

Abstract
Key points
In airway smooth muscle, tension development caused by a contractile stimulus requires phosphorylation of the 20 kDa myosin light chain (MLC), which activates crossbridge cycling and the polymerization of a pool of submembraneous actin.
The p21‐activated kinases (Paks) can regulate the contractility of smooth muscle and non‐muscle cells, and there is evidence that this occurs through the regulation of MLC phosphorylation.
We show that Pak has no effect on MLC phosphorylation during the contraction of airway smooth muscle, and that it regulates contraction by mediating actin polymerization.
We find that Pak phosphorylates the adhesion junction protein, paxillin, on Ser273, which promotes the formation of a signalling complex that activates the small GTPase, cdc42, and the actin polymerization catalyst, neuronal Wiskott–Aldrich syndrome protein (N‐WASP).
These studies demonstrate a novel role for Pak in regulating the contractility of smooth muscle by regulating actin polymerization.
Abstract
The p21‐activated kinases (Pak) can regulate contractility in smooth muscle and other cell and tissue types, but the mechanisms by which Paks regulate cell contractility are unclear. In airway smooth muscle, stimulus‐induced contraction requires phosphorylation of the 20 kDa light chain of myosin, which activates crossbridge cycling, as well as the polymerization of a small pool of actin. The role of Pak in airway smooth muscle contraction was evaluated by inhibiting acetylcholine (ACh)‐induced Pak activation through the expression of a kinase inactive mutant, Pak1 K299R, or by treating tissues with the Pak inhibitor, IPA3. Pak inhibition suppressed actin polymerization and contraction in response to ACh, but it did not affect myosin light chain phosphorylation. Pak activation induced paxillin phosphorylation on Ser273; the paxillin mutant, paxillin S273A, inhibited paxillin Ser273 phosphorylation and inhibited actin polymerization and contraction. Immunoprecipitation analysis of tissue extracts and proximity ligation assays in dissociated cells showed that Pak activation and paxillin Ser273 phosphorylation triggered the formation of an adhesion junction signalling complex with paxillin that included G‐protein‐coupled receptor kinase‐interacting protein (GIT1) and the cdc42 guanine exchange factor, βPIX (Pak interactive exchange factor). Assembly of the Pak–GIT1–βPIX–paxillin complex was necessary for cdc42 and neuronal Wiskott–Aldrich syndrome protein (N‐WASP) activation, actin polymerization and contraction in response to ACh. RhoA activation was also required for the recruitment of Pak to adhesion junctions, Pak activation, paxillin Ser273 phosphorylation and paxillin complex assembly. These studies demonstrate a novel role for Pak in the regulation of N‐WASP activation, actin dynamics and cell contractility.
Key points
In airway smooth muscle, tension development caused by a contractile stimulus requires phosphorylation of the 20 kDa myosin light chain (MLC), which activates crossbridge cycling and the polymerization of a pool of submembraneous actin.
The p21‐activated kinases (Paks) can regulate the contractility of smooth muscle and non‐muscle cells, and there is evidence that this occurs through the regulation of MLC phosphorylation.
We show that Pak has no effect on MLC phosphorylation during the contraction of airway smooth muscle, and that it regulates contraction by mediating actin polymerization.
We find that Pak phosphorylates the adhesion junction protein, paxillin, on Ser273, which promotes the formation of a signalling complex that activates the small GTPase, cdc42, and the actin polymerization catalyst, neuronal Wiskott–Aldrich syndrome protein (N‐WASP).
These studies demonstrate a novel role for Pak in regulating the contractility of smooth muscle by regulating actin polymerization.
Abbreviations
- ACh
acetylcholine
- Arf‐GAP
ADP‐ribosylation factor GTPase‐activating protein
- Arp2/3
actin related protein 2/3
- cdc42
cell division control protein 42
- DMEM
Dulbecco's Modified Eagle's medium
- FAK
focal adhesion kinase
- GEF
guanine nucleotide exchange factor
- GIT
G‐protein‐coupled receptor kinase‐interacting protein
- MLC
myosin light chain
- MYPT
myosin phosphatase targeting protein
- N‐WASP
neuronal Wiskott–Aldrich syndrome protein
- Pak
p21‐activated kinase
- PIX
Pak interactive exchange factor
- PLA
proximity ligation assay
- PSS
physiological saline solution
- Ser
Serine
- Thr
Threonine
Introduction
The p21‐activated kinase (Pak) family of serine/threonine protein kinases are recognized for their important roles in the regulation of cytoskeletal dynamics (Bokoch, 2003; Zhao & Manser, 2012). Paks have long been recognized to play a critical role in the regulation of contraction and tension development in smooth muscle and non‐muscle cells and tissues (Van Eyk et al. 1998; Dechert et al. 2001; Goeckeler et al. 2000; McFawn et al. 2003; Murthy et al. 2003; Wirth et al. 2003; Wang et al. 2007; Hoover et al. 2012; Chu et al. 2013). The role of Paks in cell contractility has been largely ascribed to their effects on the regulation of myosin light chain (MLC) phosphorylation and myosin II activation. Studies of both smooth muscle and non‐muscle tissues and cells have demonstrated roles for Pak in the regulation of MLC phosphatase and MLC kinase, and there is also evidence that Paks can directly phosphorylate MLCs (Chew et al. 1998; Goeckeler et al. 2000; Murthy et al. 2003; Wirth et al. 2003; Chu et al. 2013).
Tension development in smooth muscle and non‐muscle cells and tissues requires actomyosin crossbridge cycling, which is regulated by phosphorylation of the 20 kDa regulatory light chain of myosin on Ser19 and Thr18. Ca2+/calmodulin‐activated MLC kinase is the primary catalyst for the phosphorylation of MLC at Ser19, while other kinases have been implicated in the phosphorylation of MLCs at Thr 18 (Somlyo & Somlyo, 2003; Walsh, 2011; Sutherland & Walsh, 2012). MLC phosphorylation is also increased by the inactivation of MLC phosphatase, which can be inhibited by phosphorylation of the myosin phosphatase targeting subunit, MYPT1 (Somlyo & Somlyo, 2003; Puetz et al. 2009).
Much of the evidence from studies of both smooth muscle and non‐muscle cells has suggested that Pak regulates MLC phosphorylation and contraction by modulating the activity of either MLC kinase or MLC phosphatase (Goeckeler et al. 2000; Murthy et al. 2003; Wirth et al. 2003; Chu et al. 2013); however, conflicting effects of Paks on contractility and MLC phosphorylation have been reported. A number of studies performed in permeabilized smooth muscle tissues and cells have described an inhibitory role for Pak on MLC kinase and contraction (Murthy et al. 2003; Wirth et al. 2003). Similarly, treatment of saponin‐permeabilized endothelial cell monolayers with recombinant Pak2 caused phosphorylation of non‐muscle MLC kinase and the inhibition of MLC phosphorylation and traction tension (Goeckeler et al. 2000). Paks have also been found to regulate contractility via effects on MLC phosphatase: in cultured intestinal smooth muscle cells and tissues Pak was shown to both positively and negatively regulate MLC phosphatase activity by phosphorylating different sites on MYPT1 (Chu et al. 2013).
Evidence from other studies suggests a positive role for Pak in the regulation of contractility. The administration of recombinant Pak3 to Triton‐skinned tracheal or taenia coli smooth muscle preparations induces a Ca2+‐independent contractile response (Van Eyk et al. 1998; McFawn et al. 2003). The direct phosphorylation of non‐muscle myosin II regulatory light chain by γPak (Pak3) in vitro has also been reported (Chew et al. 1998). Furthermore, airways isolated from mice with a genetic deletion of Pak1 exhibit a markedly lower contractile response to acetylcholine (ACh) in vitro than airways from wild type (WT) mice (Hoover et al. 2012), and canine tracheal smooth muscle tissues depleted of Pak1 using antisense show a reduction in the contractile response to ACh (Wang et al. 2007). There is currently no evidence demonstrating a role for Pak in the regulation of MLC phosphorylation in smooth muscle or non‐muscle cells or tissues under physiological conditions, and thus the mechanisms by which Paks regulate cell contractility remain unclear.
Pak has been shown to be a component of a highly conserved signalling module that regulates focal adhesion turnover at the leading edge of migrating cells (Nayal et al. 2006). Pak binds with high affinity to Pak interactive exchange factor (PIX) (Frank & Hansen, 2008), a rac/cdc42 guanine nucleotide exchange factor (GEF) that binds to the Arf‐GAP scaffolding protein, G‐protein‐coupled receptor kinase‐interacting protein (GIT1) (Frank & Hansen, 2008). During cell migration, Pak can phosphorylate the focal adhesion scaffolding protein paxillin on Ser273 to promote its interaction with GIT1 and facilitate the localization of a GIT1‐PIX‐Pak signalling module to the leading edge of the cell. There is evidence that the GIT1‐Pak‐PIX signalling module regulates focal adhesion turnover at the leading edge of migrating cells by a myosin II‐dependent mechanism (Turner et al. 1999; Mishima et al. 2004; Nayal et al. 2006; Stockton et al. 2007). The roles described for Pak in the regulation of focal adhesion turnover during cell migration and cell spreading are quite distinct from those described for Pak in the regulation of contractility and tension development. There is no evidence that the Pak‐PIX‐GIT1 signalling complex plays a role in the regulation of cell contractility.
In a number of smooth muscle cell and tissue types, contractile stimulation induces the polymerization of a small pool of actin (Gunst & Zhang, 2008; Zhang et al. 2005; Zhang & Gunst, 2008). Tension development requires polymerization of this actin in addition to crossbridge cycling, and the actin polymerization is regulated independently of myosin activation and actomyosin crossbridge cycling (Zhang et al. 2005; Gunst & Zhang, 2008; Zhang & Gunst, 2008). In airway smooth muscle, actin polymerization occurs in the submembranous regions of the cells and is catalysed by neuronal Wiskott–Aldrich syndrome protein (N‐WASP) which activates the actin‐related protein complex, Arp2/3, to catalyse the formation of actin filaments (Zhang et al. 2005). N‐WASP activation is mediated specifically by its interaction with the small GTPase cdc42 (Carlier et al. 1999; Rohatgi et al. 1999; Tang & Gunst, 2004).
Our previous studies have shown that contractile stimulation activates cdc42 and N‐WASP in airway smooth muscle, and that their activation is regulated by multi‐protein signalling complexes associated with integrin adhesion junctions (adhesomes) (Tang & Gunst, 2004; Gunst & Zhang, 2008; Zhang & Gunst, 2008; Zhang et al. 2012). The adhesome protein paxillin is recruited to adhesion junction complexes by a RhoA‐dependent mechanism where it undergoes phosphorylation on tyrosine 31 and 118 by focal adhesion kinase (FAK) (Tang & Gunst, 2001; Zhang et al. 2012). Paxillin tyrosine phosphorylation is an upstream step in the activation of cdc42, which is required for N‐WASP activation and actin polymerization during contractile stimulation (Tang & Gunst, 2001; Tang et al. 2003, 2005). The contractility of airway smooth muscle is thus dependent on the assembly and activation of these multiprotein adhesion junction complexes (Zhang et al. 2012).
In the present study, we tested the hypothesis that Pak regulates airway smooth muscle contraction by regulating MLC phosphorylation, and we investigated the possibility that Pak regulates tension development in response to contractile stimulation by regulating adhesome complex assembly and actin polymerization, which are also necessary for tension development. Our results provide no evidence for a role for Pak in the regulation of MLC phosphorylation during airway smooth muscle contraction. Our results demonstrate that Pak is activated downstream of RhoA to regulate the assembly of multiprotein paxillin complexes that associate with membrane adhesomes and regulate the activation of N‐WASP by catalysing cdc42 activation. Thus, Pak regulates tension generation in response to a contractile stimulus by regulating actin polymerization. These results describe a novel mechanism by which Pak regulates smooth muscle contractility and actin dynamics that is distinct from the regulation of MLC phosphorylation.
