluation of the fetus In Fetal and Neonatal Brain Injury, 4th edn, eds Stevenson DK, Benitz WE, Sunshine P, Hintz R. & Druzin ML, pp. 174–186. Cambridge University Press, Cambridge. [Google Scholar]
- Hon EH (1958). The electronic evaluation of the fetal heart rate; preliminary report. Am J Obstet Gynecol 75, 1215–1230. [DOI] [PubMed] [Google Scholar]
- Hon EH & Quilligan EJ (1967). The classification of fetal heart rate. II. A revised working classification. Conn Med 31, 779–784. [PubMed] [Google Scholar]
- Hon EH & Quilligan EJ (1968). Electronic evaluation of fetal heart rate. IX. Further observations on ‘pathologic’ fetal bradycardia. Clin Obstet Gynecol 11, 145–167. [DOI] [PubMed] [Google Scholar]
- Huch A, Huch R, Schneider H & Rooth G (1977). Continuous transcutaneous monitoring of fetal oxygen tension during labour. Br J Obstet Gynaecol 84, 1–39. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, Goetzman BW & Rudolph AM (1982). The mechanism of late deceleration of the heart rate and its relationship to oxygenation in normoxemic and chronically hypoxemic fetal lambs. Am J Obstet Gynecol 142, 66–73. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, LaGamma EF & Rudolph AM (1983. a). The effect of reducing umbilical blood flow on fetal oxygenation. Am J Obstet Gynecol 145, 813–818. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, LaGamma EF & Rudolph AM (1983. b). Heart rate and blood pressure responses to umbilical cord compression in fetal lambs with special reference to the mechanism of variable deceleration. Am J Obstet Gynecol 147, 451–457. [DOI] [PubMed] [Google Scholar]
- Itskovitz J, LaGamma EF & Rudolph AM (1987). Effects of cord compression on fetal blood flow distribution and O2 delivery. Am J Physiol 252, H100–H109. [DOI] [PubMed] [Google Scholar]
- Itskovitz J & Rudolph AM (1982). Denervation of arterial chemoreceptors and baroreceptors in fetal lambs in utero. Am J Physiol 242, H916–H920. [DOI] [PubMed] [Google Scholar]
- Janbu T & Nesheim BI (1987). Uterine artery blood velocities during contractions in pregnancy and labour related to intrauterine pressure. Br J Obstet Gynaecol 94, 1150–1155. [DOI] [PubMed] [Google Scholar]
- Jellyman JK, Gardner DS, Edwards CM, Fowden AL & Giussani DA (2005). Fetal cardiovascular, metabolic and endocrine responses to acute hypoxaemia during and following maternal treatment with dexamethasone in sheep. J Physiol 567, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen A & Hanson MA (1995). Circulatory responses to acute asphyxia in intact and chemodenervated fetal sheep near term. Reprod Fertil Dev 7, 1351–1359. [DOI] [PubMed] [Google Scholar]
- Kennedy E (1833). Observations on Obstetric Auscultation: With an Analysis of the Evidences of Pregnancy, and an Inquiry Into the Proofs of the Life and Death of the Foetus in Utero. Hodges and Smith, Dublin. [Google Scholar]
- Keunen H & Hasaart TH (1999). Fetal arterial pressure and heart rate changes in surviving and non‐surviving immature fetal sheep following brief repeated total umbilical cord occlusions. Eur J Obstet Gynecol Reprod Biol 87, 151–157. [DOI] [PubMed] [Google Scholar]
- Kiserud T, Jauniaux E, West D, Ozturk O & Hanson MA (2001). Circulatory responses to maternal hyperoxaemia and hypoxaemia assessed non‐invasively in fetal sheep at 0.3–0.5 gestation in acute experiments. BJOG 108, 359–364. [DOI] [PubMed] [Google Scholar]
- Kunzel W, Mann LI, Bhakthavathsalan A & Airomlooi J (1980). Cardiovascular, metabolic and fetal brain function observation following total cord occlusion. J Perinat Med 8, 73–84. [DOI] [PubMed] [Google Scholar]
- Lear CA, Galinsky R, Wassink G, Mitchell CJ, Davidson JO, Westgate JA, Bennet L & Gunn AJ (2016). Sympathetic neural activation does not mediate heart rate variability during repeated brief umbilical cord occlusions in near‐term fetal sheep. J Physiol 594, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Di Loreto PC & O'Lane JM (1975). A study of fetal heart rate acceleration patterns. Obstet Gynecol 45, 142–146. [PubMed] [Google Scholar]
- Lee ST & Hon EH (1963). Fetal hemodynamic response to umbilical cord compression. Obstet Gynecol 22, 553–562. [PubMed] [Google Scholar]