Methods
Ethical approval
All procedures were in accordance with procedures approved by the Institutional Animal Care and Use Committee (IUCAC) of Indiana University School of Medicine under the National Research Council's Guide for the Care and Use of Laboratory Animals. Mongrel dogs (20–25 kg, either sex) were procured by the Indiana University Laboratory Animal Resource Centre (LARC) at Indiana University School of Medicine from LBL Kennels, Reelsville, Indiana. Animals were killed by LARC personnel in accordance with procedures approved by the Institutional Animal Care and Use Committee (IUCAC) of Indiana University School of Medicine, by i.v. injection of Fetal Plus (pentobarbital sodium, 390 mg ml−1; propylene glycol, 0.01 mg ml−1; ethyl alcohol, 0.29 mg ml−1; benzyl alcohol (preservative), 0.2 mg ml−1) at a dose of approximately 0.3 ml kg−1. Thereafter, a tracheal segment was immediately removed by laboratory personnel and placed in physiological saline solution (PSS). All investigators understand the ethical principles under which the Journal of Physiology operates and all work complies with these principles.
Preparation of smooth muscle tissues and measurement of force
A tracheal segment was immediately removed and immersed in PSS (composition in mm: 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4 and 5.6 glucose). Strips of tracheal smooth muscle (1.0 × 0.2–0.5 × 15 mm) were dissected free of connective and epithelial tissues and maintained within a tissue bath in PSS at 37°C. Force was measured during isometric contractions by attaching the tissues to Grass force–displacement transducers. Prior to the beginning of each experimental protocol, muscle length was increased to maintain a preload of approximately 0.5–1.0 g, and tissues were stimulated repeatedly with 10−5 m ACh until stable responses were obtained. The force of contraction in response to ACh was determined before and after treatment with plasmids or other reagents.
Immunoblots
For biochemical analysis, muscle tissues were rapidly frozen using liquid N2‐cooled tongs and pulverized in liquid N2 using a mortar and pestle. Pulverized muscle strips were mixed with extraction buffer containing: 20 mm Tris‐HCl at pH 7.4, 2% Triton X‐100, 0.4% SDS, 2 mm EDTA, phosphatase inhibitors (2 mm sodium orthovanadate, 2 mm molybdate and 2 mm sodium pyrophosphate) and protease inhibitors (2 mm benzamidine, 0.5 mm aprotinin and 1 mm phenylmethylsulfonyl fluoride). Each sample was centrifuged and the supernatant was then boiled in sample buffer (1.5% dithiothreitol, 2% SDS, 80 mm Tris‐HCl, pH 6.8, 10% glycerol and 0.01% bromphenol blue) for 5 min. Proteins were separated by SDS‐PAGE and transferred to nitrocellulose. The nitrocellulose membrane was blocked with 2–5% milk for 1 h and probed with primary antibodies against proteins of interest overnight followed by secondary antibodies for 1 h. Proteins were visualized by enhanced chemiluminescence using a Bio‐Rad ChemiDoc XRS detection system (Hercules, CA, USA) or by infrared fluorescence using a LiCor Odyssey imager (Lincoln, NE, USA).
Measurement of MLC phosphorylation
Frozen muscle strips were immersed in dry ice precooled in acetone containing 10% (w/v) trichloroacetic acid and 10 mm dithiothreitol. Proteins were extracted in 8 m urea, 20 mm Tris base, 22 mm glycine and 10 mm dithiothreitol. Phosphorylated and unphosphorylated MLCs were separated by glycerol–urea polyacrylamide gel electrophoresis, transferred to nitrocellulose then immunoblotted for MLC (Hathaway & Haeberle, 1985; Zhang et al. 2005, 2010). MLC phosphorylation was calculated as the ratio of phosphorylated MLC to total MLC.
Analysis of F‐actin and G‐actin
The relative proportions of F‐actin and G‐actin in smooth muscle tissues were analysed as previously described (Zhang et al. 2005, 2010). Briefly, each of the tracheal smooth muscle strips was homogenized in 200 μl of F‐actin stabilization buffer (50 mm PIPES, pH 6.9, 50 mm NaCl, 5 mm MgCl2, 5 mm EGTA, 5% glycerol, 0.1% Triton X‐100, 0.1% Nonidet P‐40, 0.1% Tween‐20, 0.1% β‐mercaptoethanol, 0.001% antifoam, 1 mm ATP, 1 μg ml−1 pepstatin, 1 μg ml−1 leupeptin, 10 μg ml−1 benzamidine and 500 μg ml−1 tosyl arginine methyl ester). Supernatants of the protein extracts were collected after centrifugation (Beckman Coulter Optima MAX Ultracentrifuge, Danvers, MA, USA) at 150,000 g for 60 min at 37°C. The pellets were resuspended in 200 μl of ice‐cold water containing 10 μm cytochalasin D and then incubated on ice for 1 h to depolymerize F‐actin. The resuspended pellets were gently mixed every 15 min. Four microlitres of supernatant (G‐actin) and pellet (F‐actin) fractions were subjected to immunoblot analysis using anti‐actin antibody (clone AC‐40; Sigma, St Louis, MO, USA). The ratios of F‐actin to G‐actin were determined using densitometry.
Transfection of smooth muscle tissues
Plasmids were introduced into tracheal smooth muscle strips by the method of reversible permeabilization (Tang et al. 2003; Zhang et al. 2005, 2010, 2012). Tissues were equilibrated and the muscle length for the generation of maximal isometric force was determined. Muscle strips were then attached to metal mounts to maintain them at constant length. The strips were incubated successively in each of the following solutions: Solution 1 (at 4°C for 120 min) containing (in mm): 10 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2 and 20 N‐tris (hydroxymethyl) methyl‐2‐aminoethanesulfonic acid (TES); Solution 2 (at 4°C overnight) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 2 MgCl2, 20 TES and 20 μg ml−1 plasmids; Solution 3 (at 4°C for 30 min) containing (in mm): 0.1 EGTA, 5 Na2ATP, 120 KCl, 10 MgCl2 and 20 TES; and Solution 4 (at 22°C for 90 min) containing (in mm): 110 NaCl, 3.4 KCl, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 dextrose. Solutions 1–3 were maintained at pH 7.1 and aerated with 100% O2. Solution 4 was maintained at pH 7.4 and was aerated with 95% O2/5% CO2. After 30 min in Solution 4, CaCl2 was added gradually to reach a final concentration of 2.4 mm. The strips were then incubated in a CO2 incubator at 37°C for 2 days in serum‐free DMEM containing 5 mm Na2ATP, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 kanamycin, 2.5 μg ml−1 antifungal and 20 μg ml−1 plasmids to allow for expression of the recombinant proteins. Expression levels of recombinant proteins in transfected muscle tissues as quantified from immunoblots averaged about 50% of the level of the endogenous proteins.
Immunoprecipitation of proteins
Pulverized muscle tissues were mixed with lysis buffer [1% Np‐40, 20 mm Tris‐HCl (pH 7.6), 0.3% NaCl, 10% glycerol, 2 mm EDTA], phosphatase inhibitors (in mm: 2 sodium orthovanadate, 2 molybdate and 2 sodium pyrophosphate) and protease inhibitors (in mm: 2 benzamidine, 0.5 aprotinin and 1 phenylmethylsulfonyl fluoride) for 2 h. Each sample was centrifuged (14,000 g) for the collection of supernatant. Muscle extracts containing equal amounts of protein (800–1200 μg) were precleared for 30 min with 30 μl of 10% protein A/G‐Sepharose and then incubated overnight with primary antibodies. Samples were then incubated for 2 h with 40 μl of a 10% suspension of protein A/G‐Sepharose beads. Immunocomplexes were washed three times in a buffer containing 50 mm Tris‐HCl, pH 7.6, 150 mm NaCl and 0.1% Triton X‐100. All procedures of immunoprecipitation were performed at 4°C. The immunoprecipitates were separated by SDS‐PAGE followed by transfer to nitrocellulose membranes. Proteins were quantified by scanning densitometry.
Assessment of cdc42 activation
The activation of cdc42 was determined using pull‐down assays for activated cdc42 (Tang & Gunst, 2004; Zhang et al. 2012). The cdc42 activation assay kits were obtained from Cytoskeleton (Denver, CO, USA). Pulverized muscle tissues were mixed with lysis buffer (see Immunoprecipitation of proteins for details) for 2 h at 4°C. The extracted proteins were reacted with GST‐Pak binding domain. Activated GTP‐bound cdc42 was affinity‐precipitated by glutathione beads and quantified by immunoblot.
Cell dissociation and immunofluorescence analysis
Freshly dissociated primary cells were used for these studies to avoid the morphological changes in cytoskeletal organization and changes in phenotype that occur during the culture of smooth muscle cells. Smooth muscle cells were enzymatically dissociated from tracheal muscle strips (Opazo Saez et al. 2004; Zhang et al. 2005). Tracheal muscle strips were minced and transferred to 5 ml of dissociation solution (in mm: 130 NaCl, 5 KCl, 1.0 CaCl2, 1.0 MgCl2, 10 Hepes, 0.25 EDTA, 10 d‐glucose and 10 taurine, pH 7.0) with collagenase (type IV, 400 U ml−1), papain (type IV, 30 U ml−1), BSA (1 mg ml−1), and dithiothreitol (1 mm). All enzymes were obtained from Sigma. The strips were then placed in a 37°C shaking water bath at 60 oscillations min–1 for 15–20 min, followed by three washes with a Hepes‐buffered saline solution (in mm: 130 NaCl, 5 KCl, 1.0 CaCl2, 1.0 MgCl2, 20 Hepes and 10 d‐glucose, pH 7.4) and trituration with a pipette to liberate individual smooth muscle cells from the tissue. The solution containing the dissociated cells was poured over glass coverslips, and the cells were allowed to adhere to the coverslips for 30–60 min at room temperature. Cells were stimulated with ACh (10−5 m) for 5 min at 37°C or left unstimulated and used as controls. Stimulated and unstimulated cells were fixed for 10 min in 4% (v/v/) paraformaldehyde in PBS (in mm: 137 NaCl, 4.3 Na2HPO4, 1.4 KH2PO4 and 2.7 KCl, pH 7.4). For immunofluorescence analysis, freshly dissociated cells were permeabilized with 0.5% Triton‐X for 5 min, washed, incubated with primary antibodies against proteins of interest and then incubated with a secondary antibody conjugated to a fluorescent green (Alexa 488) or red (Alexa 546) fluoroprobe (Molecular Probes, Eugene, OR, USA). The cellular localization of fluorescently labelled proteins was evaluated in the dissociated smooth muscle cells using a Zeiss LSM 510 laser scanning confocal microscope with an Apo 40× water‐immersion lens objective (1.2 numerical aperture). Alexa 488‐labelled (green) proteins were excited with a 488 nm argon laser light, and fluorescence emissions were collected at 500–530 nm. The fluorescence of Alexa 546‐labelled (red) proteins was excited with a helium–neon laser at 543 nm, and emissions were collected at 565–615 nm. The optical pinhole was set to resolve optical sections of ∼1 μm in cell thickness. The plane of focus was set midway between the bottom and the top of the cell.
In situ proximity ligation assay (PLA)
In situ PLAs were performed as previously described to detect protein interactions between paxillin and GIT1 and paxillin and Pak. PLA provides for the precise detection of protein–protein complexes (Soderberg et al. 2006, 2008). Target proteins are reacted with primary antibodies raised in different species, and a pair of oligonucleotide‐labelled secondary antibodies conjugated to + and − PLA probes are targeted to each pair of primary antibodies. The probes form circular DNA strands only when they are bound in very close proximity (<40 nm). These DNA circles serve as templates for localized rolling circle amplification, generating a fluorescent signal (spot) that enables individual interacting pairs of the target protein molecules to be visualized. The PLA signal thus allows for the detection of a complex between two target proteins at a very high resolution.
Smooth muscle cells were dissociated from sham‐treated or transfected canine tracheal smooth muscle tissues. Freshly dissociated smooth muscle cells were stimulated with 10−5 m ACh or left unstimulated and then fixed, permeabilized and incubated with a pair of primary antibodies against paxillin and GIT1 or paxillin and Pak1 followed by a pair of oligonucleotide‐labelled secondary antibodies conjugated to Duolink + and − PLA probes. PLA probe hybridization, ligation, amplification and detection media were administered according to the manufacturer's instructions (Olink Bioscience, Uppsala, Sweden). Randomly selected cells from both unstimulated and ACh‐stimulated groups were analysed for protein interactions by visualizing PLA fluorescent spots using a Zeiss LSM 510 confocal microscope. The total number of PLA fluorescent spots for the paxillin–GIT1 or paxillin–Pak1 interactions were counted using Olink Bioscience Image Tools software.
Reagents and antibodies
The Pak inhibitor IPA3 was generously provided by Dr Jonathan Chernoff, Fox Chase Cancer Centre, Philadelphia, PA, USA. The Duolink in situ proximity ligation assay (Olink Bioscience) was obtained from Sigma. All other chemical reagents were obtained from Sigma.
Plasmids encoding WT human Pak1, and kinase inactive human Pak1 K299R were obtained from Dr Jonathan Chernoff, Fox Chase Cancer Centre (Sells et al. 1997; Adam et al. 1998). The constructs were subcloned into the mammalian expression vector pCMV6M at BamHI/EcoRI sites. Constructs for WT chicken paxillin were generously provided by Dr Christopher Turner, SUNY Upstate Medical University, as previously described (Tang et al. 2003). Non‐phosphorylatable pcDNA3.1‐paxillin S273A mutants were constructed from WT chicken paxillin by point mutation at serine 273 using a QuikChange II site‐directed mutagenesis kit (Stratagene, La Jolla, CA, USA). Paxillin WT and paxillin S273A chicken cDNAs were subcloned into a pcDNA 3.1 vector at the BamHI/EcoRI site. Plasmids encoding RhoA T19N (Zhang et al. 2010) and plasmids encoding paxillin non‐phosphorylatable tyrosine 118/31 (Paxillin Y31/118F) (Tang et al. 2003) were previously described. Escherichia coli (Bluescript) transformed with these plasmids were grown in LB medium, and plasmids were purified by alkaline lysis with SDS using a purification kit from Qiagen Inc. (Valencia, CA, USA).
Sources of antibodies are as follows: monoclonal mouse anti‐human paxillin (cat. no. 610569, BD Transduction, Franklin Lakes, NJ, USA); polyclonal rabbit anti‐human paxillin phospho‐tyrosine 118 (cat. no. 44‐722 G, Invitrogen, Carlsbad, CA, USA); polyclonal rabbit anti‐human paxillin phospho‐serine 273 (cat. no. 44‐1028 G, Invitrogen); polyclonal rabbit anti‐human Pak1 (cat. no. 2602, Cell Signalling); polyclonal rabbit anti‐human Pak2 (cat. no. 2608, Cell Signaling, Danvers, MA, USA); polyclonal rabbit anti‐human phospho‐Pak Thr 423/402 (cat. no. 2601, Cell Signaling); polyclonal rabbit anti‐human pan‐Pak (cat. no. 2604, Cell Signaling); polyclonal rabbit anti‐human GIT1 (cat. no. 2909, Cell Signaling); polyclonal rabbit anti‐human βPIX (cat. no. 4515, Cell Signaling); mouse monoclonal anti‐human N‐WASP (cat. no. 4848, Cell Signaling); polyclonal rabbit anti‐human N‐WASP phospho‐tyrosine 256 (cat. no. ab23395, Abcam, Cambridge, MA, USA); monoclonal mouse anti‐human cdc42 (cat. no. 610929, BD Transduction); horseradish peroxidase‐conjugated IgG (cat. no. NA931 & NA934, Amersham Biosciences, Piscataway, NJ, USA); IRDye 680RD Donkey‐anti‐Mouse Antibody (cat. no. 926‐68070) and IRDye 800CW Donkey‐anti‐Rabbit Antibody (cat. no. 925‐32211, LI‐COR Biosciences, Lincoln, NE, USA); polyclonal rabbit vinculin antibody (against canine cardiac vinculin) and polyclonal rabbit myosin light chain antibody were custom made by BABCO (Richmond, CA, USA) (Huang et al. 2011; Zhang et al. 2012). All antibodies have been validated to confirm their reaction with the designated antigen in the smooth muscle tissue extracts, immunoblots or fixed cells.
Statistical analysis
Comparisons between two groups were performed using paired or unpaired two‐tailed Student's t tests. Values refer to the number of cells or tissue strips used to obtain mean values. P < 0.05 was considered statistically significant.
Results
Pak is activated by contractile stimulation and regulates tension development in smooth muscle
We analysed Pak Thr 423/402 phosphorylation as an assessment of Pak activation in smooth muscle tissues. The phosphorylation of Thr423 on Pak1 (Thr402 on Pak2) within the activation loop of the catalytic domain is required for the full catalytic function of Pak toward exogenous substrates (Yu et al. 1998; Zenke et al. 1999). The time course of Pak Thr423/402 phosphorylation was evaluated in tissues stimulated with 10−5 m ACh for up to 5 min (Fig. 1 A). Pak phosphorylation increased significantly within 1 min of contractile stimulation and remained elevated for the duration of the stimulation period. The time course of Pak activation was similar to that of tension development.
Figure 1. Pak inactivation inhibits ACh‐stimulated tension development in tracheal smooth muscle tissues .
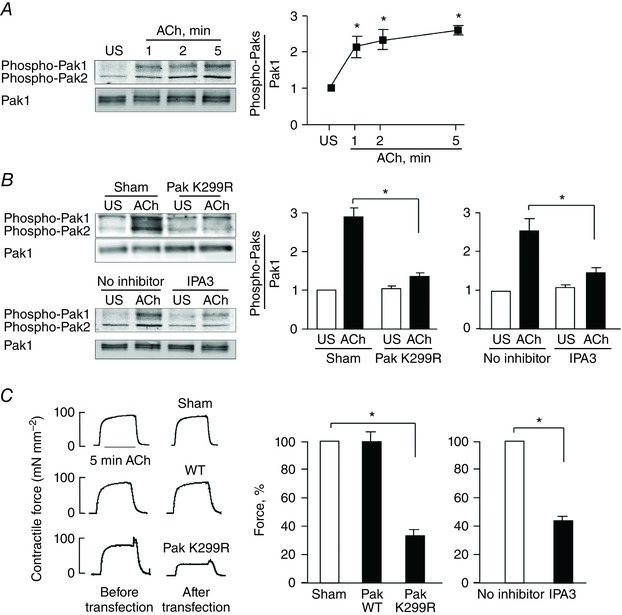
A, pak Thr423/Thr402 (T423/T402) phosphorylation was determined in tracheal smooth muscle tissues after 1, 2 or 5 min ACh stimulation or without stimulation (US). Immunoblots against phospho‐Pak T423/T402 and Pak1 were obtained using a site‐specific phospho‐Pak T423/T402 antibody and Pak1 antibody. The ratio of phospho‐Pak to Pak1 was determined for each sample (n = 11). B, Pak activation in response to 5 min ACh stimulation was inhibited in smooth muscle tissues by expressing Pak K299R (n = 9) or by treating the tissues with the Pak inhibitor, IPA3 (n = 6). C, contractile force in response to ACh is shown for tissues before and after they were transfected with plasmids encoding WT Pak, Pak1 K299R or no plasmids (Sham treated) (n = 12). Treatment with IPA3 also significantly inhibited ACh‐induced tension development compared to tissues not treated with IPA3 (No inhibitor) (n = 8). All force measurements were normalized to Sham response (91.3 ± 7.8 mN mm−2) or No inhibitor response (93.0 ± 9.0 mN mm−2). All values are means ± SEM. *Significant difference between treatment groups (P < 0.05).
The role of Pak in contractile force development in airway smooth muscle tissues was determined by expressing a kinase inactive Pak mutant, Pak1 K299R, in the tissues or by administering a Pak inhibitor, IPA3, to inhibit the activation of Pak (Fig. 1 B). IPA3 is an allosteric inhibitor that targets the activation loop of Pak and prevents the conformational change required for activation. IPA3 thus exhibits a high selectivity for Pak (Deacon et al. 2008).
The increase in Pak Thr 423/402 phosphorylation in response to 5 min stimulation with ACh was significantly inhibited by the expression of Pak K299R, indicating that Pak activation was suppressed (Fig. 1 B). Incubation of the tissues with 100 μm IPA3 for 2 h also inhibited ACh‐induced Pak activation (Fig. 1 B).
The contractile response to ACh was significantly reduced to 34 ± 6% in tissues transfected with plasmids for Pak K299R as compared to tissues transfected with plasmids for WT Pak or sham‐treated tissues (Fig. 1 C). Similarly, treatment of the tissues with IPA3 reduced the contractile response to ACh to 44 ± 4% of that in tissues without inhibitor (Fig. 1 C). These results demonstrate that Pak activation contributes to contractile tension development in airway smooth muscle tissues.
Pak regulates actin polymerization but not MLC phosphorylation in smooth muscle tissues during contractile stimulation
We evaluated the mechanism by which Pak mediates ACh‐induced tension development by expressing the catalytically inactive Pak1 mutant, Pak1 K299R, in smooth muscle tissues. MLC phosphorylation and tension development were determined in muscle tissues expressing Pak1 K299R and in sham‐treated tissues 5 min after stimulation with 10−5 m ACh. The inhibition of Pak activation did not significantly affect the increase in MLC phosphorylation in response to ACh in the smooth muscle tissues, although force was significantly inhibited (Fig. 2 A). MLC phosphorylation in response to ACh was also not significantly affected by treatment of the tissues with 100 μm IPA3 to inhibit Pak (Fig. 2 B).
Figure 2. Pak regulates actin polymerization but not MLC phosphorylation in tracheal smooth muscle tissues during contractile stimulation .