- Liggins GC (1969). Premature delivery of foetal lambs infused with glucocorticoids. J Endocrinol 45, 515–523. [DOI] [PubMed] [Google Scholar]
- McIntyre S, Blair E, Badawi N, Keogh J & Nelson KB (2013). Antecedents of cerebral palsy and perinatal death in term and late preterm singletons. Obstet Gynecol 122, 869–877. [DOI] [PubMed] [Google Scholar]
- Mann LI, Carmichael A & Duchin S (1972). The effect of head compression on FHR, brain metabolism and function. Obstet Gynecol 39, 721–726. [PubMed] [Google Scholar]
- Mark AL (1983). The Bezold‐Jarisch reflex revisited: clinical implications of inhibitory reflexes originating in the heart. J Am Coll Cardiol 1, 90–102. [DOI] [PubMed] [Google Scholar]
- Mendez‐Bauer C, Poseiro JJ, Arellano‐Hernandez G, Zambrana MA & Caldeyro‐Barcia R (1963). Effects of atropine on the heart rate of the human fetus during labor. Am J Obstet Gynecol 85, 1033–1053. [DOI] [PubMed] [Google Scholar]
- Modanlou H, Yeh SY & Hon EH (1974). Fetal and neonatal acid‐base balance in normal and high‐risk pregnancies: during labor and the first hour of life. Obstet Gynecol 43, 347–353. [PubMed] [Google Scholar]
- Naegele HF (1839). A Treatise on Obstetric Auscultation. Translated by C. West (original work published 1838). Renshaw, London. [Google Scholar]
- Nageotte MP (2015). Fetal heart rate monitoring. Semin Fetal Neonatal Med 20, 144–148. [DOI] [PubMed] [Google Scholar]
- National Institute for Health and Care Excellence (2014). Intrapartum care for healthy women and babies. Available from: http://www.nice.org.uk/guidance/cg190. (Accessed April 2016.) [PubMed]
- Nattie E (2006). Why do we have both peripheral and central chemoreceptors? J Appl Physiol (1985) 100, 9–10. [DOI] [PubMed] [Google Scholar]
- Nelson KB, Bingham P, Edwards EM, Horbar JD, Kenny MJ, Inder T, Pfister RH, Raju T & Soll RF (2012). Antecedents of neonatal encephalopathy in the Vermont Oxford Network Encephalopathy Registry. Pediatrics 130, 878–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nensi A, De Silva DA, von Dadelszen P, Sawchuck D, Synnes AR, Crane J & Magee LA (2014). Effect of magnesium sulphate on fetal heart rate parameters: a systematic review. J Obstet Gynaecol Can 36, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Nicolaides KH, Economides DL & Soothill PW (1989). Blood gases, pH, and lactate in appropriate‐ and small‐for‐gestational‐age fetuses. Am J Obstet Gynecol 161, 996–1001. [DOI] [PubMed] [Google Scholar]
- Ott WJ (1976). The current status of intrapartum fetal monitoring. Obstet Gynecol Surv 31, 339–364. [DOI] [PubMed] [Google Scholar]
- Parer JT & Ikeda T (2007). A framework for standardized management of intrapartum fetal heart rate patterns. Am J Obstet Gynecol 197, 26.e1–26.e6. [DOI] [PubMed] [Google Scholar]
- Parer JT, Ikeda T & King TL (2009). The 2008 National Institute of Child Health and Human Development report on fetal heart rate monitoring. Obstet Gynecol 114, 136–138. [DOI] [PubMed] [Google Scholar]
- Paul WM, Quilligan EJ & MacLachlan T (1964). Cardiovascular phenomenon associated with fetal head compression. Am J Obstet Gynecol 90, 824–826. [DOI] [PubMed] [Google Scholar]
- Peeters LL, Sheldon RE, Jones MD Jr, Makowski EL & Meschia G (1979). Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol 135, 637–646. [DOI] [PubMed] [Google Scholar]
- Perez R, Espinoza M, Riquelme R, Parer JT & Llanos AJ (1989). Arginine vasopressin mediates cardiovascular responses to hypoxemia in fetal sheep. Am J Physiol 256, R1011–R1018. [DOI] [PubMed] [Google Scholar]
- Pinas A & Chandraharan E (2016). Continuous cardiotocography during labour: Analysis, classification and management. Best Pract Res Clin Obstet Gynaecol 30, 33–47. [DOI] [PubMed] [Google Scholar]
- Sakai M, Kozuma S, Okai T, Kagawa H, Ryo E & Taketani Y (1997). Doppler blood flow velocity waveforms of the umbilical artery during variable decelerations in labor. Int J Gynaecol Obstet 59, 207–211. [DOI] [PubMed] [Google Scholar]
- Sameshima H & Ikenoue T (2005). Predictive value of late decelerations for fetal acidemia in unselective low‐risk pregnancies. Am J Perinatol 22, 19–23. [DOI] [PubMed] [Google Scholar]
- Schifrin BS, Soliman M & Koos B (2016). Litigation related to intrapartum fetal surveillance. Best Pract Res Clin Obstet Gynaecol 30, 87–97. [DOI] [PubMed] [Google Scholar]