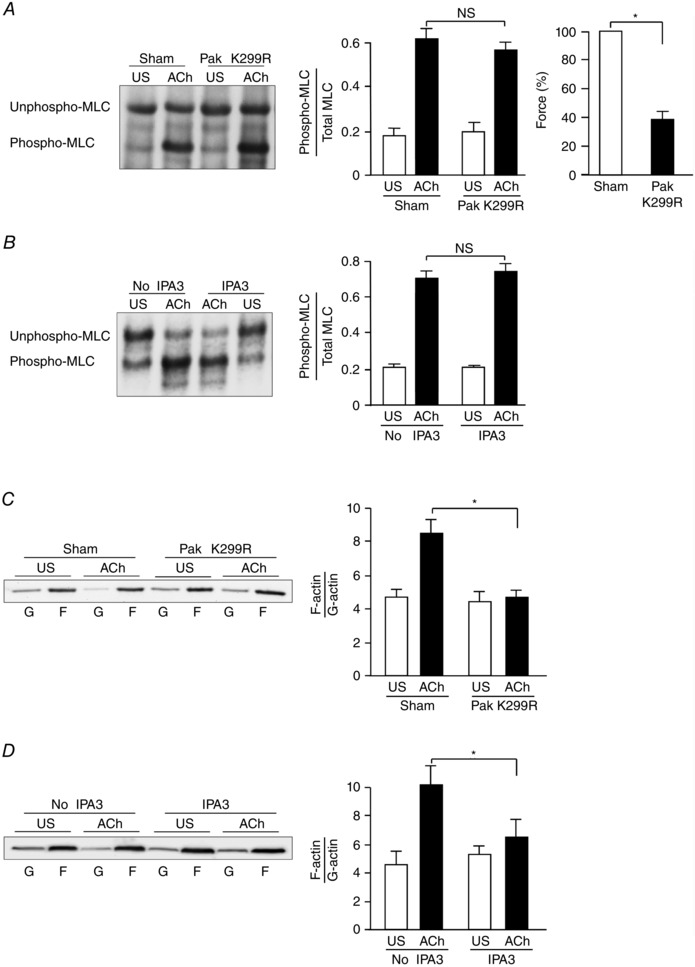
A, representative immunoblot obtained from urea gel electrophoresis of extracts of four muscle tissues treated with Pak K299R or sham‐treated and then stimulated with ACh or unstimulated (US) (left). Unphosphorylated and phosphorylated smooth muscle 20 kDa MLCs were quantified as the ratio of phosphorylated MLCs to total MLCs in each sample (middle). Expression of the Pak K299R did not significantly affect MLC phosphorylation in response to ACh, but it significantly inhibited ACh‐induced tension development of smooth muscle tissues (n = 8) (right). The absolute value of the mean force of the sham‐treated tissues was 88.2 ± 5.0 mN mm−2. B, representative immunoblot obtained from urea gel electrophoresis of extracts of four muscle tissues with or without IPA3 treatment and stimulated with ACh or unstimulated (US) (left). IPA3 did not significantly affect MLC phosphorylation in response to ACh (n = 6) (right). C, immunoblot of soluble G‐actin (globular) and insoluble F‐actin (filamentous) in fractions from extracts of unstimulated (US) or ACh‐stimulated muscle tissues treated with Pak K299R or sham‐treated. Ratios of F‐actin to G‐actin were determined by quantifying F and G actin in extracts from each muscle strip (left). Inhibition of Pak activation with Pak K299R prevented the increase in F‐actin/G‐actin ratio in response to ACh stimulation (n = 6) (right). D, inhibition of Pak activation with IPA3 prevented the increase in F‐actin/G‐actin ratio in response to ACh stimulation (n = 6). Values are means ± SEM. *Significant difference between treatments, P < 0.05. NS, not significantly different.
Actin polymerization was measured in canine tracheal smooth muscle tissues using a fractionation assay to separate soluble (G‐actin) and insoluble (F‐actin) actin in the muscle extracts. The expression of Pak K299R in the tissues or treatment of muscle tissues with IPA3 significantly inhibited the increase in actin polymerization (F‐actin/G‐actin ratio) that occurs in response to stimulation with ACh (Fig. 2 C, D). These results indicate that Pak regulates tension development in smooth muscle by regulating actin polymerization rather than by regulating MLC phosphorylation.
Pak regulates paxillin Ser273 phosphorylation in response to stimulation with ACh in tracheal smooth muscle tissues
Pak can phosphorylate paxillin at Ser273 in migrating non‐muscle cells; this facilitates the binding of paxillin to GIT and promotes the localization of GIT and PIX to adhesion junctions (Nayal et al. 2006). We assessed the role of Pak in the regulation of paxillin phosphorylation on serine and tyrosine residues during contractile stimulation by treating smooth muscle tissues with Pak K299R or IPA3 to inhibit Pak activation.
Stimulation of tracheal muscle tissues with 10−5 m ACh caused a significant increase in paxillin phosphorylation on Ser273 as well as on Tyr118 sites (Fig. 3 A and B). Expression of Pak K299R in tracheal smooth muscle tissues or treatment with IPA3 significantly inhibited the increase in paxillin Ser273 phosphorylation in response to ACh stimulation, but it had no effect on the ACh‐stimulated increase in paxillin Tyr118 phosphorylation (Fig. 3 A, B).
Figure 3. Pak regulates paxillin Ser273 phosphorylation in response to stimulation with ACh in tracheal smooth muscle tissues .
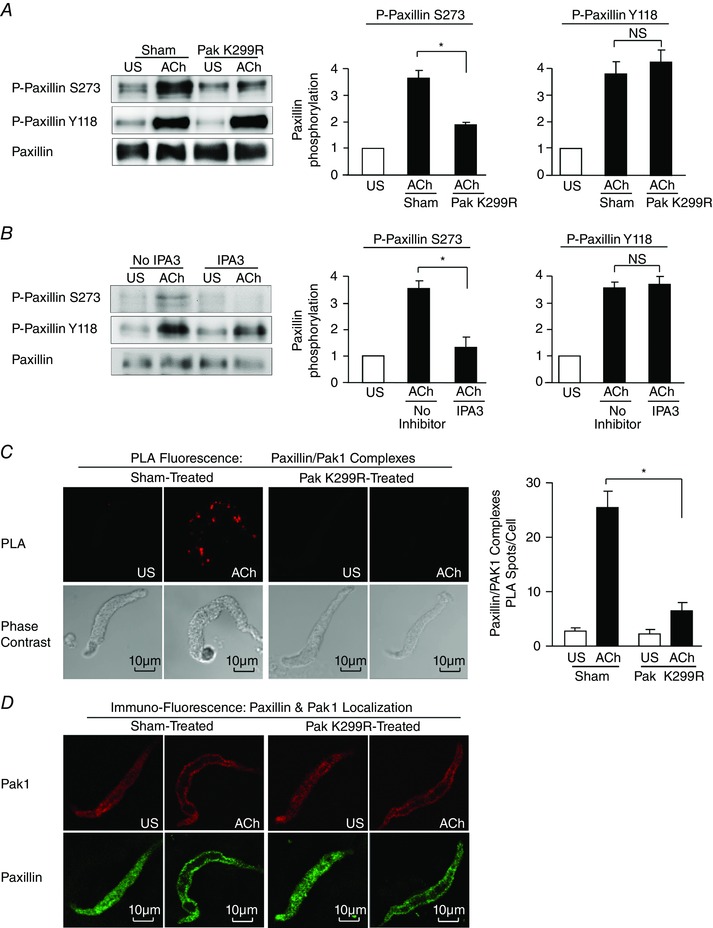
A, paxillin Tyr118 (Y118) phosphorylation and paxillin Ser273 (S273) phosphorylation were measured in tracheal smooth muscle tissues expressing the Pak K299R mutant or in sham‐treated tissues after 5 min ACh stimulation or unstimulated (US). Pak K299R significantly inhibited ACh‐induced paxillin Ser273 phosphorylation, but it did not significantly affect paxillin Tyr118 phosphorylation (n = 9). B, paxillin Tyr118 phosphorylation and paxillin Ser273 phosphorylation were measured in smooth muscle tissues treated with IPA3 or untreated (No inhibitor) tissues with or without 5 min ACh stimulation. IPA3 significantly inhibited ACh‐induced paxillin Ser273 phosphorylation, but it did not significantly affect paxillin Tyr118 phosphorylation (n = 4). C, in situ PLA shows the interaction of paxillin and Pak1 in freshly dissociated differentiated tracheal smooth muscle cells. PLA fluorescence and phase contrast images are shown for each unstimulated (US) and ACh‐stimulated cell. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 49 for ACh, n = 22 for US). In cells from Pak K299R‐treated tissues, the mean number of PLA spots was very small and did not change significantly with ACh stimulation (n = 36 for ACh, n = 20 for US). Cells were dissociated from tissues obtained from four separate experiments. D, freshly dissociated tracheal smooth muscle cells were double stained by immunofluorescence to determine the localization of Pak1 and paxillin with or without ACh stimulation. ACh stimulation caused the recruitment of both Pak1 and paxillin from the cytoplasm to the cell membrane in both sham‐treated and Pak K299R‐treated tissues. One typical cell out of eight cells of each group is shown. All values are means ± SEM. *Significant difference between ACh‐stimulated tissues, P < 0.05. NS, not significantly different.
In situ PLAs were performed on freshly dissociated smooth muscle cells to evaluate the interaction between paxillin and Pak1 in unstimulated and ACh‐stimulated tissues (Fig. 3 C). Very few spots were observed in unstimulated cells, indicating little interaction between paxillin and Pak1. Stimulation with ACh caused a dramatic increase in the number of PLA spots at the cell membrane, indicating an interaction of Pak1 with paxillin in membrane complexes. Very few spots were observed in ACh‐stimulated cells dissociated from tissues treated with the Pak K299R non‐catalytic mutant, indicating that Pak inactivation prevented the interaction of Pak1 with paxillin during contractile stimulation.
We also evaluated the effect of Pak inactivation on the localization of Pak1 and paxillin in freshly dissociated cells (Fig. 3 D). In unstimulated cells from both sham‐treated and Pak K299R‐treated tissues, both Pak1 and paxillin are observed throughout the cell cytoplasm. ACh stimulation caused the recruitment of both Pak1 and paxillin to the cell membrane; thus, the membrane recruitment of Pak and paxillin was not prevented by inhibiting Pak activation. These results demonstrate that Pak inactivation does not prevent the localization of paxillin or Pak to the membrane, and that Pak phosphorylates paxillin after both proteins localize to membrane complexes.
Paxillin Ser273 phosphorylation regulates tension development and actin polymerization in response to stimulation of smooth muscle with ACh
The role of paxillin Ser273 phosphorylation in the regulation of smooth muscle contraction was evaluated by expressing the non‐phosphorylatable paxillin mutant paxillin S273A in smooth muscle tissues. The expression of paxillin S273A in tracheal smooth muscle tissues inhibited the ACh‐induced increase in endogenous paxillin Ser273 phosphorylation, but it had no effect on paxillin Tyr118 phosphorylation (Fig. 4 A). The effect of inhibiting paxillin Ser273 phosphorylation on the activation of Pak was also evaluated by measuring Pak Thr423 phosphorylation in tissues expressing paxillin S273A. The inhibition of paxillin Ser273 phosphorylation had no effect on the activation of Pak induced by ACh (Fig. 4 B).
Figure 4. Paxillin Ser273 phosphorylation regulates tension development and actin polymerization during ACh stimulation in tracheal smooth muscle tissues .
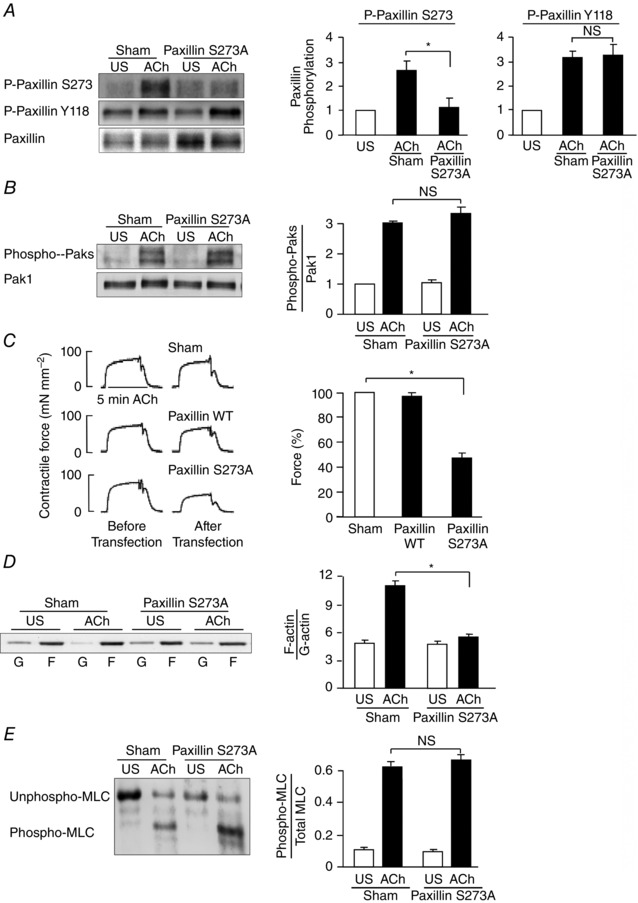
A, paxillin Tyr118 (Y118) and paxillin Ser273 (S273) phosphorylation were measured by immunoblot in extracts of tracheal smooth muscle tissues expressing the paxillin S273A mutant or in sham‐treated tissues after 5 min ACh stimulation or unstimulated (US). Paxillin S273A significantly inhibited ACh‐induced paxillin Ser273 phosphorylation, but not paxillin Tyr118 phosphorylation (n = 8). B, expression of paxillin S273A had no effect on ACh‐induced Pak activation as indicated by the increase in Thr423/Thr402 phosphorylation (n = 4). C, paxillin S273A significantly inhibited ACh‐induced tension development relative to paxillin WT or sham‐treated tissues (n = 12). All force measurements were normalized to sham response (91.3 ± 7.8 mN mm−2). D, immunoblot of soluble G‐actin (globular) and insoluble F‐actin (filamentous) from extracts of unstimulated (US) or ACh‐stimulated muscle tissues treated with paxillin S273A or sham treated. Expression of paxillin S273A significantly inhibited the increase in the F‐actin/G‐actin ratio in response to ACh stimulation (n = 5). E, MLC phosphorylation was measured in muscle strips transfected with paxillin S273A or sham‐treated after stimulation with ACh for 5 min or without stimulation (US). Paxillin S273A did not significantly affect MLC phosphorylation in response to ACh (n = 5). *Significant difference between treatments, P < 0.05. NS, not significantly different. Values are means ± SEM.
The contractile response to ACh was significantly reduced to 45.9 ± 2.9% in tissues transfected with plasmids for paxillin S273A as compared to tissues treated with plasmids for WT paxillin or sham‐treated tissues (Fig. 4 C). The inhibition of paxillin Ser273 phosphorylation also inhibited actin polymerization in response to ACh stimulation (Fig. 4 D), but it had no effect on MLC phosphorylation (Fig. 4 E).
Pak regulates the interaction of paxillin with GIT1 and βPIX at membrane adhesion complexes during contractile stimulation
The phosphorylation of paxillin at Ser273 by Pak can promote its binding to GIT1 (Nayal et al. 2006). GIT1 can form a complex with βPIX, which has been shown to have GEF activity towards cdc42 (Frank & Hansen, 2008).
ACh stimulation resulted in an increase in GIT1, βPIX and paxillin in Pak immunocomplexes relative to unstimulated tissues in sham‐treated tissues (Fig. 5 A). The inactivation of Pak by expression of Pak K299R inhibited the formation of a complex between paxillin, GIT1, βPIX and Pak1 during stimulation with ACh. Similar results were obtained in tissues in which Pak activity was inhibited by treatment with IPA3 (Fig. 5 B). The immunoprecipitation of either Pak1 or Pak2 isoforms from extracts of canine tracheal smooth muscle tissues stimulated with ACh confirmed that both isoforms form complexes with GIT1 and βPIX during contractile stimulation (Fig. 5 C).
Figure 5. Pak regulates the interaction of GIT1 and βPIX with paxillin at the cell membrane during contractile stimulation .
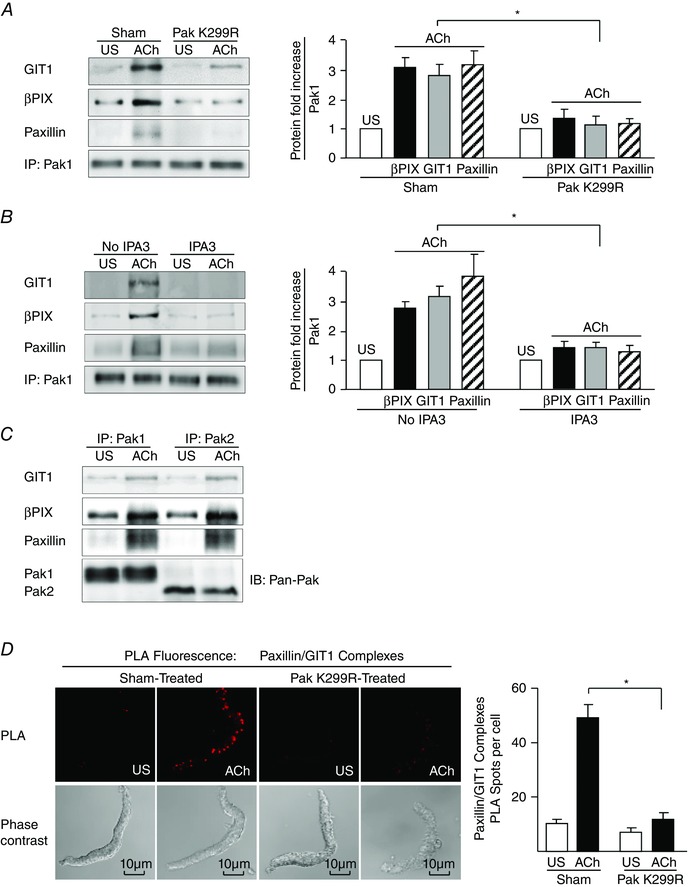
A, pak1 was immunoprecipitated from extracts of tracheal smooth muscle tissues stimulated for 5 min with ACh or unstimulated (US) and treated with Pak K299R to inhibit Pak activation or not treated (Sham) (n = 4). B, Pak1 was immunoprecipitated from extracts of canine tracheal smooth muscle tissues stimulated for 5 min with ACh or unstimulated (US) and treated with IPA3 to inhibit Pak activation or not treated (n = 7). C, Pak1 or Pak2 was immunoprecipitated from extracts of canine tracheal smooth muscle tissues stimulated for 5 min with ACh or unstimulated (US). ACh induced an increase in GIT1, βPIX and paxillin co‐immunoprecipitation with either Pak1 or Pak2. Pak1 and Pak2 were immunoblotted using a pan‐Pak antibody. D, in situ PLA shows the interaction of paxillin and GIT1 in freshly dissociated differentiated canine tracheal smooth muscle cells. PLA fluorescence and phase contrast images are shown for each cell. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 30 for US; n = 34 for ACh). In cells from Pak K299R‐treated tissues, the mean number of PLA spots was very small and did not change significantly with ACh stimulation (n = 33 for US; n = 34 for ACh). Cells dissociated from tissues obtained from four separate experiments. *Significant difference between effects of treatments on ACh‐stimulated cells, P < 0.05. Values are means ± SEM. IP, immunoprecipitate; IB, immunoblot.
The role of Pak in regulating the interaction between paxillin and GIT1 was also investigated by PLA in freshly dissociated tracheal smooth muscle cells (Fig. 5 D). In unstimulated cells, few interactions between paxillin and GIT1 were detected. Stimulation with ACh induced the interaction of paxillin and GIT1 at the cell membrane, as indicated by the dramatic increase in the number of PLA spots. The expression of Pak K299R in the tracheal muscle tissues inhibited the interaction between paxillin and GIT1 in dissociated cells stimulated by ACh (Fig. 5 D).
Paxillin Ser273 phosphorylation regulates the interaction of paxillin with GIT1 and βPIX at the membrane during contractile stimulation
The role of paxillin Ser273 phosphorylation in regulating the interaction between paxillin and GIT1 was assessed by PLA in freshly dissociated smooth muscle cells expressing the paxillin S273A mutant or WT paxillin as control (Fig. 6 A). Few interactions between paxillin and GIT1 were detected in unstimulated cells. Stimulation with ACh caused a dramatic increase in the number of PLA spots at the cell membrane in cells dissociated from tissues expressing WT paxillin, indicating that ACh induced the interaction of paxillin and GIT1 at the cell membrane. The expression of the paxillin S273A mutant inhibited the interaction between paxillin and GIT1 stimulated by ACh (Fig. 6 A). Immunoprecipitation of paxillin from smooth muscle tissue extracts also demonstrated that the interaction between paxillin and GIT1 induced by ACh stimulation was inhibited by the expression of paxillin S273A (Fig. 6 B).
Figure 6. Paxillin Ser273 phosphorylation regulates the interaction of paxillin and GIT1 at the cell membrane during contractile stimulation .
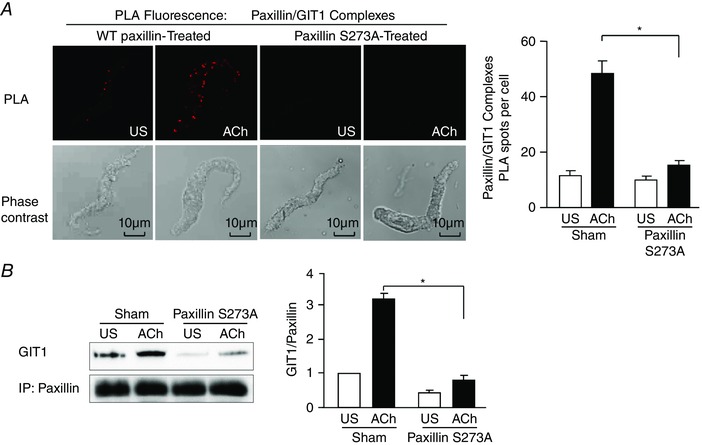
A, in situ PLA shows the interaction of paxillin and GIT1 in freshly dissociated differentiated canine tracheal smooth muscle cells. PLA fluorescence and phase contrast images are shown for each cell. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 42 cells for US; n = 46 cells for ACh). In cells from paxillin S273A‐treated tissues, the mean number of PLA spots was very small and did not change significantly with ACh stimulation (n = 40 cells for US; n = 45 cells for ACh). Cells dissociated from tissues obtained from four separate experiments. B, co‐immunoprecipitation of GIT1 with paxillin increased after contractile stimulation with ACh in sham‐treated tissues, and was significantly inhibited in tissues expressing paxillin S273A (n = 3). Values are means ± SEM. *Significant difference between treatments in ACh‐stimulated tissues, P < 0.05.
RhoA regulates the recruitment of Pak to membrane adhesomes and Pak activation during contractile stimulation
The interaction between RhoA‐ and Pak‐activated pathways during the smooth muscle contraction was determined by expressing the dominant negative mutant RhoA T19N in tracheal smooth muscle tissues to inactivate RhoA. We previously demonstrated that the expression of RhoA T19N inhibits RhoA activation in response to ACh in these tissues (Zhang et al. 2010, 2012). RhoA inhibition suppressed the activation of Pak in response to stimulation with ACh (Fig. 7 A), and inhibited paxillin phosphorylation on both Tyr118 and Ser273 (Fig. 7 B).
Figure 7. RhoA regulates the activation of Pak and its interaction with paxillin at the adhesome complex during contractile stimulation .