- Schwarcz RL, Strada‐Saenz G, Althabe O, Fernandez Funes J & Caldeyro‐Barcia R (1969). Pressure exerted by uterine contractions on the head of the human fetus during labor In Perinatal Factors Affecting Human Development, pp. 115 Pan American Health Organization, Washington, DC. [Google Scholar]
- Segar JL, Hajduczok G, Smith BA, Merrill DC & Robillard JE (1992). Ontogeny of baroreflex control of renal sympathetic nerve activity and heart rate. Am J Physiol Heart Circ Physiol 263, H1819–H1826. [DOI] [PubMed] [Google Scholar]
- Sholapurkar SL (2012). The conundrum of vanishing early decelerations in British obstetrics, a step backwards? Detailed appraisal of British and American classifications of fetal heart rate decelerations – fallacies of emphasis on waveform and putative aetiology. J Obstet Gynaecol 32, 505–511. [DOI] [PubMed] [Google Scholar]
- Sunderström A, Rosén D & Rosén KG (2000). EFM physiology In Fetal Surveillance. Noventa Medical, Mölndal. [Google Scholar]
- Thakor AS & Giussani DA (2009). Effects of acute acidemia on the fetal cardiovascular defense to acute hypoxemia. Am J Physiol Regul Integr Comp Physiol 296, R90–R99. [DOI] [PubMed] [Google Scholar]
- Thomas G (1975). The aetiology, characteristics and diagnostic relevance of late deceleration patterns in routine obstetric practice. Br J Obstet Gynaecol 82, 121–125. [DOI] [PubMed] [Google Scholar]
- Ugwumadu A (2014). Are we (mis)guided by current guidelines on intrapartum fetal heart rate monitoring? Case for a more physiological approach to interpretation. BJOG 121, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Walker D, Grimwade J & Wood C (1973). The effects of pressure on fetal heart rate. Obstet Gynecol 41, 351–354. [PubMed] [Google Scholar]
- Wassink G, Bennet L, Booth LC, Jensen EC, Wibbens B, Dean JM & Gunn AJ (2007). The ontogeny of hemodynamic responses to prolonged umbilical cord occlusion in fetal sheep. J Appl Physiol (1985) 103, 1311–1317. [DOI] [PubMed] [Google Scholar]
- Wassink G, Bennet L, Davidson JO, Westgate JA & Gunn AJ (2013). Pre‐existing hypoxia is associated with greater EEG suppression and early onset of evolving seizure activity during brief repeated asphyxia in near‐term fetal sheep. PLoS One 8, e73895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westgate JA, Bennet L, Brabyn C, Williams CE & Gunn AJ (2001. a). ST waveform changes during repeated umbilical cord occlusions in near‐term fetal sheep. Am J Obstet Gynecol 184, 743–751. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Bennet L, de Haan HH & Gunn AJ (2001. b). Fetal heart rate overshoot during repeated umbilical cord occlusion in sheep. Obstet Gynecol 97, 454–459. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Bennet L & Gunn AJ (1999. a). Fetal heart rate variability changes during brief repeated umbilical cord occlusion in near term fetal sheep. Br J Obstet Gynaecol 106, 664–671. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Gunn AJ & Gunn TR (1999. b). Antecedents of neonatal encephalopathy with fetal acidaemia at term. Br J Obstet Gynaecol 106, 774–782. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Wassink G, Bennet L & Gunn AJ (2005). Spontaneous hypoxia in multiple pregnancies is associated with early fetal decompensation and greater T‐wave elevation during brief repeated cord occlusion in near‐term fetal sheep. Am J Obstet Gynecol 193, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Westgate JA, Wibbens B, Bennet L, Wassink G, Parer JT & Gunn AJ (2007). The intrapartum deceleration in center stage: a physiological approach to interpretation of fetal heart rate changes in labor. Am J Obstet Gynecol 197, 236.e1–236.e11. [DOI] [PubMed] [Google Scholar]
- Wiberg N, Källén K & Olofsson P (2006). Physiological development of a mixed metabolic and respiratory umbilical cord blood acidemia with advancing gestational age. Early Hum Dev 82, 583–589. [DOI] [PubMed] [Google Scholar]
- Wilkening RB & Meschia G (1983). Fetal oxygen uptake, oxygenation, and acid‐base balance as a function of uterine blood flow. Am J Physiol 244, H749–H755. [DOI] [PubMed] [Google Scholar]
- Williams B & Arulkumaran S (2004). Cardiotocography and medicolegal issues. Best Pract Res Clin Obstet Gynaecol 18, 457–466. [DOI] [PubMed] [Google Scholar]