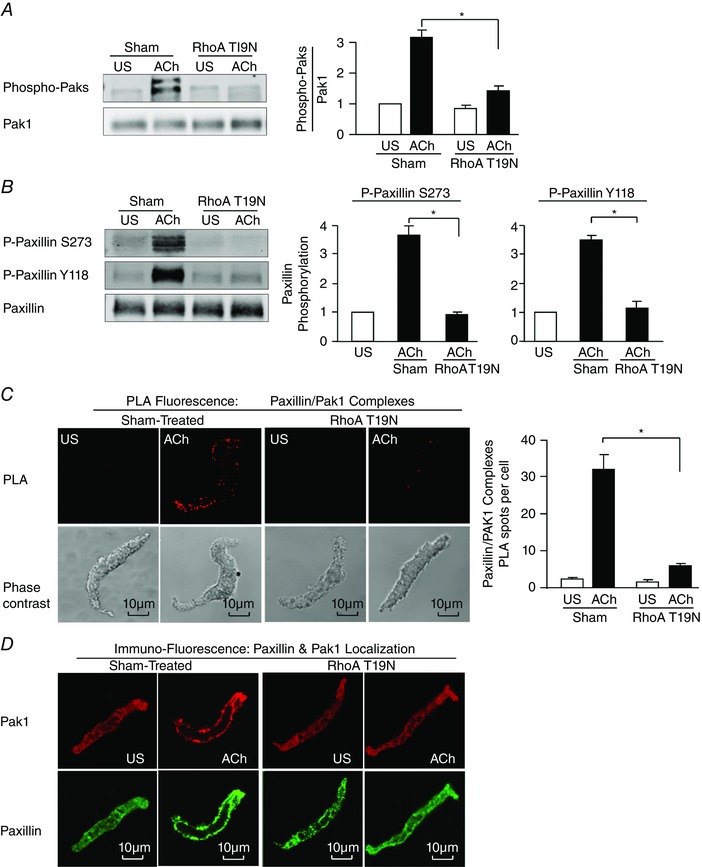
A, pak Thr423 phosphorylation was measured in tissues expressing RhoA T19N or sham‐treated tissues with or without ACh stimulation (n = 6). B, paxillin Tyr118 (Y118) phosphorylation and paxillin Ser273 (S273) were measured in tracheal smooth muscle tissues expressing the RhoA T19N mutant and in sham‐treated tissues after 5 min ACh stimulation or in unstimulated tissues (US) (n = 3). C, in situ PLA was used to analyse the interaction of paxillin and Pak1 in freshly dissociated differentiated canine tracheal smooth muscle cells. PLA fluorescence and phase contrast images are shown for each unstimulated (US) and ACh‐stimulated cell. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 20 for ACh, n = 16 for US). In cells from RhoA T19N‐treated tissues, the ACh‐induced increase in the mean number of PLA spots was very small and was significantly inhibited (n = 21 cells for ACh, n = 15 cells for US). Cells were dissociated from tissues obtained from three separate experiments. D, freshly dissociated tracheal smooth muscle cells were double stained for paxillin and Pak1 to determine the effect of ACh stimulation on the localization of Pak1 and paxillin. US, unstimulated cells. A representative cell from each group of eight cells is shown. Values are means ± SEM. *Significant difference between ACh‐stimulated tissues, P < 0.05.
The role of RhoA in regulating the interaction of paxillin with Pak was assessed using in situ PLAs on smooth muscle cells freshly dissociated from RhoA T19N‐treated tissues. RhoA inactivation prevented the interaction of Pak1 with paxillin at the membrane (Fig. 7 C).
Freshly dissociated smooth muscle cells were double stained for Pak1 and paxillin to determine whether RhoA regulated the localization of Pak1 during contractile stimulation (Fig. 7 D). In sham‐treated unstimulated cells, both Pak1 and paxillin were observed throughout the cell cytoplasm, and ACh stimulation caused the recruitment of both Pak1 and paxillin to the cell membrane. In cells from tissues treated with RhoA T19N to inactivate RhoA, the membrane recruitment of Pak and paxillin in ACh‐stimulated cells was inhibited.
These results demonstrate that RhoA regulates Pak activity by catalysing its recruitment to membrane signalling complexes, where Pak interacts with paxillin to regulate paxillin Ser273 phosphorylation.
Paxillin tyrosine phosphorylation is a prerequisite to its Ser273 phosphorylation by Pak
We expressed the paxillin double tyrosine mutant paxillin Y31/118F in tracheal muscle tissues to inhibit paxillin tyrosine phosphorylation (Tang et al. 2003), and thereby assess whether paxillin Ser273 phosphorylation by Pak can occur in the absence of paxillin tyrosine phosphorylation. Paxillin Y31/118F effectively inhibited the increase in paxillin tyrosine phosphorylation in response to ACh and suppressed paxillin phosphorylation at Ser273 (Fig. 8 A). However, the expression of paxillin Y31/118F had no effect on the activation of Pak in response to ACh (Fig. 8 B). The expression of paxillin Y31/118F also inhibited the formation of paxillin complexes with Pak1 and GIT1 (Fig. 8 C, D).
Figure 8. Paxillin tyrosine phosphorylation is a prerequisite to paxillin phosphorylation on Ser273 by Pak .
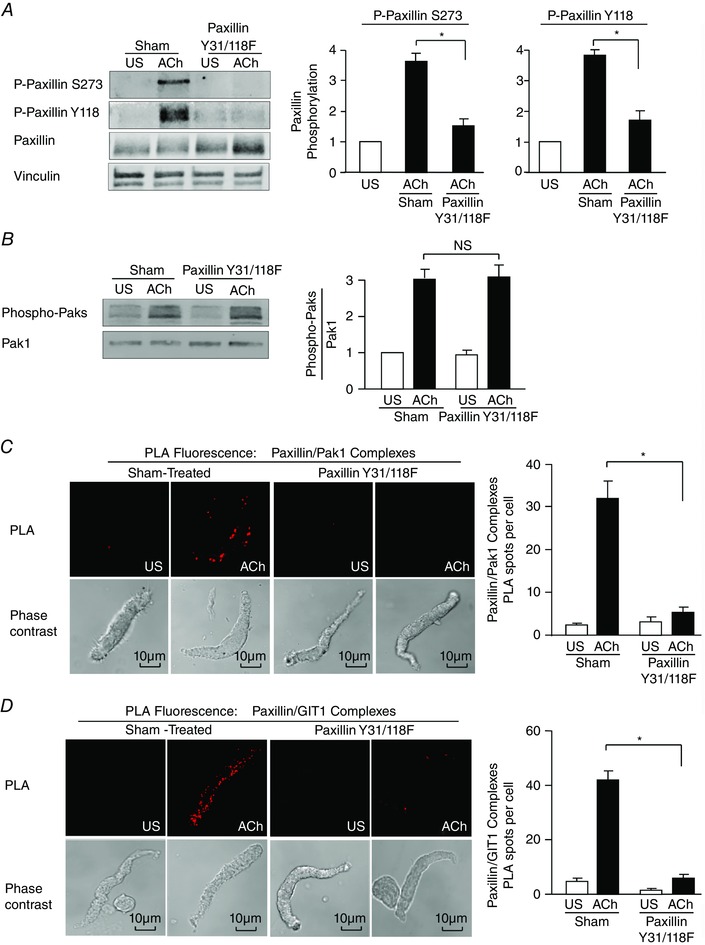
A, paxillin Tyr118 (Y118) phosphorylation and paxillin Ser273 (S273) were measured by immunoblot in extracts of tracheal smooth muscle tissues expressing the paxillin Y31/118F double mutant or sham‐treated tissues. Paxillin Y31/118F significantly inhibited ACh‐induced paxillin Tyr118 phosphorylation and also suppressed paxillin Ser273 phosphorylation (n = 4). B, expression of paxillin Y31/118F had no effect on ACh‐induced Pak activation as indicated by Thr423/Thr402 phosphorylation (n = 4). C, in situ PLA was used to analyse the interaction of paxillin and Pak1 in freshly dissociated differentiated canine tracheal smooth muscle cells. PLA fluorescence and phase contrast images are shown for each unstimulated (US) and ACh‐stimulated cell. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 20 cells for ACh, n = 16 cells for US). In cells from paxillin Y31/118F‐treated tissues, the ACh‐induced increase in the mean number of PLA spots was very small and was significantly inhibited (n = 19 cells for ACh, n = 15 cells for US). Cells were dissociated from tissues obtained from three separate experiments. D, in situ PLA was used to analyse the interaction of paxillin and GIT1 in freshly dissociated differentiated canine tracheal smooth muscle cells. In cells from sham‐treated tissues, the mean number of PLA spots was significantly higher in ACh‐stimulated cells than in unstimulated cells (n = 30 cells for ACh, n = 13 cells for US). In cells from paxillin Y31/118F‐treated tissues, the ACh‐induced increase in the mean number of PLA spots was very small and was significantly inhibited (n = 21 for ACh, n = 13 for US). Cells were dissociated from tissues obtained from three separate experiments. Values are means ± SEM. *Significant difference between ACh‐stimulated tissues, P < 0.05. NS, not significantly different.
These results demonstrate that the activation of Pak in response to ACh is independent of paxillin tyrosine phosphorylation, but the tyrosine phosphorylation of paxillin is a prerequisite to its Ser273 phosphorylation by Pak. Thus, the tyrosine phosphorylation of paxillin is also required for paxillin to form complexes with Pak1 and GIT1.
Pak activation and paxillin Ser273 phosphorylation are required for the ACh‐induced activation of cdc42 and N‐WASP during contractile stimulation of smooth muscle
In airway smooth muscle tissues, activation of the actin polymerization catalyst N‐WASP depends on its interaction with the activated GTPase cdc42 (Tang & Gunst, 2004). As βPIX is known to have GEF activity towards cdc42, we investigated the role of Pak‐mediated paxillin phosphorylation in the regulation of cdc42 and N‐WASP activation.
The role of Pak in the regulation of cdc42 was evaluated using a pull‐down assay for activated cdc42 (cdc42‐GTP) in extracts from tracheal smooth muscle tissues (Tang & Gunst, 2004; Zhang et al. 2012). When Pak was inactivated by expressing Pak K299R or by treatment with IPA3, cdc42 activation was inhibited (Fig. 9 A). N‐WASP activation, as indicated by its phosphorylation on tyrosine 256, was also suppressed by the inhibition of Pak activity (Fig. 9 B).
Figure 9. Pak activation and paxillin Ser273 phosphorylation are required for the ACh‐induced activation of cdc42 and N‐WASP during contractile stimulation of tracheal smooth muscle tissues .
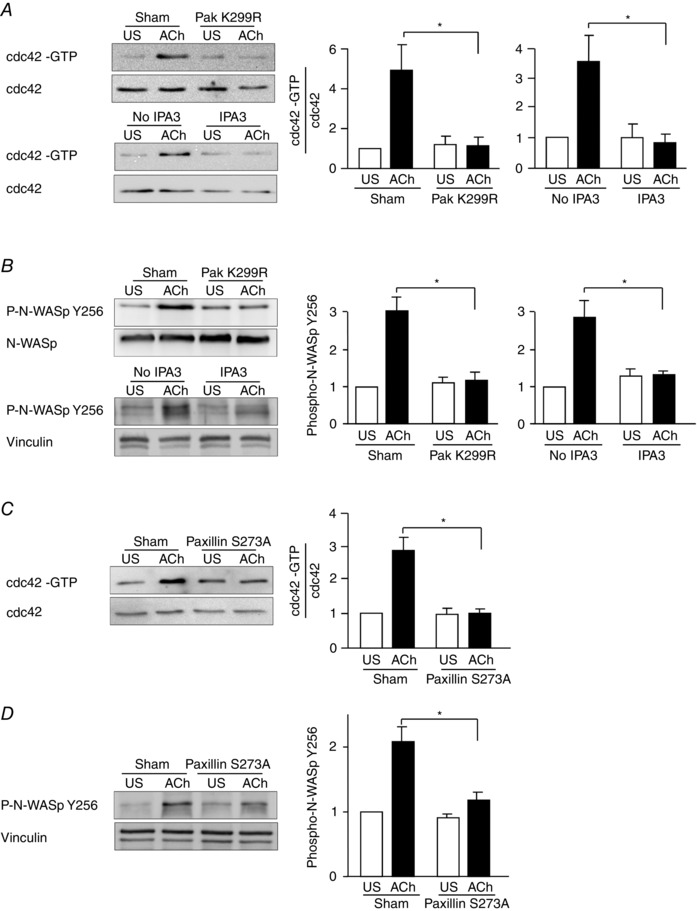
A, activated cdc42 (cdc42‐GTP) was affinity‐precipitated from extracts of unstimulated and ACh‐stimulated muscle strips, and the amount of activated cdc42 precipitated from each extract was quantified by immunoblot. Activated cdc42 was significantly higher in extracts from 10−5 m ACh‐stimulated sham‐treated tissues than from ACh‐stimulated tissues expressing Pak K299R (n = 3). IPA3 also inhibited the activation of cdc42 (n = 5). B, N‐WASP Tyr256 phosphorylation measured by immunoblot in extracts of muscle tissues transfected with Pak K299R, or sham‐treated. Pak K299R significantly inhibited ACh‐induced N‐WASP phosphorylation relative to sham‐treated tissues (n = 5). IPA3 also inhibited ACh‐induced N‐WASP phosphorylation (n = 7). C, activated cdc42 was significantly higher in extracts from 10−5 m ACh‐stimulated sham‐treated tissues than in ACh‐stimulated tissues expressing paxillin S273A (n = 6). D, N‐WASP Tyr256 phosphorylation measured by immunoblot in extracts of muscle tissues transfected with paxillin S273A or sham‐treated. Paxillin S273A significantly inhibited ACh‐induced N‐WASP phosphorylation relative to sham‐treated tissues (n = 8). *Significant difference between treatments in ACh‐stimulated tissues, P < 0.05. Values are means ± SEM.
The role of paxillin Ser273 phosphorylation in the regulation of cdc42 and N‐WASP activation was evaluated by expressing paxillin S273A in tracheal muscle tissues to inhibit paxillin Ser273 phosphorylation. The inhibition of paxillin phosphorylation on Ser273 also inhibited cdc42 and N‐WASP activation in response to stimulation with ACh (Fig. 9 C, D).
These results demonstrate that Pak activity and paxillin Ser273 phosphorylation are required for the activation of cdc42 and its effector molecule N‐WASP, a catalyst for actin polymerization in airway smooth muscle.
Discussion
In airway smooth muscle, tension development in response to a contractile stimulus triggers the polymerization of a submembranous pool of actin as well as the phosphorylation of the 20 kDa light chain of myosin, which activates crossbridge cycling (Gunst & Zhang, 2008; Zhang & Gunst, 2008). Actin polymerization and crossbridge cycling are both required for tension generation in airway smooth muscle, but they are regulated independently (Mehta & Gunst, 1999; Zhang et al. 2005; Gunst & Zhang, 2008; Zhang & Gunst, 2008). Our results demonstrate a novel role for Pak in the regulation of contractility in smooth muscle (Fig. 10). We find that during contractile stimulation, Pak is recruited to integrin adhesion junctions via a RhoA‐dependent mechanism where it undergoes activation and catalyses phosphorylation of the adhesome scaffolding protein paxillin on Ser273. The dual phosphorylation of paxillin on serine 273 residues by Pak and on tyrosine 31 and 118 residues by FAK regulates its function as a scaffold for the assembly of a multiprotein complex that situates cdc42 and N‐WASP in proximity to the cdc42 GEF, βPIX. This facilitates the activation of cdc42 and the subsequent activation of N‐WASP, which interacts with the Arp2/3 complex to catalyse the polymerization of cortical actin, which is necessary for tension development. Thus, Pak activity is critical for tension development in smooth muscle tissues during contractile stimulation because it regulates the initiation of actin polymerization.
Figure 10. Pak regulates the assembly of a multiprotein paxillin complex within smooth muscle cell adhesomes that mediates cdc42 and N‐WASP activation and actin polymerization .
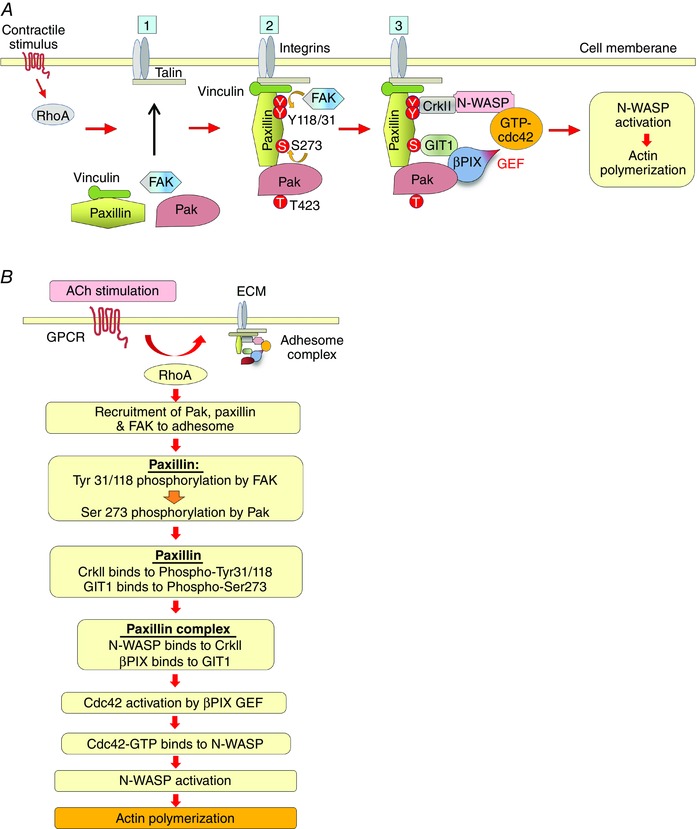
A, schematic diagram illustrating role of Pak in the assembly of a multiprotein paxillin complex that regulates actin polymerization at adhesion junctions in airway smooth muscle: ACh stimulation induces the recruitment of paxillin/vinculin complexes, FAK and Pak to adhesomes by a RhoA‐dependent mechanism. FAK then phosphorylates paxillin on Tyr 31 and Tyr118, facilitating the phosphorylation of paxillin on Ser273 by Pak. Paxillin 273 phosphorylation promotes its interaction with GIT1, and paxillin tyrosine phosphorylation regulates its interaction with the SH3/SH2 adaptor protein, CrkII. CrkII couples paxillin to N‐WASP and GIT1 couples paxillin to βPIX to form a paxillin complex that regulates the activation of cdc42 via the GEF activity of βPIX. cdc42 then catalyses the activation of N‐WASP and actin polymerization at the cortex of the smooth muscle cell. B, flow chart describing steps in the formation of the paxillin complex that regulates N‐WASP activation and actin polymerization within adhesion complexes in smooth muscle tissues. The polymerization of actin at the cortex of the cell and the fortification of the adhesome junctions facilitates the transmission of tension generated by contractile apparatus to the extracellular matrix when crossbridge cycling is activated by MLC phosphorylation (not shown).
Previous studies of both smooth muscle and non‐muscle cells and tissues have attributed Pak's role in the regulation of contractility to effects on the regulation of MLC phosphorylation and myosin II activation (Chew et al. 1998; Goeckeler et al. 2000; Murthy et al. 2003; Wirth et al. 2003; Chu et al. 2013). Our results provide no evidence that Pak participates in the regulation of MLC phosphorylation during tracheal smooth muscle contraction. We found that MLC phosphorylation in response to ACh stimulation was not affected by the inhibition of Pak or by the inhibition of paxillin Ser273 phosphorylation, although tension development was markedly inhibited (Figs 2 and 4). Thus, our results provide no evidence in support of previous findings that Pak has an inhibitory effect on MLC kinase (Goeckeler et al. 2000; Murthy et al. 2003; Wirth et al. 2003) or that it has an effect on MLC phosphatase. The inhibition of MLC kinase activity should decrease MLC phosphorylation and tension development, neither of which was observed in this study. Previous studies in smooth muscle tissues that reported an inhibitory effect of Pak on MLC kinase were performed by treating Triton‐ or saponin‐permeabilized smooth muscle tissues with recombinant Pak protein (Goeckeler et al. 2000; Murthy et al. 2003; Wirth et al. 2003). The addition of recombinant Pak protein to these smooth muscle tissue preparations may result in the phosphorylation of proteins that do not normally interact with Pak in intact tissues or cells under physiological conditions.
We found that the recruitment of Pak to membrane adhesion sites and its activation in response to contractile stimulation depends on the activation of RhoA (Fig. 7). We previously reported that RhoA catalyses the recruitment of FAK, paxillin and vinculin to membrane junctional complexes in response to a contractile stimulus (Huang et al. 2011; Zhang et al. 2012). The phosphorylation of paxillin on tyrosine residues occurs after its recruitment to membrane complexes, and is therefore dependent on RhoA activation. The tyrosine phosphorylation of paxillin enables it to couple to the SH2/SH3 adaptor protein, CrkII, which couples N‐WASP to the paxillin complex (Tang et al. 2005).
Our current results demonstrate that paxillin tyrosine phosphorylation is a prerequisite to its phosphorylation on Ser273 by Pak: the Ser273 phosphorylation of paxillin does not occur when its tyrosine phosphorylation is prevented (Fig. 8). The Ser273 phosphorylation of paxillin promotes its interaction with the Arf‐GAP activating protein, GIT1, which in turn couples the paxillin complex to the cdc42 GEF, βPIX. N‐WASP requires activation by cdc42 in airway smooth muscle (Tang & Gunst, 2004). The G‐protein binding domain (GPD) of N‐WASP is specific for cdc42; other small G‐proteins are incapable of activating N‐WASP (Rohatgi et al. 2000; Tang & Gunst, 2004). Thus, the coordinated phosphorylation of paxillin at tyrosine 118/31 by FAK and on Ser273 by Pak regulates the formation of a signalling module that catalyses cdc42 activation through the GEF activity of βPIX and positions N‐WASP for activation through its interaction with activated cdc42. All of these events occur downstream of RhoA activation through a tightly orchestrated cascade of molecular events (Fig. 10). The molecular interactions of Pak with paxillin, GIT1 and βPIX that we observed in contracting smooth muscle tissues are similar to those in migrating non‐muscle cells (Nayal et al. 2006; Rosenberger & Kutsche, 2006; Frank & Hansen, 2008; Yu et al. 2010; Delorme‐Walker et al. 2011; Zhao & Manser, 2012). However the functional role of the Pak ‐paxillin ‐GIT1 ‐βPIX complex in muscle tissues appears to be distinct from that in migrating or spreading non‐muscle cells. In migrating cells, focal adhesion complexes undergo cycles of formation and dissolution at the leading edge of the cell as it advances. In these cells, paxillin phosphorylation at Ser273 by Pak and the interaction of paxillin with GIT1 and βPIX within adhesion complexes regulate the rate of focal adhesion turnover and focal adhesion strength at the leading edge (Nayal et al. 2006; Yu et al. 2010; Delorme‐Walker et al. 2011). F‐actin flow and actin‐driven protrusion dynamics in the lamella of the cell are driven by myosin II, and Pak's role in the dynamics of cell protrusion has been attributed to its effects on myosin II activity (Nayal et al. 2006; Yu et al. 2010; Delorme‐Walker et al. 2011).
In contrast, in smooth muscle tissues, membrane adhesome complexes connect adjacent muscle cells to each other via the extracellular matrix and thereby support the structural integrity of the tissue. Contractile stimulation triggers the recruitment of structural and signalling proteins to membrane adhesome complexes, but proteins recruited to these adhesome complexes remain localized at the membrane complexes for the duration of the period of muscle contractile stimulation (Opazo Saez et al. 2004; Zhang et al. 2005, 2007, 2012; Zhang & Gunst, 2006, 2008; Gunst & Zhang, 2008; Huang et al. 2011, 2014). Adhesome complexes do not undergo repeated cycles of formation and dissolution during the course of a single contraction, which can be sustained for many minutes. Thus, the functional role that has been attributed to the Pak–paxillin–GIT1–βPIX complex in migrating cells is not directly analogous to it is role in the processes of contraction in smooth muscle tissues.
Our studies demonstrate a unique role for the Pak–paxillin–GIT1–βPIX complex in facilitating the cdc42‐mediated activation of N‐WASP and actin polymerization during the contraction of smooth muscle tissues. Although previous studies have demonstrated a role for Pak in molecular processes that regulate actin dynamics (Bokoch, 2003; Zhao & Manser, 2012), the regulation of actin polymerization catalysts such as N‐WASP by Pak has not been previously described. Thus, these studies in smooth muscle tissues provide evidence for a novel and distinct cellular function for Pak.
The large multiprotein complexes at adhesion junctions provide mechanical coupling between cells and their matrix environment and also enable cells to sense and respond to changes in the properties of their surrounding milieu (Gunst & Zhang, 2008; Zhang & Gunst, 2008). Our current observations provide further evidence that adhesome complex assembly is a fundamental step in the process of signal transduction initiated by the contractile stimulation of smooth muscle. The recruitment of Pak to the membrane of smooth muscle cells and its role in the assembly of adhesome signalling complexes may be fundamental to the process of signal transduction in response to diverse extracellular stimuli that regulate the function of other smooth muscle tissues, and may also be more broadly applicable to other types of contractile cells.
Additional information
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
W.Z. designed the experiments with S.J.G., conducted the experiments and collected the data, analysed the results, and wrote the paper with S.J.G. Y.H. constructed the paxillin S273A mutant vectors for expression of mutant paxillin protein and contributed to the critical revision of the manuscript. S.J.G. conceived of the idea for the project, designed the experiments with W.Z., supervised the data analysis and wrote the paper with W.Z. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by National Heart, Lung, and Blood Institute Grants HL029289, HL048522 and HL109629, an American Lung Association grant and an NIH T32 postdoctoral fellowship to W.Z.
This is an Editor&s Choice article from the 1 September 2016 issue.
References
- Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J & Kumar R (1998). Heregulin regulates cytoskeletal reorganization and cell migration through the p21‐activated kinase‐1 via phosphatidylinositol‐3 kinase. J Biol Chem 273, 28238–28246. [DOI] [PubMed] [Google Scholar]
- Bokoch GM (2003). Biology of the p21‐activated kinases. Annu Rev Biochem 72, 743–781. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Ducruix A & Pantaloni D (1999). Signalling to actin: the Cdc42‐N‐WASP‐Arp2/3 connection. Chem Biol 6, R235–R240. [DOI] [PubMed] [Google Scholar]
- Chew TL, Masaracchia RA, Goeckeler ZM & Wysolmerski RB (1998). Phosphorylation of non‐muscle myosin II regulatory light chain by p21‐activated kinase (gamma‐PAK). J Muscle Res Cell Motil 19, 839–854. [DOI] [PubMed] [Google Scholar]
- Chu J, Pham NT, Olate N, Kislitsyna K, Day MC, LeTourneau PA, Kots A, Stewart RH, Laine GA, Cox CS, Jr & Uray K (2013). Biphasic regulation of myosin light chain phosphorylation by p21‐activated kinase modulates intestinal smooth muscle contractility. J Biol Chem 288, 1200–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon SW, Beeser A, Fukui JA, Rennefahrt UEE, Myers C, Chernoff J & Peterson JR (2008). An isoform‐selective, small‐molecule inhibitor targets the autoregulatory mechanism of p21‐activated kinase. Chem Biol 15, 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechert MA, Holder JM & Gerthoffer WT (2001). p21‐activated kinase 1 participates in tracheal smooth muscle cell migration by signaling to p38 MAPK. Am J Physiol Cell Physiol 281, C123–C132. [DOI] [PubMed] [Google Scholar]
- Delorme‐Walker VD, Peterson JR, Chernoff J, Waterman CM, Danuser G, DerMardirossian C & Bokoch GM (2011). Pak1 regulates focal adhesion strength, myosin IIA distribution, and actin dynamics to optimize cell migration. J Cell Biol 193, 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR & Hansen SH (2008). The PIX–GIT complex: a G protein signaling cassette in control of cell shape. Semin Cell Dev Biol 19, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler ZM, Masaracchia RA, Zeng Q, Chew TL, Gallagher P & Wysolmerski RB (2000). Phosphorylation of myosin light chain kinase by p21‐activated kinase PAK2. J Biol Chem 275, 18366–18374. [DOI] [PubMed] [Google Scholar]
- Gunst SJ & Zhang W (2008). Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295, C576–C587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathaway DR & Haeberle JR (1985). A radioimmunoblotting method for measuring myosin light chain phosphorylation levels in smooth muscle. Am J Physiol 249, C345–C351. [DOI] [PubMed] [Google Scholar]
- Hoover WC, Zhang W, Xue ZC, Gao H, Chernoff J, Clapp DW, Gunst SJ & Tepper RS (2012). Inhibition of p21 activated kinase (PAK) reduces airway responsiveness in vivo and in vitro in murine and human airways. PlosOne 7, e42601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Day RN & Gunst SJ (2014). Vinculin phosphorylation at Tyr1065 regulates vinculin conformation and tension development in airway smooth muscle tissues. J Biol Chem 289, 3677–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang W & Gunst SJ (2011). Activation of vinculin induced by cholinergic stimulation regulates contraction of tracheal smooth muscle tissue. J Biol Chem 286, 3630–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFawn PK, Shen L, Vincent SG, Mak A, Van Eyk JE & Fisher JT (2003). Calcium‐independent contraction and sensitization of airway smooth muscle by p21‐activated protein kinase. Am J Physiol Lung Cell Mol Physiol 284, L863–L870. [DOI] [PubMed] [Google Scholar]
- Mehta D & Gunst SJ (1999). Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima W, Suzuki A, Yamaji S, Yoshimi R, Ueda A, Kaneko T, Tanaka J, Miwa Y, Ohno S & Ishigatsubo Y (2004). The first CH domain of affixin activates Cdc42 and Rac1 through αPIX, a Cdc42/Rac1‐specific guanine nucleotide exchanging factor. Genes Cells 9, 193–204. [DOI] [PubMed] [Google Scholar]
- Murthy KS, Zhou H, Grider JR, Brautigan DL, Eto M & Makhlouf GM (2003). Differential signalling by muscarinic receptors in smooth muscle: m2‐mediated inactivation of myosin light chain kinase via Gi3, Cdc42/Rac1 and p21‐activated kinase 1 pathway, and m3‐mediated MLC20 (20 kDa regulatory light chain of myosin II) phosphorylation via Rho‐associated kinase/myosin phosphatase targeting subunit 1 and protein kinase C/CPI‐17 pathway. Biochem J 374, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente‐Manzanares M & Horwitz AR (2006). Paxillin phosphorylation at Ser273 localizes a GIT1‐PIX‐PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 173, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD & Gunst SJ (2004). Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol 286, C433–C447. [DOI] [PubMed] [Google Scholar]
- Puetz S, Lubomirov LT & Pfitzer G (2009). Regulation of smooth muscle contraction by small GTPases. Physiology (Bethesda) 24, 342–356. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Ho HY & Kirschner MW (2000). Mechanism of N‐WASP activation by CDC42 and phosphatidylinositol 4, 5‐bisphosphate. J Cell Biol 150, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T & Kirschner MW (1999). The interaction between N‐WASP and the Arp2/3 complex links Cdc42‐dependent signals to actin assembly. Cell 97, 221–231. [DOI] [PubMed] [Google Scholar]
- Rosenberger G & Kutsche K (2006). αPIX and βPIX and their role in focal adhesion formation. Eur J Cell Biol 85, 265–274. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM & Chernoff J (1997). Human p21‐activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol 7, 202–210. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG & Landegren U (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG & Landegren U (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232. [DOI] [PubMed] [Google Scholar]
- Somlyo AP & Somlyo AV (2003). Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83, 1325–1358. [DOI] [PubMed] [Google Scholar]
- Stockton R, Reutershan J, Scott D, Sanders J, Ley K & Schwartz MA (2007). Induction of vascular permeability: βPIX and GIT1 scaffold the activation of extracellular signal‐regulated kinase by PAK. Mol Biol Cell 18, 2346–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C & Walsh MP (2012). Myosin regulatory light chain diphosphorylation slows relaxation of arterial smooth muscle. J Biol Chem 287, 24064–24076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD & Gunst SJ (2001). Depletion of focal adhesion kinase by antisense depresses contractile activation of smooth muscle. Am J Physiol Cell Physiol 280, C874–C883. [DOI] [PubMed] [Google Scholar]
- Tang DD, Turner CE & Gunst SJ (2003). Expression of non‐phosphorylatable paxillin mutants in canine tracheal smooth muscle inhibits tension development. J Physiol 553, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD, Zhang W & Gunst SJ (2005). The adapter protein CrkII regulates neuronal Wiskott–Aldrich syndrome protein, actin polymerization, and tension development during contractile stimulation of smooth muscle. J Biol Chem 280, 23380–23389. [DOI] [PubMed] [Google Scholar]
- Tang DD & Gunst SJ (2004). The small GTPase Cdc42 regulates actin polymerization and tension development during contractile stimulation of smooth muscle. J Biol Chem 279, 51722–51728. [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S & Leventhal PS (1999). Paxillin LD4 motif binds PAK and PIX through a novel 95‐kD ankyrin repeat, ARF‐GAP protein: a role in cytoskeletal remodeling. J Cell Biol 145, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak‐Kazmierczak E, Cote GP & Mak AS (1998). Different molecular mechanisms for Rho family GTPase‐dependent, Ca2+‐independent contraction of smooth muscle. J Biol Chem 273, 23433–23439. [DOI] [PubMed] [Google Scholar]
- Walsh MP (2011). Vascular smooth muscle myosin light chain diphosphorylation: mechanism, function, and pathological implications. IUBMB Life 63, 987–1000. [DOI] [PubMed] [Google Scholar]
- Wang R, Li QF, Anfinogenova Y & Tang DD (2007). Dissociation of Crk‐associated substrate from the vimentin network is regulated by p21‐activated kinase on ACh activation of airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 292, L240–L248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A, Schroeter M, Kock‐Hauser C, Manser E, Chalovich JM, de Lanerolle P & Pfitzer G (2003). Inhibition of contraction and myosin light chain phosphorylation in guinea‐pig smooth muscle by p21‐activated kinase 1. J Physiol 549, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JA, Deakin NO & Turner CE (2010). Emerging role of paxillin‐PKL in regulation of cell adhesion, polarity and migration. Cell Adh Migr 4, 342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JS, Chen WJ, Ni MH, Chan WH & Yang SD (1998). Identification of the regulatory autophosphorylation site of autophosphorylation‐dependent protein kinase (auto‐kinase). Evidence that auto‐kinase belongs to a member of the p21‐activated kinase family. Biochem J 334, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenke FT, King CC, Bohl BP & Bokoch GM (1999). Identification of a central phosphorylation site in p21‐activated kinase regulating autoinhibition and kinase activity. J Biol Chem 274, 32565–32573. [DOI] [PubMed] [Google Scholar]
- Zhang W, Du L & Gunst SJ (2010). The effects of the small GTPase RhoA on the muscarinic contraction of airway smooth muscle result from its role in regulating actin polymerization. Am J Physiol Cell Physiol 299, C298–C306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W & Gunst SJ (2008). Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc 5, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Huang Y & Gunst SJ (2012). The small GTPase RhoA regulates the contraction of smooth muscle tissues by catalyzing the assembly of cytoskeletal signaling complexes at membrane adhesion sites. J Biol Chem 287, 33996–34008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Du L, Tang DD & Gunst SJ (2005). Activation of the Arp2/3 complex by N‐WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol 288, C1145–C1160. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Wu C & Gunst SJ (2007). Integrin‐linked kinase (ILK) regulates N‐WASp‐mediated actin polymerization and tension development in tracheal smooth muscle. J Biol Chem 282, 34568–34580. [DOI] [PubMed] [Google Scholar]
- Zhang WW & Gunst SJ (2006). Dynamic association between α‐actinin and β‐integrin regulates contraction of canine tracheal smooth muscle. J Physiol 572, 659–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS & Manser E (2012). PAK family kinases: physiological roles and regulation. Cell Logist 2, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]